Back to Journals » International Journal of Nanomedicine » Volume 9 » Issue 1
Targeting human dendritic cells via DEC-205 using PLGA nanoparticles leads to enhanced cross-presentation of a melanoma-associated antigen
Authors Saluja S, Hanlon D, Sharp F, Hong E, Khalil D, Robinson E, Tigelaar R, Fahmy T, Edelson R
Received 23 April 2014
Accepted for publication 4 July 2014
Published 12 November 2014 Volume 2014:9(1) Pages 5231—5246
DOI https://doi.org/10.2147/IJN.S66639
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Sandeep S Saluja,1 Douglas J Hanlon,1 Fiona A Sharp,2 Enping Hong,2 David Khalil,1 Eve Robinson,1 Robert Tigelaar,1 Tarek M Fahmy,2,3 Richard L Edelson1
1Department of Dermatology, Yale University School of Medicine, 2Department of Biomedical Engineering, Yale University, 3Department of Immunobiology, Yale University School of Medicine, New Haven, CT, USA
Abstract: Targeting antigen to dendritic cells (DCs) is a powerful and novel strategy for vaccination. Priming or loading DCs with antigen controls whether subsequent immunity will develop and hence whether effective vaccination can be achieved. The goal of our present work was to increase the potency of DC-based antitumor vaccines by overcoming inherent limitations associated with antigen stability and cross-presentation. Nanoparticles prepared from the biodegradable polymer poly(lactic-co-glycolic acid) have been extensively used in clinical settings for drug delivery and are currently the subject of intensive investigation as antigen delivery vehicles for vaccine applications. Here we describe a nanoparticulate delivery system with the ability to simultaneously carry a high density of protein-based antigen while displaying a DC targeting ligand on its surface. Utilizing a targeting motif specific for the DC-associated surface ligand DEC-205, we show that targeted nanoparticles encapsulating a MART-127–35 peptide are both internalized and cross-presented with significantly higher efficiency than isotype control-coated nanoparticles in human cells. In addition, the DEC-205-labeled nanoparticles rapidly escape from the DC endosomal compartment and do not colocalize with markers of early (EEA-1) or late endosome/lysosome (LAMP-1). This indicates that encapsulated antigens delivered by nanoparticles may have direct access to the class I cytoplasmic major histocompatibility complex loading machinery, overcoming the need for “classical” cross-presentation and facilitating heightened DC stimulation of anti-tumor CD8+ T-cells. These results indicate that this delivery system provides a flexible and versatile methodology to deliver melanoma-associated antigen to DCs, with both high efficiency and heightened potency.
Keywords: dendritic cells, DEC-205, PLGA nanoparticles, cross-presentation, melanoma-associated antigen
Introduction
Dendritic cell (DC)-based immunotherapy for the treatment of solid tumors is premised on the possibility that tumor-associated antigens, or distinctive proteins on the tumor cell surface, can generate a clinically relevant immune response. DCs are professional antigen-presenting cells (APC) that have the capacity to process internalized antigens (Ag) for cross-presentation to anti-tumor CD8+ cytotoxic T-lymphocytes (CTLs) in the context of class I major histocompatibility complexes (MHC I), and are therefore able to initiate host T-cell responses against foreign Ag and tumors.1 As a consequence, promising strategies for priming T-cell immunity focus on manipulating the DC response by targeting Ag delivery through specific DC surface receptors, including C-type lectin receptors (CLRs).
CLRs are present on the surface of DCs and recognize sugar moieties, including mannose, fucose, and glucan expressed on the surface of mycobacteria, fungi, and viruses.2 The ability of CLRs to internalize Ag and deliver them to endocytic compartments for processing and Ag presentation has been one of the main reasons that CLRs are targeted for the induction of tumor-specific immune responses.3 Steinman et al have pioneered the field of antibody-mediated targeting of Ag to DCs through several CLRs, including DEC-205,4 Langerin,5 and DC-SIGN.6 In particular, the role of DEC-205 in targeting tumor antigens to DCs has recently garnered significant interest in the field of tumor immunology because DEC-205 is expressed at relatively high levels on myeloid blood DCs.7 In recent in vitro studies, antibody-mediated delivery of HIV gag p24 protein to DEC-205 on human monocyte-derived DCs resulted in the presentation of different MHC class I-restricted peptides,8 and DEC-205-mediated targeting of plasmacytoid DCs by nanoparticles (NPs) led to increased cross-presentation of a melanoma-associated Ag.9 Thus, DEC-205 targeting of Ag to DCs is a promising potential methodology to improve cell-based antitumor vaccines.
Melanoma is an excellent candidate for DC-based immunotherapy, as it is well recognized to be immunogenic. Recently, a number of human melanoma-associated antigens recognized by T lymphocytes have been identified. Among them, the MART-1 (melanoma antigen recognized by T-cells) gene is expressed by normal melanocytes, as well as by a majority of fresh tumor isolates and melanoma cell lines. The MART-127–35 peptide (corresponding to residues 27–35 of the non-mutated MART-1 protein) is a well characterized immune-dominant epitope for human leukocyte antigen (HLA)-A2-restricted melanoma-specific CTLs and can be used for the development of immunotherapeutic strategies.10 Thus far, however, therapeutic benefits associated with DC/peptide vaccination alone in patients with melanoma have been limited.11 And while there has been a great deal of recent excitement regarding new classes of chemotherapeutic agents, such as selective BRAF inhibitors in treatment of advanced melanoma, concerns regarding formation of secondary malignancies and rapid development of drug-resistant tumors make a clear case for combining chemotherapy agents with agents that stimulate the immune system.12 Perhaps the strongest cases for immunomodulation in the treatment of advanced melanoma were the tumor regressions observed in up to 50% of metastatic melanoma patients receiving expanded autologous melanoma-specific T lymphocytes and high-dose interleukin (IL)-2, as well as the recent effectiveness of monoclonal antibody therapies blocking the T-cell negative regulators CTLA-4 and PD-1.13 These results point to the critical role of stimulating functional antitumor T-cells in attaining durable responses in advanced disease, and if methodologies to increase the potency of DC-based vaccination strategies could be developed, they would be excellent candidates for synergistic use with these promising new agents.
Two of the most important factors limiting conventional DC vaccination are the rapid degradation of internalized protein by DC if whole protein Ag is utilized, as well as the transient nature of MHC class I epitopes at the DC surface following external loading of motif-fitting peptides.14 Encapsulation of Ag in polymeric NPs has been demonstrated in the literature to protect Ag from degradation by lysosomal proteases, potentially by facilitating endosomal escape of encapsulated Ags,15 as well as increasing and prolonging Ag release, leading to more efficient presentation of MHC-peptide complexes to CD8+ T-cells.16,17 Moreover, the particulate nature of these systems permits their rapid internalization by DCs, perhaps due to their pathogen-mimicking size and charge profile, an effect that can be heightened by surface decoration of NPs with defined PAMP.18 However, comparatively little is known about whether immune responses induced by NPs can be further enhanced by directing Ag-containing NPs via cell surface lectin receptors such as DEC-205 to human monocyte-derived DCs, the principal cell type utilized clinically in antitumor vaccination. It is understood that the presence and density of anti-DEC-205 on NPs in the mouse system can enhance a vaccine response,19 but outside of recent targeting of plasmacytoid DCs as APC,9 very little is known regarding the efficiency of this targeted approach and its mechanistic impact on cross-presentation in human DC populations. To test this, we designed experiments to determine whether targeting the DEC-205 receptor on human DCs via NPs could enhance DC Ag cross-presentation to melanoma patient-derived CD8+ antitumor T-cells. We engineered a NP-based delivery system utilizing the polymer poly(lactic-co-glycolic acid) (PLGA), which is already approved by the US Food and Drug Administration and has generated intense interest because of its favorable properties, which include excellent biocompatibility, biodegradability, and mechanical strength.20–22 PLGA-based delivery systems for a variety of pharmacological agents are already widely used in humans,23 and the possibility of targeting NP-associated Ag directly to DCs as part of anticancer or antipathogen vaccine strategies have recently been explored in a variety of preclinical models.24,25
Here we describe a versatile PLGA-based NP that combines the increased Ag delivery efficiency of particulate-based systems with specific DC targeting. In addition to quantifying antigen cross-presentation, we monitored the uptake efficiency and subcellular location of these Ag-encapsulated PLGA NPs within the DC. We report that DEC-205 targeting led to increased internalization of NPs by immature DCs, as well as endosomal release of the NP to the DC cytoplasm and enhanced levels of Ag cross-presentation of a clinically relevant melanoma epitope. To our knowledge, these data represent the first utilization of DEC-205 targeted NPs in Ag delivery experiments in human monocyte-derived DCs and indicated that these reagents show promise in potentially improving the potency of next-generation DC-based cancer vaccines.
Materials and methods
Antibodies and reagents
Antibodies
The antibodies used were anti-HLA-DR-FITC, mouse-IgG2b-FITC (both from BD Biosciences, San Jose, CA, USA); anti-CD83-PE, mouse-IgG1-PE (both from BioLegend, San Diego, CA, USA); anti-CD14-PerCP, mouse-IgG1k-PerCP, anti-CD11-APC, mouse-IgG1k-APC, biotinylated antihuman DEC-205 monoclonal antibody, biotinylated mouse IgG2bk (all from eBiosciences, San Diego, CA, USA); antihuman interferon (IFN)-γ enzyme-linked immunosorbent assay kit (BD Pharmingen, San Diego, CA, USA).
