Back to Archived Journals » Antibody Technology Journal » Volume 6
Technology advancements in antibody purification
Authors Murphy C, Devine T, O'Kennedy R
Received 9 October 2015
Accepted for publication 12 March 2016
Published 26 August 2016 Volume 2016:6 Pages 17—32
DOI https://doi.org/10.2147/ANTI.S64762
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Shixia Wang
Video abstract presented by Caroline Murphy.
Views: 4554
Caroline Murphy, Tatyana Devine, Richard O'Kennedy
School of Biotechnology and Biomedical Diagnostics Institute, Dublin City University, Glasnevin, Dublin, Ireland
Abstract: The separation of antibodies from complex mixtures can be achieved using chromatographic or non-chromatographic techniques. The purification of antibodies using chromatography involves the separation of antibodies or antibody-derived molecules present in complex mixtures by passing them through a solid phase (eg, silica resin or beads, monolithic columns, or cellulose membranes) and allowing the antibodies to bind or pass through depending on whether "bind-and-elute" or "flow-through" chromatographic methods are employed. Chromatographic methodologies can incorporate different separation techniques, such as affinity-tag binding, ion-exchange, size-exclusion chromatography, or immunoaffinity chromatography. However, in a concerted effort to become less reliant on chromatography-based separations owing to the high cost of large-scale production, non-chromatographic-based techniques such as precipitation, flocculation, crystallization, filtration, and aqueous two-phase partitioning are now becoming more popular. This review details current chromatographic and non-chromatographic methodologies used for the purification of antibodies and expands on technological advancements and practical uses that have recently been reported.
Keywords: antibody, chromatography-based purification, technology advancements, non-chromatographic purification, advances in purification technology, separation techniques
Corrigendum for this paper has been published
Introduction
The use of antibodies as therapeutics in diagnostics and in biological sensors has revolutionized modern medicine, laboratory-based science, and environmental and “in situ” monitoring. Monoclonal antibodies (mAbs), polyclonal antibodies, recombinant antibodies, and antibody fragments all have their niche uses, requiring different purification techniques. The current capabilities of antibody production by recombinant techniques has increased the need for purification on a larger scale (up to 100 kg batches),1 thus requiring innovative purification methodologies. To achieve current industry standards for antibody purification, affinity chromatographic bioaffinity ligands are commonly used. However, at present, there is a movement away from the use of bioaffinity ligands such as Protein A, because of a number of limiting factors such as the leakage of Protein A from the column with associated product contamination, high cost, low reusability of resin, and putative antibody aggregation during low pH elution.2–4
Purification can alternatively be achieved through the use of genetically fused purification tags, the most common of which is a polyhistidine tag (poly-His or His6) that is utilized in immobilized metal affinity chromatography (IMAC). The production of a poly-His tag exerts a low metabolic burden on the producing host; however, it is not as specific as affinity chromatography.5
Ion-exchange chromatography using a multicolumn countercurrent solvent gradient purification (MCSGP) system has been described as an alternative to Protein A-based chromatography.6 This system recycles partially purified fractions, therefore increasing the protein yield, and by using a simulation model and experimental setup, it was shown that ion-exchange MCSGP can achieve industrial levels of antibody yield with high purity.
To achieve lower cost and more efficient antibody purification on an industrial scale, alternative purification methods have been developed, including refinement and advancements in traditionally used methods. The production of antibodies can be divided into up- and downstream processes. This review will focus on recent innovative advances in the downstream purification of antibodies by addressing both chromatographic and non-chromatographic methods, whereby the production of active antibodies free from harmful biological or chemical contaminants is paramount.
Antibodies come in many shapes and sizes
Rodney Porter and Gerald M Edelman first elucidated the characteristic Y-shaped structure of antibodies. In 1972, they were awarded the Nobel Prize for Medicine and Physiology for their findings. These Y-shaped molecules were eventually identified as immunoglobulin G (IgG), the structure of which is composed of four polypeptide chains – two heavy (50 kDa) and two light chains (25 kDa) – linked by noncovalent bonds and disulfide bridges (Figure 1). The main functional antibody components are the antigen-binding (Fab) and the fragment crystallizable (Fc) regions. The antibody-binding region is composed of variable heavy (VH) and variable light (VL) regions, and the Fc portion (important for immune signaling and effector functions) is composed of the constant heavy (CH1, CH2, and CH3) regions. Today, naturally occurring immunoglobulin of all shapes and sizes have been discovered in mammals (IgG, IgE, IgA, IgD, and IgM), and numerous other formats occur throughout the animal kingdom (eg, IgY from birds, single-domain antibodies from camelids, and heavy-chain-only antibodies from cartilaginous fish).7 Apart from naturally occurring antibody formats, combinatorial deoxyribonucleic acid (DNA) technology has facilitated the development of recombinant antibodies such as fragment variable (Fv), disulfide-stabilized Fv antibody fragment, single-chain fragment variable (scFv), fragment antigen-binding (Fab), single-chain antibody fragment, divalent antibody formats such as minibody, diabody, F(ab′)2 and (scFv)2, and multivalent forms such as tetrabodies, triomabs, triabodies, and F(ab)3.8,9
 | Figure 1 Structural schematic of IgG. |
Overview of conventional methods
Selective antibody purification is commonly carried out using affinity chromatography or affinity-tag-based chromatography. It achieves high selectivity and purity; however, it becomes costly when utilized on a large-scale. In order to reduce cost, physicochemical separation methods are employed prior to affinity chromatography to remove larger contaminants and prevent column clogging or fouling. An overview of both methods is given in the following sections.
Affinity chromatography
MAbs are commonly used as therapeutics in the treatment of cancer, autoimmune disorders, and inflammatory diseases; these entities require the highest degree of purification before they are suitable for patient administration. MAbs are batch-purified from mammalian cells such as Chinese hamster ovary (CHO) cells. The removal of impurities such as endogenous viruses (used during antibody production), DNA, host cell proteins (HCPs), endotoxins, and aggregates is highly important as they could potentially activate the immune system of the patient. Removal can be achieved by a number of processes, the most popular of which are combinatorial approaches; for example, utilizing Protein-A affinity chromatography followed by anion-exchange chromatography.10
Affinity chromatography has been the predominant method of antibody purification because of its high selectivity. Many forms of affinity chromatography have been exploited; however, one of the most commonly used forms is affinity chromatography using bacterially derived receptors such as staphylococcal Protein-A (SpA), streptococcal Protein-G, Protein-L from Peptostreptococcus magnus and Protein-M from Mycoplasma (Table 1).11,12 Protein-A and -G are multispecific, binding the region of immunoglobulin-connecting CH2 and CH3.13 Protein-A chromatography is the method of choice for the capture and purification of therapeutic IgG and Fc-fusion proteins on an industrial scale.14 Native Protein-A (42 kDa) is a single polypeptide chain that is composed of five Fc binding domains (known as E, D, A, B, C, and finally X, a cell wall–binding domain).15–17 A hydrophobic reaction binds Protein-A and the Fc region of IgG, but this interaction is weakened at low pH, thus allowing elution of IgG.14 However, the combination of high protein concentration and the elution of Fc-fusion molecules or IgG at low pH can destabilize the protein or cause loss of function and can cause the formation of aggregate species, which are potentially harmful (immunogenic or toxic) if administered to a patient.18 Their presence also increases production costs and loss of effective material.19,20
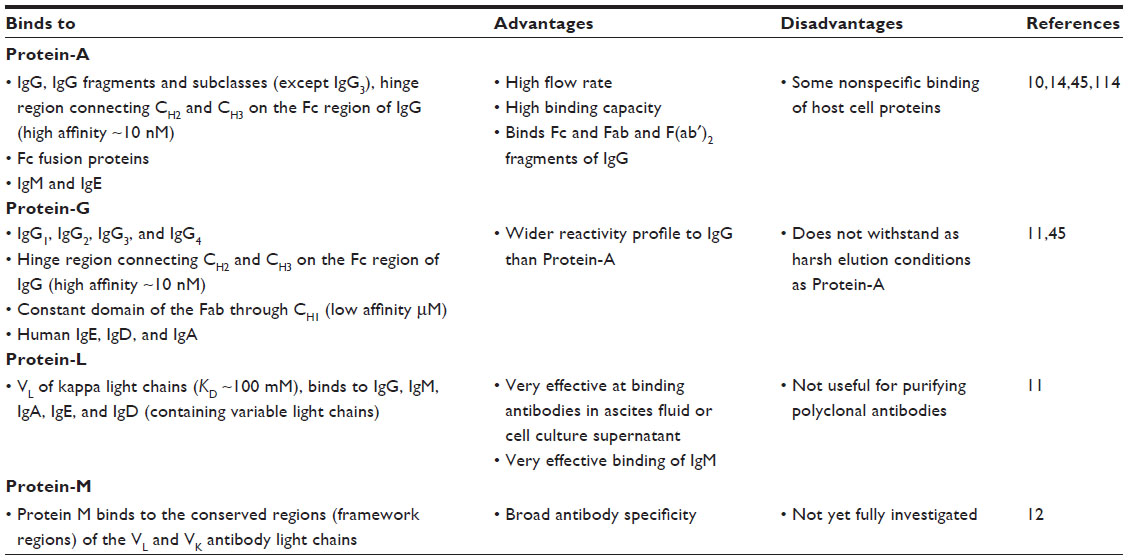 | Table 1 Overview of bioaffinity ligands Protein-A, -G, -L, and -M |
Shukla et al14 examined the binding of Fc-fusion proteins to Protein-A under various elution conditions and concluded that destabilization of the protein’s structure during interactions between Fc and Protein-A is the likely cause of increased protein aggregation, which can be reduced by adding urea (<2 M) and performing the purification at low temperatures. This was supported by a recent study by Nicoud et al,21 who carried out a kinetic study on the aggregation of IgG molecules. They found that in addition to electrostatic repulsion, van der Waals attraction forces, hydration, and depletion forces, aggregation of mAbs was also dependent on heterogeneity of the protein surface, glycosylation, and the conformational stability of the antibodies. While some researchers have focused on the prevention of aggregate formation, others have investigated the “disaggregation” or the disassociation of aggregates.18 Xu et al18 found that temperature and pH had little impact on disaggregation, whereas lowering protein concentrations did. Unfortunately, although lowering of the protein concentration promoted disaggregation, it had the negative effect of increasing sample volumes and therefore increasing production costs.
