Back to Journals » International Journal of Nanomedicine » Volume 13
Targeting prostate cancer cells with hybrid elastin-like polypeptide/liposome nanoparticles
Authors Zhang W, Song Y ![]() , Eldi P, Guo X
, Eldi P, Guo X ![]() , Hayball JD, Garg S
, Hayball JD, Garg S ![]() , Albrecht H
, Albrecht H
Received 28 September 2017
Accepted for publication 21 November 2017
Published 9 January 2018 Volume 2018:13 Pages 293—305
DOI https://doi.org/10.2147/IJN.S152485
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Thomas Webster
Wei Zhang,1 Yunmei Song,1 Preethi Eldi,1 Xiuli Guo,2 John D Hayball,1,3 Sanjay Garg,1 Hugo Albrecht1
1Centre for Pharmaceutical Innovation and Development, Centre for Drug Discovery and Development, Experimental Therapeutics Laboratory, Sansom Institute for Health Research, School of Pharmacy and Medical Sciences, University of South Australia, Adelaide, SA, Australia; 2Department of Pharmacology, School of Pharmaceutical Sciences, Shandong University, Jinan, China; 3Robinson Research Institute and Adelaide Medical School, University of Adelaide, Adelaide, SA, Australia
Abstract: Prostate cancer cells frequently overexpress the gastrin-releasing peptide receptor, and various strategies have been applied in preclinical settings to target this receptor for the specific delivery of anticancer compounds. Recently, elastin-like polypeptide (ELP)-based self-assembling micelles with tethered GRP on the surface have been suggested to actively target prostate cancer cells. Poorly soluble chemotherapeutics such as docetaxel (DTX) can be loaded into the hydrophobic cores of ELP micelles, but only limited drug retention times have been achieved. Herein, we report the generation of hybrid ELP/liposome nanoparticles which self-assembled rapidly in response to temperature change, encapsulated DTX at high concentrations with slow release, displayed the GRP ligand on the surface, and specifically bound to GRP receptor expressing PC-3 cells as demonstrated by flow cytometry. This novel type of drug nanocarrier was successfully used to reduce cell viability of prostate cancer cells in vitro through the specific delivery of DTX.
Keywords: ELP-GRP, micelle, liposome, docetaxel, prostate cancer, GRPR, GPCR
Introduction
Worldwide, prostate cancer accounts for the leading cause of cancer death in men, and the two major forms, the organ confined and the aggressive metastatic disease, are observed in the clinic.1 The current first-line treatments aim at testosterone reduction to castrate levels either by surgical removal of the testicles or by pharmacological androgen ablation using luteinizing hormone-releasing hormone (LHRH) agonists. Some additional medications (eg, anti-androgens) rely on direct androgen receptor (AR) inhibition.2,3
In most cases, these treatments are effective for limited time only, and progression to advanced castrate-refractory prostate cancer is very likely to occur within an average time span of 12–18 months. At this advanced stage, some positive effects have been achieved by inhibition of adrenal androgen synthesis using either various steroids or cytochrome P450 inhibitors such as ketoconazole and abiraterone.4–12 Prostate cancer has been considered as chemorefractory for a long time.13,14 Only in the past 10–15 years, good results have been achieved in clinic trials with taxane derivatives such as docetaxel (DTX) and cabazitaxel. However, disabling toxicities like fatigue and neuropathy limit optimal dosage, and therefore, only modest, life-prolonging effects have been achieved for prostate cancer patients. Future treatment outcomes could potentially be improved through specific delivery of chemotherapeutic compounds into cancer cells while reducing the exposure of healthy tissue. Recently, specific delivery of compounds has been demonstrated using receptor-mediated tumor targeting with immune liposomes where the enhanced permeability and retention (EPR) effect was combined with active targeting of cancer cells.15
In the past decade, elastin-like polypeptide (ELP) micelles have emerged as potential carriers for anticancer compounds. These particles contain a lipophilic core where small organic drug molecules accumulate while the surface is of hydrophilic nature and can be linked with cancer cell-specific receptor ligands. Several strategies have been reported to encapsulate anticancer drugs within ELPs.16–18 Direct hydrophobic interactions between drugs and ELP cores were not sufficient to retain the compounds for extended time as would be necessary for in vivo delivery into cancer cells.18 Alternative approaches used covalent binding of anticancer drugs to ELPs.16–18 In this case, the amount of loaded drug is limited by the number of functional groups available for covalent reaction.16–18 Recent studies described covalent conjugation of ELPs to liposome surfaces or the synthesis of ELP–lipid conjugates prior to the formation of hybrid liposomes.19–24 ELP-modified liposomes showed increased cellular uptake and anticancer drug delivery. However, the procedures for covalent conjugation are complicated and labor intense, which restrained the development of ELP-modified liposomes as drug carriers.
Herein, we report the generation of self-assembling DTX-loaded hybrid ELP/liposome nanoparticles for successful targeted DTX delivery into cancer cells (Figure 1). The gastrin-releasing peptide receptor (GRPR), which is frequently overexpressed in prostate cancer cells, was used for active targeting via gastrin-releasing peptide (GRP) displayed on the surface of ELP micelles to trigger receptor internalization and nanoparticle delivery directly into endosomal compartments.25
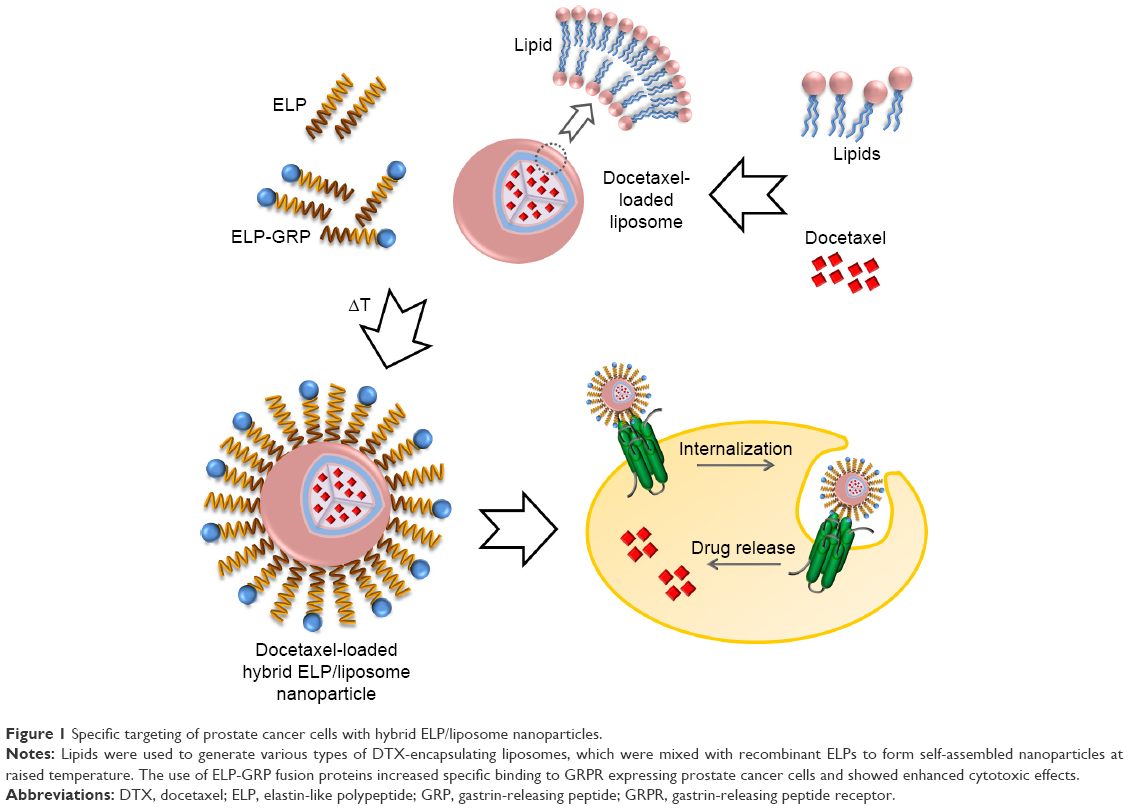 | Figure 1 Specific targeting of prostate cancer cells with hybrid ELP/liposome nanoparticles. |
Materials and methods
Materials and reagents
Presomes ACD-1, S-1, and O-1 were provided by Nippon Fine Chemical Co., Ltd (Osaka, Japan). Egg yolk lecithin was purchased from Q.P.E.C Co Ltd (Tokyo, Japan). Methanol, chloroform, acetonitrile, dimethyl sulfoxide (DMSO), and sterile 0.45 μm Millex-GP Syringe Filter Units were purchased from Merck & Co, Inc. (Melbourne, VIC, Australia). Polyethyleneimine (PEI), cholesterol, ammonium sulfate, D-lysine, glycerol, arginine, Roswell Park Memorial Institute (RPMI)-1640 medium, penicillin–streptomycin, trypsin, Atto 488 N-hydroxysuccinimide (NHS) ester, molecular weight cutoff 1.4 kDa dialysis tubing cellulose membrane, Costar 96-well tissue culture-treated plates, and PC-3 human Caucasian Prostate Adenocarcinoma cells were purchased from Sigma-Aldrich (Castle Hill, NSW, Australia). DU-145 cells originated from the American Type Culture Collection (Manassas, VA, USA) and were a generous gift from Professor Wang (University of South Australia, Adelaide, SA, Australia). Fetal bovine serum was purchased from Interpath Services Pty Ltd (Heidelberg West, VIC, Australia). Bicinchoninic acid assay (BCA) protein assay kit, 4× sodium dodecyl sulfate (SDS) loading buffer, and 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) were from Thermo Fisher Scientific (Scoresby, VIC, Australia). Protease inhibitor phenylmethylsulfonyl fluoride (PMSF) was purchased from Astral Scientific (Taren Point, NSW, Australia). Mini-PROTEAN® TGX™ Gels, 4%–20%, were from Bio-Rad Laboratories Pty Ltd (Gladesville, NSW, Australia). Disposable solvent-resistant microcuvettes were from ATA Scientific Pty Ltd (Taren Point, NSW, Australia). Folded Capillary Zeta Cells (DTS1070) were purchased from Malvern Instruments Ltd (Taren Point, NSW, Australia). DTX was ordered from Nova Pharmaceutical Co Ltd (Shandong, China). Recombinant ELP-GRP, ELP-C, and ELP-K were expressed and purified at the Centre for Pharmaceutical Innovation and Development, University of South Australia.
