Back to Journals » Clinical Pharmacology: Advances and Applications » Volume 10
Targeting EGFR and uPAR on human rhabdomyosarcoma, osteosarcoma, and ovarian adenocarcinoma with a bispecific ligand-directed toxin
Authors Oh F ![]() , Todhunter D, Taras E, Vallera DA, Borgatti A
, Todhunter D, Taras E, Vallera DA, Borgatti A ![]()
Received 23 December 2017
Accepted for publication 4 May 2018
Published 26 September 2018 Volume 2018:10 Pages 113—121
DOI https://doi.org/10.2147/CPAA.S160262
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Arthur E. Frankel
Felix Oh,1 Deborah Todhunter,1 Elizabeth Taras,1 Daniel A Vallera,1,2 Antonella Borgatti2,3
1Department of Therapeutic Radiology-Radiation Oncology, Masonic Cancer Center, University of Minnesota, Minneapolis, MN, USA; 2Animal Cancer Care and Research (ACCR) Program, University of Minnesota, St. Paul, MN, USA; 3Department of Veterinary Clinical Sciences, College of Veterinary Medicine, University of Minnesota, St. Paul, MN, USA
Purpose: Human sarcomas are rare and difficult to treat cancerous tumors typically arising from soft tissue or bone. Conversely, carcinomas are the most common cancer subtype in humans and the primary cause of mortality across all cancer patients. While conventional therapeutic modalities can prolong disease-free intervals and survival in some cases, treatment of refractory or recurrent solid tumors is challenging, and tumor-related mortality remains unacceptably high. The identification of overexpressed cell surface receptors on sarcoma and carcinoma cells has provided a valuable tool to develop targeted toxins as an alternative anticancer strategy. Recent investigation of recombinant protein-linked toxins that specifically target these cancer receptors has led to the development of highly specific, cytotoxic, and deimmunized drugs that can kill cancer cells.
Methods: This study investigated a recombinant protein called epidermal growth factor bispecific angiotoxin (eBAT), which is designed to target the epidermal growth factor receptor (EGFR) on cancer cells and the urokinase plasminogen activator receptor (uPAR) on cancer cells and associated tumor vasculature. Both receptors are expressed by a variety of human sarcomas and carcinomas. Flow cytometry techniques were used to determine binding affinity of eBAT to cancer cells, and proliferation assays were performed to calculate tumor killing ability based on half-maximal inhibitory concentrations.
Results: eBAT demonstrated cytotoxicity against a variety of sarcoma and carcinoma cells that overexpress EGFR and uPAR in vitro and showed greater cell killing ability and binding affinity to cancer cells compared with its monospecific counterparts.
Conclusion: The results of our study are promising, and further studies will be necessary to confirm the applicability of eBAT as a supplementary therapy for a variety of sarcomas, carcinomas, and possibly other refractory malignancies that express EGFR and uPAR.
Keywords: sarcoma, carcinoma, eBAT, EGFR, uPAR
Plain language summary
New therapies are urgently needed to treat drug-resistant sarcomas. To target sarcomas in a novel manner, we used an anticancer drug called epidermal growth factor bispecific angiotoxin (eBAT), which has been genetically engineered by combining two naturally occurring human ligands with deimmunized bacterial Pseudomonas exotoxin. The ligands were epidermal growth factor (EGF) that binds EGF receptors (EGFR) that is overexpressed on sarcomas and a urokinase fragment that binds urokinase receptor on tumor neovasculature. Thus, the drug simultaneously binds sarcoma cells and vascular cells in the tumor microenvironment and kills them. In these studies, flow cytometry was used to prove that bispecific eBAT was more potent in binding and killing sarcoma cells than either of its monospecific counterparts targeting only EGFR or only urokinase receptor. In addition to killing sarcoma cells, including rhabdomyosarcoma and osteosarcoma, eBAT was also effective in killing ovarian carcinoma cells indicating great promise for this reagent as a new therapeutic measure for treating solid tumors.
