Back to Journals » Therapeutics and Clinical Risk Management » Volume 14
Targeting p19 as a treatment option for psoriasis: an evidence-based review of guselkumab
Authors Wechter T, Cline A, Feldman SR ![]()
Received 14 June 2018
Accepted for publication 5 August 2018
Published 22 August 2018 Volume 2018:14 Pages 1489—1497
DOI https://doi.org/10.2147/TCRM.S177127
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Garry Walsh
Todd Wechter,1 Abigail Cline,2 Steven R Feldman2–4
1Stony Brook Medicine, Stony Brook, NY, USA; 2Center for Dermatology Research, Department of Dermatology, Wake Forest School of Medicine, Winston-Salem, NC, USA; 3Department of Pathology, Wake Forest School of Medicine, Winston-Salem, NC, USA; 4Department of Public Health Sciences, Wake Forest School of Medicine, Winston-Salem, NC, USA
Abstract: Further understanding of psoriasis pathogenesis has led to the development of effective biologic medications. Guselkumab (GUS) is a subcutaneously administered monoclonal antibody that targets the p19 cytokine subunit in IL-23 and IL-39 and is US Food and Drug Administration (FDA) approved for the treatment of moderate-to-severe psoriasis in adult patients. This review evaluates the pharmacology, safety and efficacy of GUS in patients with psoriasis. We performed a literature review by searching online databases including PubMed and Google Scholar. In clinical trials, GUS improved diseases including psoriatic arthritis (PsA) and specific areas of disease (scalp, feet, hands and fingernails). In the Phase III trials VOYAGE 1 and 2, more GUS than adalimumab (ADM) patients experienced a ≥90% reduction in Psoriasis Area and Severity Index (PASI) score (PASI90) (VOYAGE 1: 80.2% vs 53.0%; VOYAGE 2: 75.2% vs 54.8%; P<0.001 for both) and Investigator Global Assessment score of 0 or 1 (VOYAGE 1: 84.2% vs 61.7%; VOAYGE 2: 83.5% vs 64.9%; P<0.001 for both) at Week 24. GUS was also successful in treating patients unresponsive to ADM and ustekinumab in the VOYAGE 2 and NAVIGATE trials, respectively. While long-term data are necessary, GUS appears to have a favorable side effect profile with most common adverse effects including nasopharyngitis and upper respiratory tract infections. GUS is a well-tolerated and effective medication for patients with psoriasis. Continued study of GUS and the p19 subunit will help to determine GUS’s ultimate place in therapy.
Keywords: biologics, IL-23, IL-39, monoclonal antibody
Introduction
Psoriasis is an immune disease with an estimated prevalence of ~3% in the United States.1 It is typically diagnosed visually by its characteristic erythematous scaly plaques distributed on the scalp, torso, extensor surfaces and/or throughout the rest of the body.2 The disease greatly reduces patient’s quality of life and is associated with various psychiatric comorbidities including anxiety, depression and suicidality.3,4 Psoriasis can also cause debilitating arthritis and has been linked to numerous systemic pathologies including cardiovascular disease, inflammatory bowel disease and metabolic syndrome.5–7
When deciding on therapeutic options for psoriasis, considerations include disease severity, disease location, joint involvement, cost profile and patient preference.8 Treatments for psoriasis range from topical to systemic medications and include steroids, phototherapy, vitamin A and D derivatives, tars, immunosuppressants and biologics.8 Despite these numerous options, psoriasis can be a difficult disease to treat, and continued investigation is ongoing to discover additional safe and effective interventions.
Biologic medications have transformed the landscape of treatment for moderate-to-severe plaque psoriasis, allowing for better disease control.9 Cytokines and inflammatory mediators involved in the pathogenesis of plaque psoriasis can now be targeted, with some examples of biologics and their targets including adalimumab (ADM; TNF-alpha), ixekizumab (IL-17) and ustekinumab (USM; IL-12/IL-23; Figure 1).10,11 The search for better psoriasis treatments is now focusing on the IL-23/IL-17 pathway, including the two subunits of IL-23, p40 and p19.12–14 The p40 subunit is shared with IL-12, while the p19 subunit is present in IL-23 and not in IL-12 (Figure 1).14,15 USM targets the common p40 subunit, while guselkumab (GUS) targets p19 and as a result IL-23 and not IL-12 (Figure 1).15,16 IL-23 is a cytokine thought to play a significant role in the pathogenesis of the disease, as it is present at high levels both in the serum and plaques of patients with psoriasis.12,17,18 IL-23 induces the proliferation of proinflammatory Th17 cells, which are key drivers of psoriasis development.19,20 In late 2017, the US Food and Drug Administration (FDA) approved and released GUS to the market. Here, we review the pharmacology, safety and efficacy of GUS in adult patients with moderate-to-severe plaque psoriasis.