Cytokines
Recombinant human IL-4, tumor necrosis factor alpha (TNFα), IL-1β, and IL-6 were obtained from R&D Systems (Minneapolis, MN, USA). Prostaglandin E2 was obtained from MP Biomedicals (Irvine, CA, USA). Granulocyte-macrophage colony-stimulating factor was obtained from Berlex (Seattle, WA, USA).
Synthetic biomolecules
Poly(vinyl alcohol), sodium deoxycholate, palmitic acid, N-hydroxysuccinimide ester, and gelatin were all purchased from Sigma-Aldrich (St Louis, MO, USA). Methylene chloride and avidin were purchased from Thermo Fisher Scientific (Waltham, MA, USA) and Invitrogen (Carlsbad, CA, USA), respectively. Research-grade PLGA (50:50, approximately 50,000 g/mol), was purchased from Durect (Pelham, AL, USA).
Peptide and cell lines
Peptide
The HLA-A*0201-restricted peptide ELAGIGILTV (derived from the melanoma-associated differentiation antigen Melan-A/MART-1; A to L substitution at position 2 compared with its natural counterpart for higher immunogenicity and referred to as MART-127–35), as well as the peptide labeled with a COOH-terminus FITC fluorochrome were produced by the Keck Facility (Yale University).
CTL clone
The human CD8+ T-cell receptor transgenic cell line DMF5, reactive with both the MART-1(26–35) and MART-1(27L–35) epitopes, was kindly provided by John R Wunderlich, Surgery Branch, National Cancer Institute. This line, originally derived from a high avidity tumor-infiltrating lymphocyte clone that mediated tumor regression clinically, has been extensively described previously.26
Media and serum
DC cultures were maintained in serum-free DC medium (Cell-Genix, Freiburg, Germany) supplemented with 1% normal autologous human serum. Human serum was isolated in our laboratory from healthy donors after informed consent was obtained. Tumor-infiltrating lymphocyte cell lines were maintained in CTL medium: Roswell Park Memorial Institute 1640 medium supplemented with 12.5 mM, 4 mM L-glutamine, 100 U/mL penicillin, 100 μg/mL streptomycin (Invitrogen), 50 μM β-mercaptoethanol (Sigma), and 10% normal human serum. The flow cytometry wash buffer was composed of phosphate-buffered saline (Invitrogen) supplemented with 2% fetal bovine serum (Gemini BioProducts, Woodland, CA, USA).
Synthesis of NPs and DEC-205 ligand coupling to the NP surface
PLGA NPs encapsulating MART-127–35 peptide (ELAGIGILTV) (5 μg peptide/mg PLGA NP) were prepared and functionalized with avidin using a modified o/w emulsion and solvent evaporation–extraction method as described previously.27 Control NPs (blanks) were synthesized with sterile phosphate-buffered saline replacing the peptide. Fluorescent NPs were formulated by incorporating a FITC-labeled MART-127–35 peptide by identical methodologies. Biotinylated anti-human DEC-205 monoclonal antibody or isotype control antibody was added at a concentration of 10 μg/mg PLGA NPs in phosphate-buffered saline and rotated for 40 minutes at room temperature; this antibody concentration was determined to lead to near 100% incorporation on the NP surface as previously described.19 The particles were then centrifuged at 6,500 rpm for 5 minutes, washed twice in phosphate-buffered saline, and resuspended in DC medium prior to loading.
Nanoparticle characterization
Initial images of the NPs were taken by scanning electron microscopy. The NPs were analyzed using a NS500 instrument (NanoSight, Amesbury, UK) and NTA 2.0 software to determine their concentration and size distribution. The lyophilized NPs were dispersed in phosphate-buffered saline at dilute concentrations (25 μg/mL) prior to analysis. The NanoSight instrument captures the motion of individual particles in suspension using microscopy, and the software derives the concentration and hydrodynamic radii of individually tracked particles, producing a high-resolution size distribution with each measurement. The mean, mode, standard deviation, and coefficient of variation of each distribution were calculated by the software.
The dispersity of the size distribution (Ð) was calculated by the following formula:
|
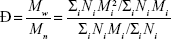
where Mw is the weight-average molecular weight and Mw is the number-average molecular weight. Taking the assumption that all NPs have equal density, we instead use the volume distribution calculated by the NanoSight to derive Ð:
|
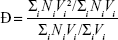
where V is the volume of each NP in the distribution.
The MART-1 peptide content of the peptide-loaded NPs was determined by high-performance liquid chromatographic analysis of degraded NPs. Specifically, 1.5 mg of the MART-1-NP complex was dissolved in 100 μL 8 M urea/0.4 M ammonium bicarbonate, followed by acidification in 20% trifluoroacetic acid and addition of 100 μL of 100% acetonitrile. Samples were spun through a 0.2 Millipore microfilter to ensure removal of residual particulate matter and a 10 μL sample was run on an HP1090 high-performance liquid chromatography system with a Vydac C18 reverse phase (1 mm ×250 mm eluted at 50 μL per minute) column. The peptide concentration was determined by monitoring of the peptide backbone at 210 nm on the instrument’s ultraviolet detector.
Generation of fast DCs from peripheral blood mononucleated cells of healthy human donors
Protocols for generating mature monocyte-derived DCs over a 48-hour period (instead of protocols 7–10 days long) have been described widely in the literature.28–31 Such shorter culture periods offer practical advantages of reduced time span and the amount of recombinant cytokines required. Briefly, after informed consent was obtained, peripheral blood mononucleated cells from healthy human donors (both HLA-A*0201+ and HLA-A* 0201-) were isolated by Ficoll-Hypaque gradient centrifugation. Peripheral blood mononucleated cells were resuspended in serum-free DC medium (Cell-Genix) supplemented with 1% normal human serum, and plated at 15×106 cells/3 mL/well of a 6-well tissue culture plate. After a 90-minute incubation at 37°C, non-adherent cells and medium were removed and replaced with 3 mL/well of fresh DC medium. After a second incubation period at 37°C for 60 minutes, non-adherent cells and medium were again removed and replaced with 3 mL/well of DC medium supplemented with 800 IU/mL granulocyte-macrophage colony-stimulating factor and 1,000 IU/mL IL-4. After one day of incubation at 37°C with 5% CO2, a cytokine cocktail of 10 ng/mL TNFα, 10 ng/mL IL-1β, 1,000 IU/mL IL-6, and 1 μg/mL prostaglandin E2 was added directly to the cultures. Cultures were incubated at 37°C for an additional day before mature DCs were harvested.
Phenotype of FastDCs and expression of DEC-205 on FastDCs
The phenotype of “fast” DCs, including the expression of DEC-205, was determined by flow cytometry. For analysis of FastDCs, the following primary antibodies and their appropriate isotype controls were used: anti-CD83-PE, anti-CD14-PerCP, anti-CD11c-APC, and anti-DEC-205-PE. Cells were analyzed on a FACS Calibur flow cytometer (Becton Dickinson, Heidelberg, Germany). The data were analyzed using FlowJo version 7.6.2 software. The gates were based on isotype controls.
MART-1-specific CD8+ T-cell responses
Human immature DCs were collected and incubated for 5 hours at 37°C with varying concentrations of anti-DEC-205 or isotype control-coated NPs containing MART-127–35 peptide (250 μg NPs/mL medium, 125 μg/mL, 62.5 μg/mL, 31.25 μg/mL). Maturation cocktail (TNFα, IL-6, prostaglandin E2, and IL-1β) was subsequently added. One day later, the mature DCs were harvested, tested by flow cytometry, and cocultured for 72 hours with the MART-1-specific CD8+ clone DMF5 (recognizing MART-127–35) at a 1:2 DC/ T-cell ratio. The efficiency of antigen presentation was evaluated in an IFN-γ enzyme-linked immunospot assay. Unstimulated CD8+ T-cells alone and T-cells stimulated using DCs loaded with blank PLGA NPs (at identical NP concentrations to those containing MART-127–35 peptide) acted as negative controls. Experiments were performed in triplicate. IFN-γ release was fitted to a sigmoidal response curve as follows:
|
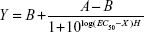
where X is the logarithm of concentration, Y is the response, A and B are the initial and final responses, respectively, H is the Hill slope, and EC50 is the half-maximal stimulation value.
Measurement and visualization of NP uptake
NP uptake was assessed by flow cytometry as well as a variety of imaging modalities. Human immature DCs were collected and incubated for 5 hours at 37°C with varying concentrations of anti-DEC-205 or isotype control-coated NPs containing fluorescent dye (FITC)-labeled MART-127–35 peptide (250 μg NPs/mL medium, 125 μg/mL, 62.5 μg/mL, 31.25 μg/mL). Maturation cocktail (TNFα, IL-6, prostaglandin E2, and IL-1β) was subsequently added. After overnight culture at 37°C, the cells were washed twice and then labeled with antihuman-CD11c-APC for 30 minutes at 4°C. After staining, the cells were washed and analyzed by flow cytometry. Differences in median fluorescence intensity for FITC (fluorescently labeled MART-1) were determined. DCs loaded with blank PLGA NPs acted as negative controls. To control for non-specific binding, a fraction of cells treated with fluorescent peptide NPs were incubated at 4°C.
