Back to Journals » Pharmacogenomics and Personalized Medicine » Volume 8
The current and future state of companion diagnostics
Authors Agarwal A, Ressler D, Snyder G
Received 24 May 2014
Accepted for publication 1 August 2014
Published 31 March 2015 Volume 2015:8 Pages 99—110
DOI https://doi.org/10.2147/PGPM.S49493
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Martin Bluth
Amit Agarwal,1 Dan Ressler,2 Glenn Snyder1
1Deloitte Consulting LLP, San Francisco, CA, USA; 2Deloitte Consulting LLP, Philadelphia, PA, USA
Abstract: Companion diagnostics are an indispensable part of personalized medicine and will likely continue to rapidly increase in number and application to disease areas. The first companion diagnostics were launched in the 1980s and in the face of significant initial skepticism from drug developers as to whether segmenting a drug's market through a diagnostic was advisable. The commercial success of drugs such as Herceptin® (trastuzumab) and Gleevec® (imatinib), which both require testing with companion diagnostics before they can be prescribed, has moved the entire companion diagnostic field forward. From an initial start of a handful of oncology drugs with corresponding diagnostics, the field has expanded to include multiple therapeutic areas, and the number of combinations has grown by 12-fold. Based on drugs in clinical trials, the rapid growth will likely continue for the foreseeable future. This expansion of companion diagnostics will also have a global component as markets in Europe will evolve in a similar but not identical pattern as the US. One of the greatest challenges to future growth in companion diagnostics is aligning the incentives of all stakeholders. A major driver of growth will continue to be the economic incentives for drug developers to pair their products with diagnostics. However, diagnostic companies are caught between the conflicting demands of two major stakeholders, pharmaceutical companies on one hand and payers/providers on the other. Regulators are also becoming more demanding in aligning development time lines between drugs and diagnostics. In order to survive and prosper, diagnostic companies will need to think more broadly about companion diagnostics than the historical match between a specific drug and a single diagnostic. They will also have to continue the process of consolidation and global expansion that the industry has already begun. Despite these potential obstacles, companion diagnostics have become one of the hottest areas of deal making in the diagnostic space in recent years, and the future trends continue to look bright.
Keywords: companion diagnostics, theranostics, personalized medicine, prognosis, monitoring
Multiple ways to provide “essential information”
The working definition of a companion diagnostics stipulated by the US Food and Drug Administration (FDA) is the basis of our analysis (see Supplementary materials). While the definition is straightforward, there are several unique aspects that warrant further comment. The first is that the FDA definition does not state the type of diagnostic tests that can be considered a companion diagnostic. While there has been the most excitement about molecular diagnostics that provide genetic information, the FDA definition does not limit the companion diagnostic space to just those tests. Diagnostic tests of proteins, metabolites, essential elements, tracers, and other categories of in vitro diagnostics are all eligible to be part of companion diagnostic combinations. The second comment is that a companion diagnostic must be included in the labeling instructions for the therapeutic product as well as the specific diagnostic test. This is a much stricter definition than the general use of the term. A well-known example of a test and therapeutic combination that is not a companion diagnostic according to the FDA definition is the combination of a cholesterol test before prescription of a statin (or more formally an HMG-CoA reductase inhibitor). For adult populations, statins do not have labeled instructions requiring a cholesterol test before prescription, and no FDA-approved cholesterol test refers to a particular statin in its label.
A consistent nomenclature for companion diagnostics
The FDA definition of companion diagnostics only encompasses two types of companion diagnostics – theranostics and monitoring tests (see Supplementary materials).1 The other three types of tests – screening and detection, prognosis, and recurrence – provide critical information but do not specify a corresponding therapeutic. While these other three categories do not fit the FDA criteria, they are essential to the growth of the companion diagnostics, as they account for the majority of revenue for companies that develop companion diagnostics. Several examples from some of the larger publicly traded diagnostic developers illustrate the point. This includes Utah-based Myriad Genetics, where companion diagnostic testing and services represent 5% of company revenues in the fiscal year 2012 and the fiscal year 2013, which was >$613 million in the last fiscal year.2 Similarly for Qiagen, a European diagnostic company, companion diagnostics accounted for 8% of the fiscal year 2012 revenues of $1.25 billion.3
In evaluating the worldwide market for all types of companion diagnostic testing, the market is split between sales of test kits/reagents and laboratory-developed testing (LDT) services. The value of test kits/reagents was $405 million in 2011 according to industry analyst Enterprise Analysis Corporation.4 With the addition of testing services for LDT of $1.17 billion, the total worldwide market is $1.57 billion.5 While this is a small component (~3%) of the roughly $50 billion worldwide in vitro diagnostic market, it is one of the fastest-growing segments.
The growth of companion diagnostics
The number of companion diagnostics on the market has grown rapidly. According to data compiled by the Personalized Medicine Coalition, in 2006 there were five drug/diagnostic combinations that met the FDA definition of a companion diagnostic.6 These included:
- Camptosar® (irinotecan) and Invader® UGT1A1 Molecular Assay
- Gleevec® (imatinib mesylate) and BCR-ABL LDT and DAKO C-KIT PharmDx® for gastrointestinal stromal tumors (GIST)
- Herceptin® (trastuzumab)and DAKO Herceptest®
- Purinethol® (mercaptopurine) and laboratory-developed thiopurine methyltransferase test
- Nolvaldex® (tamoxifen) and laboratory-developed estrogen receptor positive test.
