Back to Archived Journals » Hypoxia » Volume 4
The 2-oxoglutarate analog 3-oxoglutarate decreases normoxic hypoxia-inducible factor-1α in cancer cells, induces cell death, and reduces tumor xenograft growth
Authors Koivunen P, Fell S, Lu W, Rabinowitz J, Kung A, Schlisio S
Received 14 September 2015
Accepted for publication 11 December 2015
Published 4 March 2016 Volume 2016:4 Pages 15—27
DOI https://doi.org/10.2147/HP.S96366
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Dörthe Katschinski
Peppi Koivunen,1 Stuart M Fell,2,3 Wenyun Lu,4 Joshua D Rabinowitz,4 Andrew L Kung,5,6 Susanne Schlisio,2,7
1Biocenter Oulu, Faculty of Biochemistry and Molecular Medicine, Oulu Center for Cell-Matrix Research, University of Oulu, Oulu, Finland; 2Ludwig Institute for Cancer Research Ltd, Stockholm, Sweden; 3Department of Cell and Molecular Biology, Karolinska Institutet, Stockholm, Sweden; 4Department of Chemistry and Integrative Genomics, Princeton University, Princeton, NJ, 5Department of Medical Oncology, Dana Farber Cancer Institute, Harvard Medical School, Boston, MA, 6Department of Pediatrics, Columbia University Medical Center, New York, NY, USA; 7Department of Microbiology and Tumor and Cell Biology, Karolinska Institutet, Stockholm, Sweden
Abstract: The cellular response to hypoxia is primarily regulated by the hypoxia-inducible factors (HIFs). HIF-1α is also a major mediator of tumor physiology, and its abundance is correlated with therapeutic resistance in a broad range of cancers. Accumulation of HIF-1α under hypoxia is mainly controlled by the oxygen-sensing HIF prolyl 4-hydroxylases (EGLNs, also known as PHDs). Here, we identified a high level of normoxic HIF-1α protein in various cancer cell lines. EGLNs require oxygen and 2-oxoglutarate for enzymatic activity. We tested the ability of several cell-permeable 2-oxoglutarate analogs to regulate the abundance of HIF-1α protein. We identified 3-oxoglutarate as a potent regulator of HIF-1α in normoxic conditions. In contrast to 2-oxoglutarate, 3-oxoglutarate decreased the abundance of HIF-1α protein in several cancer cell lines in normoxia and diminished HIF-1α levels independent of EGLN enzymatic activity. Furthermore, we observed that 3-oxoglutarate was detrimental to cancer cell survival. We show that esterified 3-oxoglutarate, in combination with the cancer chemotherapeutic drug vincristine, induces apoptosis and inhibits tumor growth in vitro and in vivo. Our data imply that a novel treatment strategy targeting HIF-1α in combination with the use of existing cytotoxic agents could serve as potent, future antitumor chemotherapies.
Keywords: cancer, EGLN, HIF, hypoxia, 3-oxoglutarate, prolyl hydroxylase
Introduction
Hypoxia-inducible factors (HIFs) are key transcriptional regulators of the adaptive response when oxygen becomes limiting (hypoxia).1 The HIFα subunits, HIF-1α and HIF-2α, dimerize with HIFβ to form a transcriptionally active factor. Under normal oxygenation conditions (normoxia), HIFα is rapidly degraded via prolyl 4-hydroxylation mediated by the EGLNs (EGLNs 1–3, also known as PHDs 2, 1, and 3, respectively) and subsequent recognition and ubiquitination by the tumor suppressor von Hippel–Lindau protein (pVHL), thereby targeting hydroxylated HIFα for proteasomal degradation.2–5 In low oxygen environments, HIFα escapes hydroxylation and VHL recognition, allowing the HIFαβ dimer to transactivate target genes that are important for adaptation to a low oxygen environment, including genes regulating energy metabolism and angiogenesis.6–8
Loss or mutation of VHL causes HIFα to accumulate under normoxia, causing the so-called pseudohypoxic response in these cells.9 Furthermore, mutation of the Krebs cycle metabolic enzymes, succinate dehydrogenase (SDH) and fumarate hydratase (FH), results in the buildup of their substrates, succinate and fumarate. This, in turn, can lead to HIFα stabilization in normoxia because succinate and fumarate impair EGLN enzymatic activity by binding to EGLNs in place of the required cofactor 2-oxoglutarate.10–12 However, VHL, SDH, and FH mutations are restricted to only certain forms of cancers, despite the evidence that HIF-1α is overexpressed in many types of cancers, including breast, ovarian, renal carcinoma, glioblastoma, and leiomyoma.13–16 Thus, cancer metabolism, directly or indirectly, might account for HIFα stabilization under normoxia. Recently, on the one hand, the availability of nutrients, such as glutamine and glucose (Glc), has been shown to regulate HIF-1α translation,17 and on the other hand, HIF-1α stabilization upregulates Glc uptake and glycolytic metabolism and downregulates oxidative phosphorylation, causing potentially oncogenic positive feedback loops.18
In this regard, it has been recently suggested that oncometabolites such as succinate, fumarate and R-2-hydroxyglutarate (R-2-HG) can promote tumorigenesis by altering HIF stability and the epigenome.19–25 R-2-HG is an oncometabolite produced de novo as a result of isocitrate dehydrogenase mutations and commonly found in gliomas and acute myeloid leukemia.26,27 A common feature of these “oncometabolites” is their similarity to 2-oxoglutarate and ability to act as inhibitors for a superfamily of enzymes called the 2-oxoglutarate-dependent dioxygenases. Succinate, fumarate, and R-2-HG are able to bind to specific members of this protein family and impair normal enzymatic activity. Among the impaired enzymes are the EGLN prolyl 4-hydroxylases.