 | Figure 1 IL-12, IL-23 and IL-39 with their receptors and downstream effects. |
Pharmacodynamics, pharmacokinetics and immunogenicity
GUS is a subcutaneously (SQ) injected human monoclonal antibody that targets IL-23 by binding the IL-23 p19 subunit (Figure 1).21 IL-23 is a member of a heterodimeric family of cytokines, which also includes IL-12, IL-27, IL-35 and IL-39.22,23 IL-12 and IL-23 are proinflammatory cytokines that drive psoriasis pathogenesis, IL-27 and IL-35 are inhibitory cytokines, and the role of IL-39 in psoriasis is unclear.12,22,23 Within the IL-12 cytokine family, IL-23 and IL-39 contain the p19 subunit.15,22 Variations in the genes encoding p19 and the p19 receptor, IL-23R, are associated with an increased risk of psoriasis, thus highlighting the role of p19 in psoriasis pathogenesis.21,24,25 GUS binds to the IL-23 p19 subunit and prevents IL-23 from binding to IL-23R on the surface of various innate and adaptive immune cells.12,15,21 Through stopping the p19 subunit from binding to IL-23R, the IL-23/Th17 pathway is inhibited, thus reducing its proinflammatory effects.12,21
A Phase I randomized, placebo-controlled clinical trial examined the pharmacokinetics and pharmacodynamics of GUS in 47 healthy participants and 24 participants with moderate-to-severe plaque psoriasis.26 Healthy participants either received a single GUS intravenous (IV) administration (0.03, 0.1, 0.3, 1, 3 or 10 mg/kg), a single GUS SQ injection (3 mg/kg) or placebo treatment.26 Patients with moderate-to-severe plaque psoriasis either received a single GUS SQ injection (10, 30, 100 or 300 mg) or placebo treatment.26
In healthy participants treated with GUS IV, area under the curve for serum drug concentration vs time (AUC0–∞) and peak serum concentration (Cmax) increases were dose dependent, with median AUC0–∞ values ranging from 4.93 to 2,261.8 μg·day/mL and median Cmax values ranging from 0.47 to 200.36 μg/mL.26 In addition, in this cohort, median terminal half-life (t1/2) of the drug ranged from 12.5 to 19.5 days, median clearance ranged from 3.58 to 6.1 mL/day/kg and median volume of distribution (Vz) ranged from 97.74 to 117.88 mL/kg.26 For healthy participants receiving GUS SQ, the mean AUC0–∞ was 256.99 μg·day/mL and the mean Cmax was 9.46 μg/mL.26
SQ administration in the psoriasis cohort also had dose-dependent AUC0–∞ and Cmax increases, with median AUC0–∞ values ranging from 15.11 to 574.62 μg·day/mL and median Cmax values ranging from 0.5 to 22.7 μg/mL.26 Median t1/2 values in this cohort ranged from 15.8 to 17.8 days.26
Throughout the clinical trials examining the immunogenicity of GUS in psoriasis patients, the proportion of patients developing antibodies to GUS ranged from 4% to 9%.15,26–29 These antibodies were generally present in low titers, and their development did not affect the efficacy of the treatment or the incidence of injection site reactions.15,27–29
Treatment efficacy
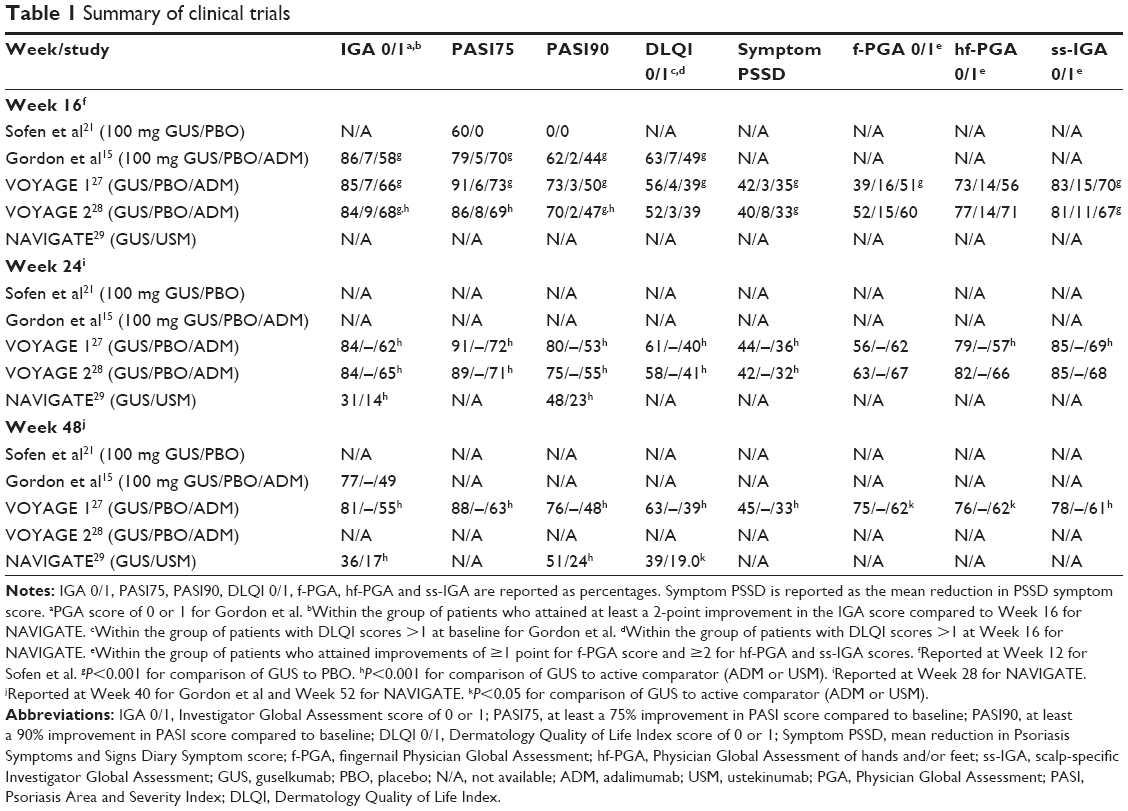
An initial Phase I clinical trial investigated a single GUS SQ administration in 24 adult patients with moderate-to-severe plaque psoriasis.21 At 12 weeks post injection, 50%, 60%, 60% and 100% of 10, 30, 100 and 300 mg GUS-treated patients, respectively, experienced at least a 75% reduction in Psoriasis Area and Severity Index (PASI) score (PASI75; Table 1).21 Comparatively, no patients in the placebo-treated cohort attained this end point (Table 1).21 These results were predominantly stable through 24 weeks post injection, although some loss of efficacy was noted in the 100 mg treated cohort.21 On immunohistochemical evaluation, GUS reduced lesional epidermal thickness and CD3 T-cell and myeloid dendritic cell infiltration (P<0.05 compared to baseline, except for myeloid dendritic cell infiltration with 10 mg GUS; P=0.072).21 Compared to baseline values, GUS reduced serum IL-17A levels in responsive patients at Week 1 (P=0.031) and Week 12 (P=0.0015).21 The placebo-treated cohort had no reduction in epidermal thickness, CD3 T-cell infiltration or serum IL-17A concentration, although there was a decrease in lesional myeloid dendritic cell infiltration compared to baseline (P=0.028).21
 | Table 1 Summary of clinical trials |
A Phase II randomized, double-blinded, placebo-controlled trial compared GUS to ADM in 293 patients with moderate-to-severe plaque psoriasis over 52 weeks.15 The ADM cohort received standard dosing, while the GUS cohort received either 5, 50, or 200 mg at 0 and 4 weeks and then every 12 weeks or 15 or 100 mg every 8 weeks.15 At Week 16, the placebo-treated cohort crossed over to 100 mg GUS with 8-week dosing intervals.15 By Week 16, 34%, 61%, 79%, 86% and 83% of 5, 15, 50, 100 and 200 mg GUS-treated cohorts reached a Physician Global Assessment (PGA) score of 0 or 1 (Table 1).15 Comparatively, 7% of placebo and 58% of the ADM cohort achieved this end point (Table 1).15 In addition, at Week 16, ≥90% PASI score improvement (PASI90) was seen in 34%, 34%, 45%, 62% and 57% of 5, 15, 50, 100 and 200 mg GUS-treated cohorts, respectively, compared to 2% of placebo and 44% of the ADM group (Table 1).15 Disease improvement with GUS persisted through Week 40, as most dosage subgroups experienced relative preservation of PGA 0 or 1 and PASI75.15 Some loss of efficacy was observed as the time of the next scheduled GUS injection approached and was seen more often when dosed every 12 weeks as opposed to every 8 weeks.15 Crossover from placebo to GUS also had improved PASI scores and an increased number of patients achieving PGA 0 or 1.15