Introduction
Human sarcomas are rare, aggressive, heterogeneous tumors of mesenchymal origin that are typically categorized as either soft tissue or bone malignancies.1 Rhabdomyosarcoma, a soft tissue sarcoma of skeletal muscle progenitor cells, and osteosarcoma, a bone sarcoma, are the two most prevalent childhood sarcomas affecting >500 children under the age of 14 every year in the United States.2 As of 2017, there are 15,650 anticipated diagnoses of soft tissue and bone sarcomas in the United States and 6,540 estimated deaths from the disease in adults and children.2 Conventional therapies for patients with sarcoma, including surgical intervention and chemotherapy, currently result in an overall 5-year survival rate of approximately 50%, but disease metastasis and tumor relapse due to treatment resistance remain the primary causes of patient mortality.3 Significant progress in treatment development has been stunted by the relatively small number of patients and the broad range of sarcoma subtypes.
In comparison, carcinomas are prevalent epithelial tumors. For example, ovarian carcinoma, which comprises just one subtype of ovarian cancer, is posed to affect 22,440 new patients in 2017 alone.2 Yet a staggering 14,080 of these patients are still expected to die from the disease and only 45% of the patients will survive 5 years after their diagnosis.2 Despite the bigger patient population, treatment advances for carcinoma face the same obstacles in terms of off-target toxicity and tumor recurrence.
Interestingly, both sarcoma and carcinoma cells have been found to overexpress receptors like the epidermal growth factor receptor (EGFR) and the urokinase plasminogen activator receptor (uPAR) on the cell surface at concentrations hundreds, if not thousands, of times greater than normal cells.5,6 The Broad-Novartis Cancer Cell Line Encyclopedia (CCLE), a genomic database of cancers and cancer cell lines, was used to determine that rhabdomyosarcomas, osteosarcomas, and adenocarcinomas were optimal targets due to their abundant expression of EGFR and uPAR.7 Our group has taken advantage of this characteristic by designing modified biological toxins that specifically target and kill the cancerous cells expressing these receptors.8
Currently, ligand-directed toxins are composed of recombinant cancer-reactive molecules that have been linked to modified bacterial toxins, such as Diphtheria Toxin (DT) or Pseudomonas exotoxin A (PE), which inhibit protein synthesis.9 We have found a way to splice two cancer-reactive cytokines along with a modified bacterial toxin into a single reagent, thereby creating a bispecific ligand-directed toxin (BLT) that can target two different overexpressed receptors on a single cell.8 Bispecificity was associated with improved specificity and cytotoxicity when a BLT was compared with its monospecific counterparts, targeting a single distinct receptor.8
Epidermal growth factor bispecific angiotoxin (eBAT) is a novel BLT that targets receptors like EGFR and uPAR and is composed of the EGF ligand and the amino terminal fragment (ATF) ligand combined with a modified truncated Pseudomonas exotoxin A (KDEL). The drug binds to EGFR or uPAR on the cell surface and induces endocytosis of the KDEL toxin which improperly modifies eukaryotic elongation factor 2 (EEF2). The modified EEF2 stops functioning and inhibits the production of new proteins, thus resulting in the death of the cell via apoptosis.10
EGFR is a commonly overexpressed receptor tyrosine kinase protein that plays a large role in the growth and division of cells and is located on the surface of a variety of solid tumors.11 uPAR, which is expressed on the sarcoma neovasculature and endothelial cells, is a glycoprotein on the cell membrane and an especially attractive target for anticancer therapeutics due its active role in the proliferation and metastasis of tumor cells.12 The expression of uPAR on the tumor neovasculature will presumably allow eBAT to destroy the blood supply that is critical to the survival of the tumor. The goal of our study was to quantitatively compare the specificity and cytotoxicity of bispecific eBAT to monospecific EGFKDEL and ATFKDEL by evaluating their tumor binding and tumor killing abilities in vitro, in the presence of EGFR- and uPAR-expressing cells.