Of equal importance to the enhancement of the affinity ligand is the development of resins used as the surface matrix. McCaw et al22 evaluated a novel methacrylate-based Protein-A resin called Amsphere (Amsphere London, UK) and compared it to the industry-standard Protein-A resins of MabSelect and MabSelect SuRe (both from GE Healthcare, Little Chalfont, UK). The Amsphere resin was specifically optimized for high IgG binding and fast mass transfer, while the accompanying Protein-A ligand was optimized for enhanced alkaline tolerance. The study found that overall, Amsphere performed similarly to MabSelect and MabSelect SuRe when tested for binding capacity and the removal of HCP impurities, although the dynamic binding capacity (DBC) of the Amsphere was lower than MabSelect resin. The authors surmised that although Amsphere may not replace current industry standards, it may be substituted during the preparative process without the need for the replacement of columns, a cycling strategy, or changes to flow rate.22 The two resins were also compared in an evaluation study of the CaptureSMB system (ChromaCon(R) AG, Zurich, Switzerland). This system utilizes affinity chromatography instead of ion-exchange in MCSGP.23 In this system, Amsphere resin showed a more stable DBC at flow rates higher than 300 cm h−1 compared to MabSelect SuRe. It was also calculated that the annual cost of Amsphere resin used in small-scale proof-of-concept experiments using the CaptureSMB system was approximately 25% less than that of MabSelect SuRe resin. However, when scaled up to commercial size, the cost difference became negligible, as more Amsphere resin was required per harvest.23 Such differences in the Amsphere resin performance between the two studies could be due to the variations in the experimental conditions and that DBC values were measured at 10% breakthrough in the study reported by McCaw et al,22 which can contribute to product losses.24 Overall, the authors showed that a multicolumn chromatography system such as CaptureSMB could save up to 30% of production cost compared to standard batch chromatography.
Affinity tags are highly useful for the purification of recombinant antibodies. They can be genetically encoded and are expressed as fusion partners along with the protein of interest. They can exist as either single amino acids, short polypeptide chains, or whole proteins.25,26 In addition to facilitating identification, affinity tags can also confer enhanced protein stability, increased expression, and facilitate native folding.27,28 Disadvantages of affinity tags include their requirement of expensive resins as display supports, in addition to possible affinity tag removal.29,30
N-terminal glutathione S-transferase (GST) is also commonly used as an affinity tag for the purification of recombinant antibodies. GST is a monomeric protein (26 kDa) originally identified in Schistosoma japonicum.31 The pGEX Escherichia coli expression vector from GE Healthcare encodes an N-terminal GST followed by a protease cleavage site. GST-fusion proteins can be purified by affinity chromatography using commercially available glutathione (γ-glutamylcysteinylglycine) sepharose as the resin-bound ligand. Elution conditions are mild, as reduced glutathione can be used in a competitive elution scheme. The purification of highly concentrated GST-fused proteins can result in the expression of aggregated proteins in inclusion bodies, which may or may not be agreeable for the chosen application.32 In addition, high-molecular-weight (>100 kDa) fusions can result in partially or fully insoluble aggregates.25,33
Epitope tags are better known for their role as antigenic determinants; however, short amino acid sequences such as poly-His (HHHHHH), C-myc (EQKLISEEDL), FLAG (DYKDDDDK), hemagglutinin (HA) (YPYDVPDYA), T7 (MASMTGGQQMG), and Glu-Glu (EYMPME) can also be used for the purification of recombinant antibodies.28,34 These short sequences are added to the end of an antibody sequence by polymerase chain reaction-based methods. Poly-His is undoubtedly the most ubiquitously used affinity tag for the purification of recombinant antibodies. Histidine forms a complex with transition metal ions such as Co2+, Ni2+, and Cu2+ (among others). The binding is pH-dependent, and this can be exploited for elution of the His-tagged protein in IMAC-based purification.32 Although poly-His-tag-based purification of proteins in E. coli can produce protein of up to 80% purity, a higher percentage of histidine residues in insect and mammalian cells can lead to a significant increase in background contaminants.32 By virtue of their smaller size, epitope affinity tags can be more favorable for protein purification as they exert less steric hindrance than their larger counterparts. The use of combination-tag affinity chromatography involves the expression of two affinity tags in tandem (tandem affinity purification), thus combining a solubility-enhancing tag and a purification tag (eg, maltose binding protein combined with poly-His). The tags are usually in frame, with a protease cleavage site between them.28
Physicochemical fractionation: precipitation and membrane filtration
During the primary recovery of antibody, it is important to minimize turbidity and facilitate impurity removal. Turbidity, arising due to the presence of cell debris during the preparation of tissue or cell lysates, must be removed before adding the solution to a chromatographic column as it may block/clog or foul the bed.
The physical methods of primary antibody recovery such as centrifugation and filtration are routinely performed at the beginning of the purification process. Purification by centrifugation involves the separation of particles based on their mass under centrifugal force. Centrifugation facilitates separation of the antibody-containing supernatant from the cells and cellular debris; however, parameters such as g-force, residence time, and discharge frequency can influence clarification efficiency and can vary greatly when moving from small-scale to large-scale centrifugation.35
Membrane filtration, such as microfiltration, ultrafiltration, and depth filtration allow separation of the small molecules by size, while charged ultrafiltration can separate proteins by both charge and size. For example, cellulose-based ultrafiltration membranes have been used to separate different therapeutic mAbs from CHO cells.1,35,36 Microfiltration membrane chromatography removes biological entities (DNA, viruses, and protein contaminants) from fluids using a microporous membrane filter, examples of which include polypropylene or polyester. The membranes are stacked ten to 15 layers deep and are packed into a small disposable cartridge. Sartorius Stedim Biotech (Concord, CA, USA) provides disposable capsules of filter membranes (Sartobind®, Sartorius AG, Göttingen, Germany), composed of either hydrophobic interaction chromatography (HIC) membranes or strong anion-exchange membranes. Ghosh and Wang37 utilized stacked microporous HIC polyvinylidine fluoride (PVDF) membrane chromatography (pore size 0.1 μm) to purify humanized mAb (hIgG1-CD4) and gave particular attention to the removal of bovine serum albumin (BSA) (which is the main protein present in the medium used during mammalian cell culture). The CHO cells produced antibody at concentrations less than 0.5 mg mL−1, and membrane chromatography was an attractive purification route to choose.37 The authors carried out the purification in the presence of concentration-optimized ammonium sulfate (1.5 M) (a precipitation agent) and produced a mAb at 97% purity. Similarly, Sadavarte et al38 reported the use of HIC membranes for purification of chimeric heavy chain mAb directly from cell culture supernatant. This technique involved the use of ten PVDF microporous membrane discs 20 mm in diameter, with the elution buffer containing 20 mM sodium phosphate buffer, pH 7, and the binding buffer consisting of an optimized concentration of ammonium sulfate salt. The authors found that the method effectively resolved monomeric antibody from antibody aggregates, as the antibody aggregates were more hydrophobic than the mAb. The authors reported that the purification yields and purity were high, but did not provide specific details.38
Depth filtration is one of the workhorses of cell medium clarification. It can be used before a complex medium is applied to a chromatography column. Depth filtration can be used when there is a high propensity for the medium to clog or plug a polishing membrane filter. Depth filters are commonly composed of cellulose fibers and filter aids (diatomaceous earth [DE]) bound together by a positively charged polymer resin. During depth filtration, the contaminant particles are retained throughout the filter as opposed to remaining at the top. Particle retention is thought to occur by a combination of size exclusion and adsorption through hydrophobic, ionic, and other interactions.35
Chemical methods for the alleviation of turbidity include flocculation, precipitation, and crystallization. Flocculation can be used to remove particulate matter of submicrometer size. It can be achieved using 30 mM calcium chloride followed by 20 mM potassium phosphate (the calcium phosphate forms a precipitate around the cellular debris).1,35 Precipitation is an effective technique used to remove antibody from its sample matrix. Antibody precipitation can be achieved using ammonium sulfate, octonic acid, or polyethylene glycol (PEG).39,40 Crystallization is a highly useful tool as it may aid concentration, purification, and stabilization of the antibody product, without the need for affinity tags or affinity tag removal.1,41,42 Advancements in these methodologies will be discussed in greater detail in the “Non-chromatographic methods” section.