To visualize internalization of NPs and to determine colocalization with the endolysosomal compartment, transmission electron microscopy (TEM), confocal microscopy, and Amnis ImageStream® were utilized. For TEM, nanospheres were cocultured with DC preparations for 90 minutes at 37°C, and washed extensively. Cells were fixed in 2% glutaraldehyde and 2% paraformaldehyde in 0.1 M cacodylate buffer, and post-fixed in 1% OsO4 for one hour at room temperature. After en bloc staining with 2% uranyl acetate, cells were dehydrated and embedded in Epon. Thin sections (50 nm) were cut with an ultramicrotome and metal stained with uranyl acetate and lead citrate, before viewing in a Tencai™ Bio-twin transmission electron microscope (FEI Company, Hillsboro, OR, USA) at 80 kV. Images were acquired by a Morada charge-coupled device camera fitted with iTEM software (Olympus, Tokyo, Japan). Confocal microscopy was carried out on DC preparations following a 3-hour internalization period and the cells were visualized on Alcian blue-coated coverslips utilizing an LSM510 META instrument (Carl Zeiss, Oberkochen, Switzerland) with a 63× objective lens, and image analysis and three-dimensional reconstructions performed with Volocity 6.0 software (Perkin-Elmer, Waltham, MA, USA). ImageStream® colocalization was determined from identical DC preparations, with image analysis performed utilizing an Amnis ImageStream instrument with the IDEAS software package and the “Similarity Score” algorithm.
Results
Nanoparticle preparation and characterization
As previously described,27,28 we utilized a strategy for surface modification of PLGA particles by introducing functionally active avidin coupled to palmitic acid during the emulsion preparation of the particles. The addition of avidin-palmitic acid results in incorporation of avidin at high density on the surface of the NP. The surface of these particles may then be further modified by addition of biotinylated proteins that bind avidin with high affinity. This system allows for prolonged presentation of these proteins over the course of several weeks while release of encapsulated proteins from the PLGA matrix is unhindered.27
In the experiments reported here, we have used these PLGA NPs, which contain encapsulated MART-127–35 peptide as an Ag source, with either biotinylated isotype control monoclonal antibody (immunoglobulin G2b) or biotinylated anti-DEC-205 monoclonal antibody-targeting antibody decorating the surface (Figure 1A). Scanning electron microscopic images revealed that the NPs were spherical in shape, with a smooth surface and a size distribution in the 200 nm range (Figure 1B). NanoSight analysis confirmed this size distribution, with very similar values obtained for particles with or without attached monoclonal antibody; PLGA particles alone had a modal diameter of 193 nm, while DEC-205-coated particles had a modal diameter of 196 nm (Figure 1C). Variations in size distribution and dispersity due to DEC-205 binding were minimal (Figure 1D). The MART-127–35 peptide content of NP preparations was determined to be 1.1 μg peptide/mg polymer, representing an encapsulation efficiency of 22%. This concentration was quantified from peptide released from degraded NP preparations, with analysis performed on a C18 reverse phase high-performance liquid chromatography system and ultraviolet absorbance determined by monitoring the peptide backbone at 210 nm, since no chromophoric residues such as tryptophan or tyrosine were present in the sequence.
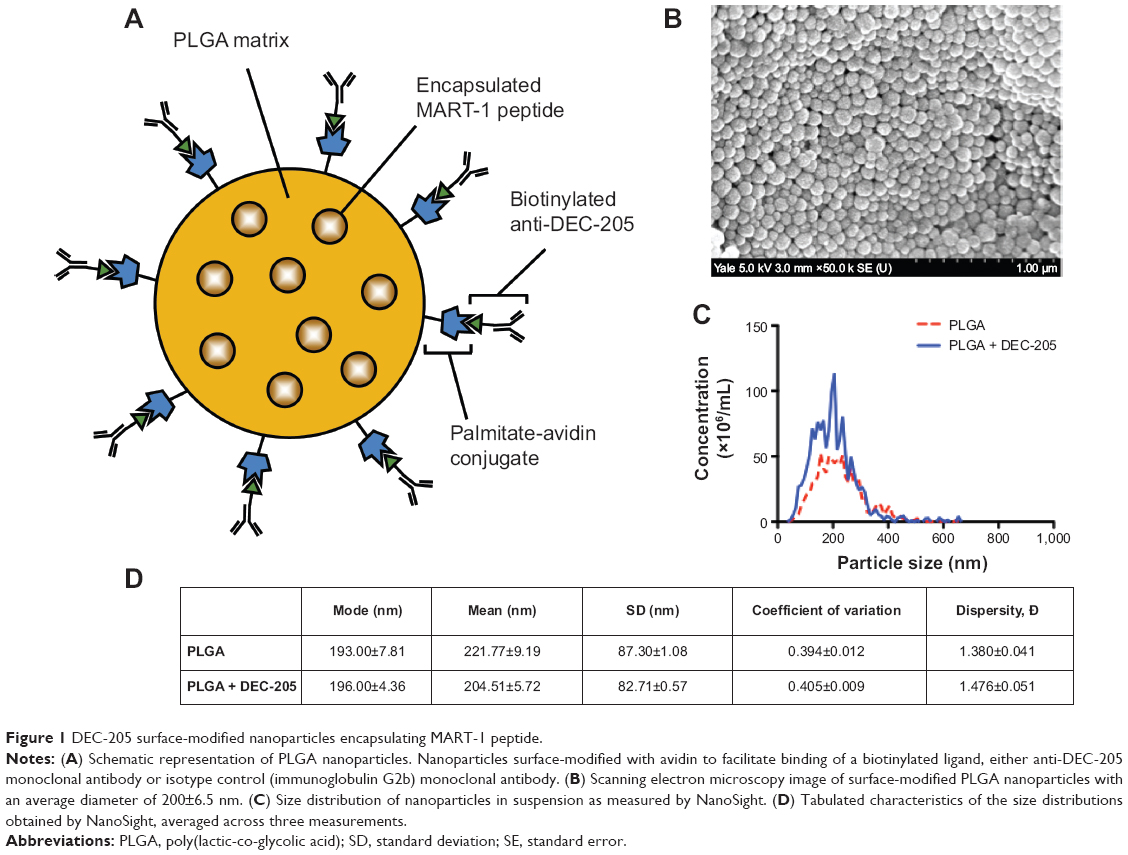 | Figure 1 DEC-205 surface-modified nanoparticles encapsulating MART-1 peptide. |
FastDC protocol generates mature monocyte-derived DCs
In the recently developed FastDC protocol,29,30 monocytes are incubated with granulocyte-macrophage colony-stimulating factor and IL-4 for 24 hours and are subsequently activated with a combination of proinflammatory mediators (TNFα, IL-1β, IL-6, and prostaglandin E2) for another 24 hours to rapidly generate mature and fully functional DCs. This differentiation protocol has been confirmed to generate immunostimulatory DCs with equal potency of those derived from 7–10-day cultures.31,32 To confirm this in our system, cells were monitored for DC differentiation by staining for the monocyte marker CD14 and maturation marker CD83 during the differentiation and maturation period. Before administration of the maturation cocktail, the cells displayed an immature DC phenotype (CD83low CD14high; Figure 2, left panel), whereas just 24 hours after adding the maturation cocktail, the majority of the cells had differentiated into fully mature DCs (CD83high CD14low; Figure 2, right panel). Phenotypic characterization of three additional donor DC cultures of differing HLA subtypes confirmed the FastDC protocol uniformly generated mature DC phenotypes (Figure S1).