Materials and methods
eBAT production
eBAT was synthesized by genetic recombination of competent Escherichia coli bacteria.13 The genes for EGF, ATF, and modified Pseudomonas exotoxin were linked onto a pET28c plasmid vector and then transfected into E. coli bacteria. The bacteria were then exposed to antibiotics that killed any bacterium that had not undergone transformation. Inclusion bodies, or aggregates of bacterial proteins, were extracted from the bacteria. Our target protein was then isolated, refolded, dialyzed, purified, and analyzed via gel electrophoresis as shown in Figure 1. Monospecific EGFKDEL and ATFKDEL and negative control CD3CD3KDEL were synthesized using similar methods.
 | Figure 1 Construction and production of eBAT. Notes: In order to construct eBAT, an NcoI/XhoI gene fragment was cloned encoding the genes for the EGF and ATF ligands and the modified KDEL toxin (A). The eBAT gene was inserted into the protein expression vector pET28c and then transfected into E. coli bacteria. The bacteria were then exposed to antibiotics that killed any bacterium that had not undergone transformation. Inclusion bodies, or aggregates of bacterial proteins, were extracted from the bacteria. Our target protein, eBAT, was then isolated, refolded, dialyzed, and purified. The monospecific counterparts EGFKDEL and ATFKDEL and the negative control CD3CD3KDEL were synthesized using similar methods. Gel electrophoresis was used to confirm the size of the reagents (B). Abbreviations: ATF, amino terminal fragment; eBAT, epidermal growth factor bispecific angiotoxin; EGF, epidermal growth factor. |
Cell cultures
Five cancer cell lines, originally derived from patients and obtained from American Type Culture Collection (ATCC, Rockville, MD, USA), were cultured to emulate human sarcoma, carcinoma, and lymphoma growth. Human rhabdomyosarcoma cells (RD) originated in muscle spindle tissue and expressed EGFR and uPAR.14,15 Human osteosarcoma cells (Saos2) originated in a primary bone sarcoma and expressed EGFR and uPAR.16,17 Human ovarian adenocarcinoma (SKOV3) originated in ovarian tissue and expressed EGFR and uPAR.18,19 Raji and Daudi were human B-cell lymphomas that negatively expressed EGFR and uPAR.8 All these cell lines were maintained as adherent cultures at 37°C in 5% CO2 atmosphere. Cells were dissociated twice a week into single-cell suspensions using 2 mL of Trypsin EDTA to detach adherent cells from culturing surfaces. Cells were allowed to reattach under the same conditions for maintenance or plated as single cells for experiments. Raji and Daudi human B-cell lymphoma cells were maintained as nonadherent cultures at 37°C in 5% CO2 atmosphere.
Flow cytometry protocol
We studied the binding ability of eBAT, EGFKDEL, and ATFKDEL when exposed to an EGFR- and uPAR-expressing cell line. CD3CD3KDEL was used as an irrelevant negative control toxin that only targets the cluster of differentiation 3 (CD3), a marker that is almost exclusively overexpressed on T cells.20
To determine the binding ability of our experimental reagents to EGFR and uPAR, we performed a flow cytometry technique known as fluorescence-activated cell sorting (FACS) using Saos2 osteosarcoma cells. eBAT, EGFKDEL, ATFKDEL, and CD3CD3KDEL were labeled with fluorescein isothiocyanate (FITC) and were serially diluted from concentrations of 1,000 nM at 500, 100, 50, and 10 nM increments. The FITC-labeled reagents were then incubated with 5×105 Saos2 cells in 500 µL of PBS and 2% fetal bovine serum (FBS) on ice for 1 hour to allow binding. After incubation, the cells were washed three times and analyzed using FACS Calibur and CellQuest software (BD Biosciences, San Jose, CA, USA). The percent of positive control cells was determined by gating control cells which were not incubated with FITC-labeled targeted toxin. The equilibrium binding constant (Kd) value was calculated to comparatively measure the concentration of reagents needed to achieve half-maximal binding. The FACS Calibur at the University of Minnesota’s Flow Cytometry Core Facility was used for all flow experiments.