In the present study, we show that diethyl-3-oxoglutarate (DE-3-oxo), a cell-permeable 2-oxoglutarate analog, decreases the abundance of HIF-1α protein in cancer cells in normoxia independent of EGLN hydroxylase activity. Furthermore, we demonstrate that cell-permeable 3-oxoglutarate in combination with the cancer chemotherapeutic drug vincristine induces apoptosis and inhibits tumor growth in vivo.
Materials and methods
Reagents
DE-3-oxo was purchased from Sigma-Aldrich (St Louis, MO, USA) or Alfa Aesar (Ward Hill, MA, USA). DE-2-oxo and vincristine were purchased from Sigma-Aldrich. FG-0041 was a gift from FibroGen Inc (San Francisco, CA, USA).
Cell culture
HeLa, MDA-MB-231, MDA-MB-453, MCF7, U87, LN2306, H1299, HCT116, NHA, H460, RCC4, and T47D cell lines were purchased from the American Type Culture Collection (ATCC) (Manassas, VA, USA) and cultured in Dulbecco’s Modified Eagle’s Medium supplemented with 10% fetal bovine serum (FBS), while SK-N-F1, NB1, NLF, SK-N-AS, LAN6, IMR32, SK-N-SH, SM-SK-AN, CHP212, and SK-N-DZ cell lines were cultured in Roswell Park Memorial Institute-1640 supplemented with 10% FBS. When indicated, the cells were exposed to hypoxia/anoxia in an InVivo2 400 (Ruskinn Technology Ltd, Bridgend, UK) or in a Coy Laboratory Products Inc’s hypoxic glove box. No ethics statement was required from the institutional review board for the use of these cell lines.
Immunoblot analysis
Cells grown in culture were rinsed with ice-cold phosphate-buffered saline (PBS), scraped, and lysed with 1× EBC buffer (50 mM Tris [pH 8.0], 120 mM NaCl, 0.5% NP-40) supplemented with complete protease inhibitor mixture (Hoffman-La Roche Ltd., Basel, Switzerland). Equal amounts of protein extract, as determined by the Bradford method, were resolved by sodium dodecyl sulfate–polyacrylamide gel electrophoresis and blotted onto polyvinylidene difluoride membranes. After blocking in Tris-buffered saline with 5% nonfat dry milk, the membranes were probed with the following primary antibodies: anti-HA (HA-11; Covance Research Products Inc., Denver, CO, USA), anti-HIF-1α (NB100-479; Novus International Inc, St Louis, MO, USA), anti-cleaved Caspase 3 (9661, Cell Signaling Technology, Inc, Danvers, MA, USA), anti-p53 (Calbiochem, Darmstadt, Germany), vinculin (Sigma-Aldrich), and anti-tubulin (B-512, Sigma-Aldrich). Horseradish-peroxidase-conjugated secondary antibodies were from Pierce, and immobilon Western chemiluminescent horseradish peroxidase substrate was purchased from EMD Millipore, Billerica, MA, USA.
RNA extraction and quantitative polymerase chain reaction
NLF cells were treated with 1 mM DE-3-oxo or vehicle (dimethyl sulfoxide) for 20 hours. Total RNA was isolated and purified with an EZNA Total RNA Kit (Omega Bio-Tek Inc, Norcross, GA, USA), and reverse transcription was performed with an iScript cDNA synthesis kit (Bio-Rad Laboratories Inc., Hercules, CA, USA). TATA-binding protein was used to equalize the amount of template in quantitative polymerase chain reaction, which was performed with iTaq SYBR Green Supermix in a Touch Thermal Cycler and a CFX96 Real-Time System (Bio-Rad). The sequences for the primers used in quantitative polymerase chain reaction are available upon request.
Crystal violet staining
Cell staining with crystal violet has been previously described.28 In short, the medium was removed, and the cells were washed with PBS and fixed and stained with 20% methanol and 0.5% crystal violet for 30 minutes at room temperature and then washed twice with PBS.
In vivo tumor growth assay
Xenografts and in vivo imaging using luciferase-based reporter of oxygen-dependent degradation domain (ODD) from HIF-1α has been previously described.29 In short, HCT116 cells were stably transfected to express a fluorescent and a luminescent marker for visualizing live animals, that is, mCherry together with either wild-type (WT) luciferase or luciferase fused to HIF-1α ODD containing the P564 hydroxylatable residue (ODD-luc [ODD-luciferase]). A total of 106 cells were injected into the flanks of female nude mice, and 14 tumors for each treatment group were analyzed. All animal-handling procedures were performed according to the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health and followed the 21 guidelines of the Animal Welfare Act. All animal experiments were approved by the local Experimental Animal Ethical Committee. When tumors of equal size, as evaluated by mCherry fluorescence detected with IVIS 100 (Caliper Life Sciences, Hopkinton, MA, USA), had developed, DE-3-oxo (100 mg/kg) or vehicle was administered via intraperitoneal injection. Bioluminescence of the tumors was detected 2 hours post intraperitoneal luciferin injection. To study the effect of DE-3-oxo on tumor growth, nude mice injected with 106 HCT116 mCherry cells on both flanks were left to develop measurable tumors as evaluated by fluorescence detected with IVIS 100 (Caliper Life Sciences). The mice were thereafter treated for 10 days, with DE-3-oxo (100 mg/kg twice a day), vincristine (0.5 mg/kg 1× weekly), or a combination of DE-3-oxo and vincristine, or vehicle. Tumor sizes were measured at the start of the treatment and at 6 days and 10 days after starting the treatment. Relative tumor size was calculated by normalizing to tumor size at the beginning of the treatment.