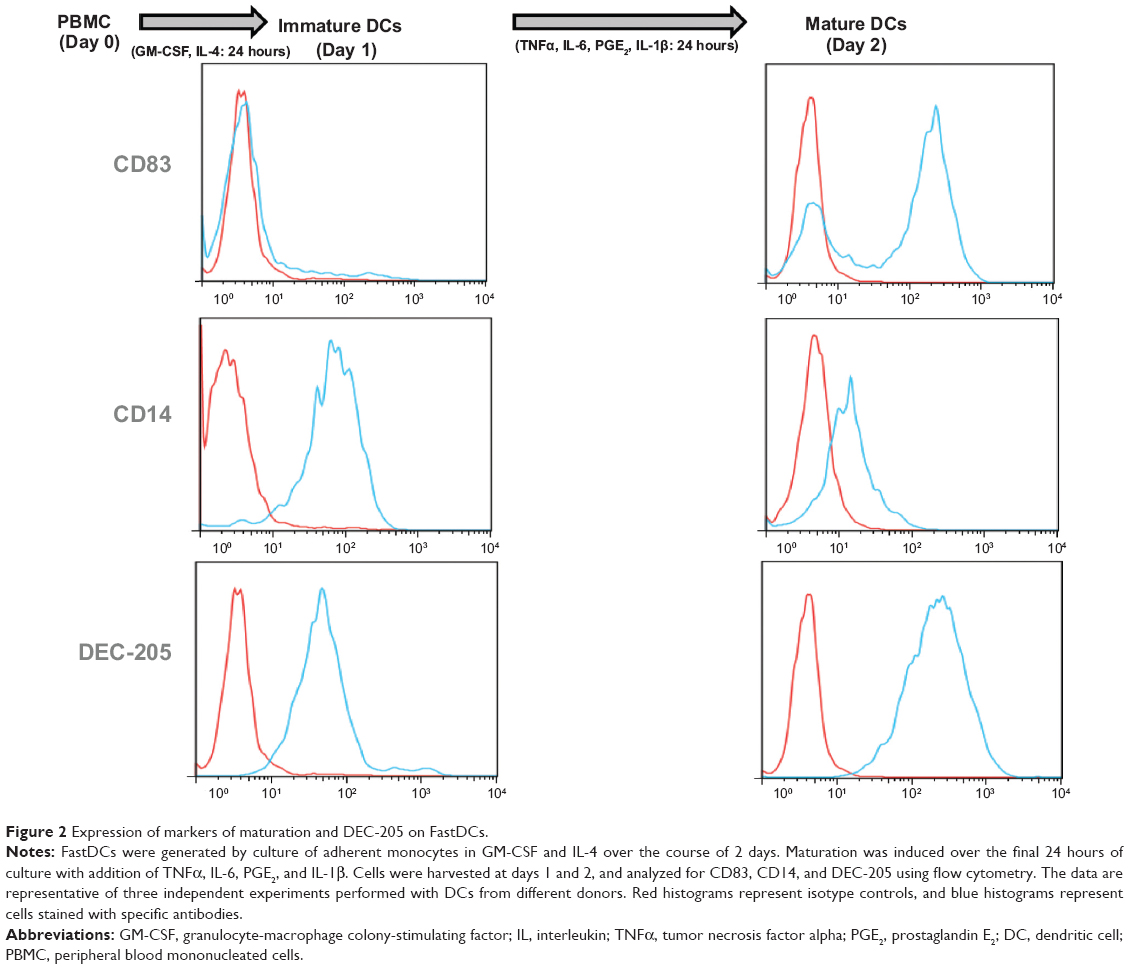 | Figure 2 Expression of markers of maturation and DEC-205 on FastDCs. |
DEC-205 is expressed on both immature and mature human monocyte-derived DCs
DEC-205 is highly expressed on a variety of murine and human DC populations and is therefore an excellent candidate for specific targeting of Ag through receptor-mediated endocytosis of DEC-205 antibody-coated NPs. To assess the potential efficacy of using DEC-205 targeting with monocyte-derived DCs for vaccination, we followed the expression of DEC-205 during the development and maturation of human monocyte-derived DCs (Figure 2, left and right panel bottom). DEC-205 expression increased consistently during FastDC development, as indicated by the increasing specific median fluorescence intensity. The majority of immature DCs were DEC-205-positive (Figure 2, left panel, day 1); however, the expression level further increased during maturation (Figure 2, right panel, day 2). Having established that the majority of both immature DCs and mature DCs were DEC-205-positive in the human system, we next sought to investigate whether these FastDC populations could serve as targets for NP-mediated tumor Ag delivery.
Tumor Ag delivered via DEC-205 to human monocyte-derived DCs are more efficiently cross-presented to CD8+ T-cells
Cross-presentation of endocytosed tumor-associated antigens to cytotoxic CD8+ T-cells is essential for the induction of antitumor immunity.17,33 To investigate whether antigens delivered to monocyte-derived DCs via DEC-205 are more efficiently cross-presented to CD8+ T-cells, we used anti-DEC-205-coated PLGA NPs with encapsulated MART-127–35 peptide, and compared them with MART-127–35 peptide-loaded NPs coated with isotype control antibody.
As shown in the flow chart represented in Figure 3A, we first prepared fast monocyte-derived DCs from an HLA-A2-positive donor, and these DCs were subsequently exposed for 24 hours to either DEC-205-targeted MART-127–35 NPs, control immunoglobulin G-coated MART-127–35 NPs, or blank NPs with no encapsulated Ag. To determine the effects of targeting NPs to DEC-205 on DCs, the different groups of NP-loaded DCs were cocultured with an HLA-A2 restricted CD8+ CTL line recognizing MART-127–35 (DMF5) for 72 hours at a DC to T-cell ratio of 1:2. IFN-γ production from the supernatants was compared as a measure of cross-presentation of MART-127–35 peptide to CD8+ T-cells.
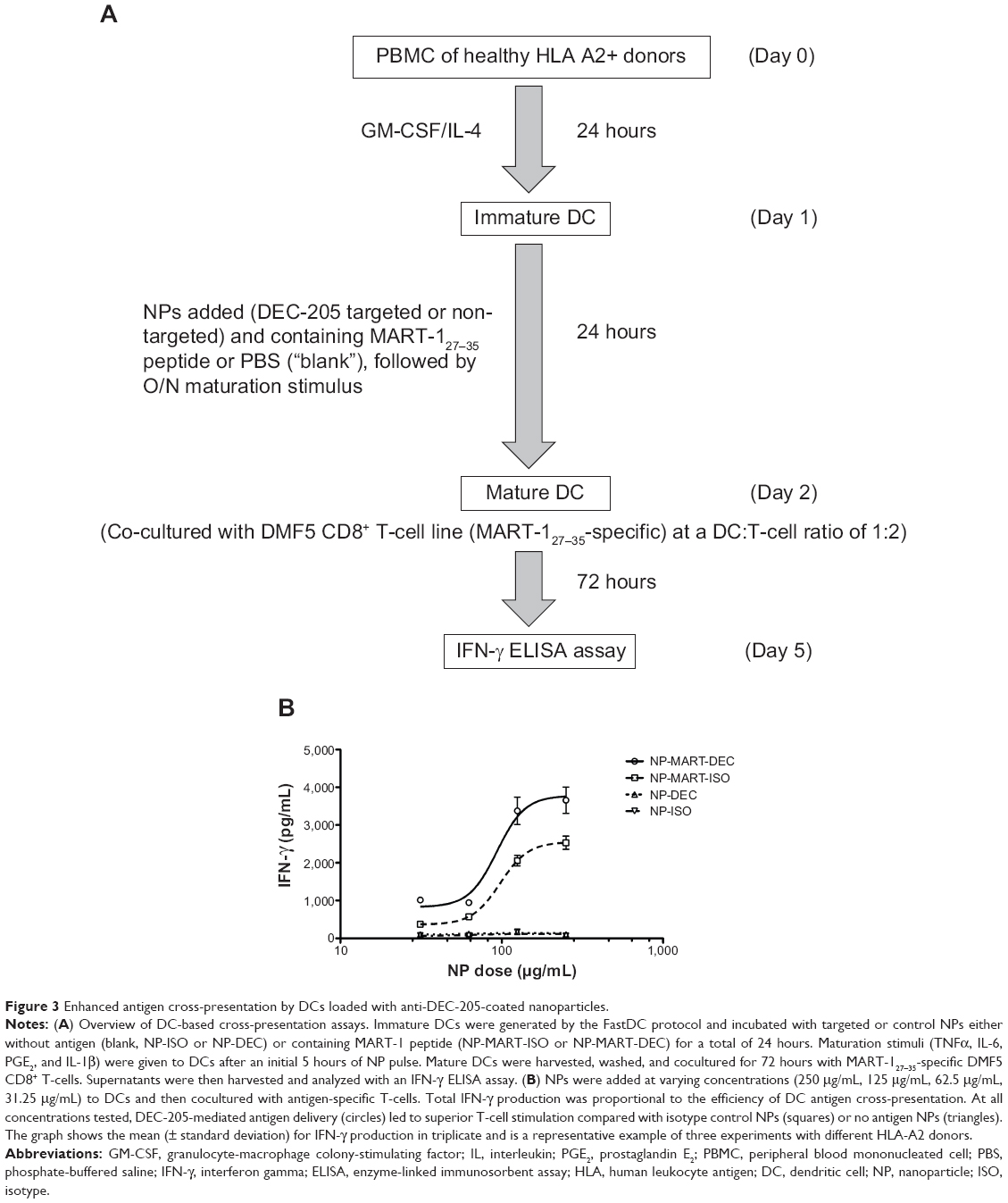 | Figure 3 Enhanced antigen cross-presentation by DCs loaded with anti-DEC-205-coated nanoparticles. |
DCs exposed to either preparation of MART-127–35-containing NPs produced significant quantities of IFN-γ (Figure 3B). However, we observed that compared with isotype-coated NPs, DCs exposed to DEC-205 targeted NP had a greater than 1.5-fold increase in IFN-γ production by MART-1-specific CD8+ T-cells. These results indicate that DEC-205-mediated delivery to DCS led to more efficient CD8+ T-cell stimulation, a result that was observed at all NP concentrations tested. DCs stimulated with blank NPs (containing no Ag) and coated with either DEC-205 or isotype control produced negligible levels of IFN-γ compared with MART-127–35- containing NPs under these conditions (Figure 3B, triangles), indicating minimal levels of non-specific stimulation. In addition, DCs loaded with irrelevant soluble peptide (gp-100) or the soluble MART-127–35 peptide were included as additional negative and positive controls, respectively (data not shown). These results indicated that specific targeting of Ag through the DEC-205 receptor, rather than non-specific binding and internalization of antibody-coated NPs through DC Fc receptors, was responsible for the increase in DC Ag cross-presentation observed.
To confirm that no significant toxicity was associated with antibody-coated PLGA NPs, immature DCs treated identically to those described above (with highest NP concentration) were incubated with maturation cocktail or maturation cocktail along with either isotype control-coated or anti-DEC-205-coated NPs for 24 hours. Using a propidium iodide staining solution, the CD11c+ cell death percentage was measured with flow cytometry after overnight incubation. The results showed that NPs were not toxic to DCs and more than 90% of cells were still viable following this extended incubation, only slightly more than DCs that received no NPs (Figure S2).