Advances in chromatography-based purification technologies
Chromatographic methods may involve the reversible adsorption of a target molecule onto a solid support. They are often the method of choice for antibody processing and more specifically, affinity-based methods are routinely used in the biopharmaceutical industry owing to the high selectivity and purity that can be achieved to meet regulatory and quality-control standards.9 Innovations to improve the traditional methods such as biological-ligand-based methods are consistently investigated, while the use of complementary chromatographic techniques are now starting to achieve purities to match affinity-based techniques.
As previously mentioned, MCSGP is a relatively new technology that was shown to significantly reduce the cost of antibody production, by reducing the volume of the resin utilized and by increasing the loading and binding capacity of the resin.23 The countercurrent movement of the liquid and the stationary phase, called simultaneous moving bed (SMB), is the basic principle in various continuous and semicontinuous countercurrent sequential loading systems that can utilize up to 12 columns simultaneously.23,43,44 The smaller columns used in these systems reduce affinity resin and buffer consumption by 40%–50%, while sequential loading of the columns with the flow-through from the previous column ensures maximum product capture.23,43 Also, having all the columns in operation at any given point of time, albeit at different stages of the process, significantly reduces production times.
Improvements in Protein-A/-G-based chromatography
Protein-G chromatography is well known for its ability to bind the Fc region of IgG with high affinity (Table 1). However, it binds to the Fab region with much lower affinity (low μM affinity). To enable purification of both the Fc and Fab in a single-step process, Bailey et al44 employed a combinatorial phage display approach to create a Protein-G mutant expressing eight amino acid changes that displayed a 100-fold increase in affinity for Fab, and subsequently, employed a rational approach to create a Protein-G with a pH switch (when moving from pH 7.4 to 4, induces a KD change of 1,000-fold) that could be advantageously used during Fab purification. The need for milder IgG elution conditions during Protein-A chromatography was facilitated by Pabst et al,46 who engineered a novel SpA ligand that incorporated a double mutant (histidine to serine [H18S] and an asparagine to alanine [N28A]) in the synthetic Z domain (formerly the B domain of the native protein) of SpA. The binding and elution capacity of the SpA containing the double mutant was tested against six antibodies and one Fc-fusion protein and displayed DBC and selectivity similar to that of the parental ligand. It successfully facilitated the elution of IgG from SpA under milder conditions (>0.5 pH units), thus enabling the elution of proteins that are sensitive to aggregation under acidic conditions.46
Novel tags and self-cleaving tags
The FLAG tag (DYKDDDDK) is a hydrophilic peptide that is commonly used for the purification of recombinant mAbs. In the presence of calcium, Einhauer and Jungbauer47 described how an antibody (denoted here as M1) recognizes and binds FLAG-tagged recombinant antibody, which can be subsequently eluted under mild conditions, using metal chelators such as ethylenediaminetetraacetic acid (EDTA) or ethylene glycol tetraacetic acid.25 In addition, they describe an anti-FLAG antibody (M2) that binds and elutes FLAG-tagged antibodies in a pH-dependent manner. In 2001, Zhang et al48 used a triple FLAG-epitope (3XFLAG tags in a row) to significantly enhance the sensitivity and detection of a mammalian expressed fusion protein using the anti-FLAG antibody M2. The triple FLAG-epitope has yet to be used for the purification of recombinant antibodies, and in the future it will be interesting to see if it will be of benefit. A further advantage of using FLAG is the presence of an inherent enterokinase cleavage site, which may prove advantageous for the purification of FLAG-tagged therapeutic antibodies.28
In some cases, the removal of the carrier protein or tag may be necessary, for example, during structural or functional studies, if the antibody is to be used as a therapeutic or if the tag interferes with the biological activity. Unfortunately, tag removal introduces another step, thus further increasing production costs. Tags can be removed by two methods – first, by using harsh chemicals such as cyanogen bromide or hydroxylamine.25 However, these harsh conditions may have adverse effects on the structure and/or consequently on biological activity. Secondly, a more elegant method of tag removal is the use of enzymes such as endoproteases. They do not require the same harsh regimens as they are more selective. However, the high cost of proteases and their inability to work in certain buffers limit both their widespread use and their applicability to large-scale protein production.30 Recently, a new method of tag removal has emerged. Self-cleaving tags can facilitate purification of target protein and cleavage of the fusion protein in one step. Site-specific cleavage is initiated by either low-molecular-weight compounds or by a conformational change in the fusion protein.49 Examples of self-cleaving tags include intein, sortase-A, N-terminal protease, Fc-regulated protein C (FrpC), and cysteine protease domain.30,50 Inteins are self-splicing proteins that are capable of excising themselves from a translated protein. Depending on the intein used, cleavage can be achieved using pH-dependent cleavage or thio-induced N- and C-terminal cleavage.30 The intein cleavage tag was used many times for protein purification; however, its use has been confined to laboratory scale, mainly because the intein tag does not promote protein solubility or protein expression but merely acts as an affinity tag.49 In addition, it is thought that the intein tag may cause uncontrolled cleavage, thus lowering protein yield.49 In the “Thermoresponsive elastin-like polypeptide tag” section of this review, a description of the use of an intein self-cleaving tag for the isolation of target protein in a temperature-dependent purification using elastin-like protein (ELP) is given.29 FrpC is an alternative self-cleavage tag from Neisseria meningitidis that is capable of mediating Ca2+-dependent self-cleavage.51 While Ca2+-dependent cleavage mediates mild elution of target protein, the introduction of EDTA to remove Ca2+ introduces an additional step.
Multimodal (mixed-mode) affinity chromatography
Over the past decade, substantial attention has focused the development of improved techniques for antibody purification utilizing HIC. This approach involves the binding of proteins to chromatographic supports via hydrophobic residues/patches on their surface. The addition of salts drives the binding of the protein’s hydrophobic areas onto hydrophobic areas on the solid surface. Consequently, desorption is initiated by the reduction of the salt content. HIC is a well-established antibody polishing step post Protein-A purification; however, as yet, it has not seen widespread use due to two limiting factors.52 First, HIC resins exhibit lower binding capacities compared to other chromatographic steps (eg, Protein-A chromatography); second, the high salt content that is sometimes required for binding mAbs can result in a high salt content in the eluted product and can cause denaturation of adsorbed proteins, inducing their aggregation and precipitation.52–54 In a push to achieve higher binding capacities at lower salt concentrations, Burton and Harding55 developed hydrophobic charge induction (HCIC) mixed-mode chromatography. HCIC relates to the interaction between target protein (antibody) and an ionizable ligand (eg, 4-mercaptoethyl-pyridine [MEP]). At neutral pH, a hydrophobic interaction occurs between the antibody and the MEP ligand, thus adsorbing the antibody onto the column; desorption occurs under acidic conditions, whereupon, electrostatic (positively charged antibody and positively charged MEP ligand) repulsion elutes the antibody from the solid support.56 Multimodal or mixed-mode chromatography is a separation technique that makes use of complementary chromatographic (eg, hydrophobic, ionic, or thiophilic interactions) techniques and combines them for use in a single chromatographic process.55,57 HCIC combines hydrophobic and electrostatic interactions. Its use can reduce the number of columns needed for purification, thereby reducing overall cost and time.56,58 Lin et al58 tested the capacity of MEP to purify IgG and found that it could achieve approximately 25–35 mg mL−1, and displayed purity comparable with Protein-A chromatography.
Bio-Rad Laboratories Inc. (Hercules, CA, USA) provide two types of mixed-mode chromatographic media (ceramic hydroxyapatite) (Ca5(PO4)3OH)2 that can be used for antibody purification. Type I aids in the removal of virus particles, DNA, endotoxin, and aggregate and is suitable for Fab purification, while type II is capable of removing albumin and is also suitable for Fab purification.56
In 2010, Bresolin et al59 outlined a new method for the purification of IgG (IgG1, IgG2, IgG3, and IgG4) by negative chromatography using mixed-mode interactions (electrostatic and hydrophobic) from human serum. Negative chromatography (flow-through method) entails the addition of feed solution onto the column; the protein of interest (in this case IgG) flows through the column while the contaminants attach to the column and are removed during elution.59 In this case, Bresolin et al59 succeeded in separating polyclonal IgG from both human serum and plasma. The experiments were carried out using ω-aminodecyl-agarose as the solid phase. A separate study evaluated four different commercially available mixed-mode ligands – N-benzyl-N-methyl ethanol amine (Bestarose Diamond MMA), 2-benzamiodo-4-mercaptobutanoic acid (Bestarose Diamond MMC), 4-mercapto ethyl-pyridine (MEP HyperCel), and phenylpropylamine (PPA HyperCel) – for their ability to separate IgG from serum albumin under pH gradient elution.4 The study showed that Bestarose Diamond MMA (Diamond MMA, Northbrook, IL, USA) outperformed the other ligands, achieving an IgG purity of 99.0% and recovering 89.5%.4 The use of mixed-mode ligands facilitated the control of elution by controlling pH. Tong et al60 found that the addition of caprylate as an albumin-selective moiety improved IgG yield during HCIC. Caprylate was added to the mobile phase to reduce nonspecific binding of serum albumin to the HCIC column. The authors found that addition of caprylate enhanced separation efficiency. However, a question arises as to whether this may add another layer of complexity to the purification process as it increases time and cost.