These drugs were all indicated for oncology. By 2012 the number of combinations of companion diagnostics with corresponding drugs had increased to 63 (Figure 1).7–9
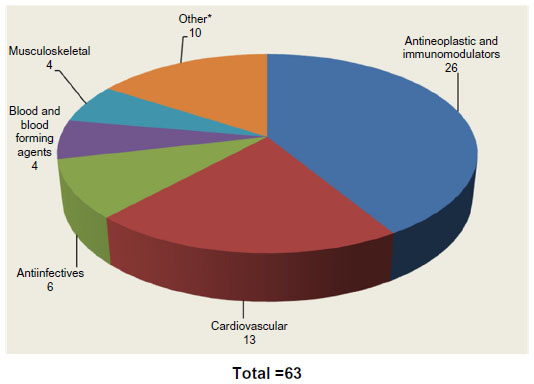 | Figure 1 US Food and Drug Administration-approved companion diagnostic drugs (2012). |
Companion diagnostics are not just for cancer
Oncology remains the largest segment for FDA-defined marketed theranostic drugs based on our analysis of drugs with companion diagnostics that are on the market. Oncology drugs still comprise >40% of all marketed products. The well-known examples of companion diagnostics have all come from the oncology segment, including Herceptin® and Gleevec®. These companion diagnostic-enabled drugs have been labeled as “niche busters” – drugs targeted to smaller patient populations but commanding premium prices, which allows them to achieve annual sales >$1 billion. Despite the reduction of eligible patient populations through companion diagnostic testing, Herceptin® and Gleevec® had worldwide sales of $6.5 billion and $4.7 billion, respectively, in 2012.10,11
While oncology will remain the therapeutic area at the forefront of companion diagnostics for the foreseeable future based on currently approved drugs, other therapeutic areas are beginning to emerge, including cystic fibrosis, human immunodeficiency virus (HIV), and severe growth failure. Within these therapeutic areas, three new drugs have been approved with corresponding diagnostics as part of their labels:
- Kalydeco® (ivacaftor) for cystic fibrosis is paired with COBAS 4800 BRAF V600® from Roche Molecular Systems
- Selzentry® (maraviroc) for HIV has Trofile®, an HIV coreceptor tropism assay from Monogram Biosciences (now Labcorp)
- Increlex® (mecasermin) for severe growth failure requires an insulin-like growth factor 1 reference LDT.
Of these drugs, Kalydeco® could become the next niche buster with consensus analyst projections of sales >$2 billion by 2018.12
Evaluating the longer-term revenue potential for companion diagnostics paired with drugs requires evaluating compounds in clinical trials. Presently, there are roughly 100 drugs in Phase II, III, and IV with diagnostic information listed as part of the primary and secondary outcome measures as well as the inclusion/exclusion eligibility criteria in clinicaltrials.gov.13 The number is constantly shifting as trials are discontinued and new ones are started. A snapshot of these trials nets only nine examples that meet the strict FDA definition of companion diagnostic in which a specific diagnostic is included as part of the clinical trial. Clinicaltrials.gov is not a comprehensive registry of all clinical trials being conducted across the globe, and there is no regulatory requirement that drug developers list the specific diagnostic being utilized in the trial summaries. However, the nine drug and diagnostic combinations presented in Table 1 provide a sampling of drugs that may be launched with companion diagnostics.
 | Table 1 US Food and Drug Administration Definition of Companion Diagnostic Drugs in Clinical Trials |
However, the number of drugs that require companion diagnostics will continue to expand as many of the approximately 100 drugs in which nonspecific diagnostic information is included will make the decision to pursue development of diagnostics as they progress through the clinical development process. While the clinical trials for all these drugs will not be successful, those drug developers that can identify patient segments in which their drug is more likely to be efficacious, or identify segments that are at greater risk for adverse events or nonresponsiveness, have higher chances of obtaining regulatory approval. Companion diagnostics are one of the few tools available to drug developers that can generate this information.
The growth in companion diagnostics is not just a US phenomenon. If we broaden the lens and include drugs that have been approved by the European Medicines Agency (EMA) with required testing, we see a similar but not exact match to the US. Some of the differences in required pharmacogenetic testing between the EU and US are presented in Figure 2. The vast majority of drugs (78%) have similar label requirements for diagnostic testing in both geographies.14 However, there are a few instances where the EMA requires testing where it is only recommended in the FDA-approved label (eg, Tegretol® [carbamazepine] and Ziagen® [abacavir sulfate]). In both cases, testing is for hypersensitivity caused by a genetic mutation. Another situation arises for Iressa® (gefitinib), which was approved by the EMA in 2009 but has not been available to US patients since 2005 when the FDA took action to severely restrict its access based on a clinical trial that demonstrated limited efficacy. The only instance in which the FDA requires testing but is only recommended by the EMA is Revlimid® (lenalidomide). The FDA label highlights particular issues in using Revlimid® for myelodysplastic syndrome patients with a particular chromosomal abnormality (eg, del 5q). Despite these limited instances of differing regulation between the US and Europe, for most products the companion diagnostic requirements are very similar if not identical.