Preparation of DTX-loaded liposomes
A thin-film hydration method was used to prepare drug-loaded liposomes.26,27 Two hundred milligrams of ACD-1, S-1, or O-1 presomes were mixed with 10 mg of DTX and placed into a 200 mL round-bottom flask and dissolved in 20 mL of a 1:1 (v/v) methanol:chloroform solvent system. The organic solvents were removed using a rotating evaporator (BUCHI, Victoria, Australia) for 30 minutes at 40°C and 80 rpm, followed by overnight drying at room temperature (RT). Lecithin liposomes were formulated with the same procedure starting with 200 mg of egg yolk lecithin, 30 mg of cholesterol, and 10 mg of DTX. The described protocol resulted in the formation of thin lipid/DTX films on the wall of the round-bottom flasks which were hydrated with 5 mL of prewarmed PBS (35°C) on a rotating evaporator (4 hours, 80 rpm, 35°C). Subsequently, a Q-500 sonicator (Adelab Scientific, Thebarton, SA, Australia) was employed to generate small unilamellar vesicles with a 4 second on/off sonication cycle (5 minutes, on ice, 45% amplitude).28 Free DTX precipitated instantly, due to its low solubility in aqueous solution, and was removed from the preparations by filtration with a 0.45 μm pore-size sterile syringe filter.29 Drug-loaded ACD-1, S-1, O-1, or lecithin liposomes were stored in PBS at 4°C until further use.
Analysis of encapsulation efficiency
The encapsulation efficiency of drug-loaded liposomes was analyzed with a high-performance liquid chromatography (HPLC) system (LC-20AD liquid chromatograph, SIL-20A HT autosampler and SPDM20A DAD detector, Shimadzu, NSW, Australia). A Zorbax Eclipse XDB C-18 column (Agilent Technologies, Cheshire, UK) was employed.30 The mobile phase consisted of 20 mM ammonium sulfate (adjusted to pH 5.0 with HCl) and acetonitrile at a ratio of 43:57 (v/v), and degassing was carried out by 15 minutes of sonication prior to use. The flow rate was set at 0.7 mL/minute. Fifty microliters of DTX-loaded liposomes were dissolved with 950 μL of methanol and incubated at RT for 10 minutes to disrupt liposome structures. Aliquots of 20 μL were introduced into the HPLC system by an autoinjector, with 10 minutes run time for each sample, and compound elution was monitored by absorption at 230 nm. Reference standards were injected into the HPLC system using above conditions and encapsulation efficiencies were calculated according to the following formula:
|
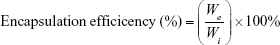
where We was the total amount of encapsulated DTX and Wi was the initial amount of DTX added for the preparation of drug-loaded liposomes.31
Drug-loading rates were calculated according to the formula below:
|
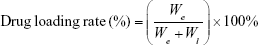
where We was the total amount of encapsulated DTX in the preparation and Wl was the total amount of the excipients in the liposome preparations.
Production of ELPs
Recombinant ELP-C, ELP-GRP, and ELP-K proteins were expressed in Rosetta (DE3) pLysS Escherichia coli bacteria using the pET-31b(+) expression system (Novagen Inc., Madison, WI, USA). Plasmid vectors that contained the ELP-C, ELP-GRP, and ELP-K genes were prepared with standard molecular biology techniques as described previously.25 Transformed bacteria were harvested from liquid cultures (400 mL LB medium, 50 μg/mL ampicillin, 225 rpm, 37°C, 24 hours) by centrifugation at 2,300 g (4°C, 15 minutes), resuspended in 20 mL PBS (pH 7.5), and complemented with 1 mM PMSF. The cell membranes were disrupted by sonication in an ice bath for 18 minutes at 35% amplitude, using a 4 second on/off cycle. The samples were cleared from bacterial debris by 15 minutes of centrifugation at 16,000 g and 4°C. Bacterial DNA was removed from the supernatants by 40 minutes precipitation on ice with 0.5% (w/w) PEI. The PEI/DNA complexes were removed by centrifugation at 16,000 g, 4°C for 15 minutes and the supernatant was collected for purification of recombinant ELPs by inverse transition cycling.32,33 The phase transition of ELPs was triggered by addition of 5 M NaCl to an approximate final concentration of 2.5 M and incubation at 37°C for 30 minutes. Formed ELP aggregates were harvested by centrifugation at 2,300 g, 37°C for 10 minutes. The supernatant was discarded, and the polypeptide aggregates were gently dissolved in 8 mL of cold PBS. The sample was incubated on ice for 30 minutes, and insoluble aggregates were removed by centrifugation at 2,300 g, 4°C for 10 minutes. A second round of inverse transition cycling (ITC) was performed to increase polypeptide purity, and the ELPs were stored in 15% glycerol/PBS (v/v) at −20°C until further use. Polypeptide concentrations were routinely determined using a BCA assay kit. Purity and molecular weight of the polypeptides were examined by standard sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) using a Mini-PROTEAN Tetra Cell system (Bio-Rad Laboratories, Gladesville, NSW, Australia).
Preparation of DTX-loaded hybrid ELP/liposome nanoparticles
Stock solutions containing ELP-C or a mixture of ELP-GRP and ELP-C in a ratio of 3:2 (referred to as 1.5× ELP-GRP/C in this report) were diluted with ice-cold PBS to 25 μM total protein concentration and kept on ice. Subsequently, 10 volumes of the freshly diluted ELP solutions were mixed with 1 volume of DTX-loaded liposomes, which have been prepared as described above, and diluted with PBS to obtain final DTX concentrations of 40 or 80 μg/mL (under consideration of encapsulation rates). Mixtures were then incubated at 37°C for 15 minutes to form DTX-loaded polypeptide/liposome nanoparticles and used immediately for the studies described in the “Results” section.