In a study that incorporated recent innovations in antibody purification, Maria et al61 carried out a study that investigated antibody purification capabilities when using two successive mixed-mode chromatographic steps 1) using HEA HyperCel (Pall Life Science, Saint Germain en Laye, France), followed by 2) Capto MMC ImpRes ligand (GE Healthcare), and thereafter, 3) anion-exchange chromatography was used as a polishing step to remove remaining DNA and HCPs. The study also incorporated the use of membrane adsorbers (Sartobind Q75 and Sartobind STIC PA Nano from Sartorius Stedim Biotech) that have recently witnessed increased use as chromatographic matrices of choice. The three-step process was chosen, as it mirrors current purification technologies in industrial IgG production, and therefore, could be easily used as a substitute for Protein-A chromatography.61 A final mAb purity of 99.9% and yield of 88% was achieved, and impurities were removed to meet regulatory levels required for therapeutic products. In an effort to gain a greater understanding of the complexities of mixed-mode chromatography during antibody purification, Wolfe et al62 investigated the influence of various mobile phase modulators (including urea, ethylene glycol, arginine, ammonium sulfate, and sodium thiocyanate) on antibody binding and the removal of contaminants during mixed-mode chromatography using Capto MMC resin. The binding capacities and separation characteristics of four antibodies in the presence of mobile phase and mobile phase modulators were tested. The experiment examined the amount of NaCl that was required to elute each protein, thus gaining an understanding of the alterations in the antibodies’ and proteins’ binding affinities. The modulators were found to decrease the antibodies’ binding affinity for the resin by reducing electrostatic and hydrophobic interactions or both.
Synthetic ligand affinity chromatography
In an effort to reduce purification costs and move away from the use of biological ligands such as Protein-A, synthetic affinity ligands have been manufactured. To date, synthetic ligands have not achieved the same commercial success as MabSelect SuRe from GE Healthcare, a derivative of Protein-A. However, Lund et al2 have found that the synthetic ligand DAAG that exerts mixed-mode characteristics in its binding capacity for IgG achieved IgG concentrations of >48 mg mL−1, and >90% purity. In addition, the authors described the separation of aggregated IgG from nonaggregated IgG and claimed that DAAG would be a very useful polishing step.
Bresolin and Bueno63 also attempted to provide a replacement for bioaffinity ligands (Protein-A, -G, and -L) by using amino acids. A number of amino acid affinity ligands had been previously examined including histidine, arginine, tryptophan, and phenylalanine.64–68 Bresolin and Bueno63 evaluated the binding ability of an amino acid derivative orthophosphoserine (OPS) for polyclonal IgG in human serum. OPS is a negatively charged molecule, and so it can be used to purify positively charged molecules using electrostatic interactions. Untreated human serum was used as the feed solution diluted in low-ionic-strength buffer (25 mM sodium phosphate buffer [pH 6.5]) and was added to the column. A purity of almost 90% was reached upon elution of IgG with 0.4 mol L−1 NaCl and was comparable to past attempts in purifying IgG using amino acids.64–68 The mild elution conditions used have many advantages, including a reduced loss of antibody function and reduced aggregation.
Anion/cation-exchange chromatography
Ion-exchange chromatography relies on the pI value of the protein to achieve separation. In cation-exchange chromatography, a positively charged protein is attracted to a negatively charged solid support, while in anion-exchange chromatography, a negatively charged protein is attracted to a positively charged support. Elution is generally achieved by altering the salt or pH content of the buffer in a stepwise or gradient elution.69 Ion-exchange is a rapid and inexpensive method of antibody purification and is commonly used as a polishing step subsequent to Protein-A affinity chromatography. In addition, for the purification of antibody fragments that do not possess Fc domains, Protein-A chromatography is redundant; hence, ion-exchange chromatography is commonly used. Recently, ion-exchange-based methods using membrane technology have become popular, and it is thought that they may soon replace anion-exchange resins.70 In another study, Hall et al71 employed cation-exchange chromatography for the resolution of a bispecific antibody and the removal of a misformed mAb-diabody species as well as aggregate products. Furthermore, Müller-Späth et al6 showed that ion-exchange chromatography could be used to fully substitute a costly Protein-A resin for mAb purification. Using a lumped kinetic model and experimental setup, they showed that a productivity of 25 g mAb L−1 h−1 with up to 97% purity could be achieved using cation-exchange chromatography in a MCSGP system.6
Monolith chromatography
Chromatographic monoliths were first envisaged by Nobel Prize winner Richard Synge, who in 1952 imagined a method of separating protein from particulates using a “continuous block of porous gel”. In chromatographic terms, a monolith is defined as a porous, single-unit material incorporated into a chromatographic device.72,73 Manufacturers and analytical laboratories have been interested in the use of monolithic supports owing to their associated low cost, high porosity, and ease of use.72 Since its discovery, a number of different materials, including organic monoliths, silica monoliths, agarose monoliths, and cryogels have been tested for utilization in affinity monolith chromatography, and some of the materials have been tested for antibody purification.74 Glycidyl methacrylate (GMA) and ethylene glycol dimethacrylate have been used for affinity (using ligands Protein-A, -L, and -G and poly-His) and ion-exchange purification of antibodies. IgG, IgM, IgA, and antibody fragments have all been purified in varying purities using monolithic supports.72,75 Feng et al76 used immobilized lectin (Concanavalin A) monolithic chromatography to successfully purify glycoproteins using an IMAC-based mechanism. A comprehensive review of monolithic supports used for antibody purification was recently published by Barroso et al.72
Immunoaffinity-based chromatography
Immunoaffinity-based chromatography (IAC) is a highly selective approach used to separate target components from their matrix using specific antibodies.77 It is commonly used in the isolation and extraction of fungal toxins (mycotoxins) from contaminated foodstuff.78 IgG or IgG-derived fragments are most commonly utilized as the resin-bound ligand during IAC; however, different immunoglobulin formats have also been used for various purification procedures. However, it should be noted that conditions used for elution need to be carefully tested. When McMahon and O’Kennedy79 investigated the binding of monoclonal IgM antibodies to their associated antigens, the use of low pH for elution affected the specificity of some antibodies and these findings have been widely confirmed.
The emergence of recombinant antibody development through phage display has led to the discovery of high-affinity antibodies in various formats that are highly conducive to act as ligands in IAC.77,80,81 However, due to the highly specific nature of the antibodies’ binding ability, elution can be a problem, and often the temporary denaturation of the binding ligand (antibody) and/or target protein is required.82 However, as mentioned previously in this review, elution could be Ca2+-dependent if using a CaM-based system.82 In a novel use of CaM, Kobatake et al82 fused the heavy and light chain of an anti-hen egg lysozyme (HEL) scFv (antibody heavy and light chain only) to the N-terminal and C-terminal of CaM. In the absence of Ca2+, the VH and VL chains of the scFv were separated and could not bind HEL; however, when Ca2+ was added, the conformation of the CaM changed, thus bringing the VH and VL chains into proximity, whereupon they could bind HEL. The experiments displayed how an antibody construct could be generated to respond to Ca2+, thus providing a mild elution method. This method was used to purify HEL; however, this same method could be used in the future to purify antibody constructs.