A Phase IIa, randomized trial investigated GUS for the treatment of psoriatic arthritis (PsA) in 149 patients.30 Patients received either GUS 100 mg at 0 and 4 weeks and then every 8 weeks or placebo.30 At Week 24, more GUS-treated patients experienced 20% (58.0% vs 18.4%; P<0.001), 50% (34.0% vs 10.2%; P=0.002) and 70% (14.0% vs 2.0%; P=0.023 [post hoc]) American College of Rheumatology score improvements compared to the placebo group.30 Furthermore, greater improvements in multiple psoriatic arthropathy measures – such as Leeds Enthesitis Index (median percentage change from baseline: −100.0% vs −33.3%; P=0.009), Dactylitis Score (median percentage change from baseline: −100.0% vs −33.3%; P<0.001) and Psoriatic Arthritis Disease Activity Score (mean change from baseline: −2.50 to −0.49; P<0.001) – were observed in the GUS-treated cohort compared to placebo.30
In VOYAGE 1, the first of three Phase III randomized, double-blinded, placebo-controlled GUS clinical trials, GUS was generally more effective than placebo and ADM.27 A total of 837 patients were randomized to one of three treatment groups, including GUS (100 mg at 0 and 4 weeks and then every 8 weeks), ADM (standard dosing) or placebo, followed over 48 weeks.27 The placebo group was crossed over to GUS 16 weeks into the study.27
At Week 16, 85.1% of GUS patients had an Investigator Global Assessment (IGA) score of 0 or 1, compared to 65.9% and 6.9% of ADM- and placebo-treated groups, respectively (P<0.001 compared to the placebo group; Table 1).27 Furthermore, 73% of GUS patients experienced PASI90 at Week 16 compared to 3% of the placebo group and 50% of the ADM group (P<0.001 compared to the placebo group; Table 1).27 At Week 24, more GUS patients than ADM patients had an IGA score of 0 or 1 (84.2% vs 61.7, respectively; P<0.001) and attained PASI90 (80.2% vs 53.0%, respectively; P<0.001; Table 1).27 GUS treatment response was persistent, with improved PASI and IGA scores through Week 48.27 Crossover from placebo to GUS improved disease measures comparable to measures seen in the original GUS cohort.27
VOYAGE 2 differed from the VOYAGE 1 study as it examined noncontinuous GUS treatment and GUS treatment for patients unresponsive to ADM injection.28 The multicenter, randomized, double-blinded, placebo-controlled study included 992 subjects.28 At Week 28, the “randomized withdrawal and retreatment” segment began.28 Responsive patients were those who attained PASI90.28 Unresponsive patients in the original GUS cohort continued their current regimen, while responders were re-randomized into one of two groups – one group that continued GUS 100 mg every 8 weeks or another group that received placebo until a defined loss of efficacy, at which point they were reinitiated on GUS.28 In the placebo-to-GUS crossover cohort, unresponsive patients continued their current regimen, while responders received placebo until a defined loss of efficacy was observed and were then reinitiated on GUS.28 Patients unresponsive to ADM were converted to GUS, while patients responsive to ADM received placebo until a defined loss of efficacy was observed and were then initiated on GUS.28 At Week 16, 84% of patients in the initial GUS cohort had IGA scores of 0 or 1 compared to 68% and 9% of the ADM- and placebo-treated cohorts, respectively (P<0.001 for both; Table 1).28 Similarly, at Week 16, 70.0% of patients in the GUS cohort reached PASI90 compared to 46.8% of ADM-treated group or 2.4% of placebo-treated group (P<0.001 for both; Table 1).28 Of patients who started on and continued GUS to Week 28, 19.9% were classified as nonresponders compared to 47.3% of the ADM cohort.28 Overall, continuous GUS attained better clinical responses than noncontinuous treatment.28 At Week 48, 89% of patients receiving continuous GUS experienced PASI90 compared to 37% of patients in the placebo re-randomized cohort (P<0.001).28 For patients responsive to GUS and re-randomized to placebo, loss of PASI90 was observed in a median time of 15.2 weeks.28 GUS also effectively treated patients unresponsive to ADM.28 At the end of the study, 66.1% of patients initially unresponsive to ADM who were converted to GUS attained PASI90.28