Measurement of intracellular 2-oxoglutarate, 3-oxoglutarate, and acetoacetate
Metabolite levels in samples were determined by negative-mode electrospray liquid chromatography–mass spectrometry (LC–MS) as previously decribed.26 In short, the LC method uses a Synergi Hydro column (4 μm particle size, 150 mm × 2 mm, from Phenomenex Inc, Torrance, CA, USA), with solvent A being 10 mM tributylamine + 15 mM acetic acid in the ratio of 97:3 (water:methanol), and solvent B being 100% methanol. The flow rate is 200 μL/min, and the running time is 50 minutes. The gradient is t=0 minutes, 0% B; t=5 minutes, 0% B; t=10 minutes, 20% B; t=20 minutes, 20% B; t=35 minutes, 65% B; t=38 minutes, 95% B; t=42 minutes, 95% B; t=43 minutes, 0% B; and t=50 minutes, 0% B. Compounds were detected using selected reaction monitoring on a Thermo Quantum Discovery Max triple quadrupole mass spectrometer operating in negative ion mode.
Enzymatic assays
Human EGLN1 and EGLN3 were produced and purified as Flag-His-tagged recombinant proteins in insect cells. The catalytic activity of EGLN1 and EGLN3 in the presence of 3-oxoglutarate was determined using
Reactive oxygen species analysis
Reactive oxygen species (ROS) analysis has been previously described.32 In short, NLF cells were incubated for 1 hour with 5 μM CM-H2DCFDA (Molecular Probes), harvested, resuspended at 106 cells/mL in PBS supplemented with 7% FBS, and analyzed by FACS (fluorescence-activated cell sorting).
Statistical analyses
Pairwise comparisons were performed using Student’s two-tailed t-test. The data are shown as mean ± standard deviation. Error bars represent standard error of the mean. Values of P<0.05 were considered statistically significant (*P<0.05; **P<0.01; ***P<0.005).
Results
Cell-permeable 3-oxoglutarate, but not 2-oxoglutarate, decreases HIF-1α in several cancer cell lines
We observed that HIF1α levels vary across cancer cell lines, with some having high normoxic levels of HIF-1α, such as the neuroblastoma cell lines: NLF, SK-N-AS, IMPR32, and SK-N-SH (Figure S1A). Elevated HIF-1α is associated with high glycolytic activity along with the suppression of Krebs cycle activity and oxidative phosphorylation – common traits of most cancer cell lines.33 HIF-1α protein levels are regulated by the EGLN proteins that require 2-oxoglutarate as an enzymatic cofactor. We investigated whether the observed high normoxic HIF-1α is a consequence of inadequate cellular 2-oxoglutarate concentrations leading to insufficient HIF-1α degradation by EGLNs in cancer cells. We treated neuroblastoma cell line NLF, which expresses abundant HIF-1α protein even in normoxia, with a cell membrane permeable form of 2-oxoglutarate (DE-2-oxo). At low concentrations (10–80 μM) of DE-2-oxo, no effect on the abundance of HIF-1α was detected (Figure S1B), whereas at millimolar concentrations DE-2-oxo increased the abundance of HIF-1α protein (Figure 1A). Intracellular levels of nonesterified 2-oxoglutarate were increased under these conditions (Figure 1B). The increased abundance of HIF-1α observed is consistent with the previous observations demonstrating that dimethyl-2-oxoglutarate can transiently stabilize HIF-1α.34 In the course of studying the effects of several cell-permeable 2-oxoglutarate analogs on HIF-1α, we identified DE-3-oxo as a downregulator of HIF-1α in several cancer cell lines (Figures 1A and C and Figure S1C). Intracellular levels of nonesterified 2-oxoglutarate and 3-oxoglutarate were increased in DE-2-oxo- and DE-3-oxo-treated cells, respectively (Figure 1B). DE-3-oxo-mediated regulation of HIF-1α was dose and time dependent (Figures 1A and D), and complete downregulation of HIF-1α in NLF cells was observed 6 hours post treatment with 1 mM DE-3-oxo (Figure 1D). DE-3-oxo treatment significantly decreased the expression of the HIF-1α target genes GLUT1 and PDK1 (Figure 1E). HIF-1α mRNA abundance was not altered in NLF cells by DE-3-oxo treatment, while HIF-2α mRNA was slightly increased (Figure 1F). In addition, metabolite profiling using LC-MS scanning revealed increased cellular acetoacetate levels in DE-3-oxo-treated cells (Figure S1D). Furthermore, DE-3-oxo did not alter the stability of another short-lived protein, p53, in these cell lines (Figure S1E).