Dynamic light scattering analysis
Hydrodynamic diameters of DTX-loaded liposomes and polypeptide/liposome nanoparticles were studied at 37°C using dynamic light scattering (DLS). Freshly prepared DTX-loaded liposomes were diluted with 10 volumes of PBS and incubated at 37°C for 15 minutes prior to DLS analysis. DTX-loaded hybrid polypeptide/liposome nanoparticles were formed by mixing 10 volumes of 25 μM ELP-C or 1.5× ELP-GRP/C with 1 volume of DTX-loaded liposomes followed by filtration through a sterile 0.45 μm Millex-GP Syringe Filter Unit to remove microaggregates. The samples were incubated at 37°C for 15 minutes to form hybrid nanoparticles. DTX-loaded liposomes or polypeptide/liposome nanoparticles were transferred into disposable solvent-resistant microcuvettes followed by measurement of hydrodynamic diameters with a DLS apparatus (Zetasizer Nano ZS, Malvern Instruments Ltd). The samples were equilibrated for 2 minutes at 37°C prior to measurements.
Zeta potential analysis
The zeta potentials of DTX-loaded liposomes and polypeptide/liposome nanoparticles were evaluated at 37°C to assess surface charges. One hundred microliters of freshly prepared DTX-loaded liposomes were diluted with 1 mL of PBS and filtered through a 0.45 μm Millex-GP Syringe Filter Unit. Samples were incubated at 37°C for 15 minutes before measurement. DTX-loaded polypeptide/liposome nanoparticles were prepared in PBS by mixing 100 μL of freshly prepared DTX-loaded liposomes with 1 mL of 5 μM 1.5× ELP-GRP/C or ELP-C in PBS on ice. Samples were filtered through a 0.45 μm Millex-GP Syringe Filter Unit and incubated at 37°C for 15 minutes. All samples were transferred into Folded Capillary Zeta Cells for measurement in a zeta potential analyzer (Zetasizer Nano ZS, Malvern Instruments Ltd). All experiments were carried out in triplicate at 37°C.
Release rate analysis
The release rates of DTX from drug-loaded carriers into release buffer (PBS with 10% glycerol [v/v] and 5% arginine [w/w], pH 7.4) were analyzed by a dialysis method using an HPLC protocol for quantification.34,35 Freshly prepared DTX-loaded liposomes or polypeptide/liposome nanoparticles were diluted into release buffer at a final polypeptide concentration of 25 μM and a liposome to polypeptide ratio of 1:10 (v/v). Final volumes of 550 μL were placed into a rinsed dialysis tubing cellulose membrane with 1.4 kDa molecular weight cutoff.
The dialysis tube was sealed and placed into a 20 mL glass vial bottle containing 15 mL of prewarmed release medium (37°C) followed by incubation on an orbital shaker at 37°C, 80 rpm (IKA Works GmbH & Co KG, Selangor, Malaysia). The release medium was replaced after 0.5, 1, 1.5, 2, 3, 4, and 5 hours to keep sink condition.36 The collected release medium was diluted with 15 mL of ethanol to dissolve DTX prior to analysis using the HPLC system as outlined above. All measurements were performed in triplicate, and the averages were recorded as the mean release rates.
Flow cytometry analysis
Flow cytometry was used to study specific binding of drug-loaded polypeptide/liposome nanoparticles to the surface of GRPR expressing PC-3 and DU-145 prostate cancer cells. For this purpose, recombinant ELP-K protein (which has been designed with 3 C-terminal lysine residues for enhanced NHS ester-mediated conjugation) was chemically linked with Atto 488 NHS ester. In brief, 50 μM of ELP-K (in PBS, pH 7.4) was supplemented with 10% (v/v) of 1 M sodium bicarbonate buffer (pH 8.3) to adjust the pH value to approximately 8.0, followed by the addition of 5% (v/v) 2 mM Atto 488 NHS ester solution (in DMSO) to reach a 2× molar excess of the Atto 488 dye.37 The mixtures were protected from light and incubated for 45 minutes at RT on an orbital shaker (50 rpm). Precipitation of ELP-K-Atto was triggered by the addition of 40% (v/v) of 5 M NaCl solution, followed by 15 minutes incubation at 37°C in a water bath. The ELP-K-Atto aggregates were recovered by 5 minutes centrifugation (5,000 g, 37°C), and the excess of the Atto 488 NHS ester was discarded with the supernatant. The precipitates were washed three times with prewarmed PBS (37°C) and finally dissolved at 10 μM final protein concentration in cold PBS. ELP-GRP or ELP-C were combined with ELP-K-Atto at a molar ratio of 3:2 to form mixed micelles referred to as 1.5× ELP-GRP/K-Atto or 1.5× ELP-C/K-Atto, respectively. Hybrid ELP/liposome nanoparticles were generated by adding 80 μL of drug-loaded ACD-1 liposomes into 800 μL of 10 μM ELP mixtures on ice, followed by incubation in a 37°C water bath for 15 minutes to form drug-loaded 1.5× ELP-GRP/K-Atto/ACD-1 or 1.5× ELP-C/K-Atto/ACD-1 nanoparticles.
The prostate cancer cells were harvested by centrifugation and resuspended in 880 μL of freshly prepared, drug-loaded polypeptide/liposome nanoparticle solution at 5×105 cells/mL followed by incubation in an orbital shaker at 50 rpm and 37°C for 1 hour to allow specific binding and uptake. The cells were washed three times in PBS to remove free nanoparticles and harvested by centrifugation as described above. Cells were resuspended in 200 μL of PBS followed by loading onto a BD FACSARIA™ fusion flow cytometer using FACS Diva software for analysis. 293T cells which do not express the GRPR were used as negative control.38
Cell viability assay
The anticancer effects of drug-loaded liposomes and hybrid polypeptide/liposome nanoparticles were evaluated with PC-3 prostate cancer cells using the MTT assay.18 For this purpose, cells were seeded in tissue culture-treated 96-well plates at a density of 5,000 cells/well and cultured in RPMI 1640 medium plus 10% FBS (v/v) at 37°C and 5% CO2 for 24 hours. DTX and DTX-loaded liposomes were placed in RPMI supplemented with 5% glycerol and 5% D-lysine. The pH value was adjusted to 7.4, and the final DMSO concentration was 0.1% in all wells. The applied volume was 100 μL per well, and the final DTX concentration was 40 or 80 μg/mL (adjusted according to encapsulation efficiency).
Preparations that contained ELP/liposome particles (eg, 1.5× ELP-GRP/C or ELP-C) were prepared in the same medium containing 10 μM total protein with DTX concentration of 40 or 80 μg/mL.
The culture medium was replaced by 100 μL of freshly prepared DTX-containing solutions and the cells were incubated at 37°C and 5% CO2 for 4 hours. After 4 hours incubation, cells were gently rinsed three times with PBS to remove unbound DTX, DTX-loaded liposomes, or polypeptide/liposome nanoparticles. The cells were kept in culture (RPMI 1640, 10% FBS) for an additional 20 hours and then supplemented with 0.5 mg/mL of MTT. The incubation was continued for another 4 hours, prior to replacement of the culture medium with 150 μL of DMSO. The samples were transferred to an orbital shaker and agitated at 500 rpm for 5 minutes to dissolve MTT-formazan crystals. The absorbance was then determined with an EnVision® Multilabel Plate Reader (Perkin Elmer PTY Ltd, Gladesville, New South Wales, Australia) at 570 nm. Cell viability was evaluated according to the following formula:31
|
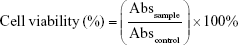
where Abssample is the absorbance of samples, Abscontrol represents absorbance of control (cells grown in RPMI without treatment) at 570 nm.
Statistical analysis
All data were analyzed and presented as mean ± SD. The significance of differences was compared by unpaired and two-tailed t-test analysis (Sigma plot 12.5) and significance was confirmed with p<0.05.