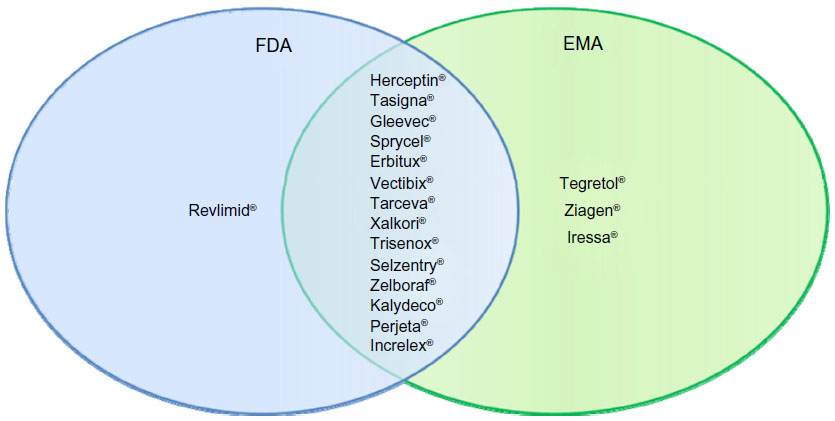 | Figure 2 Drugs with required pharmacogenetic companion diagnostic testing. |
Timing of drug and diagnostic approvals
Another implication of the FDA definition is that there is no requirement that the drug and diagnostic are approved at the same time. While much of the interest in companion diagnostics has been spurred by examples of targeted therapies and their corresponding diagnostics, the timing even in well-known cases has rarely been simultaneous. Only a handful of drug diagnostic combinations have had the drug and diagnostic approved simultaneously, with the pace of approvals increasing in the last year. These were Herceptin®, Erbitux® (cetuximab), Vectibix® (pantiumumab), Zelboraf® (vemurafenib), Xalkori® (crizotinib), Mekinist® (trametinib), and Tafinlar® (dabrafenib).15 In each of these cases, the diagnostic was critical to identifying patient populations in which the corresponding drug would be effective. While only seven of the 63 current drugs with required companion diagnostics were approved simultaneously (only ~11%), as more drug developers conduct clinical trials with diagnostics as part of the inclusion/exclusion criteria as well as primary and secondary outcome measures, this number will continue to increase.
The challenges of early integration of companion diagnostics into drug development time lines for simultaneous approval are highlighted by a current drug in development, MetMab® (onartuzumab), which is a humanized monovalent monoclonal antibody directed against the hepatocyte growth factor receptor (c-Met) with potential anticancer activity. Drug developers must assess if there are subpopulations of patients that could benefit from a drug early in the development cycle even before clinical efficacy of the drug is established. This requires the drug developer to have strong hypotheses or beliefs as to the mechanism of action or specific biomarkers that could lead to a diagnostic. In practical terms, this means a prototype diagnostic must be available by Phase II clinical trials. In the case of MetMab®, which was initially developed for advanced non-small-cell lung cancer (NSCLC), the 120 patient Phase II trial showed no statistical evidence of increased efficacy of a MetMab® and Tarceva® combination across all intent-to-treat patients.16 In contrast, when Phase II patients were split into two groups based on c-Met levels, there were significant improvements in progression-free and overall survival in the high c-Met expression group. While these were encouraging results, the next hurdle for Roche/Genentech was to improve the prototype diagnostic assay to a sufficient technical validity so that by the end of Phase II, the data could be unblinded to compare safety and efficacy in the two patient groups. This time line permits creation of an in vitro diagnostic assay before Phase III, allowing prospective enrollment of patients for a Phase III trial. Roche/Genentech was able to follow this pathway and time line with Metmab®, with a Roche diagnostic subsidiary, Ventana, creating an immunohistochemistry test for c-Met that was utilized in the Phase II trial. However, despite the potential value and investment in a companion diagnostic, drug development is still a risky business. On March 2014, Roche/Genentech reported that Phase III trials of Metmab® with Tarceva® had been discontinued due to a lack in response in the high c-Met patient population over a placebo.17
In general, however, the FDA has permitted a therapy and its required companion diagnostic to be approved at different times. In the case of Gleevec®, it was initially approved for patients with chronic myeloid leukemia (CML) with very few alternatives available. The specific criteria were CML in blast crisis, accelerated phase, or chronic phase after failure of interferon-α therapy. In these instances, no diagnostic was required. Diagnostics only became part of Gleevec’s® label with subsequent approval for earlier phases of CML and for GIST. The original diagnostics to test for chromosomal mutations of CML and GIST were LDTs that existed before Gleevec® was on the market. Only later did FDA-approved testing kits appear.
Based on a review of currently approved drugs with a companion diagnostic presented earlier, about 90% of all drugs with companion diagnostic have followed Gleevec’s® path rather than the Metmab® approach. The diagnostic test that is added after the drug is on the market can be either laboratory developed or an FDA-approved testing kit. An example of LDTs being added to the label after drug approval is abacavir sulfate, which was on the market for 10 years before the companion diagnostic testing was added to the label. Abacavir was approved in the US in 1998 and in the EU in 1999. As part of the approval process, the drug developer was asked by the EMA to develop a diagnostic test for hypersensitivity, which occurs in 2%–9% of the patient population and is potentially life threatening. The drug developer undertook 8 years of retrospective and prospective studies to identify, validate, and confirm the clinical utility of the biomarker HLA-B*57:01 allele to predict hypersensitivity. While undertaking these studies, major clinical testing laboratories had already launched screening assays for the HLA-B*57:01 allele in 2004 and 2005, respectively.18 The change to abacavir’s drug label occurred later in 2008.
Elitek® (rasburicase) is another example but with a 510K approved diagnostic test that was available a decade before testing became required. Elitek® is for the initial management of plasma uric acid levels in pediatric and adult patients with leukemia, lymphoma, and solid tumor malignancies who are receiving anticancer therapy expected to result in tumor lysis and subsequent elevation of plasma uric acid. The drug was initially approved in 2002 with recommended testing for glucose-6-phosphate dehydrogenase (G6PD) deficiency because patients with this deficiency could develop severe hemolysis. The drug’s labeling was updated with a black box warning requiring pretreatment screening of patients in 2009. Screening tests for G6PD deficiency received 510k clearance from the FDA in 1995 >14 years prior to their mandatory use with Elitek®.15 The earlier approval for G6PD test kits was due to the long-standing recognition that G6PD deficiency could lead to hemolysis upon taking certain drugs, which had first been demonstrated with an antimalaria drug in the 1950s. Many of the original diagnostic tests were laboratory developed but had been replaced with more accurate FDA-approved rapid testing kits by 1995.