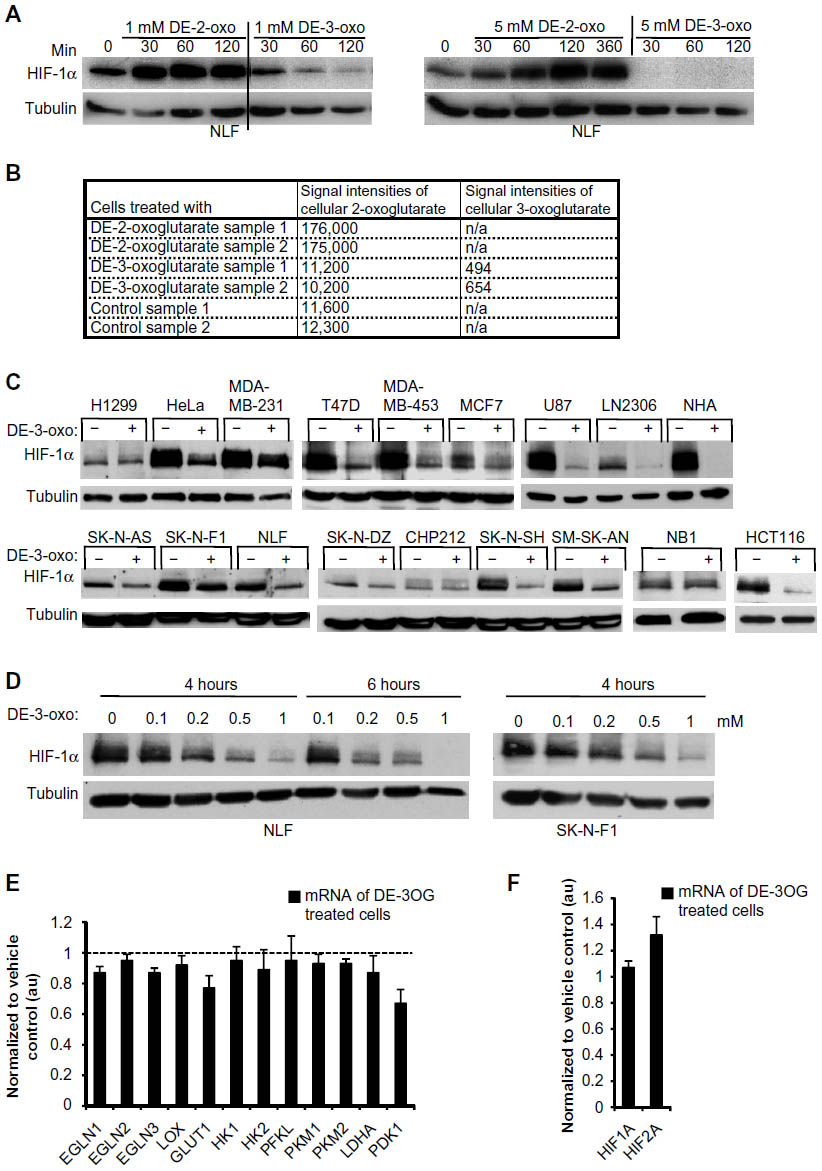 | Figure 1 Cell-permeable 3-oxoglutarate, but not 2-oxoglutarate, decreases HIF-1α in several cancer cell lines. |
3-oxoglutarate regulates HIF-1α abundance independent of EGLN enzymatic activity
To understand mechanistically how 3-oxoglutarate impacts the abundance of HIF-1α protein, we studied the effect of DE-3-oxo under moderate (1%) and severe (0.1%) hypoxia. The effect of DE-3-oxo on the abundance of HIF-1α was not impaired under 1% O2 in NLF or HCT116 cells (Figure 2A). Furthermore, DE-3-oxo reduced the abundance of HIF-1α in NLF cells even in severe hypoxia (0.1% O2; Figure 2B), implying that the regulation of HIF-1α by 3-oxoglutarate might be independent of the oxygen-requiring activity of the EGLNs. To further study whether 3-oxoglutarate acts on HIF-1α independently of EGLN enzymatic activity, we stably transfected NLF cells with vectors encoding either hemagglutinin (HA)-tagged WT HIF-1α or a mutant HIF-1α that escapes prolyl hydroxylation and subsequent degradation (HA-dbPA-HIF-1α), due to point mutation of the two proline residues (P402A and P402A) otherwise hydroxylated by the EGLN proteins. The treatment of stably transfected NLF cells with DE-3-oxo reduced the abundance of both HA-dbPA-HIF-1α protein and WT HA-HIF-1α protein, by a similar magnitude to the observed decreases in endogenous HIF-1α protein (Figure 2C). This indicates that the effect of DE-3-oxo on HIF-1α is independent of prolyl hydroxylation. In addition, we tested whether the EGLN inhibitor FG-0041 could revert the effect of DE-3-oxo on HIF-1α. Despite simultaneous FG-0041 treatment up to 25 μM and 50 μM, DE-3-oxo treatment still significantly reduced the HIF-1α protein in NLF and HCT116 cells, respectively (Figures 2D and E). Taken together, this indicates that the effect of DE-3-oxo on HIF-1α is independent of prolyl hydroxylation.