Results and discussion
Liposome production and characterization
Four types of DTX-loaded liposomes were formulated as the basis to generate self-assembling hybrid nanoparticles with different ELPs. All formulations contained cholesterol to attain decreased water and ion permeability of the lipid bilayers.39–41 The following, mainly phosphocholine-based, components were selected to generate liposomes: egg yolk lecithin, hydrogenated soy L-phosphatidylcholine (HSPC), and the synthetic lipids 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC), 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[amino(polyethylene glycol)-2000] (MPEG-DSPE), and 1,2-distearoyl-sn-glycero-3-phosphocholine (DSPC). One of the synthetic lipids contained unsaturated side chains and one was PEG modified, the latter to prevent nonspecific cellular uptake and to potentially obtain prolonged systemic circulation for future in vivo studies. Detailed compositions are given in Table 1 and the chemical structures of lipids are shown in Figure 2. The four liposomes are referred to as lecithin, O-1, ACD-1, and S-1 liposomes throughout this study.
 | Table 1 Composition of lecithin, O-1, ACD-1, and S-1 liposomes |
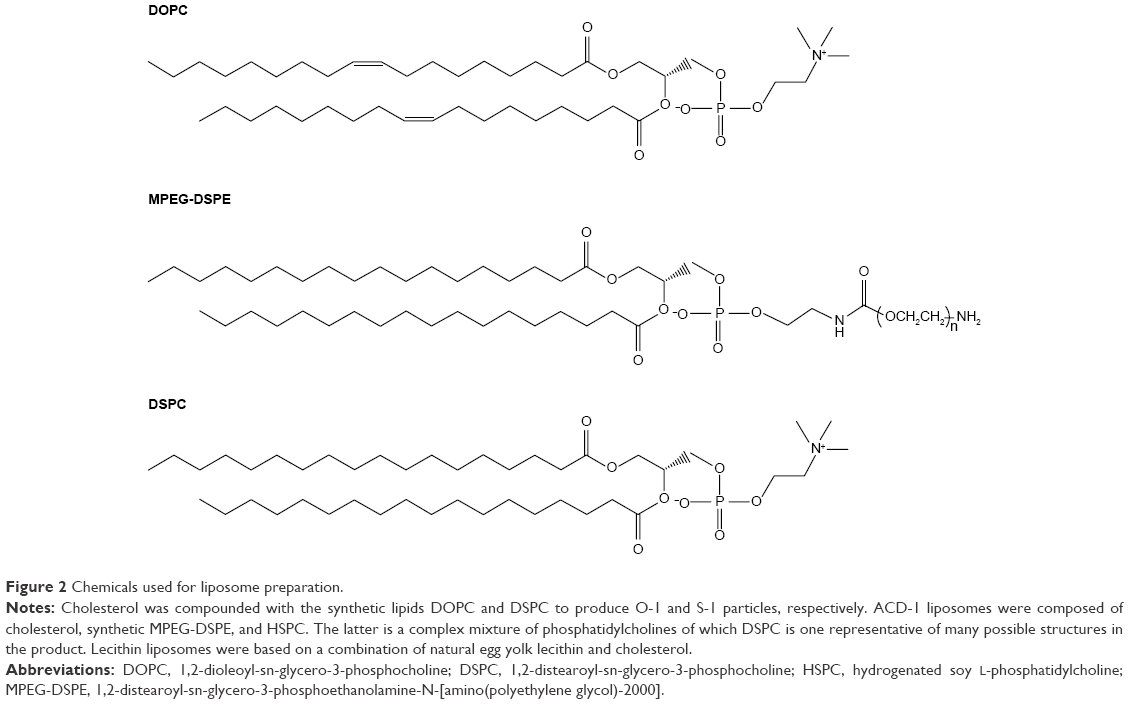 | Figure 2 Chemicals used for liposome preparation. |
DTX-loaded liposomes were generated by thin-film hydration using an initial DTX content of 5% (w/w) as described in the “Methods” section. Hydrodynamic diameters of the nanoparticles were determined with DLS analysis.34 All DTX-loaded liposomes showed monodisperse size distributions and exhibited mean diameters ranging from 23.5 to 154.8 nm (Figure 3). Noteworthy, DTX-loaded S-1 liposomes with 33% cholesterol formed the largest particles with diameters of ~155 nm, while DTX-loaded liposomes with lecithin, O-1, and ACD-1, and cholesterol contents of 13%, 9%, and 20% possessed smaller diameters of ~23, 37, and 61 nm, respectively. Earlier studies indicated a potential correlation between cholesterol content and liposome size.42
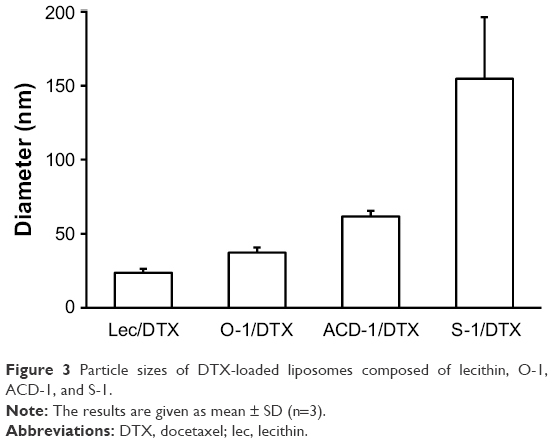 | Figure 3 Particle sizes of DTX-loaded liposomes composed of lecithin, O-1, ACD-1, and S-1. |
The encapsulation capacities were evaluated for all DTX liposomal preparations. The bilayer structures of drug-loaded liposomes were disrupted with methanol followed by separation and quantification on a C-18 reversed-phase column as outlined in the “Materials and methods” section (Figure 4). DTX-loaded O-1 liposomes contained the lowest cholesterol content (9%) and showed the highest DTX-loading capacity with an encapsulation rate of almost 90%.The remaining DTX-loaded liposomes (lecithin, ACD-1, and S-1) with cholesterol contents of 13%, 20%, and 33% exhibited lower drug-loading efficiencies, ranging from 20% to 40%. The total drug-loading rates for these formulations were calculated to be 1.6%, 4.0%, 1.1%, and 1.9% for lecithin, O-1, ACD-1, and S-1 liposomes, respectively. Cholesterol has been reported previously to affect liposomal encapsulation rates, and in addition to this, the unsaturated character of DOPC in O-1 liposomes may contribute to higher DTX entrapment due to decreased packing density of lipids, thereby providing more space for the loaded drug.34,43 The lowest encapsulation efficiency was obtained with ACD-1 at 20% cholesterol content. This may be caused by interactions between hydrophobic tails of DSPE-PEG and phospholipids occupying potential drug-loading areas.35,44
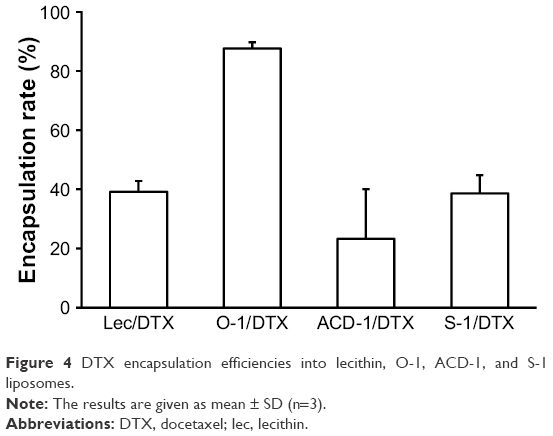 | Figure 4 DTX encapsulation efficiencies into lecithin, O-1, ACD-1, and S-1 liposomes. |
The encapsulation rates were sufficient to deliver equivalents of 40 and 80 μg/mL of DTX into PC-3 cell cultures. Liposome stocks were diluted in PBS to obtain 400 μg/mL of DTX, followed by a subsequent 1:10 or 1:5 dilution into RPMI medium to achieve final concentrations of 40 and 80 μg/mL, respectively. PC-3 cells were exposed to 100 μL of the final liposome dilutions in 96-well plates to evaluate effects on cell viability. Empty liposomes were included in the experiments as negative controls. Free DTX with concentrations of 40 and 80 μg/mL in 100 μL of RPMI were used as positive controls. After an initial incubation for 4 hours, DTX-loaded liposomes, blank liposomes, and free DTX were removed by gentle washing with PBS followed by additional 24 hours incubation in fresh cell culture medium prior to cell viability testing with the MTT assay. The data shown in Figure 5 have been calculated as relative cellular viabilities in comparison to negative controls (cells without treatment). DTX-loaded liposomes exhibited reduced cytotoxicity compared to free DTX. This might be due to the slow release of DTX from the liposomes and the removal of some unbound liposomes after 4 hours of incubation. Control experiments with blank liposomes did not reveal any effect on cellular viability with lecithin, ACD-1, and S-1 formulations, therefore confirming suitable use for future in vitro and in vivo studies. Unexpectedly, blank O-1 liposomes reduced the cellular viability of PC-3 cells to 80% indicating potential cytotoxic effects. Therefore, the use of O-1 liposomes was excluded from further investigations.