Targeting NPs to DEC-205 on DCs leads to enhanced NP uptake
To determine whether differential IFN-γ production by MART-1-specific CD8+ T-cells was associated with more efficient NP uptake mediated by DEC-205, we measured particle internalization as a function of NP concentration. Flow cytometry-based monitoring of NP uptake was facilitated by fabrication of fluorescently labeled MART-127–35 NPs, and these fluorescent nanospheres were then surface-modified with either anti-DEC-205 antibody or isotype control monoclonal antibody. Immature fast DCs were exposed to varying concentrations (250 μg/mL, 125 μg/mL, 62.5 μg/mL, 31.25 μg/mL, or 15.62 μg/mL) of isotype control-coated NPs or anti-DEC-205-coated NPs for 5 hours at 4°C and 37°C and then matured overnight in maturation cocktail. Analysis of particle uptake was carried out by flow cytometry, with total fluorescence proportional to the total number of NPs internalized by DCs. Numbers represent median fluorescence intensity following subtraction of the control 4°C signal (non-specific surface binding) from 37°C values (internalized NPs). As shown in Figure 4, although isotype-coated NPs were taken up by DCs to some degree (dashed squares), significantly increased fluorescence intensity was associated with anti-DEC-205-coated NPs (solid line circles) indicated that the specific internalization mediated through the DEC-205 receptor was driving higher levels of receptor-mediated endocytosis. And although the difference in uptake could be observed at low NP concentrations, it was much more pronounced at higher concentrations (>100 μg/mL), indicating that antibody-coated NPs in general were internalized by Fc-mediated mechanisms; however, specifically targeted DEC-205 NPs were associated with additional endocytic or phagocytic activity that did not plateau even at high particle concentrations. These results also indicate that the previously observed increases in Ag cross-presentation efficiency (Figure 3B) associated with DEC-205-mediated delivery could be explained largely by increased levels of NP internalization. DCs loaded with blank NPs containing no fluorescent Ag were used as controls (represented by dashed triangles directly on x-axis in Figure 4). No significant uptake was detected when DCs were exposed to particles and kept at 4°C (data not shown).
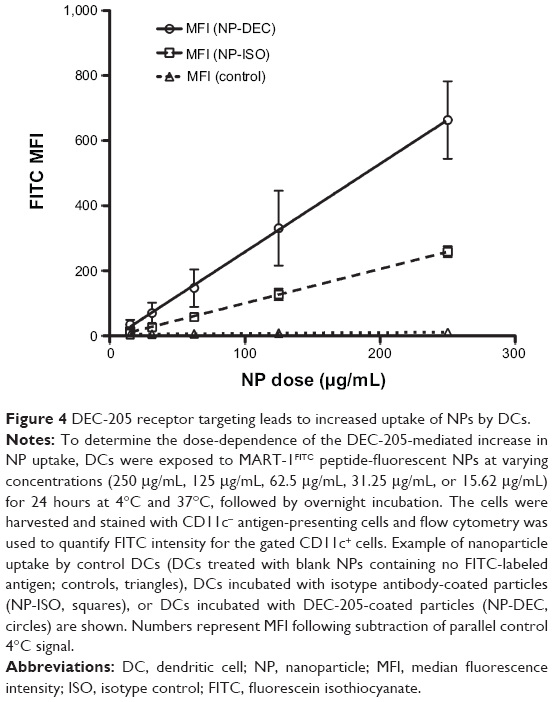 | Figure 4 DEC-205 receptor targeting leads to increased uptake of NPs by DCs. |
Intracellular localization of targeted NP
To determine whether targeted NPs deliver increased Ag to the traditional endolysosomal pathway or whether this Ag has increased access to the cytoplasmic MHC class I loading machinery, internalization and subcellular localization of DEC-205-targeted NPs was also assessed using TEM, confocal microscopy, and ImageStream analysis (Figure 5A–C), to access NP delivery and residence in the endolysosomal compartment. Previous work by our group and others15 (reviewed by Dahnier et al23) indicated that PLGA NPs escape the endosomal compartment and degrade in the cytoplasm of the DC, providing prolonged and enhanced Ag cross-presentation. Here we provide evidence from three different imaging modalities that targeted PLGA NPs, surface-labeled with a DEC-205 targeting antibody, also appear to “escape” lysosomal degradation and reside in the DC cytoplasm. TEM images of DCs that had internalized DEC-205-labeled PLGA NPs (Figure 5A, left panel, arrows and inset) confirm that the NPs are not surrounded by a classical endolysosomal double membrane, indicating that, following receptor-mediated endocytosis, a full membrane either does not form or is quickly destabilized by the NP, leading to endosomal rupture and release to the cytoplasm. By comparison, when a non-PLGA NP of identical size, in this case a non-biodegradable, unlabeled latex bead (5A, right panel, arrows and inset), is internalized by identical DCs, a clear double membrane surrounds each particle. This indicates that DEC-205-coated PLGA NPs are preferentially excluded from the endolysosomal compartment, and instead reside (and presumably degrade) within the DC cytoplasm.
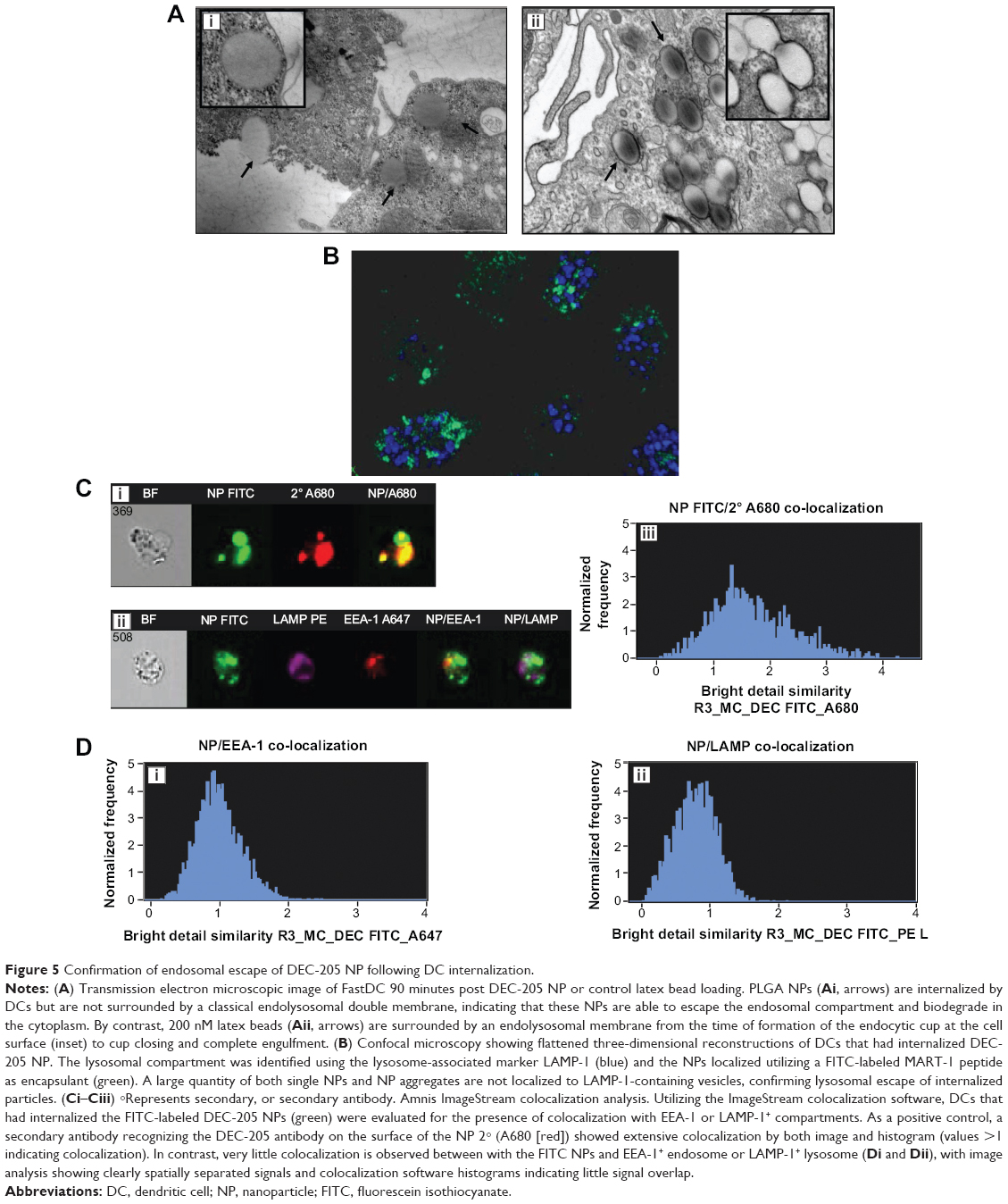 | Figure 5 Confirmation of endosomal escape of DEC-205 NP following DC internalization. |
This observation was confirmed by confocal microscopy (Figure 5B), as three-dimensional reconstructions of DC that had internalized fluorescently labeled DEC-205 NP (green), show extremely limited colocalization with a marker of the lysosomal compartment, LAMP-1 (blue). Single NPs or areas of accumulated multiple NPs are spatially separated from DC lysosomes, with little or no colocalized signal. These data indicate that the DEC-205 NPs are not shuttled through the standard endolysosomal pathway, an observation that differs from larger particulate Ag which are targeted to the lysosomes for degradation and Ag presentation.34 Finally, ImageStream analysis confirmed the results obtained by TEM and confocal microscopy, indicating low levels of intracellular colocalization with markers of the endolysosome (Figure 5C and D). The ImageStream system is designed to image cells under flow conditions and combines the capabilities of microscopy and flow cytometry in a single platform, allowing visualization of individual cells and simultaneous spatial localizations of intracellular signals. Each cell is represented by a bright field image and up to five fluorescent images (Figure 5C), and utilizing the instrument’s IDEAS software and “similarity” algorithm it is possible to quantitate colocalization of signals from different spectral channels and provide similarity histograms (Figure 5D). Thus, detection of fluorescent signal from an internalized FITC NP (Figure 5Cii) which is in the same spatial register as signal from the endolysosomal markers EEA-1 and LAMP-1 (Figure 5Cii) would be visualized as completely overlapping signal in overlays (Figure 5Cii). As a positive control for signal overlap, a labeled secondary antibody recognizing the DEC-205 antibody on the NP surface (Figure 5Ci), was included as an indicator of complete colocalization. As shown in the upper fluorescent panel (Figure 5Ci) and associated histogram (Figure 5Ciii), this control exhibited near 100% colocalization (where histogram values >1 indicate increasing degrees of overlap). This is in sharp contrast to the localization associated with the endolysosomal panel (Figures 5Ci and 5D), where a comparatively limited degree of colocalization is observed.