 | Figure 2 3-oxoglutarate regulates the abundance of HIF-1α protein independent of EGLN enzymatic activity. |
Furthermore, we tested the ability of 3-oxoglutarate to substitute for 2-oxoglutarate as an EGLN cofactor using an in vitro EGLN enzyme activity assay that measures the generation of 4-hydroxyproline in HIF-1α. In this assay, 3-oxoglutarate failed to substitute for 2-oxoglutarate as a cofactor for either EGLN1 or EGLN3 activity (Figure 2F), further implying that the effect of 3-oxoglutarate on HIF-1α downregulation is prolyl hydroxylase independent. An alternative possibility is that DE-3-oxo acts as a ROS scavenger and thereby promotes the downregulation of HIF-1α. However, the measurement of ROS in NLF cells treated with either DE-3-oxo or DE-2-oxo did not support this hypothesis (Figure S2). Instead, we observed rather the opposite effect that ROS was increased after DE-2-oxo and DE-3-oxo treatment.
Combination treatment of DE-3-oxoglutarate and vincristine synergistically induced apoptosis in cancer cells
We next studied whether DE-3-oxo would impair the growth of cancer cells or sensitize them to apoptosis when administered over a longer period of time. Daily administration of DE-3-oxo for 96 hours to NLF cells induced cell death as judged by crystal violet staining, whereas in HCT116 cells DE-3-oxo had only a minor effect after 7 days of administration (Figure 3A). However, a single-dose treatment of DE-3-oxo in combination with the cancer chemotherapeutic drug vincristine caused induction of apoptosis within 24 hours, as evaluated by caspase-3 cleavage and crystal violet staining in NLF, HCT116, and H460 cells (Figures 3B–D). The effect of the combination treatment of DE-3-oxo and vincristine on cell death appeared to be p53 independent (Figure 3E).
 | Figure 3 Combination treatment of DE-3-oxo and vincristine synergistically induces apoptosis in cancer cells. |
DE-3-oxoglutarate decreased HIF-1α protein in vivo and reduced tumor xenograft growth in combination with vincristine
To evaluate the potential of 3-oxoglutarate as a cancer therapeutic, we first studied its ability to decrease HIF-1α in vivo. A plasmid encoding a protein chimera of the HIF-1α ODD fused to luciferase (ODD-luc), and mCherry was stably transfected into HCT116 cells. HCT116-ODD-luc cells or control cells expressing WT luciferase and mCherry were injected into the flanks of nude mice. Mice with tumors of equal size, as evaluated by mCherry fluorescence, were administered with DE-3-oxo (100 mg/kg) or vehicle via intraperitoneal injection. Bioluminescence of the tumors was detected 2 hours post treatment following a luciferin injection (Figure 4A). Significant decrease in the luminescence from the ODD-luc, but not WT luciferase, was detected after DE-3-oxo treatment (Figures 4A and B). Encouraged by these results, we set out to study the effect of DE-3-oxo on tumor xenograft growth in vivo. Nude mice bearing equal size of HCT116 mCherry-expressing tumors on both flanks were divided into four groups which received either vehicle, DE-3-oxo, vincristine, or both DE-3-oxo and vincristine. Tumor size was assessed at 6 days and 10 days after treatment by measuring xenograft fluorescence. Significantly reduced tumor fluorescence, indicating smaller tumors, was detected at 6 days and 10 days after treatment in mice that received the combination therapy when compared with vehicle-treated control mice (Figures 4C and D). Treatment with DE-3-oxo or vincristine alone showed only marginal decrease in tumor growth relative to vehicle control (Figure 4D and Figures S3A and B).
 | Figure 4 DE-3-oxo decreases HIF-1α protein in vivo and reduces tumor xenograft growth in combination with vincristine. |
Discussion
Normoxic HIF-1α stabilization has been linked to tumor malignancy in several tissues.12–15 Our data here support these findings as we observed high HIF-1α levels in cancer cell lines originating from breast, brain, colon, cervix, and lung under normoxia. The abundance of HIF-1α is chiefly regulated by oxygen and 2-oxoglutarate-dependent EGLN prolyl hydroxylases.2–5 We explored whether the increased HIF-1α expression observed is due to a deficit in 2-oxoglutarate that potentially impairs EGLN activity. The administration of cell-permeable 2-oxoglutarate to the cells increased intracellular 2-oxoglutarate concentration but failed to attenuate the normoxic stabilization of HIF-1α, suggesting that 2-oxoglutarate is not limiting for EGLN activity in these cancer cell lines. In the course of studying the effects of several cell-permeable 2-oxoglutarate analogs on HIF-1α, we identified 3-oxoglutarate as a potent downregulator of HIF-1α in nearly all (or in 13 out of 16) cancer cell lines studied.