Hexa- and octapeptide ligand-based chromatography
The purification of IgM has become more widely investigated owing to its potential future role in anticancer treatment, and hexameric peptide HWRGWV was identified by Liu et al83 to exhibit IgM binding capacity. IgM is a large protein (pentameric form is 950 kDa) whose purification has had little success using traditional methods such as Protein-A chromatography. Liu et al83 previously identified HWRGWV through a combinatorial library screening and found that it displayed good affinity toward the Fc domain of immunoglobulin, and on further investigation, they found that it also bound well to IgM displaying 86% purity and 75.7% recovery from complete minimum essential medium. The method of using dual-affinity octapeptide ligand systems was found to be beneficial for the purification of human polyclonal IgG and mAb by Zhao et al.84 They identified three IgG binding octapeptides 1) FYWHCLDE, 2) FYCHWALE, and 3) FYCHTIDE by using a biomimetic design strategy. They found that all three bound IgG through its Fc domain. However, the use of a combination of peptides (1) and (3) mediated a synergistic effect, increasing the purification capacity while achieving 95% pure hIgG with a recovery of over 90%.84
Chromatography-based membrane filtration
Filter membranes have not yet achieved widespread application for the purification of antibodies as they have not yet reached the same binding capacity as their bead-based counterparts.85 However, membrane filtration is among some of the alternative methods that have been investigated in order to avoid the high costs associated with the purification of antibodies on a large-scale using Protein-A chromatographic methods. Membrane filtration also addresses issues related to use of traditional chromatographic column methods such as flow distribution, limitations to flow rates, and problems associated with cleaning in situ.61 Boi et al85 tested a novel Protein-A affinity membrane (from Sartorius Stedim Biotech) for its binding capacity to IgG. Affinity membrane chromatography differs from the traditional resin-based methods in that the active area of the ligand is more accessible to the antibody (as the ligand is not required to be tethered to a resin or bead). The target proteins move through the membrane primarily by convective transport which brings them into very close proximity with the ligands. Hence, the diffusion limitations associated with bead-based chromatography are avoided.85–87 Membrane filtration techniques can also withstand higher flow-through rates, thus significantly reducing processing time. A new high-capacity affinity membrane was tested and compared to commercially available Sartobind Protein-A membranes. The novel membrane was composed of a regenerated cellulose microporous matrix immobilized with recombinant Protein-A, and it displayed significantly improved binding capacity when compared to its commercially available counterpart.85,88
Use of hydroxyapatite mixed matrix membranes
Hydroxyapatite (HA) is a naturally occurring mixed-mode material that is commonly used for the purification of antibodies.89 HA (Ca10(PO4)6(OH)2) is a ceramic, crystal-forming inorganic material. It is composed of functional groups of positively charged pairs of calcium ions and clusters of negatively charged phosphate groups.90 It is essentially made up of the same materials that constitute bones and teeth, and so it is of “biosimilar” composition and is highly biocompatible.91 The advantages of HA are that it is cheap, and biocompatible solid phases are available; cleaning in situ is feasible and it demonstrates good stability under alkaline conditions.91,92 It can be used in conjunction with chromatographic methods, such as Protein-A affinity chromatography, to achieve high levels of purity. HA chromatography is very well suited for IgA and IgM purification, and this is thought to be as a result of their multimeric structure.89,91
Advances in non-chromatographic methods
Non-chromatographic purification methods can be affinity-tag-based or nonaffinity-tag-based. Non-tag-based methods rely on physicochemical attributes of the target protein and the surrounding environment. Non-chromatographic methods have been much sought after owing to their potential use in large-scale purification platforms. However, first, they must overcome the low purities that have historically been associated with these methods.25
Thermoresponsive elastin-like polypeptide tag
The elastin-like polypeptide (ELP) tag facilitates the purification of ELP-fused proteins without the use of chromatography. The ELP tag is an artificial polymer of five repeating peptides. Upon increasing salt concentration or temperature, the ELP tag induces the reversible aggregation of itself and the fusion protein.93,94 The fusion protein can then be removed by centrifugation and resolubilized in suitable buffer at a lower temperature. However, for next-stage structural or biochemical analysis, the removal of the ELP tag may be necessary. Lan et al93 successfully developed a novel strategy by incorporating a protease to facilitate the removal of the ELP tag without the need for chromatographic techniques. The use of ELP as a fusion partner is still a work in progress, as the ELP fusion proteins have reduced overall expression and cause reduced cell growth.29,94–96 However, in 2012, Liu et al29 developed an approach that incorporated a mini intein self-cleavage domain (that was fused to the target protein) to facilitate affinity tag removal, along with separately expressed ELP that contained a capture domain for the affinity tag (Figure 2). The affinity pair that was used in this purification technique was a naturally occurring cohesion-dockerin (Coh-Doc) from Clostridium thermocellum that mediates Ca2+-dependent binding.97 In essence, this purification technique incorporated temperature-dependent precipitation by ELP, pH-dependent cleavage of intein for the removal of the affinity tag, and finally, Ca2+-dependent binding of the affinity tag to the captured protein (Figure 2).
 | Figure 2 Non-chromatographic purification of target protein, incorporating pH-dependent intein cleavage, Ca2+-dependent affinity binding, and temperature-dependent purification by ELP aggregation. |
Aqueous two-phase partitioning
Aqueous two-phase partitioning systems (ATPS) have been widely used as a mild method of protein separation.98 ATPS involves the formation of two immiscible liquid phases, which can be achieved when polymers such as PEG are mixed with dextran or other polymers or salts (eg, phosphate, sulfate, or citrate) under particular conditions.98–100 The partitioning property of a protein depends on its surface properties (charge, hydrophilicity, and hydrophobicity) and on the physicochemical properties of the two liquid phases.98 ATPS has increased in popularity owing to its associated mild purification capabilities, and in addition, associated denaturation and loss of biological activity are rare.100 Furthermore, the polymers used can promote protein stability, and the ability to separate the protein of interest in one step is highly attractive.100 The two phases have different hydrophobic properties and hence, cellular debris, particulate matter, and contaminants such as BSA and HCPs tend to move to the lower, more polar phase, while the protein of interest moves to the upper, less polar layer, which is more hydrophobic.99,100 A successful example of antibody purification by ATPS was achieved by Mao et al101 using an aqueous-citrate two-phase system. A design of experiment (DOE) screening strategy was used to identify the percentages of PEG, citrate, and NaCl required to achieve optimal separation. The system consisted of 14% (w/v) PEG, 8% (w/w) citrate, and 7.2% (w/w) NaCl solution at pH 7.2 and resulted in a recovery of 89% of mAb in the upper phase and an overall purity of 70%, achieving a 7.6-fold reduction in HCPs. The authors found that although the ATPS technique alone produced a modest clearance of HCP, in combination with Protein-A chromatography and size-exclusion chromatography, a purity of 99.5% was achieved.101 In addition, Platis and Labrou102 applied the same technique for the isolation of anti-human immunodeficiency virus (HIV) mAbs 2G12 and 4E10 from a transgenic tobacco crude extract. In a similar fashion to the previous ATPS example, Platis and Labrou102 observed phase partitioning of plant debris/mAb using 13.1 (w/w) PEG 1,500 (kDa), 12.5% (w/w) phosphate at pH 5. Both antibodies transitioned to the lower phosphate phase and were purified with 85% and 84% purity, respectively. The authors found that the use of the ATPS protocol was highly useful for initial protein recovery and partial purification of mAb before proceeding to Protein-A affinity chromatography. Wu et al103 reported the use of ATPS for the purification of mAbs in cell culture supernatant. The authors found that using the salt/polymer method of ATPS (which involved increasing the concentration of NaCl) caused the formation of IgG precipitants, an undesirable end product contaminant.
Examples of antibody purification using ATPS have reported antibody yields from 88% to 97%.101,103,104 Cao et al104 investigated the purification of monoclonal anti-clenbuterol (CL) antibody using ATPS with differing PEG concentrations, pH values, and NaCl concentrations. Optimum conditions achieved antibody recovery yields of 87.86%. Interestingly, Cao et al104 introduced polystyrene microspheres (PSMSs) into the PEG phase to recover the antibodies. The utilization of the PSMSs saw the retention of 95% antibody activity compared to 25% activity retention without microspheres. Efforts are continuously ongoing to optimize ATPS purification efficiencies because of the associated low cost and biocompatibility of ATPS. Its ease of use and scale-up potential is ensuring continued attention in experimental research and in industrial settings.
Flocculation and depth filtration
Flocculation is a method in which dispersed particles clump together. The clumped particles are then removed either by sedimentation, filtration, or centrifugation, thereby clarifying the medium and reducing turbidity, in preparation for subsequent product removal by chromatography (or alternative methods) thus avoiding column fouling or blocking.105 Depth filtration is emerging as a highly suitable method for target protein purification following flocculation, because of the availability of disposable cassettes, thus removing the need for cleaning and avoiding cross contamination.105–107 Flocculation is highly useful for the removal of cell debris, HCPs, and DNA. Flocculation can be induced by charged flocculation polymers (or flocculants) (Figure 3). Flocculants act to bridge dispersed particles. They bind to oppositely charged particles, but the length of the polymer must extend beyond the length of the particle so as to bridge more particles. The process continues with the binding of the next polymer, and until all of the particles have clumped together (Figure 3). Flocculation can be influenced by many factors, including shear rate, pH and conductivity of the medium, size, surface charge and concentration of the suspended particles, and the charge, length, concentration and molecular mass of the polymer.108–110 The recovery of target protein must also be tested to ensure that it is does not succumb to flocculation. Following flocculation, the larger particles can be removed by inexpensive methods of purification, for example, bag filters,108 leaving the removal of the antibody of interest by more selective methods such as depth filtration or affinity chromatography.
 | Figure 3 Schematic overview of the process of flocculation. |
Kang et al111 developed a method of flocculation during antibody purification using a novel stimulus responsive polymer called benzylated poly(allylamine) followed by depth filtration. Buyel and Fischer105 achieved the clarification of media expressing HIV-neutralizing mAb (2G12) and fluorescent marker protein (DsRed), both transgenically expressed in tobacco leaves. They tested 23 different flocculants in a range of buffers and varying pHs as well as optimized temperatures and incubation times. They found that Polymin P (BASF, Ludwigshafen, Germany), a branched cationic polyethylenimine, produced the greatest reduction in turbidity. The authors showed that Polymin P demonstrated the greatest compatibility with plant tissue homogenates and therefore may be a suitable matrix for the production of other plant-derived biopharmaceuticals in the future. Concurrently, the authors demonstrated that flocculation is a cost-effective method for the clarification of medium before commencing finer target protein purification techniques such as chromatography or depth filtration.