Taken together, these data indicate that increased Ag presentation associated with DEC-205-mediated Ag delivery could result from a combination of more efficient NP internalization (Figure 4) as well as differential delivery of NP conjugates to the DC cytoplasm (Figure 5).
Discussion
Activating the immune system to trigger a sufficient cytotoxic T lymphocyte-mediated response is a major challenge in the development of successful therapeutic vaccines against pathogens and cancer. Currently, there are no human peptide-based cancer vaccines on the market, mostly because of the difficulties associated with their poor immunogenicity, stability, and delivery. Our group15,17,35–38 and others39–42 have described strategies to enhance peptide/protein immunogenicity and stability by directing Ag to DCs using particulate delivery systems. The particulate nature of these delivery vehicles permits their rapid internalization by DCs and their composition can provide adjuvant properties required for immunogenicity.18 Ag encapsulated in NPs and targeted to APC are protected from enzymatic degradation, and processed and presented to T lymphocytes after their delivery to the APC.
Receptors previously used for DC targeting, such as DEC-205 as a protein Ag conjugate, have the ability to deliver antigens to late endosomal elements that serve as efficient sites for the formation of immunogenic peptides and their loading onto MHC class II molecules (which are needed for CD4 T-cell and antibody responses).43,44 Effective vaccination, however, will also often require the production of CD8 cytotoxic T-cell responses that occur only when antigen is present in the cytoplasm. DCs are adept at this function via the process of “cross-presentation”, whereby exogenous antigens escape endocytic vesicles and enter the cytoplasm where they are cleaved into peptides by the proteasome, imported into the endoplasmic reticulum, and loaded onto newly synthesized MHC class I molecules (which are required for stimulation of CD8 T-cells). In previous work, we have found that it is possible to greatly enhance the efficiency of cross-presentation by artificially causing the limited disruption of endosome-lysosome membranes during antigen uptake.15 Our results indicate that this can be accomplished in vitro with DEC-205-targeted PLGA NPs. This approach is directly analogous to that taken by many pathogens, including viruses, such as adenovirus, reovirus, and papillomavirus, that effect endosome disruption in order to gain access to the cytosol for purposes of infection.45–48
Here we have shown that surface modification of Ag-loaded NPs to direct them to DCs affects the outcome of the immune response towards the delivered antigen. Several studies have demonstrated that targeting soluble Ag to DCs via DEC-205 fusion proteins leads to improved receptor-mediated endocytosis and Ag presentation.4,8,19 Furthermore, the concept of targeting DEC-205 via particulate delivery to DCs has been introduced in mice, with Kwon et al using pH-responsive microparticles surface-conjugated with the anti-DEC-205 monoclonal antibody to demonstrate that targeting DEC-205 enhances particle uptake and Ag presentation,49 as well as our groups’ demonstration that the density of DEC-205 on the NP surface is crucial to controlling murine DC antigenicity.19 However, these findings have not yet been extended to relevant human in vitro experiments that mimic current DC vaccinations utilizing monocyte-derived DCs, although data exist suggesting possible utility in DEC-205 targeting of plasmacytoid DCs.9 For the first time, we demonstrate here that targeting human monocyte-derived DCs in vitro via DEC-205 using biodegradable PLGA NPs leads to enhanced stimulation of relevant antitumor CD8+ T-cells.
To target DCs in vitro and to initiate a directed CTL-mediated immune response, PLGA NPs coated with anti-DEC-205 monoclonal antibodies were used as a delivery system to load clinically relevant, melanoma-associated antigen-derived peptide, MART-127–35, into human DCs (Figure 1A). A beneficial feature of the use of anti-DEC-205 monoclonal antibody-conjugated particles is that mature DCs as well as immature DCs are targeted. Birkholz et al reported that mature DCs express high levels of DEC-205 on their cell surface and are able to stimulate naïve T-cells with high efficiency.8 As DEC-205-expression was observed on both immature and mature monocyte-derived DCs in our experiments (Figure 2A), in vitro DEC-205 targeting of DCs was carefully analyzed, and its impact on DC phenotype and functionality was investigated. Four different DC populations were studied (Figure 3A): DCs loaded with MART-127–35 containing anti-DEC-205-coated NPs; DCs loaded with MART-127–35 containing isotype-coated NPs; and DCs loaded with blank NPs similarly labeled. We observed that the MART-127–35 peptide delivered via anti-DEC-205-coated NPs was more efficiently cross-presented to MART-127–35-specific CD8+ T-cells (Figure 3B), and also determined that increased levels of Ag presentation were paralleled by increased levels of NP internalization (Figure 4). Importantly, in this study, we used patient-derived CD8+ CTL lines and peptides previously utilized in a large group of clinical trials, ensuring the translation relevance of these reagents.
Having demonstrated the increased Ag cross-presentation capability of anti-DEC-205-coated NPs, we wished to investigate the underlying mechanism driving these findings. We suggest two possibilities for this enhancement of Ag presentation: increased uptake of the anti-DEC-205-coated NPs by DCs and/or that uptake via DEC-205 results in transport of NPs to intracellular compartments involved in antigen processing for MHC I cross-presentation.
DCs are known to uptake soluble and particulate Ag by means of macropinocytosis, so can internalize relatively small particles compared with macrophages. Therefore, to further enhance DC-specific uptake, it is important to target these cells by using NP surface moieties which stimulate through DC endocytic receptors. As DEC-205 is a well-defined DC-associated lectin receptor, previously shown to facilitate Ag delivery and increased MHC I and II DC presentation,4,8 NPs conjugated with anti-DEC-205 monoclonal antibody are a logical next step in improving DC vaccine potency. We show here that human DCs more efficiently internalize DEC-205-labeled NPs than particles conjugated to isotype monoclonal antibody, indicating that DEC-205 NPs can signal through both DC Fc receptors and DC lectin receptors (Figure 4). These results clearly indicate that enhanced DC-specific endocytosis of particles achieved through their conjugation with anti-DEC-205 antibody is one of the likely drivers of the enhanced Ag presentation observed.
Besides simple increases in Ag internalization, another potential mechanism explaining the increased cross-presentation associated with DEC-205 NP-mediated Ag delivery is the potential access of these NP conjugates to the cytoplasmic MHC class I machinery. This access would circumvent the need for “traditional” cross-presentation through the vacuolar or phagosome-to-cytosol pathways currently proposed to explain DC cross-presentation,50,51 which was a possibility proposed previously by our group following biochemical and microscopic analysis of DCs loaded with unmodified NPs.15 Here we provide additional confirmatory evidence that the phenomenon of “endosomal escape” of PLGA NPs can also be extended to NPs surface-modified with DEC-205. Escape of NPs from the endolysosomal compartment has become an increasingly accepted mechanism for PLGA NP utility in drug and Ag delivery,23 although some conflicting reports exist,34 most likely because of the use of microparticles versus NPs in DC loading and the inherent differences in the way in which large particles are routed intracellularly. Utilizing evidence from three different imaging modalities, ie, TEM, confocal microscopy, and ImageStream analysis, we show that DEC-205-targeted NPs appear to “escape” lysosomal degradation by avoiding routing to the endolysosomal pathway, and instead reside in the cytoplasm following internalization. TEM images of DCs that had internalized DEC-205-labeled NPs (Figure 5A, left panel) confirm that the NPs are not surrounded by a classical endolysosomal double membrane, indicating that a traditional membrane either does not form properly or is quickly ruptured by the NPs, leading to NP release to the cytoplasm. In contrast, when a non-biodegradable particle, in this case latex beads of identical size (Figure 5A, right panel), is internalized by a DC, a phagosomal membrane is clearly present from the formation of the phagocytic cup to complete engulfment of the internalized particle. This cytosolic localization of DEC-205-labeled PLGA NPs was confirmed by confocal microscopy (Figure 5B), as internalized FITC-labeled NPs show limited colocalization with the lysosomal marker LAMP-1, with clear spatial separation between the DC lysosomes and single or aggregated NPs. Finally, ImageStream analysis confirmed these results, indicating low levels of intracellular colocalization with markers of the endolysosome (Figure 5C and D). Taken together, these data indicate that DEC-205 NPs degrade within DCs in fundamentally different subcellular compartments than other particulate Ag, which are generally targeted to the lysosomes for degradation and traditional MHC class I and II presentation. Further studies are underway to determine whether DEC-205-labeled NPs are shunted to intracellular compartments in a mechanistically similar or different way when compared with control antibody-labeled NPs or untargeted particles; this will help define whether the increase in T-cell stimulation associated with targeted NPs is the result of simple increases in incorporation of DEC-205-labeled NPs into DCs or the differential transport of DEC-205 Ag once internalized by the cell, a phenomenon previously described for this ligand.52
In summary, the development of DC-targeting NP vaccine reagents derived from clinically relevant tumor antigens holds great promise. The results presented here indicate that DEC-205 receptor-mediated targeting of tumor Ag to DCs is a safe and effective methodology to heighten antigen uptake and subsequent CD8+ T-cell stimulation by DCs, which are features essential to improving DC vaccine potency. Recent advances in NP technology allow for many variations in the design of NPs, including simultaneous delivery of antigens, immunomodulators, or other compounds and targeting moieties, creating a potent multivalent therapeutic strategy. The reagents described here, which remain focused on utilizing only reagents approved by the US Food and Drug Administration and real-world fabrication techniques, are another example of these advances that potentially represent a significant step forward in vaccine development.