3-oxoglutarate is a structural analog of 2-oxoglutarate, however, not a physiologically relevant metabolite that is normally present in cells. Because it is a structural analog of 2-oxoglutarate, we considered whether it might substitute for the latter as an EGLN cosubstrate. Our data showed that 3-oxoglutarate could downregulate HIF-1α under hypoxia, even in the presence of a 2-oxoglutarate analogous EGLN inhibitor, FG-0041, and also when the prolyl residues in HIF-1α targeted by EGLNs were mutated to alanines. Moreover, 3-oxoglutarate failed to substitute for 2-oxoglutarate in an in vitro enzymatic assay studying prolyl hydroxylation in HIF-1α ODD. These data collectively indicate that 3-oxoglutarate effects HIF-1α independently of EGLNs. Moreover, the decrease in ectopically expressed HIF-1α in cells treated with 3-oxoglutarate suggests that 3-oxoglutarate affects posttranscriptional regulation of HIF-1α. In support of these findings, hydroxylation and VHL-independent degradation of HIF-1α has been previously reported.35,36
High HIF-1α levels in tumors are associated with treatment resistance.37–40 We therefore asked whether lowering HIF-1α with 3-oxoglutarate would predispose cancer cells to cell death. Repeated administration of DE-3-oxo to NLF cells resulted in complete cell death in 96 hours, whereas a single-dose treatment within 24 hours had no effect. We therefore explored whether the potency of 3-oxoglutarate could be enhanced in combination with another, complementary cytotoxic agent. When combined with the anti-mitotic chemotherapeutic agent vincristine, 3-oxoglutarate caused total or almost complete apoptosis in NLF, HCT116 and H460 cell lines. These cancer cell lines originate from the nervous system, colon and lung, respectively, demonstrating the chemotherapeutic potential of this combined treatment on cancer cells of diverse origins. Moreover, the cell killing by 3-oxoglutarate was independent of p53, which is significant since at least 50% of cancers harbor mutations in TP53,40 making them resistant to TP53-dependent cell death.
Targeting elevated HIF-1α levels in tumors has become an attractive area for the development of novel cancer therapeutics during the recent years. A large number of small molecule compounds, both extant and novel, are now known to downregulate HIF-1α and several of these compounds have been studied in preclinical settings.41 For instance, the cardiac glycoside digoxin, that inhibits HIF-1α protein synthesis decreases the growth of P493-Myc tumor xenografts.42 More recently, BAY87-2243, an inhibitor of hypoxia-inducible gene activation demonstrated antitumoral activity under Glc deprivation and reduced the growth of H460 tumor xenografts.43 DE-3-oxo exhibits therapeutic potential as it is well tolerated and downregulates HIF-1α in vivo in HCT116 tumor xenografts. Therapeutic treatment of combining agents that target HIF-1α with traditional cytostatic chemotherapeutics has been suggested as beneficial for those subsets of patients otherwise resistant to traditional chemotherapeutics alone.40 Our data here provide the first evidence to support this, since combination of DE-3-oxo with vincristine reduced the growth of HCT116 tumor xenografts, which neither of these compounds did alone.
HIF-1α transcriptional targets include Glc transporters, several enzymes involved in glycolysis, and pyruvate dehydrogenase kinase, which inhibits the conversion of pyruvate to acetyl coenzyme A (acetyl-CoA) and its subsequent entry into the Krebs cycle. Thus, cells with high normoxic HIF-1α are both pseudohypoxic and glycolytic. Increased glycolysis and decreased oxidative phosphorylation are considered to promote cancer cell growth and survival.44 Our metabolic analysis suggested that DE-3-oxo treatment increased the level of cellular acetoacetate, a ketone body. Acetoacetate can be converted into acetyl-CoA, can enter the Krebs cycle, and can be oxidized in the mitochondria for energy. Therefore, treatment with DE-3-oxo may potentially feed the Krebs cycle. An abundance of intracellular acetyl-CoA might inhibit glycolysis and lead to the so-called Randle cycle, the use of fatty acid originating metabolites in preference to Glc-related molecules, which is then accompanied by decreased levels of HIF-1α to slow down glycolysis. This is supported by recent observations demonstrating that the treatment of cancer cells with acetoacetate inhibited cell growth by reducing the use of Glc as an energy source.45 Moreover, a reduction in glycolytic flux, induction of apoptosis, and diminished cachexia was seen in pancreatic cancer cells with ketone body supplementation.46 Ketone supplementation was also shown to decrease tumor cell viability and increase life span in mice with metastatic cancer.47 An alternative hypothesis is that acetyl-CoA functions as a signaling molecule that regulates the abundance of HIF-1α.
Cancer cells often have an altered energy metabolism, with high intake and use of Glc, the so-called Warburg effect. HIF-1 is a potent regulator of Glc metabolism and is often highly expressed in cancer. Here, we identified 3-oxoglutarate as a novel potential cancer therapeutic, targeting normoxic HIF-1α in vitro and in vivo. Furthermore, 3-oxoglutarate in combination with the cytotoxic chemotherapeutic vincristine reduced the growth of tumor xenografts in vivo. These data support the treatment of resistant tumors with a therapeutic combination of a cytotoxic compound and an agent that can downregulate HIF-1α and revert tumor metabolism, which may prime the cells for cytotoxicity.
Acknowledgments
PK was supported by the Academy of Finland Grants 120156, 140765, 218129, and 266719, and by grants from the Sigrid Jusélius Foundation, the Emil Aaltonen Foundation, the Jane and Aatos Erkko Foundation, and the Finnish Cancer Organizations. SS is supported by grants from the Swedish Children Cancer Foundation and the Swedish Cancer Society, and he is an LICR Assistant Member.
Disclosure
The authors report no conflicts of interest in this work.