In both VOYAGE 1 and VOYAGE 2, GUS improved specific areas of disease, including the scalp, feet, hands and fingernails.27,28 At Week 16 of both studies, more GUS-treated patients than placebo-treated patients attained a fingernail Physician Global Assessment (f-PGA) score of 0 or 1 (39.1%/52.0% vs 15.9%/14.6%), Physician Global Assessment of hands and/or feet (hf-PGA) score of 0 or 1 (73.3%/77.2% vs 14.0%/14.3%) and scalp-specific Investigator Global Assessment (ss-IGA) score of 0 or 1 (83.4%/80.6% vs 14.5%/10.9%; P<0.001 for both; Table 1).27,28 At Week 24 of both studies, more GUS patients than ADM patients had an hf-PGA score of 0 or 1 (78.9%/81.6% vs 56.8%/66.1%) and ss-IGA score of 0 or 1 (84.5%/85.3% vs 69.2%/67.5%; Table 1).27,28 At Week 24 of VOYAGE 1, less GUS patients than ADM patients attained f-PGA scores of 0 or 1 (56.3% vs 62.4%), although at Week 48, more GUS patients than ADM patients achieved this measure (74.7% vs 61.8%; P=0.038; Table 1).27 Similarly, at Week 24 of VOYAGE 2, less GUS patients than ADM patients had an f-PGA score of 0 or 1 (62.6% vs 66.9%; Table 1).28
A subsequent study combined the data from VOYAGE 1 and VOYAGE 2 to determine if specific patient populations had distinctive responses to GUS.31 Patients were stratified by age, gender, weight, body mass index, ethnicity, pretreatment disease features and previous treatments.31 Overall, in almost all the examined subgroups, GUS had greater efficacy than placebo and ADM.31
The final Phase III trial, NAVIGATE, examined GUS safety and efficacy for patients unresponsive to USM.29 In the initial 16-week open-label segment of the study, patients received USM at Weeks 0 and 4.29 At Week 16, patients with IGA scores of 0 or 1 were considered responsive and remained on USM, while patients with IGA scores of ≥2 were considered unresponsive and randomized to either receive GUS 100 mg at Weeks 16 and 20 and then every 8 weeks or continue USM.29 Overall, patients on GUS had better clinical outcomes than patients in the randomized USM group.29 The primary end point of the study was the number of patient visits with recorded IGA scores of 0 or 1 and with a minimum 2-point IGA score improvement from Week 16 scores.29 From Week 28 to Week 40, the GUS-treated cohort achieved a greater average number of these visits than the randomized USM cohort (1.5 vs 0.7 visits; P≤0.001).29 In addition, from Week 28 to Week 40, GUS-treated patients had a greater average number of visits with PASI90 compared to randomized USM patients (2.2 vs 1.1 visits; P≤0.001).29 GUS remained more effective than randomized USM through Week 52, with 51.1% of GUS patients compared to 24.1% of randomized USM patients reaching PASI90 (P<0.001; Table 1).29
Safety and adverse events (AEs)
Side effects of GUS are typically mild, and the medication is well tolerated.15,21,26–29 A total of 48%–74% of patients treated with GUS experience at least one AE, similar to the frequency of AE seen in ADM treatment.15,21,26–29 In any clinical trial, <3% of patients receiving GUS discontinued their treatment due to AEs and <7% experienced serious AEs.15,21,26–29 The most common AE of GUS is infection, most frequently nasopharyngitis and upper respiratory tract infections.15,21,26–29 The majority of infections observed did not require treatment, and serious infections were generally rare.15,21,26–29 No opportunistic infections, active tuberculosis (TB) infections or hypersensitivity reactions were noted for GUS patients in any reported studies, and of 105 patients with latent TB who were administered both TB prophylaxis and GUS, none experienced disease reactivation.15,21,26–29,32 Other less common AEs included injection site reaction, headache, arthralgia, pruritus, cardiovascular events and cancer.15,21,26–29 In the NAVIGATE trial, a higher rate of musculoskeletal AE, such as PsA, was noted with GUS treatment compared to USM treatment.29 Throughout all the described clinical trials, <2% of GUS patients were noted to have cardiovascular events compared to none in the placebo groups.15,21,26–29 Rates of cardiovascular events were generally comparable between GUS and ADM.15,21,27,28 Malignancies observed in GUS-treated groups included nonmelanoma skin cancer, prostate cancer, breast cancer, bladder cancer, grade 3 cervical intraepithelial neoplasia and squamous cell carcinoma of the head and neck.15,27–29
Patient-focused perspectives
GUS improved quality of life end points.15,27–30,33 A Phase II clinical trial measured GUS treatment on patients’ quality of life with the Dermatology Quality of Life Index (DLQI).15 At Week 16, GUS at any of the five administered dosages (5, 15, 50, 100 and 200 mg) improved DLQI scores compared to placebo (P≤0.01).15 DLQI scores of 0 or 1 through the first 16 weeks of the study were observed in 26%, 34%, 42%, 63% and 70% of the 5, 15, 50, 100 and 200 mg GUS cohorts, respectively, compared to 7% of placebo and 49% of the ADM cohort (P<0.001 for comparison to placebo; Table 1).15 In the Phase IIa study examining GUS for the treatment of PsA, GUS improved more quality of life measures than placebo, including mean change from baseline in Medical Outcomes Study 36-Item Short Form (SF-36) Physical (6.59 vs 0.46, respectively; P<0.001) and Mental (4.95 vs 0.42, respectively; P=0.002) Component Scores and Health Assessment Questionnaire without Disability Index (HAQ-DI) scores (−0.42 to −0.06; P<0.001).30