Acknowledgments
This project was funded in part by the Albert Einstein College of Medicine Research Fellowship. The authors would like to thank Jeffrey Futterleib, Hao Feng, Kristina Liu, Renata Filler, and Julia Lewis for their support and advice. The authors would also like to thank Ewa Menet (ImageStream) and Al Menone (confocal microscopy) for imaging technical assistance.
Disclosure
The authors report no conflicts of interest in this work.
References
Banchereau J, Steinman RM. Dendritic cells and the control of immunity. Nature. 1998;392:245–252. | ||
Figdor CG, van Kooyk Y, Adema GJ. G. C-type lectin receptors on dendritic cells and Langerhan cells. Nat Rev Immunol. 2002;2:77–84. | ||
Tacken PJ, de Vries IJ, Torensma R, Figdor CG. Dendritic-cell immunotherapy: from ex vivo loading to in vivo targeting. Nat Rev Immunol. 2007;7:790–802. | ||
Bonifaz LC, Bonnyay DP, Charalambous A, et al. In vivo targeting of antigens to maturing dendritic cells via the DEC-205 receptor improves T-cell vaccination. J Exp Med. 2004;199:815–824. | ||
Idoyaga J, Cheong C, Suda N, et al. Cutting edge: Langerin/CD207 receptor on dendritic cells mediates efficient antigen presentation on MHC I and II products in vivo. J Immunol. 2008;180:3647–3650. | ||
Singh S, Stephani J, Schaefer M, et al. Targeting glycan modified OVA to murine DC-SIGN transgenic dendritic cells enhances MHC class I and II presentation. Mol Immunol. 2009;47:164–174. | ||
Kato M, McDonald KJ, Khan S, et al. Expression of human DEC-205 (CD205) multilectin receptor on leukocytes. Int Immunol. 2006; 18:857–869. | ||
Birkholz K, Schwenkert M, Kellner C, et al. Targeting of DEC-205 on human dendritic cells results in efficient MHC class II-restricted antigen presentation. Blood. 2010;116:2277–2285. | ||
Tel J, Sittig SP, Blom RA, et al. Targeting uptake receptors on human plasmacytoid dendritic cells triggers antigen cross-presentation and robust type I IFN secretion. J Immunol. 2013;191:5005–5012. | ||
Kawakami Y, Eliyahu S, Sakaguchi K, et al. Identification of the immunodominant peptides of the MART-1 human melanoma antigen recognized by the majority of HLA-A2 restricted tumor infiltrating lymphocytes. J Exp Med. 1994;180:347–352. | ||
Marincola FM. A balanced review of the status T-cell-based therapy against cancer. J Transl Med. 2005;3:16. | ||
Eggermont AM, Robert M. Melanoma in 2011: a new paradigm tumor for drug development. Nat Rev Clin Oncol. 2012;9:74–76. | ||
Postow MA, Harding J, Wolchok JD. Targeting immune checkpoints: releasing the restraints on anti-tumor immunity for patients with melanoma. Cancer J. 2012;18:153–159. | ||
Stewart TJ, Greeneltch KM, Lutsiak ME, Abrams SI. Immunological responses can have both pro-antitumour effects: implications for immunotherapy. Expert Rev Mol Med. 2007;9:1–20. | ||
Shen H, Ackerman AL, Cody V, et al. Enhanced and prolonged cross-presentation following endosomal escape of exogenous antigens encapsulated in biodegradable nanoparticles. Immunology. 2006;117:78–88. | ||
Scholl I, Boltz-Nitulescu G, Jensen-Jarolim E. Review of novel particulate antigen delivery systems with special focus on treatment of type I allergy. J Control Release. 2005;104:1–27. | ||
Fahmy TM, Demento SL, Caplan MJ, Mellman I, Saltzman WM. Design opportunities for actively targeted nanoparticle vaccines. Nanomedicine (Lond). 2008;3:343–355. | ||
Demento SL, Siefert AL, Bandyopadhyay A, Sharp FA, Fahmy TM. Pathogen-associated molecular patterns on biomaterials: a paradigm for engineering new vaccines. Trends Biotechnol. 2011;29:294–306. | ||
Bandyopadhyay A, Fine RL, Demento S, Bockenstedt LK, Fahmy TM. The impact of nanoparticle ligand density on dendritic-cell targeted vaccines. Biomaterials. 2011;32:3094–3105. | ||
Sharp FA, Ruane D, Claass B, et al. Uptake of particulate vaccine adjuvants by dendritic cells activates the NALP3 inflammasome. Proc Natl Acad Sci U S A. 2009;106:870–875. | ||
Rokkanen P, Böstman O, Vainionpää S, et al. Biodegradable implants in fracture fixation: early results of treatment of fractures of the ankle. Lancet. 1985;1:1422–1424. | ||
Bercovy M, Goutallier D, Voisin MC, et al. Carbon-PGLA prostheses for ligament reconstruction. Experimental basis and short-term results in man. Clin Orthop Relat Res. 1985;196:159–168. | ||
Dahnier F, Ansorena E, Silva JM, et al. PLGA-based NP: an overview of medical applications. J Control Release. 2012;161:505–522. | ||
Hamdy S, Haddadi A, Hung RW, Lavasanifar A. Targeting DC with nano-particulate PLGA cancer vaccine formulations. Adv Drug Deliv Rev. 2011;63:943–955. | ||
De Koker S, Lambrecht BN, Willart MA, et al. Designing polymeric particles for antigen delivery. Chem Soc Rev. 2011;40:320–339. | ||
Johnson LA, Heemskerk B, Powell DJ Jr, et al. Gene transfer of tumor-reactive TCR confers both high avidity and tumor reactivity to nonreactive peripheral blood mononuclear cells and tumor-infiltrating lymphocytes. J Immunol. 2006;177:6548–6559. | ||