References
Semenza GL. HIF-1: mediator of physiological and pathophysiological responses to hypoxia. J Appl Physiol. 2000;88(4):1474–1480. | |
Kaelin WG Jr. The von Hippel-Lindau tumour suppressor protein: O2 sensing and cancer. Nat Rev. 2008;8(11):865–873. | |
Ivan M, Kondo K, Yang H, et al. HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: implications for O2 sensing. Science. 2001;292(5516):464–468. | |
Jaakkola P, Mole DR, Tian YM, et al. Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science. 2001;292(5516):468–472. | |
Epstein AC, Gleadle JM, McNeill LA, et al. C. elegans EGL-9 and mammalian homologs define a family of dioxygenases that regulate HIF by prolyl hydroxylation. Cell. 2001;107(1):43–54. | |
Kaelin WG Jr, Ratcliffe PJ. Oxygen sensing by metazoans: the central role of the HIF hydroxylase pathway. Mol Cell. 2008;30(4):393–402. | |
Wenger RH. Mammalian oxygen sensing, signalling and gene regulation. J Exp Biol. 2000;203(pt 8):1253–1263. | |
Hickey MM, Simon MC. Regulation of angiogenesis by hypoxia and hypoxia-inducible factors. Curr Top Dev Biol. 2006;76:217–257. | |
Schlisio S. Neuronal apoptosis by prolyl hydroxylation: implication in nervous system tumours and the Warburg conundrum. J Cell Mol Med. 2009;13(10):4104–4112. | |
Selak MA, Armour SM, MacKenzie ED, et al. Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF-alpha prolyl hydroxylase. Cancer Cell. 2005;7(1):77–85. | |
Isaacs JS, Jung YJ, Mole DR, et al. HIF overexpression correlates with biallelic loss of fumarate hydratase in renal cancer: novel role of fumarate in regulation of HIF stability. Cancer Cell. 2005;8(2):143–153. | |
Lee S, Nakamura E, Yang H, et al. Neuronal apoptosis linked to EglN3 prolyl hydroxylase and familial pheochromocytoma genes: developmental culling and cancer. Cancer Cell. 2005;8(2):155–167. | |
Semenza GL. Oxygen sensing, hypoxia-inducible factors, and disease pathophysiology. Annu Rev Pathol. 2014;9:47–71. | |
Semenza GL. Defining the role of hypoxia-inducible factor 1 in cancer biology and therapeutics. Oncogene. 2010;29(5):625–634. | |
Zhong H, De Marzo AM, Laughner E, et al. Overexpression of hypoxia-inducible factor 1alpha in common human cancers and their metastases. Cancer Res. 1999;59(22):5830–5835. | |
Zhong H, Semenza GL, Simons JW, et al. Up-regulation of hypoxia-inducible factor 1alpha is an early event in prostate carcinogenesis. Cancer Detect Prev. 2004;28(2):88–93. | |
Kwon SJ, Lee YJ. Effect of low glutamine/glucose on hypoxia-induced elevation of hypoxia-inducible factor-1alpha in human pancreatic cancer MiaPaCa-2 and human prostatic cancer DU-145 cells. Clin Cancer Res. 2005;11(13):4694–4700. | |
Semenza GL. HIF-1: upstream and downstream of cancer metabolism. Curr Opin Genet Dev. 2010;20(1):51–56. | |
Shim EH, Livi CB, Rakheja D, et al. L-2-Hydroxyglutarate: an epigenetic modifier and putative oncometabolite in renal cancer. Cancer Discov. 2014;4(11):1290–1298. | |
Nowicki S, Gottlieb E. Oncometabolites: tailoring our genes. FEBS J. 2015;282(15):2796–2805. | |
Schulze A, Harris AL. How cancer metabolism is tuned for proliferation and vulnerable to disruption. Nature. 2012;491(7424):364–373. | |
Noushmehr H, Weisenberger DJ, Diefes K, et al. Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. Cancer Cell. 2010;17(5):510–522. | |
Xu W, Yang H, Liu Y, et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of alpha-ketoglutarate-dependent dioxygenases. Cancer Cell. 2011;19(1):17–30. | |
Letouze E, Martinelli C, Loriot C, et al. SDH mutations establish a hypermethylator phenotype in paraganglioma. Cancer Cell. 2013;23(6):739–752. | |
Xiao M, Yang H, Xu W, et al. Inhibition of alpha-KG-dependent histone and DNA demethylases by fumarate and succinate that are accumulated in mutations of FH and SDH tumor suppressors. Genes Dev. 2012;26(12):1326–1338. | |