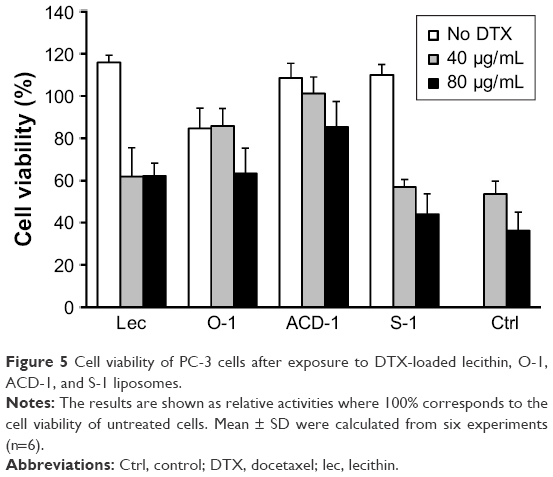 | Figure 5 Cell viability of PC-3 cells after exposure to DTX-loaded lecithin, O-1, ACD-1, and S-1 liposomes. |
Generation of hybrid nanoparticles
The aim of the next step was to display GRP as the active targeting ligand on the surface of DTX-loaded lecithin, ACD-1, and S-1 liposomes. In our previous work, we established self-assembling micelles based on an ELP-GRP fusion protein (Table 2), which specifically bound to GRPR expressing prostate cancer cells and triggered internalization into endosomal compartments.25 The presented report is an extension of this work by the generation of hybrid ELP/liposome nanoparticles with the aim to deliver DTX into GRPR expressing prostate cancer cells.
 | Table 2 ELPs used in this study |
Ice-cold ELP-GRP and ELP-C were mixed at a ratio of 3:2 in a volume of 100 μL PBS. Using this ratio has been shown to form stable micelles at 37°C in PBS without aggregation, and this type of micelle will be referred to as 1.5× ELP-GRP/C throughout this report. The final total ELP concentration was adjusted to 25 μM, and 10 μL of freshly prepared DTX-loaded liposomes were added on ice. The mixtures were then incubated in a 37°C water bath for 15 minutes to form DTX-loaded polypeptide/liposome nanoparticles followed by particle size determination. Micelles formed by ELPs only, such as ELP-C and 1.5× ELP-GRP/C, displayed particle sizes ranging from 40 to 50 nm (Figure 6), which correlated well with previous results.25 Liposome particle sizes increased after incorporation into ELP micelles and were found to be monodispersed and ranged from approximately 40 to 180 nm. These results provided a proof of concept for the rapid assembly of hybrid nanoparticles with successful encapsulation of DTX-loaded liposomes into the hydrophobic cores of polypeptide micelles.
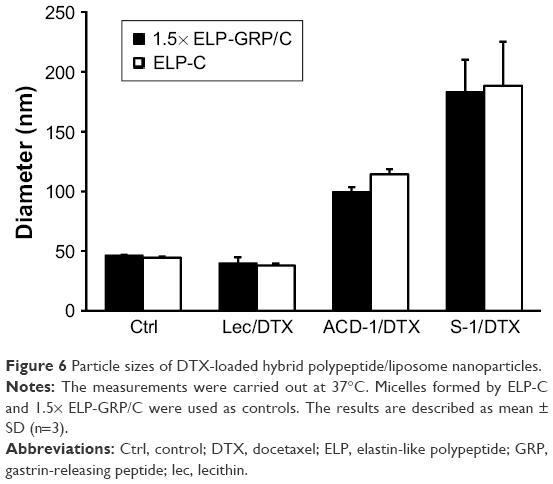 | Figure 6 Particle sizes of DTX-loaded hybrid polypeptide/liposome nanoparticles. |
For effective drug delivery, it is essential to balance the nanoparticle size between maximal EPR effects and minimal nonspecific clearance. Particles >300 nm are considered too large for tumor infiltration and best results have been seen with particles of sizes ≤100 nm. From this perspective, ELP hybrids with ACD-1 and lecithin seemed most suitable. The vascular fenestration in liver is estimated at 50–100 nm and, therefore, ELP/ACD-1 hybrid particles were considered the most optimal, followed by ELP/lecithin. ELP/S-1 particles with a size of ~200 nm and no surface PEGylation are expected to bind serum proteins to a large extent, which is assumed to further increase the size and might be a disadvantage for future in vivo experiments. Due to this, further studies were limited to ACD-1- and lecithin-containing carriers.
In vitro drug release
It is desirable to achieve low drug release in circulation with nanoparticle-mediated cancer cell targeting. In contrast, efficient intracellular drug release is necessary after internalization into cancer cells. The DTX release kinetics from lecithin and ACD-1 liposomes and hybrid polypeptide/liposome nanoparticles were assessed with an in vitro dialysis protocol. Five hundred microliters of 25 μM 1.5× ELP-GRP/C or ELP-C in PBS were mixed on ice with 50 μL of freshly prepared DTX-loaded liposomes. The mixtures were incubated at 37°C for 15 minutes and then loaded into a dialysis tube. The release medium was replaced, and samples were withdrawn after 0.5, 1, 1.5, 2, 3, 4, and 5 hours. The profiles are presented as cumulative percentages of DTX released versus time (Figure 7).
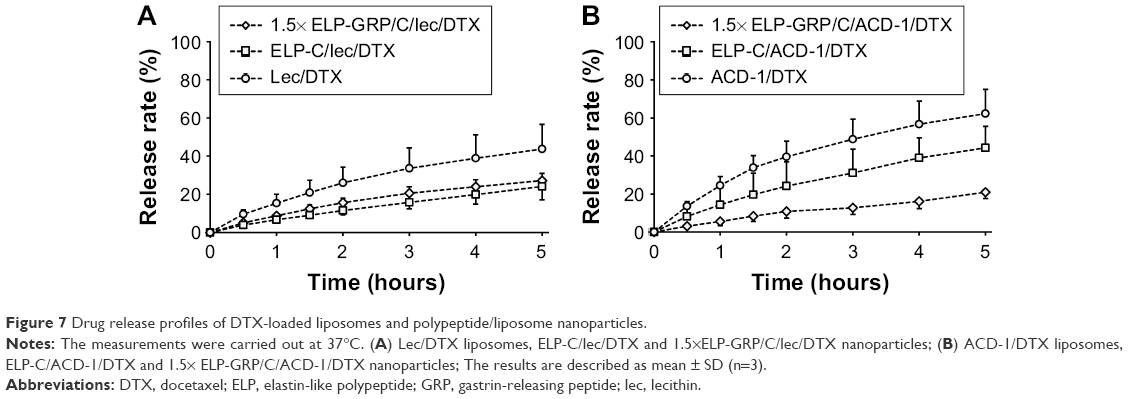 | Figure 7 Drug release profiles of DTX-loaded liposomes and polypeptide/liposome nanoparticles. |
The DTX release rates after 5 hours were 44% and 62% from lecithin and ACD-1 liposomes, respectively. The relatively large difference between these two formulations was assumed to be caused by weaker interactions between DTX and PEGylated bilayers in ACD-1 liposomes compared with lecithin.34,35,39–41,43,44
The release profiles significantly changed when DTX-loaded liposomes with lecithin or ACD-1 were incorporated into ELP-C or 1.5× ELP-GRP/C micelles. The cumulative percentage of drug release over 5 hours decreased to 24% and 27% for lecithin liposomes with ELP-C and ELP-GRP, respectively. The corresponding effects for ACD-1 liposomes were reductions to 44% and 21%. The encapsulation of the entire liposomes into hydrophobic ELP cores led to significantly reduced DTX release rates, most likely due to the additional polypeptide barriers outside of the liposomes. All over, prolonged retention times were achieved for DTX through the combination of polypeptide micelles with drug-loaded liposomes. These results were in line with earlier reports, which described ELPs as suitable carriers of hydrophobic drugs in vitro and in vivo.18
Intracellular ELP biodegradation by lysosomal elastases has been suggested earlier.46 In line with this, our results indicate that faster partitioning out of liposomes will occur in cells, once the ELP-GRP and ELP-C components have been degraded in lysosomal compartments.
Zeta potential
The surface net charges were determined by zeta potential (ζ) measurement to assess pharmacokinetic and biodistribution properties for future in vivo experiments.18,45 Generally, negative particles (ζ<−10 mV) exhibit strong reticuloendothelial system (RES) uptake triggered by plasma protein binding and opsonization causing rapid clearance from the blood circulation by Kupffer cells. Positive particles (ζ>+10 mV) are known to form aggregates in serum, and neutral nanoparticles (within ±10 mV) exhibit the least RES interaction, longest circulation, and reduced nonspecific cellular uptake.45 For the presented study, we aimed to produce neutral nanoparticles to avoid RES clearance and nonspecific cellular uptake.