Depth filtration, as previously mentioned, is routinely used as a secondary clarification method or for affinity purification. However, depth filtration was tested by Singh et al112 as a method for primary recovery. The authors suggest that it is a method that could be used to purify recombinant proteins that are currently being produced in high titer numbers in mammalian cells.111 By optimizing particle size and pore size and testing various pretreatment conditions, Singh et al112 have designed new and improved “ClarisolveTM” (Merck Millipore, Billerica, MA, USA) depth filters that could be used for primary clarification of flocculated material or as disposable filters for single-step filtration.111
Crystallization
Smejkal et al41 recently purified a therapeutic full-length mAb using a crystallization-based process. A purification yield of 88%–90% was obtained with 98% purity. The mAb was produced in CHO cells and secreted into complex sample medium. The mAb solution was dialyzed against 10 mM histidine/acetate buffer, pH 5.0, to remove HCP, peptides, DNA, RNA, and salts, the presence of which may interfere with the crystallization process. The crystallization was initiated, a purity of 92.9% was achieved for the first round of crystallization, and 98.5% purity was achieved after a second round of crystallization (or recrystallization). The authors reported that they achieved the same level of purification that was previously achieved using the same antibody and purifying it by Protein-A chromatography. The crystallization method was also successfully scaled-up from 6 mL to 1 L and was found to be reproducible.41
Use of copolymers for antibody precipitation
Antibody purification by precipitation is an accepted method of crude antibody purification (eg, using ammonium sulfate or PEG precipitation). However, to produce antibodies of higher purity using a method that may be suitable for large-scale production, customized copolymers composed of 2-acrylamido-2-methylpropane sulfonic acid (AMPS) and 4-(acryloylamino)benzoic acid (ABZ) were tested for their antibody precipitation abilities.113 AMPS–ABZ copolymers facilitated mAb precipitation yields of up to 90%–100% using a model mAb. It was found that increasing the polymer length caused an increase in antibody precipitation, but it also caused the increased precipitation of the model impurity protein, BSA. Therefore, a balance must be sought. Subsequently, a separate study tested the capacity of copolymers of AMPS and ABZ to purify a therapeutic antibody.113 The study found therapeutic mAb recovery of >90%, with >80% clearance of HCP. Precipitation did not negatively affect mAb binding capacity, and the precipitated mAb was easily redissolved in a small buffer volume. Unfortunately, the copolymers did not achieve the same level of purity that can be obtained by Protein-A chromatography (which displayed 93% purity in this study). However, due to the high level of mAb recovered, in the future, it may become an attractive choice owing to its ease of up-scaling and low cost.113
Conclusion
Currently, the most cost-effective and practical strategy for antibody cleanup seems to be initial separation of larger contaminants using physiochemical approaches including aqueous two-phase separations, followed by a finer, more specific separation involving chromatography. The new advances in Protein-A affinity chromatography, such as use of improved resins like Amsphere and MabSelect and optimization of MCSGP technology, may assist in reducing the costs of antibody purification by reducing the amount of resin, buffers and time required. However, to perform the most efficient purification possible it is vital to have good knowledge of the structure of the antibody (or antibody derived structures) and its cognate antigen and ideally the affinity of their interaction.
An ideal scenario would be to produce a generic platform for antibody purification. This would require less time and less expense for validation. However, this approach may be fundamentally unfeasible or prove to be quite a significant challenge due to inherent antibody diversity and the ongoing generation of even more complex antibody-based derivatives.
Acknowledgments
This material was produced with the support of Beaufort Marine Research Award, which is carried out under the Sea Change Strategy and the Strategy for Science and Technology and Innovation (2006–2013), with the support of the Marine Institute, funded under the Marine Research Sub-Program of the National Development Plan 2007–2013. We would also like to thank the Biomedical Diagnostics Institute, Dublin City University, and Science foundation Ireland for their financial support, through grants (No 10/CE/B1821 and 08/US/I1512).
Disclosure
The authors report no conflicts of interest in this work.
References
Thommes J, Etzel M. Alternatives to chromatographic separations. Biotechnol Prog. 2007;23:42–45. | |
Lund LN, Gustavsson PE, Michael R, et al. Novel peptide ligand with high binding capacity for antibody purification. J Chromatogr A. 2012;1225:158–167. | |
Li R, Dowd V, Stewart DJ, Burton SJ, Lowe CR. Design, synthesis, and application of a Protein A mimetic. Nat Biotechnol. 1998; 16:190–195. | |
Wang RZ, Lin DQ, Tong HF, Lu HL, Yao SJ. Evaluation of mixed-mode chromatographic resins for separating IgG from serum albumin containing feedstock. J Chromatogr B Analyt Technol Biomed Life Sci. 2013;936:33–41. | |
Nilvebrant J, Alm T, Hober S. Orthogonal protein purification facilitated by a small bispecific affinity tag. J Vis Exp. 2012;59:e3370:1–6. | |
Muller-Spath T, Ströhlein G, Aumann L, et al. Model simulation and experimental verification of a cation-exchange IgG capture step in batch and continuous chromatography. J Chromatogr A. 2011;1218:5195–5204. | |
Feige M, Gräwert MA, Marcinowski M, et al. The structural analysis of shark IgNAR antibodies reveals evolutionary principles of immunoglobulins. Proc Natl Acad Sci U S A. 2014;111:8155–8160. | |
Arora S, Ayyar BV, O’Kennedy R. Affinity chromatography for antibody purification. Methods Mol Biol. 2014;1129:497–516. | |
Ayyar BV, Arora S, Murphy C, O’Kennedy R. Affinity chromatography as a tool for antibody purification. Methods. 2012;56:116–129. | |
Liu HF, Ma J, Winter C, Bayer R. Recovery and purification process development for monoclonal antibody production. MAbs. 2010;2:480–499. | |
Graille M, Stura EA, Housden NG, et al. Complex between Peptostreptococcus magnus protein L and a human antibody reveals structural convergence in the interaction modes of Fab binding proteins. Structure. 2001;9:679–687. | |
Grover RK, Zhu X, Nieusma T, et al. A structurally distinct human mycoplasma protein that generically blocks antigen-antibody union. Science. 2014;343:656–661. | |
Kato K, Lian LY, Barsukov IL, et al. Model for the complex between protein G and an antibody Fc fragment in solution. Structure. 1995;3:79–85. | |
Shukla AA, Gupta P, Han X. Protein aggregation kinetics during protein A chromatography. Case study for an Fc fusion protein. J Chromatogr A. 2007;1171:22–28. | |
Starovasnik MA, O’Connell MP, Fairbrother WJ, Kelley RF. Antibody variable region binding by Staphylococcal protein A: thermodynamic analysis and location of the Fv binding site on E-domain. Protein Sci. 1999;8:1423–1431. | |
Deisenhofer J. Crystallographic refinement and atomic models of a human Fc fragment and its complex with fragment B of protein A from Staphylococcus aureus at 2.9- and 2.8-A resolution. Biochemistry. 1981;20:2361–2370. | |
Graille M, Stura EA, Corper AL, et al. Crystal structure of a Staphylococcus aureus protein A domain complexed with the Fab fragment of a human IgM antibody: structural basis for recognition of B-cell receptors and superantigen activity. Proc Natl Acad Sci U S A. 2000;97:5399–5404. | |
Xu X, Didio DM, Leister KJ, Ghose S. Disaggregation of high-molecular weight species during downstream processing to recover functional monomer. Biotechnol Prog. 2010;26:717–726. | |
Fuglistaller P. Comparison of immunoglobulin binding capacities and ligand leakage using eight different protein A affinity chromatography matrices. J Immunol Methods. 1989;124:171–177. | |
Vijayalakshmi MA. Pseudobiospecific ligand affinity chromatography. Trends Biotechnol. 1989;7:71–76. | |