Incentives for drug developers in companion diagnostic development
A major driver of growth for companion diagnostic deals is the potential economic benefits for drug developers early in drug development. If a companion diagnostic is used to select patients for clinical trials, drug developers can reduce the costs and shorten the time to approval. While the presence of a companion diagnostic does not in itself improve the probability of technical success of a corresponding drug, companion diagnostics can help improve the economics. An example of three drugs that all have companion diagnostics as part of their label today but took different paths for NSCLC illustrates the potential benefits (see Table 2).19 It is clear from the data that the ability to selectively segment the patients enrolling in clinical trials through a companion diagnostic has the potential to reduce the number of patients required as well as the time to conduct trials, which has a dramatic impact on overall development costs.
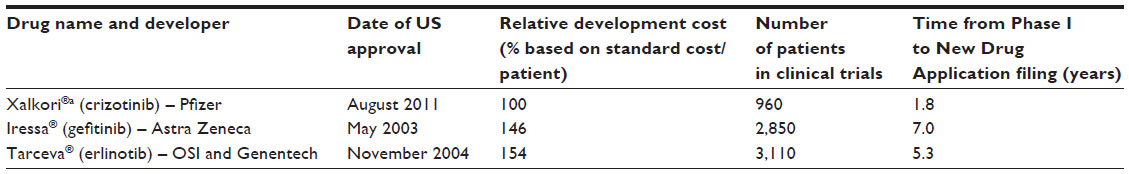 | Table 2 Comparison of development costs, patient enrollment, and time for non-small-cell lung cancer drugs |
Only Xalkori® (crizotinib) was initially codeveloped with a companion diagnostic (Abbott’s Vysis ALK Break Apart FISH Probe® Kit), which was available upon approval of the drug. The other two drugs – Iressa® and Tarceva® – were initially approved without a labeled companion diagnostic. In Iressa’s® case, the companion diagnostic was added to the label 2 years after launch, when academics were able to demonstrate that mutations in the epidermal growth factor receptor for tyrosine kinase (EGFR-TK) were the reason for variable response to the drug in patient populations. Tarceva® took even longer for EGFR testing to become part of the label. Genzyme Genetics (now part of LabCorp) launched an LDT in 2005 for EGFR mutations. However, it was not until specific recommendations for EGFR testing were adopted by the American Society of Clinical Oncology in 2010 that an FDA-approved testing kit was developed by Roche Diagnostics. The label for Tarceva® remained unchanged until 2013 when it was approved as a first-line therapy for NSCLC.
The Tarceva® example also demonstrates the potential value of companion diagnostics in taking a therapy that was a second- or third-line treatment option for the general population and making it first line in a select group of patients even later in the drug’s life cycle. Tarceva’s® worldwide sales in 2013 were $1.4 billion, with the US accounting for $640 million.20 The impact of moving to first-line treatment is seen by the uptick in growth forecasts for an 8-year-old drug. Tarceva® had grown by a 2.7% cumulative annual growth rate over the preceding 5 years. With an ability to garner first-line patients through use of a companion diagnostic, analyst projections show an increase in growth to 3.2% over the next 5 years.21 While this sales growth is modest, it is very unusual for a drug 8 years after initial launch to experience faster growth.
The downside for drug developers with companion diagnostics is when companion diagnostics are added to the drug label after a drug is on the market and it has no impact on the line of therapy. In those instances, the companion diagnostic decreases the potential patient population without an upside for the drug developer.
Companion diagnostic deals
Given the incentives for drug developers to have companion diagnostics for their products in development and on the market, it is not surprising that there has been a recent uptick in deals. While the number of companion diagnostic deals is still a small percentage of the total diagnostic deals (ranging from 3% to 14%) completed over the last 5 years, the last 3 years have seen companion diagnostic deals average 12% of the total deals (Figure 3).22
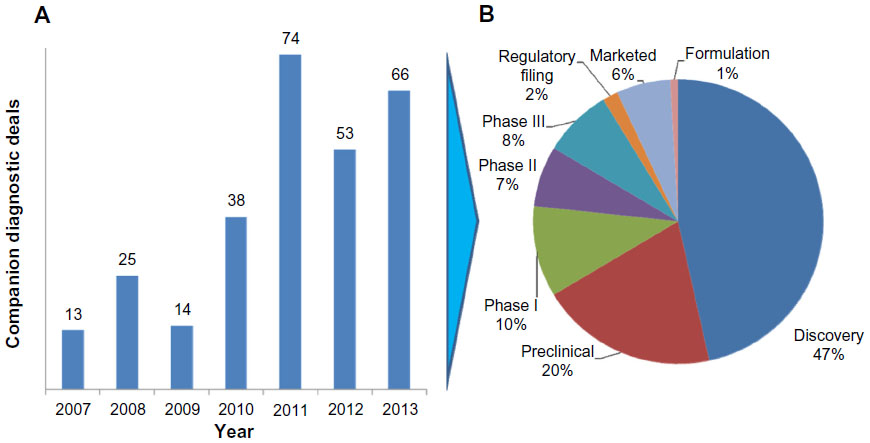 | Figure 3 Companion diagnostic deals signed by year (A) and stage of companion diagnostic deal (B). |
One of the interesting characteristics of deals in which information was publicly provided was that nearly half are for discovery stage efforts. Many of the discovery deals include both drug compounds and companion diagnostics, as two of the largest headline value deals demonstrate. Roche/Genentech and Xenon Pharmaceuticals signed a collaborative research and development agreement for compounds and companion diagnostics for the treatment of pain in January 2012. The headline value of the deal was $646 million. The second-largest deal reported during this time was the collaborative research and development agreement between Jannssen Pharmaceuticals and Metamark Genetics for $365 million in December 2011.22 The deal included a licensing agreement for a discovery platform used to characterize and identify proprietary targets in oncology that predict tumor progression and spread. While the headline values do not always reflect the realized economics, they do provide a useful gauge on the potential value the two parties envision.