Freshly prepared DTX-loaded ACD-1 or lecithin liposomes in PBS were mixed with ELP-C or 1.5× ELP-GRP/C preparations on ice. After incubation in a 37°C water bath for 15 minutes, samples were transferred to a Zetasizer Nano ZS for the analysis. The results are shown in Table 3. Zeta potentials of all nanoparticles were within a small range from −13.23 to 0.05 mV at 37°C, which was in correlation with the reported zeta potentials of lecithin, HSPC/MPEG-DSPE, DSPC, and DOPC lipids.47 The results indicated that the surface charges of DTX-loaded lipid nanoparticles were only marginally affected by the introduction of polypeptides. All further testings were conducted with the ACD-1-containing formulations based on the fact that the measured zeta potentials were almost neutral and the formulations contained PEGylated lipids. The expected benefits will be good stability in the circulation, limited nonspecific uptake, and optimal EPR effects due to the size of ~100 nm.
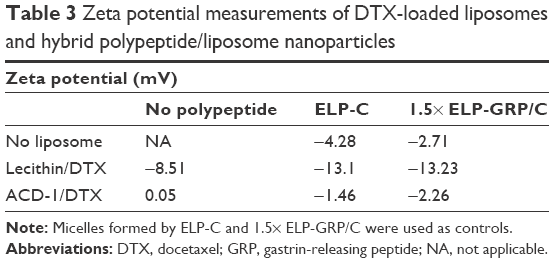 | Table 3 Zeta potential measurements of DTX-loaded liposomes and hybrid polypeptide/liposome nanoparticles |
Specific binding of drug-loaded ELP/liposome nanoparticles to prostate cancer cells
Specific binding of DTX-loaded polypeptide/liposome nanoparticles to PC-3 cells was analyzed by flow cytometry. For this purpose, fluorescence-labeled ELP-K (ELP-K-Atto) was mixed with ELP-GRP and ELP-C in a ratio of 2:3 to form detectable 1.5× ELP-GRP/K-Atto and 1.5× ELP-C/K-Atto nanoparticles, respectively. DTX-loaded ACD-1 liposomes were used in the next step to form ELP/liposome hybrid carriers (eg, 1.5× ELP-GRP/K-Atto/ACD-1/DTX and 1.5× ELP-C/K-Atto/ACD-1/DTX). Subsequently, PC-3 cells were harvested and resuspended in 880 μL of drug-loaded ELP/liposome nanoparticle solutions at 5×105 cells/mL followed by incubation in an orbital shaker at 37°C and 50 rpm for 1 hour. The cells were washed after the incubation and resuspended in 200 μL of PBS for flow cytometry analysis (Figure 8A and B). Untreated cells were used as controls to set an arbitrary fluorescence intensity cutoff for distinction between positive and negative cells with and without nanoparticle binding, respectively. Exposure to 1.5× ELP-GRP/K-Atto led to increased fluorescence in 7.5% of all PC-3 cells. In comparison, only 3.4% of cells were detected as positive when 1.5× ELP-C/K-Atto micelles were used. In correlation with this, 11.3% of PC-3 cells showed increased signals with 1.5× ELP-GRP/K-Atto/ACD-1/DTX nanoparticles, whilst only 4.6% of PC-3 cells were detected after treatment with 1.5× ELP-C/K-Atto/ACD-1/DTX nanoparticles. The results proved that the enhanced binding of nanoparticles to prostate cancer cells was mediated by GRP ligands. DU-145 prostate cancer cells express lower levels of GRPR than PC-3 cells, and in correlation with this, elevated fluorescence signals were detected in 4.2% and 3.9% of cells after exposure to 1.5× ELP-GRP/K-Atto and 1.5× ELP-GRP/K-Atto/ACD-1/DTX, respectively. In comparison, the controls showed only 0.4% of positive cells (Figure 8C and D). Finally, no significant differences were detected with GRPR-negative 293T cells when treated with ELP-GRP- and ELP-C-modified nanoparticles (Figure 8E and F). These results confirmed the specific binding of 1.5× ELP-GRP/K-Atto/ACD-1/DTX nanoparticles to GRPR expressing cell lines.
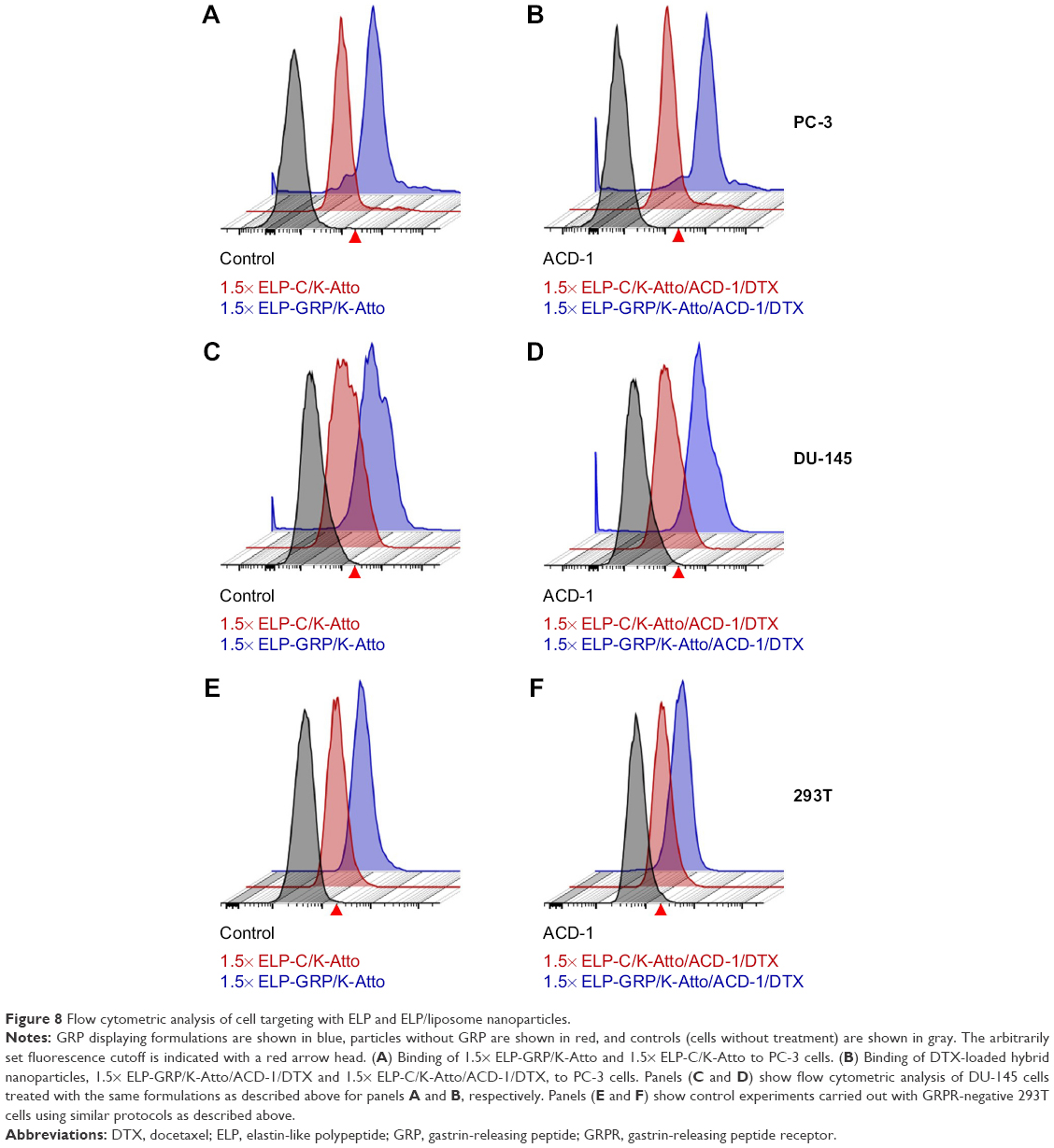 | Figure 8 Flow cytometric analysis of cell targeting with ELP and ELP/liposome nanoparticles. |
In vitro cytotoxicity study
The anticancer effects of DTX-loaded drug carriers were evaluated in cell viability studies with GRPR expressing PC-3 prostate cancer cells. The cells were seeded in 96-well plates for 24 hours and then exposed to 100 μL of DTX-loaded hybrid ELP/liposome nanoparticles at final polypeptide concentrations of 10 μM and DTX concentrations equivalent to 80 μg/mL. According to the above data (Figure 7), 21%–62% of encapsulated DTX was expected to be released within 5 hours. Therefore, to avoid the influence of free drug, the cells were washed with PBS after 4 hours and kept in fresh medium for an additional 20 hours. The viability of PC-3 cells was studied by MTT assay comparing free DTX, ACD-1 liposome encapsulated DTX or hybrid ELP/liposome encapsulated DTX (ELP-C/ACD-1 vs 1.5× ELP-GRP/C/ACD-1).