Nicoud L, Arosio P, Sozo M, Yates A, Norrant E, Morbidelli M. Kinetic analysis of the multistep aggregation mechanism of monoclonal antibodies. J Phys Chem B. 2014;118:10595–10606. | |
McCaw TR, Koepf EK, Conley L. Evaluation of a novel methacrylate-based protein a resin for the purification of immunoglobulins and Fc-fusion proteins. Biotechnol Prog. 2014;30:1125–1136. | |
Angarita M, Müller-Späth T, Baur D, Lievrouw R, Lissens G, Morbidelli M. Twin-column CaptureSMB: a novel cyclic process for protein A affinity chromatography. J Chromatogr A. 2015;1389:85–95. | |
Subramanian G. Continuous Processing in Pharmaceutical Manufacturing. Illustrated edition. Vol 232. Hoboken, NJ: John Wiley & Sons; 2015. | |
Pina AS, Lowe CR, Roque AC. Challenges and opportunities in the purification of recombinant tagged proteins. Biotechnol Adv. 2014;32:366–381. | |
Arnau J, Lauritzen C, Petersen GE, Pedersen J. Current strategies for the use of affinity tags and tag removal for the purification of recombinant proteins. Protein Expr Purif. 2006;48:1–13. | |
Walls D, Loughran ST. Tagging recombinant proteins to enhance solubility and aid purification. Methods Mol Biol. 2011;681:151–175. | |
Young CL, Britton ZT, Robinson AS. Recombinant protein expression and purification: a comprehensive review of affinity tags and microbial applications. Biotechnol J. 2012;7:620–634. | |
Liu F, Tsai SL, Madan B, Chen W. Engineering a high-affinity scaffold for non-chromatographic protein purification via intein-mediated cleavage. Biotechnol Bioeng. 2012;109:2829–2835. | |
Fong BA, Wu WY, Wood DW. The potential role of self-cleaving purification tags in commercial-scale processes. Trends Biotechnol. 2010;28:272–279. | |
Boyer TD. The glutathione S-transferases: an update. Hepatology. 1989;9:486–496. | |
Kimple ME, Brill AL, Pasker RL. Overview of affinity tags for protein purification. Curr Protoc Protein Sci. 2013;73:Unit-9.9. | |
Frangioni JV, Neel BG. Solubilization and purification of enzymatically active glutathione S-transferase (pGEX) fusion proteins. Anal Biochem. 1993;210:179–187. | |
Hedhammar M, Gräslund T, Hober S. Protein engineering strategies for selective protein purification. Chem Eng Technol. 2005;28:1315–1325. | |
Roush DJ, Lu Y. Advances in primary recovery: centrifugation and membrane technology. Biotechnol Prog. 2008;24:488–495. | |
van Reis R. Charged filtration membranes and uses therefor. United States patent. 2006. Patent no. US 7001550. | |
Ghosh R, Wang L. Purification of humanized monoclonal antibody by hydrophobic interaction membrane chromatography. J Chromatogr A. 2006;1107:104–109. | |
Sadavarte R, Spearman M, Okun N, Butler M, Ghosh R. Purification of chimeric heavy chain monoclonal antibody EG2-hFc using hydrophobic interaction membrane chromatography: an alternative to protein-A affinity chromatography. Biotechnol Bioeng. 2014;111:1139–1149. | |
Gagnon P. Purification Tools for Monoclonal Antibodies. 1st ed. Vol 254. Tucson, AZ: Validated Biosystems, Inc; 1996. | |
Geise G, Myroid A, Gorrell J, Persson, J. Purification of antibodies by precipitating impurities using Polyethylene Glycol to enable a two chromatography step process. J Chromatogr B Analyt Technol Biomed Life Sci. 2013;1;938:14–2. | |
Smejkal B, Agrawal NJ, Helk B, et al. Fast and scalable purification of a therapeutic full-length antibody based on process crystallization. Biotechnol Bioeng. 2013;110:2452–2461. | |
Etzel MR. Bulk protein crystallization – principles and methods. Biotechnol Bioproc Ser. 2007;31:159–178. | |
Mahajan E, George A, Wolk B. Improving affinity chromatography resin efficiency using semi-continuous chromatography. J Chromatogr A. 2012;1227:154–162. | |
Bailey LJ, Sheehy KM, Hoey RJ, Schaefer ZP, Ura M, Kossiakoff AA. Applications for an engineered Protein-G variant with a pH controllable affinity to antibody fragments. J Immunol Methods. 2014;415:24–30. | |
Pollock J, Bolton G, Coffman J, Ho SV, Bracewell DG, Farid SS. Optimising the design and operation of semi-continuous affinity chromatography for clinical and commercial manufacture. J Chromatogr A. 2013;1284:17–27. | |
Pabst TM, Palmgren R, Forss A, et al. Engineering of novel Staphylococcal Protein A ligands to enable milder elution pH and high dynamic binding capacity. J Chromatogr A. 2014;1362:180–185. | |
Einhauer A, Jungbauer A. The FLAG peptide, a versatile fusion tag for the purification of recombinant proteins. J Biochem Biophys Methods. 2001;49:455–465. | |
Zhang L, Hernan R, Brizzard B. Multiple tandem epitope tagging for enhanced detection of protein expressed in mammalian cells. Mol Biotechnol. 2001;19:313–321. | |
Li Y. Self-cleaving fusion tags for recombinant protein production. Biotechnol Lett. 2011;33:869–881. | |
Thompson SA, Wang LL, West A, Sparling PF. Neisseria meningitidis produces iron-regulated proteins related to the RTX family of exoproteins. J Bacteriol. 1993;175:811–818. | |
Osicka R, Procházková K, Sulc M, Linhartová I, Havlícek V, Sebo P. A novel “clip-and-link” activity of repeat in toxin (RTX) proteins from gram-negative pathogens. Covalent protein cross-linking by an Asp-Lys isopeptide bond upon calcium-dependent processing at an Asp-Pro bond. J Biol Chem. 2004;279:24944–24956. | |
Chen J, Tetrault J, Ley A. Comparison of standard and new generation hydrophobic interaction chromatography resins in the monoclonal antibody purification process. J Chromatogr A. 2008;1177:272–281. | |
Wu SL, Figueroa A, Karger BL. Protein conformational effects in hydrophobic interaction chromatography. Retention characterization and the role of mobile phase additives and stationary phase hydrophobicity. J Chromatogr. 1986;371:3–27. | |
Ueberbacher R, Haimer E, Hahn R, Jungbauer A. Hydrophobic interaction chromatography of proteins V. Quantitative assessment of conformational changes. J Chromatogr A. 2008;1198–1199:154–163. | |
Burton SC, Harding DR. Hydrophobic charge induction chromatography: salt independent protein adsorption and facile elution with aqueous buffers. J Chromatogr A. 1998;814:71–81. | |
Cheng F, Li MY, Wang HQ, Lin DQ, Qu JP. Antibody–ligand interactions for hydrophobic charge-induction chromatography: a surface plasmon resonance study. Langmuir. 2015;31:3422–3430. | |
Multimodal or Mixed-Mode Chromatography. 2015. Available from: http://www.bio-rad.com/en-ie/applications-technologies/liquid-chromatography-principles/multimodal-or-mixed-mode-chromatography. Accessed March 31, 2016. | |
Lin DQ, Tong HF, Wang HY, Shao S, Yao SJ. Molecular mechanism of hydrophobic charge-induction chromatography: interactions between the immobilized 4-mercaptoethyl-pyridine ligand and IgG. J Chromatogr A. 2012;1260:143–153. | |
Bresolin IT, de Souza MC, Bueno SM. A new process of IgG purification by negative chromatography: adsorption aspects of human serum proteins onto omega-aminodecyl-agarose. J Chromatogr B Analyt Technol Biomed Life Sci. 2010;878:2087–2093. | |
Tong HF, Lin DQ, Gao D, Yuan XM, Yao SJ. Caprylate as the albumin-selective modifier to improve IgG purification with hydrophobic charge-induction chromatography. J Chromatogr A. 2013;1285:88–96. | |
Maria S, Joucla G, Garbay B, et al. Purification process of recombinant monoclonal antibodies with mixed mode chromatography. J Chromatogr A. 2015;1393:57–64. | |
Wolfe LS, Barringer CP, Mostafa SS, Shukla AA. Multimodal chromatography: characterization of protein binding and selectivity enhancement through mobile phase modulators. J Chromatogr A. 2014;1340:151–156. | |
Bresolin IT, Bueno SM. Evaluation of amino acid O-phosphoserine as ligand for the capture of immunoglubulin G from human serum. Appl Biochem Biotechnol. 2012;167:632–644. | |
Yakup Arica M, Akin-Öktem G, Denizli A. Novel hydrophobic ligand-containing hydrogel membrane matrix: preparation and application to γ-globulins adsorption. Colloids Surf B Biointerfaces. 2001;21:273–283. | |
Arica MY, Yilmaz M, Yalçin E, Bayramogğlu G. Affinity membrane chromatography: relationship of dye-ligand type to surface polarity and their effect on lysozyme separation and purification. J Chromatogr B. 2004;805:315–323. | |
Bayramogğlu G, Senel AU, Arica MY. Adsorption of IgG on spacer-arm and L-arginine ligand attached poly(GMA/MMA/EGDMA) beads. J Appl Polym Sci. 2007;104:672–679. | |