Dang L, White DW, Gross S, et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature. 2009;462(7274):739–744. | |
Ward PS, Patel J, Wise DR, et al. The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting alpha-ketoglutarate to 2-hydroxyglutarate. Cancer Cell. 2010;17(3):225–234. | |
Chen ZX, Wallis K, Fell SM, et al. RNA helicase A is a downstream mediator of KIF1Bbeta tumor-suppressor function in neuroblastoma. Cancer Discov. 2014;4(4):434–451. | |
Kimbrel EA, Davis TN, Bradner JE, et al. In vivo pharmacodynamic imaging of proteasome inhibition. Mol Imaging. 2009;8(3):140–147. | |
Juva K, Prockop DJ. Modified procedure for the assay of H-3-or C-14-labeled hydroxyproline. Anal Biochem. 1966;15(1):77–83. | |
Koivunen P, Hirsila M, Kivirikko KI, et al. The length of peptide substrates has a marked effect on hydroxylation by the hypoxia-inducible factor prolyl 4-hydroxylases. J Biol Chem. 2006;281(39):28712–28720. | |
Gerald D, Berra E, Frapart YM, et al. JunD reduces tumor angiogenesis by protecting cells from oxidative stress. Cell. 2004;118(6):781–794. | |
Semenza GL. HIF-1 mediates metabolic responses to intratumoral hypoxia and oncogenic mutations. J Clin Invest. 2013;123(9):3664–3671. | |
Hou P, Kuo CY, Cheng CT, et al. Intermediary metabolite precursor dimethyl-2-ketoglutarate stabilizes hypoxia-inducible factor-1alpha by inhibiting prolyl-4-hydroxylase PHD2. PLoS One. 2014;9(11):e113865. | |
Flugel D, Gorlach A, Kietzmann T. GSK-3beta regulates cell growth, migration, and angiogenesis via Fbw7 and USP28-dependent degradation of HIF-1alpha. Blood. 2012;119(5):1292–1301. | |
Flugel D, Gorlach A, Michiels C, et al. Glycogen synthase kinase 3 phosphorylates hypoxia-inducible factor 1alpha and mediates its destabilization in a VHL-independent manner. Mol Cell Biol. 2007;27(9):3253–3265. | |
Brown JM. Exploiting the hypoxic cancer cell: mechanisms and therapeutic strategies. Mol Med Today. 2000;6(4):157–162. | |
Patiar S, Harris AL. Role of hypoxia-inducible factor-1alpha as a cancer therapy target. Endocr Relat Cancer. 2006;13(suppl 1):S61–S75. | |
Samanta D, Gilkes DM, Chaturvedi P, et al. Hypoxia-inducible factors are required for chemotherapy resistance of breast cancer stem cells. Proc Natl Acad Sci USA. 2014;111(50):E5429–E5438. | |
Amelio I, Melino G. The p53 family and the hypoxia-inducible factors (HIFs): determinants of cancer progression. Trends Biochem Sci. 2015;40(8):425–434. | |
Xia Y, Choi HK, Lee K. Recent advances in hypoxia-inducible factor (HIF)-1 inhibitors. Eur J Med Chem. 2012;49:24–40. | |
Zhang H, Qian DZ, Tan YS, et al. Digoxin and other cardiac glycosides inhibit HIF-1alpha synthesis and block tumor growth. Proc Natl Acad Sci USA. 2008;105(50):19579–19586. | |
Ellinghaus P, Heisler I, Unterschemmann K, et al. BAY 87-2243, a highly potent and selective inhibitor of hypoxia-induced gene activation has antitumor activities by inhibition of mitochondrial complex I. Cancer Med. 2013;2(5):611–624. | |
Dang CV. Links between metabolism and cancer. Genes Dev. 2012;26(9):877–890. | |
Fine EJ, Miller A, Quadros EV, et al. Acetoacetate reduces growth and ATP concentration in cancer cell lines which over-express uncoupling protein 2. Cancer Cell Int. 2009;9:14. | |
Shukla SK, Gebregiworgis T, Purohit V, et al. Metabolic reprogramming induced by ketone bodies diminishes pancreatic cancer cachexia. Cancer Metab. 2014;2:18. | |
Poff AM, Ari C, Arnold P, et al. Ketone supplementation decreases tumor cell viability and prolongs survival of mice with metastatic cancer. Int J Cancer. 2014;135(7):1711–1720. |
Supplementary Materials
 | Figure S1 Cell-permeable 3-oxoglutarate, but not 2-oxoglutarate, decreases HIF-1α in several cancer cell lines. |
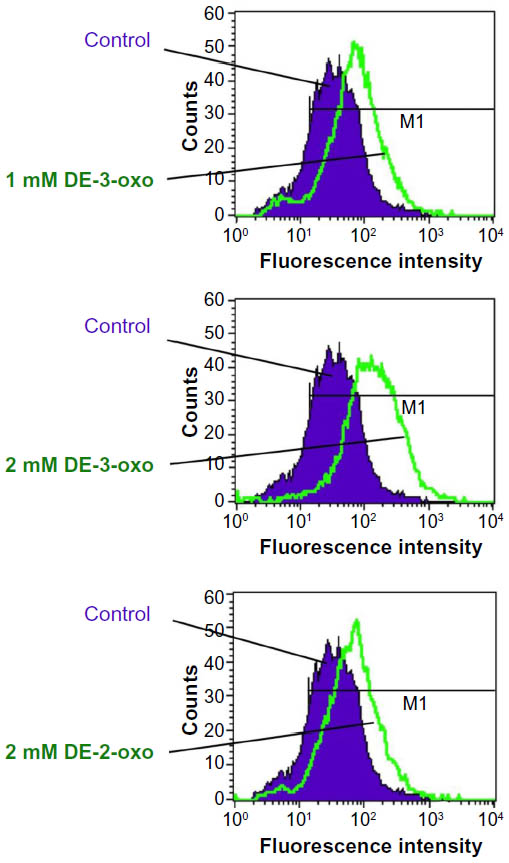 | Figure S2 DE-3-oxo does not act as an ROS scavenger. |
 | Figure S3 DE-3-oxo reduces tumor xenograft growth in combination with vincristine. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.