The size of deals in the preclinical and later stages declines rapidly from the discovery headline values. The value of establishing biomarkers that lead to potential drug targets is substantial for both drug developers and diagnostic test developers, but once a lead compound has been established by a drug developer, economic value starts to diverge. The potential revenue from a “blockbuster” companion diagnostic is rarely >$100 million, while the annual sales of the corresponding drug can easily reach ten times that amount or more. One of the reasons drug developers are reluctant to pay premiums to companion diagnostics partners in later stages of drug development is that diagnostics rarely enjoy the same level of intellectual property protection as drugs. In the Herceptin® case, within 10 years of drug launch there were six different FDA-approved companion diagnostic assays that utilized different testing technologies. Drug developers see little reason to pay a premium to the initial diagnostic test developer when numerous diagnostics players may enter the market.
Diagnostic companies, on the other hand, would like to receive royalties on sales of the drug or sales-based milestones to share some of the long-term value of the combination and compensate them for the risk of the drug not being approved or lackluster sales. Up to now, drug development partners have preferred to structure payments to test developers as fee for service or through fixed milestone payments. These payments are usually not enough to ensure a profitable outcome for the diagnostic developer and require them to obtain reimbursement from payers in order to generate sufficient returns on investment.
While the fundamental structure of payments has remained unchanged, in recent years drug developers have been willing to consider cross-subsidizing diagnostics. The impetus for these types of deals has been in Europe where universal insurance and national policies have required a different approach. In Spain, for example, the multiyear delays in obtaining reimbursement from the national insurance program for companion diagnostic testing have forced pharmaceutical developers to pay for Her2/neu and K-RAS testing for drugs that require testing before prescription.23 Similarly, in France, Pfizer offered a €3 million grant to the French National Cancer Institute to allow 28 laboratories across France to conduct ALK testing for NSCLC, which is required before Xalkori® can be prescribed. Pfizer also obtained cooperation from Abbott Molecular, which developed the companion diagnostic test, to provide training and materials, including the ALK lymphoma probes, to testing laboratories in France.24 It is not too great of a leap to predict how in certain instances drug developers would also be willing to fund a voucher program to cover the cost of the test in the US, particularly as the impact of health care reform and the rise of accountable care organizations take hold.
Misaligned incentives between partners
Once the decision has been made to move forward with a companion diagnostic, drug developers face the quandary of who should pay to bring the companion diagnostic to market. While almost everyone can agree with the personalized medicine mantra of the right medicine at the right dose to the right patient at the right time, it is in the implementation details where interests start to diverge.
In addition to the obvious costs inherent in developing a companion diagnostic, there are additional expenses in educating health care providers on the need for companion diagnostic testing, ensuring that appropriate clinical laboratories are available to conduct the testing, and obtaining reimbursement. While the ideal answer from a drug developer’s perspective is to throw these issues into the laps of diagnostic companies that are competing to launch a companion diagnostic, the real-world implementation of companion diagnostics is rarely this straightforward and incentives are not aligned.
Starting with the development of the companion diagnostic, the drug developer has different interests from those of the diagnostic company. The drug developer wants the most accurate test available to the greatest number of physicians at the lowest cost in the shortest period of time, with all attention focused on selling the greatest volume of pharmaceuticals. For chronic diseases, it does not require many patients for drug volumes to increase substantially. The companion diagnostic is the key gatekeeper in determining which patients will benefit.
Diagnostic developers, on the other hand, may not have an interest in developing a companion diagnostic test, especially if the test is for a limited patient population. Unlike the drug developer that can count on a lifetime of revenue from chronic treatment of a patient, the diagnostic developer only gets paid per test. Even in cases where the market for diagnostic tests is sufficiently large, resource constraints can create roadblocks. Few diagnostic companies have a sales force to educate health care providers on ordering the appropriate diagnostic test. Diagnostic companies may also seek to bring tests to market under the quicker analyte-specific reagent (ASR) pathway rather than undertaking the expense of developing an FDA-approved test kit. While the ASR test may be able to enter the market faster and generate revenue for the diagnostic company, the restricted number of laboratories that can run ASR tests and variation in results could negatively impact sales of the corresponding drug.
These issues have led pharmaceutical companies to evaluate four different approaches to obtaining companion diagnostics: internal development of companion diagnostics, partnering with an established diagnostic company, acquisitions of diagnostic companies, and hybrids of the first three options. The first approach of internal development of companion diagnostics is most applicable to drug developers that have significant diagnostic division underneath the corporate umbrella. A well-known example of this structure is Roche, which has a significant presence in drug development through Roche Pharmaceuticals/Genentech and diagnostic development through Roche Diagnostics. Roche’s collection of multiple diagnostic platforms such as molecular diagnostics (Roche Molecular), anatomical pathology (Ventana Medical Systems), and immunology (Roche Professional) allow it to meet the needs of Roche Pharmaceuticals as well as other drug developers. Until recently, there were two other groups that had similar corporate structures: Novartis and Abbott. With the recent divestiture of Novartis Diagnostics and the spinoff of Abbvie from Abbott Diagnostics, the drug development groups at Novartis and Abbvie will pursue collaboration with diagnostic companies in the future, much like other biopharmaceutical companies have done.