Treatment of PC-3 cells with free DTX resulted in a cell viability of 47% relative to untreated cells (Figure 9), indicating relative efficient nonspecific uptake of the free drug. In contrast, DTX encapsulation into ACD-1 liposomes and ELP-C/ACD-1 nanoparticles seemed to protect the cells from DTX and resulted in higher viabilities of 85% and 102%, respectively. These data strongly indicated that nanoparticles without GRP display were not taken up into the cells, and the effect seen with ACD-1 liposomes was most likely due to the release of DTX directly into the medium. Exposure to ELP-C/ACD-1/DTX particles did not show reduced cell viability and, in correlation with the results from Figure 7, this can be explained by the slower drug release from the ELP/liposome formulation in comparison with liposomes only. However, most importantly, the cell viabilities decreased to 76% when cells were treated with DTX-loaded 1.5× ELP-GRP/C/ACD-1 nanoparticles (p-value =0.0001), therefore, proving specific DTX delivery into PC-3 cells, whilst hollow liposomes and empty polypeptide micelles did not exhibit growth inhibition effects (data not shown). These relatively moderate, nevertheless, very significant effects correlated well with the flow cytometry data shown in Figure 8, leading to the assumption that only a small fraction of the PC-3 cells will actively take up the GRP displaying particles. It will be important for future in vivo studies to assess the GRPR expression in xenograft and allograft tumor models. Finally, this will also have potential implications for translational research. A detailed analysis of GRPR expression in tumor biopsies will be necessary. All over, the presented results demonstrated increased cytotoxicity to be linked to specific GRP-mediated binding and uptake of DTX-loaded 1.5× ELP-GRP/ELP-C/liposome nanoparticles into PC-3 cells. These findings further support the future development of GRP displaying nanoparticles as anticancer drug carriers to target GRPR- expressing prostate cancer cells.
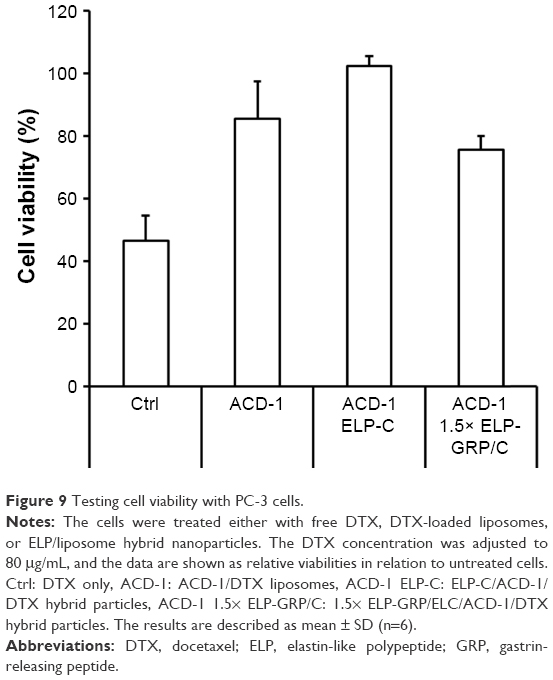 | Figure 9 Testing cell viability with PC-3 cells. |
Conclusion
Various hybrid ELP/liposome nanoparticles were developed which showed rapid self-assembly in PBS at physiological temperature, and DTX was successfully entrapped into the nanoparticles at high concentrations. The sizes of DTX-loaded nanoparticles ranged from ~40 to 200 nm. Interestingly, all hybrid nanoparticles showed monodisperse size distributions. This suggested that all liposomes were assembled at even distribution into the protein micelles during phase transition.
Various factors have to be taken into consideration for the optimal design of drug nanocarriers. For example, the RES consists of macrophages in the liver, spleen, and bone marrow. Nanoparticles are mainly cleared in liver and spleen at a rate that is strongly affected by size. Nanoparticles with a hydrodynamic diameter of ~100 nm are considered optimal to leverage between long serum time (eg, avoidance of RES clearance >50 nm) and sufficient tumor permeation. In addition to this, opsonization of nonspecifically adsorbed serum proteins (IgG and complement proteins) has been shown to drive rapid clearance from serum. To prevent this, nonspecific protein binding can be reduced by PEGylation and neutral zeta potential (ζ=±10 mV). Under these criteria, our ELP/ACD-1 hybrid particles were of most optimal size (~100 nm), contained PEGylated lipid components, and showed almost neutral zeta potential. Therefore, cell-binding studies were carried out with 1.5× ELP-GRP/C/ACD-1/DTX particles, and the potential for tumor retention by displayed GRP was demonstrated in vitro using PC-3 cells in flow cytometry experiments. In correlation with this, the same nanoparticles also significantly reduced cell viability of PC-3 cells. The presented report provides the proof of concept for active GRP-mediated targeting of PC-3 prostate cancer cells with self-assembling hybrid ELP/liposome nanoparticles.
Acknowledgment
This study was supported by China-Australia Centre for Health Sciences Research.
Disclosure
The authors report no conflicts of interest in this work.
References
Australian Institute of Health and Welfare (AIHW) 2017. Australian Cancer Incidence and Mortality (ACIM) books. Canberra: AIHW. Available from: http://www.aihw.gov.au//reports/cancer/acim-books>. Accessed June 15, 2017. | ||
Seidenfeld J, Samson DJ, Hasselblad V, et al. Single-therapy androgen suppression in men with advanced prostate cancer: a systematic review and meta-analysis. Ann Intern Med. 2000;132(7):566–577. | ||
Suzuki H, Okihara K, Miyake H, et al; Nonsteroidal Antiandrogen Sequential Alternation for Prostate Cancer Study Group. Alternative nonsteroidal antiandrogen therapy for advanced prostate cancer that relapsed after initial maximum androgen blockade. J Urol. 2008;180(3):921–927. | ||
Sternberg CN, Whelan P, Hetherington J, et al; Genitourinary Tract Group of the EORTC. Phase III trial of satraplatin, an oral platinum plus prednisone vs. prednisone alone in patients with hormone-refractory prostate cancer. Oncology. 2005;68(1):2–9. | ||
Kantoff PW, Halabi S, Conaway M, et al. Hydrocortisone with or without mitoxantrone in men with hormone-refractory prostate cancer: results of the cancer and leukemia group B 9182 study. J Clin Oncol. 1999;17(8):2506–2506. | ||
Storlie JA, Buckner JC, Wiseman GA, Burch PA, Hartmann LC, Richardson RL. Prostate specific antigen levels and clinical response to low dose dexamethasone for hormone-refractory metastatic prostate carcinoma. Cancer. 1995;76(1):96–100. | ||
Nishimura K, Nonomura N, Yasunaga Y, et al. Low doses of oral dexamethasone for hormone-refractory prostate carcinoma. Cancer. 2000;89(12):2570–2576. | ||
Saika T, Kusaka N, Tsushima T, et al; Okayama Urological Cancer Collaborating Group. Treatment of androgen-independent prostate cancer with dexamethasone: A prospective study in stage D2 patients. Int J Urol. 2001;8(6):290–294. | ||
Small EJ, Baron A, Bok R. Simultaneous antiandrogen withdrawal and treatment with ketoconazole and hydrocortisone in patients with advanced prostate carcinoma. Cancer. 1997;80(9):1755–1759. | ||
Keizman D, Huang P, Carducci MA, Eisenberger MA. Contemporary experience with ketoconazole in patients with metastatic castration-resistant prostate cancer: clinical factors associated with PSA response and disease progression. Prostate. 2012;72(4):461–467. | ||
Reid AH, Attard G, Danila DC, et al. Significant and sustained antitumor activity in post-docetaxel, castration-resistant prostate cancer with the CYP17 inhibitor abiraterone acetate. J Clin Oncol. 2010;28(9):1489–1495. | ||