Fahmy T, Samstein R, Harness C, Saltzman W. Surface modification of biodegradable polyesters with fatty acid conjugates for improved drug targeting. Biomaterials. 2005;26:5727–5736. | ||
Park J, Mattessich T, Jay SM, Agawu A, Saltzman WM, Fahmy TM. Enhancement of surface ligand display on PLGA nanoparticles with amphiphilic ligand conjugates. J Control Release. 2011;156:109–115. | ||
Dauer M, Obermaier B, Herten J, et al. Mature dendritic cells derived from human monocytes within 48 hours: a novel strategy for dendritic cell differentiation from blood precursors. J Immunol. 2003;170:4069–4076. | ||
Obermaier B, Dauer M, Herten J, Schad K, Endres S, Eigler A. Development of a new protocol for 2-day generation of mature dendritic cells from human monocytes. Biol Proced Online. 2003;5:197–203. | ||
Ho WY, Nguyen HN, Wolfl M, Kuball J, Greenberg PD. In vitro methods for generating CD8+ T-cell clones for immunotherapy from the naïve repertoire. J Immunol Methods. 2006;310:40–52. | ||
Ramadan G. Fast monocyte-derived dendritic cell-based immunotherapy. Methods Mol Biol. 2014;1139:131–144. | ||
Kurts C, Robinson BW, Knolle PA. Cross-priming in health and disease. Nat Rev Immunol. 2010;10:403–414. | ||
Schliehe C, Schliehe C, Thiry M, et al. Microencapsulation of inorganic nanocrystals into PLGA microsphere vaccines enables their intracellular localization in dendritic cells by electron and fluorescence microscopy. J Control Release. 2011;151:278–285. | ||
Prasad S, Cody V, Saucier-Sawyer JK, et al. Polymer nanoparticles containing tumor lysates as antigen delivery vehicles for dendritic cell-based antitumor immunotherapy. Nanomedicine. 2011;7:1–10. | ||
Solbrig CM, Saucier-Sawyer JK, Cody V, Saltzman WM, Hanlon DJ. Polymer nanoparticles for immunotherapy from encapsulated tumor-associated antigens and whole tumor cells. Mol Pharm. 2007;4:47–57. | ||
Demento SL, Cui W, Criscione JM, et al. Role of sustained antigen release from nanoparticle vaccines in shaping the T-cell memory phenotype. Biomaterials. 2012;33:4957–4964. | ||
Demento S, Steenblock ER, Fahmy TM. Biomimetic approaches to modulating the T-cell immune response with nano- and micro- particles. Conf Proc IEEE Eng Med Biol Soc. 2009;2009:1161–1166. | ||
Moon JJ, Huang B, Irvine DJ. Engineering nano- and microparticles to tune immunity. Adv Mater. 2012;24:3724–3746. | ||
Nembrini C, Stano A, Dane KY, et al. Nanoparticle conjugation of antigen enhances cytotoxic T-cell responses in pulmonary vaccination. Proc Natl Acad Sci U S A. 2011;108:E989–E997. | ||
Ma W, Smith T, Bogin V, et al. Enhanced presentation of MHC class Ia, Ib and class II-restricted peptides encapsulated in biodegradable nanoparticles: a promising strategy for tumor immunotherapy. J Transl Med. 2011;9:34. | ||
Zhang Z, Tongchusak S, Mizukami Y, et al. Induction of anti-tumor cytotoxic T-cell responses through PLGA-nanoparticle mediated antigen delivery. Biomaterials. 2011;32:3666–3678. | ||
Mellman I. Antigen processing and presentation by dendritic cells: cell biological mechanisms. Adv Exp Med Biol. 2005;560:63–67. | ||
Mellman I, Steinman RM. Dendritic cells: specialized and regulated antigen processing machines. Cell. 2001;106:255–258. | ||
Li C, He Y, Nicolson S, et al. Adeno-associated virus capsid antigen presentation is dependent on endosomal escape. J Clin Invest. 2013;123:1390–1401. | ||
Maier O, Marvin SA, Wodrich H, Campbell EM, Wiethoff CM. Spatiotemporal dynamics of adenovirus membrane rupture and endosomal escape. J Virol. 2012;86:10821–10828. | ||
Danthi P, Guglielmi KM, Kirchner E, Mainou B, Stehle T, Dermody TS. From touchdown to transcription: the reovirus cell entry pathway. Curr Top Microbiol Immunol. 2010;343:91–119. | ||
Smith JL, Campos SK, Wandinger-Ness A, Ozbun MA. Caveolin-1-dependent infectious entry of human papillomavirus type 31 in human keratinocytes proceeds to the endosomal pathway for pH-dependent uncoating. J Virol. 2008;82:9505–9512. | ||
Kwon YJ, James E, Shastri N, Fréchet JM. In vivo targeting of dendritic cells microparticles. Proc Natl Acad Sci U S A. 2005;102: 18264–18268. | ||
Hoffmann E, Kotsias F, Visentin G, Bruhns P, Savina A, Amigorena S. Autonomous phagosomal degradation and antigen presentation in dendritic cells. Proc Natl Acad Sci U S A. 2012;109:14556–14561. | ||
Joffre OP, Segura E, Savina A, Amigorena S. Cross-presentation by dendritic cells. Nat Rev Immunol. 2012;12:557–569. | ||
Mahnke K, Guo M, Lee S, et al. The dendritic cell receptor for endocytosis, DEC-205, can recycle and enhance antigen presentation via major histocompatibility complex class II-positive lysosomal compartments. J Cell Biol. 2000;151:673–684. |
Supplementary materials
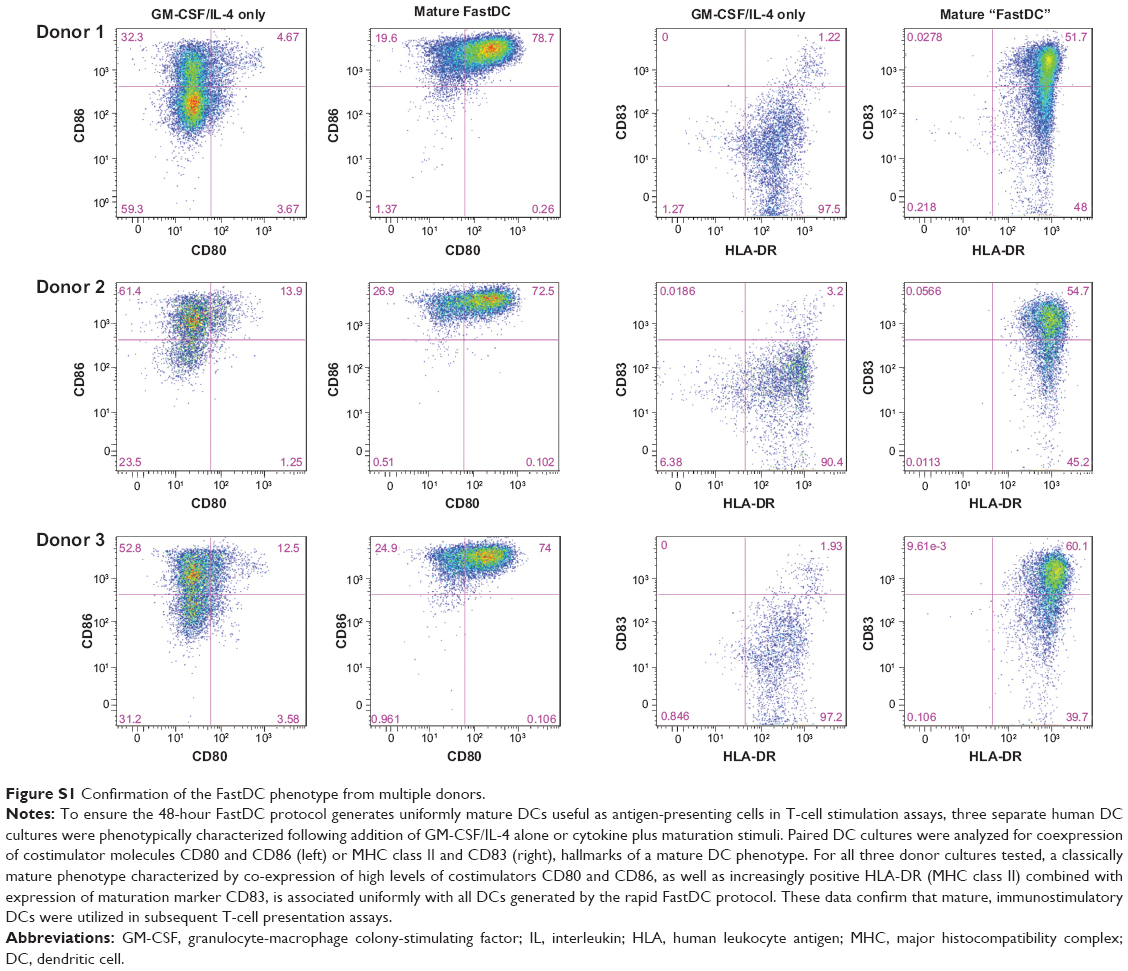 | Figure S1 Confirmation of the FastDC phenotype from multiple donors. |
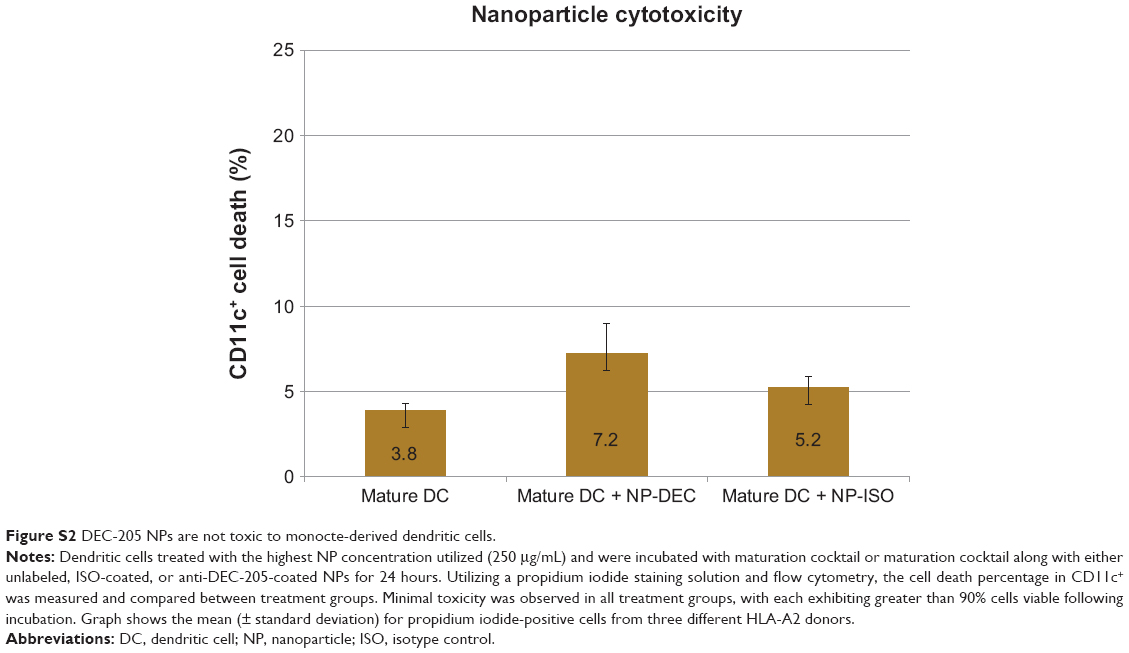 | Figure S2 DEC-205 NPs are not toxic to monocte-derived dendritic cells. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.