VOYAGE 1 added the Psoriasis Symptoms and Signs Diary (PSSD) as a quality of life measure.27 Through Week 16, 56.3% of the GUS group attained a DLQI score of 0 or 1 compared to 4.2% of the placebo group (P<0.001; Table 1).27 GUS improved PSSD symptom scores compared to placebo at Week 16 (41.9±24.6 vs 3.0±19.6, respectively; P<0.001; Table 1).27 Furthermore, GUS had a greater impact on these measures compared to ADM through Week 48 (62.5% vs 38.9%, respectively, achieving DLQI scores of 0 or 1 and mean PSSD symptom score reductions of 45.3±25.5 vs 32.5±31.1, respectively; P<0.001 for both; Table 1).27
In VOYAGE 2, GUS improved quality of life measures more than placebo and ADM treatments.28 At Week 16, 51.7% of GUS patients achieved a DLQI score of 0 or 1, with a mean PSSD symptom score reduction of 40.4±26.5 (Table 1).28 Comparatively, also at Week 16, 3.3% of the placebo group and 39.0% of the ADM group attained a DLQI score of 0 or 1, with mean PSSD symptom score reductions of 8.3±23.7 and 32.8±24.9, respectively (Table 1).28 Through Week 24, more GUS-treated patients than ADM-treated patients attained DLQI scores of 0 or 1 (57.6% vs 41.1%, respectively; P<0.001; Table 1).28 Mean PSSD symptom score reductions at Week 24 were also greater for GUS than ADM (42.1±26.8 vs 31.9±27.0, respectively; P<0.001; Table 1).28 Through Week 48, continuous GUS treatment improved DLQI and PSSD scores more than noncontinuous treatment (P<0.001 for both).28
Another VOYAGE 2 analysis investigated the impact of GUS on anxiety and depression by utilizing the Hospital Anxiety and Depression Scale (HADS).33 Through 16 weeks of the study, GUS improved both mean anxiety (HADS-A) and depression (HADS-D) HADS subcomponent scores than placebo (HADS-A: −1.7±3.4 to −0.2±2.9; HADS-D: −1.6±3.6 to −0.1±2.9; P<0.001 for both).33 Through Week 24, GUS reduced mean HADS-A and HADS-D scores more than ADM (HADS-A: −2.0±3.6 to −1.0±3.6; P<0.001; HADS-D: −1.7±3.8 to −1.1±3.5; P=0.6), indicating greater improvements in anxiety and depression, although the difference in mean HADS-D scores between the two groups was not statistically significant (P=0.06).33 Furthermore, at Week 16, a higher percentage of GUS-treated patients with HADS scores consistent with anxiety and depression attained HADS scores below the defined threshold for mild anxiety and depression compared to placebo-treated patients (HADS-A: 51.4% vs 25.9%, respectively; HADS-D: 59.2% vs 27.0%, respectively; P<0.001 for both).33 Similarly, at Week 24, a higher percentage of GUS-treated patients compared to ADM-treated patients reached HADS scores below this threshold (HADS-A: 58.4% vs 42.9%, respectively; P=0.028; HADS-D: 59.8% vs 46.4%, respectively; P=0.079), although the difference between the two groups was not statistically significant for the depression specific HADS-D measure (P=0.079).33
In NAVIGATE, GUS had better DLQI and PSSD score improvements compared to randomized USM.29 At Week 52, within the group of patients with DLQI scores >1 at Week 16, 38.8% of GUS patients achieved DLQI scores of 0 or 1 compared to 19% of patients in the randomized USM group (P=0.002; Table 1).29 Similarly, at Week 52, among patients who had PSSD symptom scores >0 at Week 16, 20.3% of patients in the GUS cohort achieved a PSSD symptom score of 0 compared to 9.5% of patients in the randomized USM cohort (P<0.05).29
Discussion
Overall, GUS is an excellent treatment option for adult patients with moderate-to-severe plaque psoriasis. It targets the p19 cytokine subunit, which is present in the IL-23 and IL-39 members of the IL-12 heterodimeric cytokine family, but not in IL-12 (Figure 1).14,15,22,23 For comparison, USM targets the p40 cytokine subunit, which is present in both IL-23 and IL-12 (Figure 1).16 IL-23 plays a crucial role in psoriasis pathogenesis, and targeting IL-23 through the p19 subunit is an effective treatment strategy.15,21,26–29
GUS provides effective disease control and is more effective than the established TNF-alpha inhibitor ADM.15,27,28 Furthermore, GUS’s success in treating patients with incomplete responses to ADM and USM highlights its role as an alternative treatment option for patients failing initial biologic medications.28,29 The effectiveness of GUS in treating scalp, hand, feet and fingernail diseases also suggests that it is a good option for patients with refractory disease in these difficult treat areas.27,28 In the Phase IIa clinical trial, GUS was effective in treating PsA.30 For patients with PsA, TNF-alpha inhibitors are generally first-line options, with IL-17 inhibitors and USM as alternatives.34 GUS is a good option for moderate-to-severe psoriasis whether patients have PsA or not and may be a reasonable treatment option for PsA patients failing TNF-alpha inhibitors.