Danila DC, Morris MJ, de Bono JS, et al. Phase II multicenter study of abiraterone acetate plus prednisone therapy in patients with docetaxel-treated castration-resistant prostate cancer. J Clin Oncol. 2010;28(9):1496–1501. | ||
O’Hanlon Brown C, Waxman J. Current management of prostate cancer: dilemmas and trials. Br J Radiol. 2012;85 Spec No 1:S28–S40. | ||
Yagoda A, Petrylak D. Cytotoxic chemotherapy for advanced hormone-resistant prostate cancer. Cancer. 1993;71(3 Suppl 1):1098–1109. | ||
Huwyler J, Drewe J, Krähenbühl S. Tumor targeting using liposomal antineoplastic drugs. Int J Nanomedicine. 2008;3(1):21–29. | ||
Dreher MR, Raucher D, Balu N, Michael Colvin O, Ludeman SM, Chilkoti A. Evaluation of an elastin-like polypeptide–doxorubicin conjugate for cancer therapy. J Control Release. 2003;91(1–2):31–43. | ||
Furgeson DY, Dreher MR, Chilkoti A. Structural optimization of a “smart” doxorubicin-polypeptide conjugate for thermally targeted delivery to solid tumors. J Control Release. 2006;110(2):362–369. | ||
Shi P, Aluri S, Lin YA, et al. Elastin-based protein polymer nanoparticles carrying drug at both corona and core suppress tumor growth in vivo. J Control Release. 2013;171(3):330–338. | ||
Kim MS, Lee DW, Park K, et al. Temperature-triggered tumor-specific delivery of anticancer agents by cRGD-conjugated thermosensitive liposomes. Colloids Surf B Biointerfaces. 2014;116:17–25. | ||
Lee H, Kim HR, Larson RG, Park JC. Effects of the size, shape, and structural transition of thermosensitive polypeptides on the stability of lipid bilayers and liposomes. Macromolecules. 2012;45(17):7304–7312. | ||
Lee H, Kim HR, Park JC. Dynamics and stability of lipid bilayers modulated by thermosensitive polypeptides, cholesterols, and PEGylated lipids. Phys Chem Chem Phys. 2014;16(8):3763–3770. | ||
Na K, Lee SA, Jung SH, Hyun J, Shin BC. Elastin-like polypeptide modified liposomes for enhancing cellular uptake into tumor cells. Colloids Surf B Biointerfaces. 2012;91:130–136. | ||
Park SM, Cha JM, Nam J, et al. Formulation optimization and in vivo proof-of-concept study of thermosensitive liposomes balanced by phospholipid, elastin-like polypeptide, and cholesterol. PLoS One. 2014;9(7):e103116. | ||
Park SM, Kim MS, Park SJ, et al. Novel temperature-triggered liposome with high stability: formulation, in vitro evaluation, and in vivo study combined with high-intensity focused ultrasound (HIFU). J Control Release. 2013;170(3):373–379. | ||
Zhang W, Garg S, Eldi P, et al. Targeting prostate cancer cells with genetically engineered polypeptide-based micelles displaying gastrin-releasing peptide. Int J Pharm. 2016;513(1):270–279. | ||
Dua JS, Rana AC, Bhandari AK. Liposome: methods of preparation and applications. Int J Pharm Stud Res. 2012;3:14–20. | ||
Ding T, Xue Y, Lu H, Huang Z, Sun J. Effect of particle size of hydroxyapatite nanoparticles on its biocompatibility. IEEE Trans Nanobioscience. 2012;11(4):336–340. | ||
Kalepu S, Kumar TS, Betha S, Mohan M. Liposomal drug delivery system – a comprehensive review. Int J Drug Dev Res. 2013;5(4):62–75. | ||
Yang ZZ, Li JQ, Wang ZZ, Dong DW, Qi XR. Tumor-targeting dual peptides-modified cationic liposomes for delivery of siRNA and docetaxel to gliomas. Biomaterials. 2014;35(19):5226–5239. | ||
Liu Z, Liu D, Wang L, Zhang J, Zhang N. Docetaxel-loaded pluronic p123 polymeric micelles: in vitro and in vivo evaluation. Int J Mol Sci. 2011;12(3):1684–1696. | ||
Sanna V, Roggio AM, Posadino AM, et al. Novel docetaxel-loaded nanoparticles based on poly(lactide-co-caprolactone) and poly(lactide-co-glycolide-co-caprolactone) for prostate cancer treatment: formulation, characterization, and cytotoxicity studies. Nanoscale Res Lett. 2011;6(1):260–268. | ||
Yeboah A, Cohen RI, Rabolli C, Yarmush ML, Berthiaume F. Elastin-like polypeptides: a strategic fusion partner for biologics. Biotechnol Bioeng. 2016;113(8):1617–1627. | ||
Meyer DE, Chilkoti A. Protein purification by inverse transition cycling. In: Golemis EA, Adams PD (Editors). Protein-Protein Interactions: A Molecular Cloning Manual. Cold Spring Harbor, N.Y.: Cold Spring Harbor Laboratory Press. 2002; Chapter 18: 329–343. | ||
Immordino ML, Brusa P, Arpicco S, Stella B, Dosio F, Cattel L. Preparation, characterization, cytotoxicity and pharmacokinetics of liposomes containing docetaxel. J Control Release. 2003;91(3):417–429. | ||
Pereira S, Egbu R, Jannati G, Al-Jamal WT. Docetaxel-loaded liposomes: The effect of lipid composition and purification on drug encapsulation and in vitro toxicity. Int J Pharm. 2016;514(1):150–159. | ||
Naguib YW, Rodriguez BL, Li X, Hursting SD, Williams RO 3rd, Cui Z. Solid lipid nanoparticle formulations of docetaxel prepared with high melting point triglycerides: in vitro and in vivo evaluation. Mol Pharm. 2014;11(4):1239–1249. | ||
Ravi S, Krishnamurthy VR, Caves JM, Haller CA, Chaikof EL. Maleimide-thiol coupling of a bioactive peptide to an elastin-like protein polymer. Acta Biomater. 2012;8(2):627–635. | ||
Cassinello J, Carballido Rodriguez J, Anton Aparicio L. Role of taxanes in advanced prostate cancer. Clin Transl Oncol. 2016;18(10):972–980. | ||
Raffy S, Teissie´ J. Control of lipid membrane stability by cholesterol content. Biophys J. 1999;76(4):2072–2080. | ||
Soema PC, Willems GJ, Jiskoot W, Amorij JP, Kersten GF. Predicting the influence of liposomal lipid composition on liposome size, zeta potential and liposome-induced dendritic cell maturation using a design of experiments approach. Eur J Pharm Biopharm. 2015;94:427–435. | ||
Zhang JA, Anyarambhatla G, Ma L, et al. Development and characterization of a novel Cremophor EL free liposome-based paclitaxel (LEP-ETU) formulation. Eur J Pharm Biopharm. 2005;59(1):177–187. | ||
Liu DZ, Tseng LP, Liang HJ, Chung TW, Huang YY. Liposomes incorporated with cholesterol for drug release triggered by magnetic field. J Med Biol Eng. 2007;27:29–34. | ||
Funakoshi Y, Iwao Y, Noguchi S, Itai S. Effect of alkyl chain length and unsaturation of the phospholipid on the physicochemical properties of lipid nanoparticles. Chem Pharm Bull (Tokyo). 2015;63(9):731–736. | ||
Naik S, Patel D, Surti N, Misra A. Preparation of PEGylated liposomes of docetaxel using supercritical fluid technology. J Supercritical Fluids. 2010;54(1):110–119. | ||
Ernsting MJ, Murakami M, Roy A, Li SD. Factors controlling the pharmacokinetics, biodistribution and intratumoral penetration of nanoparticles. J Control Release. 2013;172(3):782–794. | ||
Shah M, Hsueh PY, Sun G, Chang HY, Janib SM, MacKay JA. Biodegradation of elastin-like polypeptide nanoparticles. Protein Sci. 2012;21(6):743–750. | ||
Szoka F Jr, Papahadjopoulos D. Comparative properties and methods of preparation of lipid vesicles (liposomes). Ann Rev Biophys Bioeng. 1980;9:467–508. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.