The number of potential collaborators for the second option of partnering with established diagnostic players that are not in drug development has narrowed as the diagnostic industry has consolidated. The large players have undertaken a series of acquisitions, creating multiplatform diagnostics companies. These include Agilent–Dako, ThermoFisher–Life Techologies, Labcorp–Genzyme Genetics, and Qiagen–DxS. These players have the financial muscle and global reach to negotiate broader, longer-term deals with drug developers. An example of such a deal is the “master agreement” between Eli Lilly and Qiagen. The agreement arose from a successful partnership to develop a K-RAS test for Erbitux® (cetuximab), which was approved in 2012 and has grown to include other therapeutic areas, including developing a test for a clinical stage Janus kinase 2 inhibitor for blood cancer and a companion diagnostic for a novel undisclosed Lilly lung cancer compound.25
The last remaining option is acquisition of companion diagnostic capability by a drug developer. We have seen several instances of this in the last few years, including Eli Lilly’s acquisition of Avid Radiopharmaceuticals in 2010 to develop a diagnostic for Alzheimer’s disease for $300 million and Novartis Pharmaceutical’s acquisition of Genoptix’s clinical testing laboratory business in 2011 for $470 million.26
As can be seen from the examples provided, drug developers have not stayed true to a single option but have attempted multiple strategies. As long as the incentives between drug developers and diagnostic developers are not completely aligned, the market should expect continued market fermentation and experimentation in business models.
Increasingly important role of payers and reimbursement
While pharmaceutical companies are most interested in companion diagnostics that are theranostics and monitoring types of tests, payers appear to be more interested in the other types of companion diagnostic tests. This has already been demonstrated by the rapid acceptance and commercialization of Genomic Health’s Oncotype Dx® for breast cancer. While the test does not specify a particular drug that will be successful in a patient, it does provide valuable information on the potential efficacy of standard chemotherapy treatment for breast cancer patients. As the standard course of chemotherapy may cost >$100,000 per patient and have significant associated adverse events, insurers and clinicians are very much interested in obtaining a priori information on its potential efficacy. Genomic Health’s Oncotype Dx®’s list price of $4,290 has not hindered its acceptance with payers, with >90% of US lives covered.27
Payers are also much more interested in diagnostic tests that provide information on multiple potential treatment options in a therapeutic area. The current model of a single diagnostic test tied to a single pharmaceutical agent is unlikely to survive payer pressure for greater efficiency and cost-effectiveness. As multiple therapeutic agents are approved for similar clinical indications, payers want to identify the best product for a particular patient and not pay for multiple similar tests. This push to multiplex assays, which has already occurred in most other diagnostics categories (eg, blood work panels, metabolic panels), will become increasingly common in companion diagnostics. Given the high costs of many of the drugs with companion diagnostics, which can easily exceed $100,000 per course of therapy, getting the optimal drug to a patient is more important than ever. With health care reform on a global basis pushing clinical provider payments based on patient outcomes rather than the number/type of clinical interventions, there is a significant upside to selecting the optimal drug based on diagnostic testing that compares similar therapeutics against one another. This is an area where the interests of payers and drug developers diverge. No drug developer wants a companion diagnostic that could potentially point to the prescription of a competitor’s product. Companion diagnostic companies that often seek alliances with drug developers to defray the costs of developing a companion diagnostic will face the unenviable position of being caught in the middle of drug developers and payers.
The move to multiplex assays has significant implications for the business models of diagnostic developers. Today, many companion diagnostics are initially developed under the ASR approval pathway and conducted in Clinical Laboratory Improvement Amendment of 1988 (CLIA) laboratories. The diagnostic developer often establishes a dedicated laboratory to which all diagnostic samples must be shipped, and results are sent back to the ordering physician. While this business model can be successful in a market environment where a single diagnostic is tied to a single therapeutic, it breaks down if payers and health care providers start to demand panels that compare companion diagnostics against each other. In this new multiplex environment, it is unlikely that a single test companion diagnostic laboratory can provide the needed tests.
There are at least two potential solutions to supply the multiplex assays, but both have implications for companion diagnostic companies’ business models. The first is to create testing kits and obtain FDA 510K approval and European CE marks. Creation of multiplex kits will require cross-licensing of intellectual property from multiple companion diagnostic companies, which could delay their introduction while contract negotiations are ongoing. The other option is clinical testing laboratories that are not affiliated with the development of the initial test. Once the initial companion diagnostic is approved, clinical testing laboratories can create their own ASR-driven tests. The recent Supreme Court decision disallowing Myriad Genetics patents on BRCA testing opens the possibility of third-party clinical laboratories creating multiplex assays based on naturally occurring genetic variations.
The future of companion diagnostics
The future growth of companion diagnostics will rely upon a thriving ecosystem. The ecosystem needs multiple players to be vibrant. The major players must include drug developers that continue to develop targeted therapies based on biomarkers, diagnostic firms with the capability to create new diagnostic tests, regulators that are willing to approve combination products of drugs and diagnostics, and payers who see the value in reimbursing targeted therapies. The economic incentives for the continued development of companion diagnostics are compelling. Drug developers can achieve faster time to market with less expensive clinical trials for drugs with significant revenue potential. Regulators see the potential for more directed regulatory submissions with fewer adverse events based on targeted therapies. Payers see the potential reduced cost of unnecessary treatments. However, diagnostic firms are the one group for which the business model of companion diagnostics is still uncertain. This is particularly true for those diagnostic companies seeking to develop theranostics. As demonstrated in this review, those diagnostics companies that develop for all types of companion diagnostics – screening and detection, prognosis, recurrence, as well as theranostics and monitoring – can leverage their portfolio of tests to create a sustainable business model. Those companies that seek to focus only on theranostics and monitoring diagnostics will face a more uncertain future.