While there are no clinical trials that directly compare GUS to IL-17 inhibitors, they appear to have similar efficacies. At Week 12 of Phase III clinical trials, the IL-17A antagonists ixekizumab and secukinumab had 70.7% and 59.2% PASI90 response rates, respectively, while the IL-17A receptor antagonist brodalumab had a 70.3% PASI90 response rate.35–37 These efficacies are comparable to GUS, which had a 73.3% PASI90 response rate at Week 16 of the Phase III VOYAGE 1 trial.27
Even though both GUS and USM block IL-23, they have different efficacies.29 One potential explanation is that IL-12 may have protective effects in psoriasis.38 In one study, imiquimod-treated mice lacking IL-12 signaling components developed worse psoriasis compared to wild-type mice.38 Therefore, inhibition of IL-12 may impair therapeutic efficacy in psoriasis.38 Another possibility is that other p19 cytokines, such as IL-39, play a role in psoriasis pathogenesis. GUS’s effects might extend beyond inhibition of IL-23 through the p19 subunit and include effects from blocking other p19 cytokines. While more complete blockage of p19 may have increased efficacy, it may also produce additional side effects.
USM is a very safe medication even when used long term, which suggests that inhibiting the IL-12/IL-23 pathway produces a favorable side effect profile.39 GUS appears to also have a favorable side effect profile, although long-term data are necessary to fully evaluate its safety.15,21,26–29 Considering that GUS inhibits IL-23 without blocking IL-12, many people believe that it should be at least as safe as USM; such assumptions are not warranted, as the immune system is inscrutable. Moreover, because GUS inhibits IL-39, a cytokine not inhibited by USM, in addition to IL-23, it seems inappropriate to assume that USM’s excellent safety profile necessarily extends to GUS. The natural experiment of genetic defects in p19 expression is reassuring. In one study, mice without IL-23 p19 had impaired T-cell immune responses with otherwise normal development.40
While IL-17 antagonists exacerbate inflammatory bowel disease,41 biologics antagonizing IL-23 (including an IL12/23 blocker and an IL23/39 blocker) improve inflammatory bowel disease.42,43 As a result, GUS may be a good choice for patients with both moderate-to-severe plaque psoriasis and concurrent inflammatory bowel disease. Finally, GUS did not cause disease reactivation in patients with latent TB who also received TB prophylaxis.32 In clinical trials evaluating USM, there were similarly no reported reactivations of latent TB in patients receiving concurrent isoniazid prophylaxis.34,44 USM is currently the preferred biologic agent for patients with concomitant latent TB, with IL-17 inhibitors as second-line options.34 Considering their similar mechanisms, GUS is a reasonable alternative to USM for patients with both psoriasis and latent TB.
In addition to GUS, other p19 inhibitors are increasingly becoming the focus of clinical trials.45–47 Risankizumab and tildrakizumab are both humanized monoclonal antibodies that similarly target p19.45,46 In a Phase II clinical trial, more risankizumab-treated patients than USM-treated patients attained PASI90 at Week 12 (77% vs 40%, respectively; P<0.001).46 These results were corroborated in two recent Phase III trials, UltlMMa-1 and UltlMMa-2, in which more risankizumab-treated patients attained PASI90 at Week 16 compared to USM-treated patients (UltlMMa-1: 75.3% vs 42.0%, respectively; UltlMMa-2: 74.8% vs 47.5%, respectively; P<0.001 for both).47 At Week 12 of another Phase III clinical trial, more patients treated with tildrakizumab than with etanercept attained PASI75 (66% for 200 mg tildrakizumab vs 61% for 100 mg tildrakizumab vs 48% for etanercept; P<0.0001 and P<0.001 for 200 and 100 mg tildrakizumab vs etanercept, respectively).45 Based on these studies, p19 inhibition may be a more effective treatment strategy than TNF-alpha and IL-12/IL-23 inhibition, although not all p19 inhibitors appear to have the same degree of efficacy. Further understanding of psoriasis pathogenesis will clarify the exact roles and mechanisms of IL-23 and the p19 subunit.
The FDA has instructed that additional studies be reported on long-term malignancy risk, the safety of GUS for pregnant patients and long-term risk of other AEs.32 Prolonged follow-up and large long-term clinical trials will ultimately determine the safety and efficacy of GUS.
Conclusion
GUS is an effective novel monoclonal antibody that is FDA approved for the treatment of moderate-to-severe plaque psoriasis in adult patients. It targets the p19 subunit of IL-23 and IL-39, which appears to play a critical role in psoriasis pathogenesis.21 The medication is well tolerated, and side effects are generally mild and most commonly include infections.15,21,26–29 In the reported clinical trials, GUS markedly improved disease with corresponding improvements in quality of life measures.15,21,27–29 The medication also successfully treated difficult to treat areas of the body, including the scalp, hands, feet and fingernails.27,28 In addition to its impact on cutaneous lesions, GUS may be effective in treating PsA.30 Furthermore, GUS was more effective than ADM and was successful in treating patients with incomplete responses to ADM and USM.15,27–29 Long-term investigation of both side effects and efficacy is required.