Gan HY, Shang ZH, Wang JD. New affinity nylon membrane used for adsorption of γ-globulin. J Chromatogr A. 2000;867:161–168. | |
Türkmen D, Öztürk N, Akgöl S, Elkak A, Denizli A. Phenylalanine containing hydrophobic nanospheres for antibody purification. Biotechnol Prog. 2008;24:1297–1303. | |
GE Lifescience. Antibody Purification Handbook. Little Chalfont, UK: Amersham Pharmacia Biotech; 2015. Available from: http://proteins.gelifesciences.com/knowledge-library/protein-handbooks/. Accessed March 31, 2016. | |
Hou Y, Brower M, Pollard D, et al. Advective hydrogel membrane chromatography for monoclonal antibody purification in bioprocessing. Biotechnol Prog. 2015;31:974–982. | |
Hall T, Wilson JJ, Brownlee TJ, Swartling JR, Langan SE, Lambooy PK. Alkaline cation-exchange chromatography for the reduction of aggregate and a mis-formed disulfide variant in a bispecific antibody purification process. J Chromatogr B Analyt Technol Biomed Life Sci. 2015;975:1–8. | |
Barroso T, Hussain A, Roque AC, Aguiar-Ricardo A. Functional monolithic platforms: chromatographic tools for antibody purification. Biotechnol J. 2013;8:671–681. | |
Iberer G, Hanh R, Jungbauer A, Majors RE. Stationary-phase technology in separation science. LC-GC Eur. 2000;13:88–93. | |
Pfaunmiller EL, Paulemond ML, Dupper CM, Hage DS. Affinity monolith chromatography: a review of principles and recent analytical applications. Anal Bioanal Chem. 2013;405:2133–2145. | |
Luo Q, Zou H, Zhang Q, Xiao X, Ni J. High-performance affinity chromatography with immobilization of protein A and L-histidine on molded monolith. Biotechnol Bioeng. 2002;80:481–489. | |
Feng S, Yang N, Pennathur S, Goodison S, Lubman DM. Enrichment of glycoproteins using nanoscale chelating concanavalin A monolithic capillary chromatography. Anal Chem. 2009;81:3776–3783. | |
Fitzgerald J, Leonard P, Darcy E, O’Kennedy RJ. In: Protein Chromatography. Vol 35–59. Berlin, Germany: Springer; 2011. | |
Moran KLM, Loftus JH, Murphy C., O’Kennedy R. Current and emerging technologies for the analysis of fungal and marine toxin contaminants in food. Submitted to book Nano-Inspired Biosensors for Improved Healthcare. Submitted for review, 2016. | |
McMahon MJ, O’Kennedy R. Polyreactivity as an acquired artefact, rather than a physiologic property, of antibodies: evidence that monoreactive antibodies may gain the ability to bind to multiple antigens after exposure to low pH. J Immunol Methods. 2000;241:1–10. | |
Hoogenboom HR. Overview of antibody phage-display technology and its applications. Methods Mol Biol. 2002;178:1–37. | |
Hoogenboom HR. Selecting and screening recombinant antibody libraries. Nat Biotech. 2005;23:1105–1116. | |
Kobatake E, Kosaku C, Hanzawa S, Mie M. Construction of affinity changeable antibody in response to Ca2+. Biotechnol Lett. 2012;34:1019–1023. | |
Liu Z, Gurgel PV, Carbonell RG. Affinity chromatographic purification of human immunoglobulin M from human B lymphocyte cell culture supernatant. Biochem Eng J. 2013;70:63–70. | |
Zhao WW, Shi QH, Sun Y. Dual-ligand affinity systems with octapeptide ligands for affinity chromatography of hIgG and monoclonal antibody. J Chromatogr A. 2014;1369:64–72. | |
Boi C, Dimartino S, Sarti GC. Performance of a new Protein A affinity membrane for the primary recovery of antibodies. Biotechnol Prog. 2008;24:640–647. | |
Roper DK, Lightfoot EN. Separation of biomolecules using adsorptive membranes. J Chromatogr A. 1995;702:3–26. | |
Ghosh R. Protein separation using membrane chromatography: opportunities and challenges. J Chromatogr A. 2002;952:13–27. | |
Boi C. Membrane adsorbers as purification tools for monoclonal antibody purification. J Chromatogr B Analyt Technol Biomed Life Sci. 2007;848:19–27. | |
Luellau E, von Stockar U, Vogt S, Freitag R. Development of a downstream process for the isolation and separation of monoclonal immunoglobulin A monomers, dimers and polymers from cell culture supernatant. J Chromatogr A. 1998;796:165–175. | |
Yu S, Geng J, Zhou P, Wang J, Chen X, Hu J. New hydroxyapatite monolithic column for DNA extraction and its application in the purification of Bacillus subtilis crude lysate. J Chromatogr A. 2008;1183:29–37. | |
Hilbrig F, Freitag R. Isolation and purification of recombinant proteins, antibodies and plasmid DNA with hydroxyapatite chromatography. Biotechnol J. 2012;7:90–102. | |
Gagnon P, Ng P, Zhen J, et al. A ceramic hydroxyapatite-based purification platform: Simultaneous removal of leached Protein A, aggregates, DNA and endotoxins from MABs. BioProcess Int. 2006;4:50–60. | |
Lan D, Huang G, Shao H, Zhang L, Ma L, Chen S. An improved non-chromatographic method for the purification of recombinant proteins using elastin-like polypeptide-tagged proteases. Anal Biochem. 2011;415:200–202. | |
Banki MR, Feng L, Wood DW. Simple bioseparations using self-cleaving elastin-like polypeptide tags. Nat Methods. 2005;2:659–661. | |
Kostal J, Mulchandani A, Chen W. Tunable biopolymers for heavy metal removal. Macromolecules. 2001;34:2257–2261. | |
Shimazu M, Mulchandani A, Chen W. Thermally triggered purification and immobilization of elastin-OPH fusions. Biotechnol Bioeng. 2003;81:74–79. | |
Bayer EA, Belaich JP, Shoham Y, Lamed R. The cellulosomes: multienzyme machines for degradation of plant cell wall polysaccharides. Annu Rev Microbiol. 2004;58:521–554. | |
Balasubramaniam D, Wilkinson C, Van Cott K, Zhang C. Tobacco protein separation by aqueous two-phase extraction. J Chromatogr A. 2003;989:119–129. | |
Asenjo JA, Andrews BA. Aqueous two-phase systems for protein separation: a perspective. J Chromatogr A. 2011;1218:8826–8835. | |
Asenjo JA, Andrews, BA. Aqueous two-phase systems for protein separation: phase separation and applications. J Chromatogr A. 2012;1238:1–10. | |
Mao LN, Rogers JK, Westoby M, Conley L, Pieracci J. Downstream antibody purification using aqueous two-phase extraction. Biotechnol Prog. 2010;26:1662–1670. | |
Platis D, Labrou NE. Application of a PEG/salt aqueous two-phase partition system for the recovery of monoclonal antibodies from unclarified transgenic tobacco extract. Biotechnol J. 2009;4:1320–1327. | |
Wu Q, Lin DQ, Zhang QL, Gao D, Yao SJ. Evaluation of a PEG/hydroxypropyl starch aqueous two-phase system for the separation of monoclonal antibodies from cell culture supernatant. J Sep Sci. 2014;37:447–453. | |
Cao H, Yuan M, Wang L, Yu J, Xu F. Coupling purification and in situ immobilization process of monoclonal antibodies to clenbuterol for immunosensor application. Anal Biochem. 2015;476:59–66. | |
Buyel JF, Fischer R. Flocculation increases the efficacy of depth filtration during the downstream processing of recombinant pharmaceutical proteins produced in tobacco. Plant Biotechnol J. 2014;12:240–252. | |
Laukel M, Rogge P, Dudziak G. Disposable downstream processing for clinical manufacturing. BioProcess Int. 2011;9:14–21. | |
Whitford WG. Single-use systems as principal components in bioproduction. BioProcess Int. 2010;8:34–44. | |
Buyel JF, Fischer R. Downstream processing of biopharmaceutical proteins produced in plants: the pros and cons of flocculants. Bioengineered. 2014;5:138–142. | |
Gregory J, Barany S. Adsorption and flocculation by polymers and polymer mixtures. Adv Colloid Interface Sci. 2011;169:1–12. | |
Zhou Y, Franks GV. Flocculation mechanism induced by cationic polymers investigated by light scattering. Langmuir. 2006;22:6775–6786. | |
Kang YK, Hamzik J, Felo M, et al. Development of a novel and efficient cell culture flocculation process using a stimulus responsive polymer to streamline antibody purification processes. Biotechnol Bioeng. 2013;110:2928–2937. | |
Singh N, Pizzelli K, Romero JK, et al. Clarification of recombinant proteins from high cell density mammalian cell culture systems using new improved depth filters. Biotechnol Bioeng. 2013;110:1964–1972. | |
Capito F, Skudas R, Stanislawski B, Kolmar H. Customization of copolymers to optimize selectivity and yield in polymer-driven antibody purification processes. Biotechnol Prog. 2013;29:1484–1493. | |
Jaoko WG, Lund M, Michael E, Simonsen PE. A simple and quick method for enhanced detection of specific IgE in serum from lymphatic filariasis patients. Acta Trop. 2001;80:51–57. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.