One of the key indicators of the future viability of the entire companion diagnostic market is the robustness of the financing environment for diagnostic and research tools companies. Without access to capital, the entire companion diagnostic marketplace will be unable to survive and prosper. The sources of funding for companion diagnostics are similar to other parts of life sciences and include the usual suspects: venture capital, corporate debt, initial public offerings of equity, follow-on public offerings of equity, and corporate spinoffs. These sources of funding include both public and private sources, as shown in Figure 4.28 Mergers and acquisitions have been excluded from the analysis despite being critical in providing profitable exits for entrepreneurs and venture capitalists, because they do not contribute to ongoing funding for the enterprise.
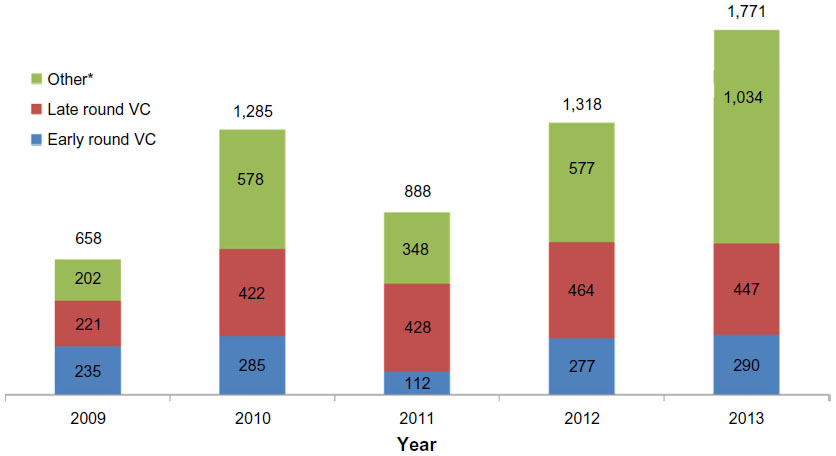 | Figure 4 Annual value of financing for diagnostics and research tools companies (USD millions). |
As Figure 4 clearly shows, there has been considerable volatility through the public financing channels available to diagnostic firms. The greatest volatility has been driven by the closing and opening of initial public offerings windows into the public markets. However, private investment in the form of early- and late-round venture capital investment has been a growing source of funding, from $450 million in 2009 to nearly $750 million in 2013. While many life science segments have experienced declining venture capital interest, the increase in venture capital funding promises that emerging diagnostic companies will continue to be formed, providing the backbone for the future growth in the companion diagnostic market. While the companion diagnostic market will continue to evolve and certainly experience bumps in the road, the venture capital funding engine will keep the market moving forward into the foreseeable future.
Disclosure
The authors report no conflicts of interest in this work.
References
US Food and Drug Administration. Companion diagnostics [webpage on the Internet]. Available from: http://www.fda.gov/MedicalDevices/ProductsandMedicalProcedures/InVitroDiagnostics/ucm407297.htm. Accessed August 26, 2014. | |
Myriad Genetics [webpage on the Internet]. 2013 Annual Report. Available from: http://files.shareholder.com/downloads/MYGN/3422924703x0x699241/42B88246-0E90-4F5C-9697-3DF98027C46D/2013_Annual_Report.pdf. Accessed August 26, 2014. | |
Dutton G. Companion Dx and personalized medicine [webpage on the Internet]. Genetic Engineering and Biotechnology News. 2012;32(19). Available from: http://www.genengnews.com/gen-articles/companion-dx-and-personalized-medicine/4577. Accessed August 26, 2014. | |
Hughes MD. Molecular Diagnostics Market Trends and Outlook [webpage on the Internet]. Enterprise Analysis Corporation; 2013. Available from: http://www.eacorp.com/images/PDFS/Molecular%20Diagnostics%20IVD%20Article%20v21%20MEK%20-%20Reprint%20FINAL.pdf. Accessed August 26, 2014. | |
Visiongain [webpage on the Internet]. Companion Diagnostics: World Market Outlook 2011–2021. Available from: https://www.visiongain.com/Report/675/Companion-Diagnostics-World-Market-Outlook-2011-2021. Accessed August 26, 2014. | |
Personalized Medicine Coalition. The Case for Personalized Medicine. Washington, DC: Personalized Medicine Coalition; 2006. | |
Analysis by authors utilizing FDA labels at Daily Med (http://dailymed.nlm.nih.gov). Accessed August 26, 2014. | |
Personalized Medicine Coalition. The Case for Personalized Medicine. Washington, DC: Personalized Medicine Coalition; 2011. | |
Singh V. Companion diagnostics poised for a breakout. The Burrill Report [webpage on the Internet]. 2012;2(9). Available from: http://www.burrillreport.com/content/BR_0912%20final-v3.pdf. Accessed August 26, 2014. | |
Roche [webpage on the Internet]. Annual Reports Archive; 2012 [updated July 28, 2014]. Available from: http://www.roche.com/investors/annual_reports/annual_reports_archive.htm. Accessed August 26, 2014. | |
Novartis [webpage on the Internet]. Annual Report 2012; 2012. Available from: http://www.novartis.com/downloads/investors/reports/novartis-annual-report-2012-en.pdf. Accessed August 26, 2014. | |
Surveying Tomorrow’s BioPharma Landscape [webpage on the Internet]. Evaluate Pharma; 2012. Available from: http://info.evaluatepharma.com/rs/evaluatepharmaltd/images/EvaluatePharma_NBI_Up_Close_2012.pdf. Accessed August 26, 2014. | |
Review of clinical trials listed at http://www.clinicaltrials.gov. Accessed on August 26, 2014. | |
Review of EMA drugs labels at http://www.ema.europa.eu/ema/index.jsp?curl=pages/regulation/document_listing/document_listing_000267.jsp. Accessed August 26, 2014. | |