Disclosure
Dr Feldman has received research, speaking and/or consulting support from Galderma, GSK/Stiefel, Almirall, LEO Pharma, Boehringer Ingelheim, Mylan, Celgene, Pfizer, Valeant, AbbVie, Samsung, Janssen, Lilly, Menlo, Merck, Novartis, Regeneron, Sanofi, Novan, Qurient, National Biological Corporation, Caremark, Advance Medical, Sun Pharma, Suncare Research, Informa, UpToDate and National Psoriasis Foundation. The authors report no other conflicts of interest in this work.
References
Helmick CG, Lee-Han H, Hirsch SC, Baird TL, Bartlett CL. Prevalence of psoriasis among adults in the U.S.: 2003–2006 and 2009–2010 National Health and Nutrition Examination Surveys. Am J Prev Med. 2014;47(1):37–45. | ||
Kim WB, Jerome D, Yeung J. Diagnosis and management of psoriasis. Can Fam Physician. 2017;63(4):278–285. | ||
Kurd SK, Troxel AB, Crits-Christoph P, Gelfand JM. The risk of depression, anxiety, and suicidality in patients with psoriasis: a population-based cohort study. Arch Dermatol. 2010;146(8):891–895. | ||
Weiss SC, Kimball AB, Liewehr DJ, Blauvelt A, Turner ML, Emanuel EJ. Quantifying the harmful effect of psoriasis on health-related quality of life. J Am Acad Dermatol. 2002;47(4):512–518. | ||
Kimball AB, Gladman D, Gelfand JM, et al. National Psoriasis Foundation clinical consensus on psoriasis comorbidities and recommendations for screening. J Am Acad Dermatol. 2008;58(6):1031–1042. | ||
Oliveira MF, Rocha BO, Duarte GV. Psoriasis: classical and emerging comorbidities. An Bras Dermatol. 2015;90(1):9–20. | ||
Langan SM, Seminara NM, Shin DB, et al. Prevalence of metabolic syndrome in patients with psoriasis: a population-based study in the United Kingdom. J Invest Dermatol. 2012;132(3 Pt 1):556–562. | ||
Canadian Psoriasis Guidelines Addendum C. 2016 Addendum to the Canadian Guidelines for the Management of Plaque Psoriasis 2009. J Cutan Med Surg. 2016;20(5):375–431. | ||
Smith CH, Anstey AV, Barker JN, et al. British Association of Dermatologists’ guidelines for biologic interventions for psoriasis 2009. Br J Dermatol. 2009;161(5):987–1019. | ||
Bonifati C, Ameglio F. Cytokines in psoriasis. Int J Dermatol. 1999;38(4):241–251. | ||
Carrascosa JM, Jacobs I, Petersel D, Strohal R. Biosimilar drugs for psoriasis: principles, present, and near future. Dermatol Ther. 2018;8(2):173–194. | ||
di Cesare A, di Meglio P, Nestle FO. The IL-23/Th17 axis in the immunopathogenesis of psoriasis. J Invest Dermatol. 2009;129(6):1339–1350. | ||
Oppmann B, Lesley R, Blom B, et al. Novel p19 protein engages IL-12p40 to form a cytokine, IL-23, with biological activities similar as well as distinct from IL-12. Immunity. 2000;13(5):715–725. | ||
Lupardus PJ, Garcia KC. The structure of interleukin-23 reveals the molecular basis of p40 subunit sharing with interleukin-12. J Mol Biol. 2008;382(4):931–941. | ||
Gordon KB, Duffin KC, Bissonnette R, et al. A Phase 2 trial of guselkumab versus adalimumab for plaque psoriasis. N Engl J Med. 2015;373(2):136–144. | ||
Luo J, Wu SJ, Lacy ER, et al. Structural basis for the dual recognition of IL-12 and IL-23 by ustekinumab. J Mol Biol. 2010;402(5):797–812. | ||
Lee E, Trepicchio WL, Oestreicher JL, et al. Increased expression of interleukin 23 p19 and p40 in lesional skin of patients with psoriasis vulgaris. J Exp Med. 2004;199(1):125–130. | ||
Michalak-Stoma A, Bartosińska J, Kowal M, Juszkiewicz-Borowiec M, Gerkowicz A, Chodorowska G. Serum levels of selected Th17 and Th22 cytokines in psoriatic patients. Dis Markers. 2013;35(6):625–631. | ||
Bettelli E, Oukka M, Kuchroo VK. T(H)-17 cells in the circle of immunity and autoimmunity. Nat Immunol. 2007;8(4):345–350. | ||
Raychaudhuri SP. Role of IL-17 in psoriasis and psoriatic arthritis. Clin Rev Allergy Immunol. 2013;44(2):183–193. | ||
Sofen H, Smith S, Matheson RT, et al. Guselkumab (an IL-23-specific mAb) demonstrates clinical and molecular response in patients with moderate-to-severe psoriasis. J Allergy Clin Immunol. 2014;133(4):1032–1040. | ||
Wang X, Wei Y, Xiao H, et al. A novel IL-23p19/Ebi3 (IL-39) cytokine mediates inflammation in Lupus-like mice. Eur J Immunol. 2016;46(6):1343–1350. | ||
Vignali DA, Kuchroo VK. IL-12 family cytokines: immunological playmakers. Nat Immunol. 2012;13(8):722–728. | ||
Cargill M, Schrodi SJ, Chang M, et al. A large-scale genetic association study confirms IL12B and leads to the identification of IL23R as psoriasis-risk genes. Am J Hum Genet. 2007;80(2):273–290. | ||
Nair RP, Duffin KC, Helms C, et al. Genome-wide scan reveals association of psoriasis with IL-23 and NF-kappaB pathways. Nat Genet. 2009;41(2):199–204. | ||
Zhuang Y, Calderon C, Marciniak SJ, et al. First-in-human study to assess guselkumab (anti-IL-23 mAb) pharmacokinetics/safety in healthy subjects and patients with moderate-to-severe psoriasis. Eur J Clin Pharmacol. 2016;72(11):1303–1310. | ||