Carver HC. Companion diagnostics: evolving FDA regulation and issues for resolution. In Danzis SD, Flannery EJ, editors. In Vitro Diagnostics: The Complete Regulatory Guide. Washington, DC: Food and Drug Law Institute; 2010:149–184. | |
Titus K. Two for the road with companion diagnostics [webpage on the Internet]. CAP Today; 2012. Available from: http://www.cap.org/apps//cap.portal?_nfpb=true&cntvwrPtlt_actionOverride=%2Fportlets%2FcontentViewer%2Fshow&_windowLabel=cntvwrPtlt&cntvwrPtlt{actionForm.contentReference}=cap_today%2F0412%2F0412b_diagnostics.html&_state=maximized&_pageLabel=cntvwr. Accessed August 26, 2014. | |
Roche. Roche provides update on Phase III study of onartuzumab in people with specific type of lung cancer [press release]. March 3, 2014. Available from: http://www.roche.com/media/media_releases/med-cor-2014-03-03.htm. Accessed August 26, 2014. | |
The Academy of Medical Sciences. “Realising the Potential of Stratified Medicine” Case Studies; 2012. Available from: http://www.acmedsci.ac.uk/viewFile/51f28620d5ec6.pdf. Accessed August 26, 2014. | |
Smith B, Ransom J, Spinner D, Faulkner E. Future Best Practices in Oncology Development. Quintiles White Paper. Available from: http://www.quintiles.com/library/white-papers/future-best-practices-in-oncology-development-critical-aspects-of-codeveloping-and-launching-an. Accessed November 21, 2014. | |
Roche. 2013 Annual Report. Available from: http://www.roche.com/investors/annual_reports.htm. Accessed August 26, 2014. | |
Datamonitor PharmaVitae Database. https://service.datamonitorhealthcare.com/drug/drug-search/. Accessed March 20, 2014. | |
Wildwood Ventures Ltd. Current Agreements Database. Available from: http://www.currentagreements.com/. Accessed Nov 2013. | |
Garfield S. Advancing Access to Personalized Medicine: A Comparative Assessment of European Reimbursement Systems. Personalized Medicine Coalition. Bridgehead International; 2011; Available from: http://www.personalizedmedicinecoalition.org/Userfiles/PMC-Corporate/file/pmc_bridgehead_issue_brief_european_reimbursement.pdf. Accessed November 21, 2014. | |
Crook A. Better access to molecular tests: the INCa/Pfizer partnership. EuropaBio 2011: Workshop on Market Access to Personalized Medicine. Feb 9 Brussels, Belgium. Available from: http://www.europabio.org/sites/default/files/3_-_access_to_molecular_tests_europabio_pers_med_workshop_090211.pd. Accessed March 20, 2014. | |
Merrill J, Bonanos P, Lash A, LaMotta L, Haas J, Goodman M. Deals of the week wonders: will pharma ever pay more for companion diagnostics? The Pink Sheet, November 25, 2013. Available from: http://invivoblog.blogspot.com/2013/11/deals-of-week-wonders-will-pharma-ever.html. Accessed August 26, 2014. | |
Leamon CP, Sherman MA. The rise of companion diagnostics: a step towards truly personalized medicine. Oncology Business Review. May 2012. Available from: https://obroncology.com/obrgreen/article/The-Rise-of-Companion-Diagnostics-A%20Step-Towards-Truly-Personalized-Medicine. Accessed August 26, 2014. | |
Genomic Health. 2012 Genomic Health Annual Report. Available from: http://investor.genomichealth.com/annuals.cfm. Accessed August 26, 2014. | |
Informa Strategic Transactions Database for In Vitro Diagnostics and Research Tools. https://www.pharmamedtechbi.com/mkt/db/transactions/st-lp. Accessed March 20, 2014. |
Supplementary materials
Companion diagnostic definition
A companion diagnostic device can be an in vitro diagnostic device or an imaging tool that provides information that is essential for the safe and effective use of a corresponding therapeutic product. The use of an in vitro diagnostic companion device with a particular therapeutic product is stipulated in the instructions for use in the labeling of both the device and the corresponding therapeutic product, as well as in the labeling of any generic equivalents and biosimilar equivalents of the therapeutic product.
Types of companion diagnostic testing
Screening and detection
Tests that screen for familial genetic patterns (eg, BRCA for aggressive breast cancer) and difficult to diagnose conditions (eg, CupPrint® test from Agendia, which identifies cancers of unknown primary origin).
Prognosis
Tests that predict the future course of a disease (eg, Genomic Health’s Oncotype Dx® for breast cancer).
Theranostics
Tests that indicate a patient’s response to a prescribed therapy (eg, HER2/Neu test for Herceptin®).
Monitoring
Tests that evaluate the effectiveness and appropriate dosing of a prescribed therapy (eg, CYP2C9 and VKORC1 testing for warfarin sensitivity).
Recurrence
Tests that analyze the patient’s risk for a recurrence of the disease (eg, Agendia Mammaprint® for recurrence of breast cancer).
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.