Blauvelt A, Papp KA, Griffiths CE, et al. Efficacy and safety of guselkumab, an anti-interleukin-23 monoclonal antibody, compared with adalimumab for the continuous treatment of patients with moderate to severe psoriasis: results from the phase III, double-blinded, placebo- and active comparator-controlled VOYAGE 1 trial. J Am Acad Dermatol. 2017;76(3):405–417. | ||
Reich K, Armstrong AW, Foley P, et al. Efficacy and safety of guselkumab, an anti-interleukin-23 monoclonal antibody, compared with adalimumab for the treatment of patients with moderate to severe psoriasis with randomized withdrawal and retreatment: results from the phase III, double-blind, placebo- and active comparator-controlled VOYAGE 2 trial. J Am Acad Dermatol. 2017;76(3):418–431. | ||
Langley RG, Tsai TF, Flavin S, et al. Efficacy and safety of guselkumab in patients with psoriasis who have an inadequate response to ustekinumab: results of the randomized, double-blind, phase III NAVIGATE trial. Br J Dermatol. 2018;178(1):114–123. | ||
Deodhar A, Gottlieb A, Boehncke WH, et al. Efficacy and safety results of guselkumab, an anti-Il23 monoclonal antibody, in patients with active psoriatic arthritis over 24 weeks: a phase 2a, randomized, double-blind, placebo-controlled study. Ann Rheum Dis. 2017;76:142–143. | ||
Gordon KB, Blauvelt A, Foley P, et al. Efficacy of guselkumab in subpopulations of patients with moderate-to-severe plaque psoriasis: a pooled analysis of the phase III VOYAGE 1 and VOYAGE 2 studies. Br J Dermatol. 2018;178(1):132–139. | ||
Center for Drug Evaluation and Research. Multi-discipline review/summary application number. Research CfDEa; 2016761061Orig1s000. | ||
Gordon KB, Armstrong AW, Han C, et al. Anxiety and depression in patients with moderate-to-severe psoriasis and comparison of change from baseline after treatment with guselkumab vs. adalimumab: results from the Phase 3 VOYAGE 2 study. J Eur Acad Dermatol Venereol. (Suppl 1). Epub 2018 April 28. | ||
Amin M, No DJ, Egeberg A, Wu JJ, Dj N, Jj W. Choosing first-line biologic treatment for moderate-to-severe psoriasis: what does the evidence say? Am J Clin Dermatol. 2018;19(1):1–13. | ||
Griffiths CE, Reich K, Lebwohl M, et al. Comparison of ixekizumab with etanercept or placebo in moderate-to-severe psoriasis (UNCOVER-2 and UNCOVER-3): results from two phase 3 randomised trials. Lancet. 2015;386(9993):541–551. | ||
Langley RG, Elewski BE, Lebwohl M, et al. Secukinumab in plaque psoriasis – results of two phase 3 trials. N Engl J Med. 2014;371(4):326–338. | ||
Papp KA, Reich K, Paul C, et al. A prospective phase III, randomized, double-blind, placebo-controlled study of brodalumab in patients with moderate-to-severe plaque psoriasis. Br J Dermatol. 2016;175(2):273–286. | ||
Kulig P, Musiol S, Freiberger SN, et al. IL-12 protects from psoriasiform skin inflammation. Nat Commun. 2016;7:13466. | ||
Papp KA, Griffiths CE, Gordon K, et al. Long-term safety of ustekinumab in patients with moderate-to-severe psoriasis: final results from 5 years of follow-up. Br J Dermatol. 2013;168(4):844–854. | ||
Ghilardi N, Kljavin N, Chen Q, Lucas S, Gurney AL, de Sauvage FJ. Compromised humoral and delayed-type hypersensitivity responses in IL-23-deficient mice. J Immunol. 2004;172(5):2827–2833. | ||
Hohenberger M, Cardwell LA, Oussedik E, Feldman SR. Interleukin-17 inhibition: role in psoriasis and inflammatory bowel disease. J Dermatolog Treat. 2018;29(1):13–18. | ||
Sandborn WJ, Gasink C, Gao LL, et al. Ustekinumab induction and maintenance therapy in refractory Crohn’s disease. N Engl J Med. 2012;367(16):1519–1528. | ||
Feagan BG, Sandborn WJ, D’Haens G, et al. Induction therapy with the selective interleukin-23 inhibitor risankizumab in patients with moderate-to-severe Crohn’s disease: a randomised, double-blind, placebo-controlled phase 2 study. Lancet. 2017;389(10080):1699–1709. | ||
Tsai TF, Ho V, Song M, et al. The safety of ustekinumab treatment in patients with moderate-to-severe psoriasis and latent tuberculosis infection. Br J Dermatol. 2012;167(5):1145–1152. | ||
Reich K, Papp KA, Blauvelt A, et al. Tildrakizumab versus placebo or etanercept for chronic plaque psoriasis (reSURFACE 1 and reSURFACE 2): results from two randomised controlled, phase 3 trials. Lancet. 2017;390(10091):276–288. | ||
Papp KA, Blauvelt A, Bukhalo M, et al. Risankizumab versus ustekinumab for moderate-to-severe plaque psoriasis. N Engl J Med. 2017;376(16):1551–1560. | ||
Gordon KB. Efficacy and safety of risankizumab: results from two double-blind, placebo- and ustekinumab-controlled, phase 3 trials in moderate-to-severe plaque psoriasis. Paper presented at: American Academy of Dermatology; 2018; San Diego, CA. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.