Proliferation assay protocol
Proliferation assays were conducted to quantify the cell killing ability of eBAT in the presence of its target receptors. Saos2 osteosarcoma, RD rhabdomyosarcoma, SKOV3 ovarian carcinoma, and Raji lymphoma cell lines with distinct expression levels of EGFR and uPAR were used to determine the half-maximal inhibitory concentrations (IC50), or the concentration of the reagent at which 50% of the cell population was killed, for each of the four reagents.14–20
RD and SKOV3 cells were cultured in DMEM medium and Saos2 and Raji cells in RPMI 1640 medium at 37°C, 5% CO2 for approximately 48 hours to allow the cells to adhere. Trypsin EDTA was used to detach the cells before FBS-containing media was added to the flask to inactivate the Trypsin EDTA. The single suspension was then counted using a hemocytometer and Trypan blue. Cells were then diluted to 10,000 cells per mL and 100 µL was pipetted into each well of a 96-well plate. Plated cells were then allowed to incubate for 24 hours. The drug-specific volume of medium was calculated and allocated to four 5 mL tubes; 900 µL of medium was placed in 32 5 mL tubes. The reagents were serially diluted from concentrations of 102 nM to 10–6 nM in 10 nM increments. The dilutions were then pipetted into the 96-well plate (100 µL per well) in triplicate, leaving a toxin-free control for each cell line. After 48 hours of incubation, RD rhabdomyosarcoma, Raji, and Daudi Human B-cell lymphoma cells were pulsed with radioactive [3H] thymidine (proliferation assay) while Saos2 osteosarcoma and SKOV3 ovarian adenocarcinoma cells were pulsed with radioactive [3H] leucine (protein synthesis assay) with 1 µCi per well, before being incubated again for 24 hours. Both [3H] thymidine and [3H] leucine were used as most eukaryotic cells have been found to successfully incorporate at least one of the two radioactive nucleosides.21 The plate was placed at –80°C for 20 minutes in order to detach the cells and was harvested on glass fiber filters before being counted by standard scintillation techniques. Reagents that demonstrated greater cytotoxicity killed more target cells at lower concentrations and thus produced lower counts of [3H] thymidine or [3H] leucine. Control cells that were not exposed to reagent were used to provide a normalized sample of [3H] thymidine or [3H] leucine counts and indicate possible contamination. All data were reported as percentages of the untreated control cell counts. When applicable, IC50 values of the reagents were calculated.
Statistical analysis
All statistical analyses were performed using Prism 5 (Graphpad Software, San Diego, CA, USA) and Microsoft Excel (Microsoft Corporation, Redmond, WA, USA). The equilibrium-binding constant (Kd) was calculated using exponential regression to find the ligand concentration that bound at 50% affinity to compare the binding affinity of the reagents in flow cytometry analysis. IC50 values were calculated using linear regression and the line of best fit to determine the potency of the drugs in [3H] thymidine and [3H] leucine incorporation assays. Fold changes were calculated as the ratio of the difference between two IC50 values divided by the smaller IC50 value. Student’s t-test calculations were used to determine the significance of differences between single data points. P-values <0.05 were considered significant.
Results
eBAT binds to human sarcoma cells with high affinity
FACS was performed to determine the functional binding ability of eBAT, EGFKDEL, ATFKDEL, and CD3CD3KDEL to their respective receptors on Saos2 and RD cells as shown in Figure 2A and B. Fluorescently labeled toxins were incubated with target cells and sorted based on the distinct fluorescent and light-scattering properties of reagent-bound and reagent-unbound cells. A graph was produced based on the percentage of fluorescent reagent that bound to the cell when compared with normal, unbound cells.
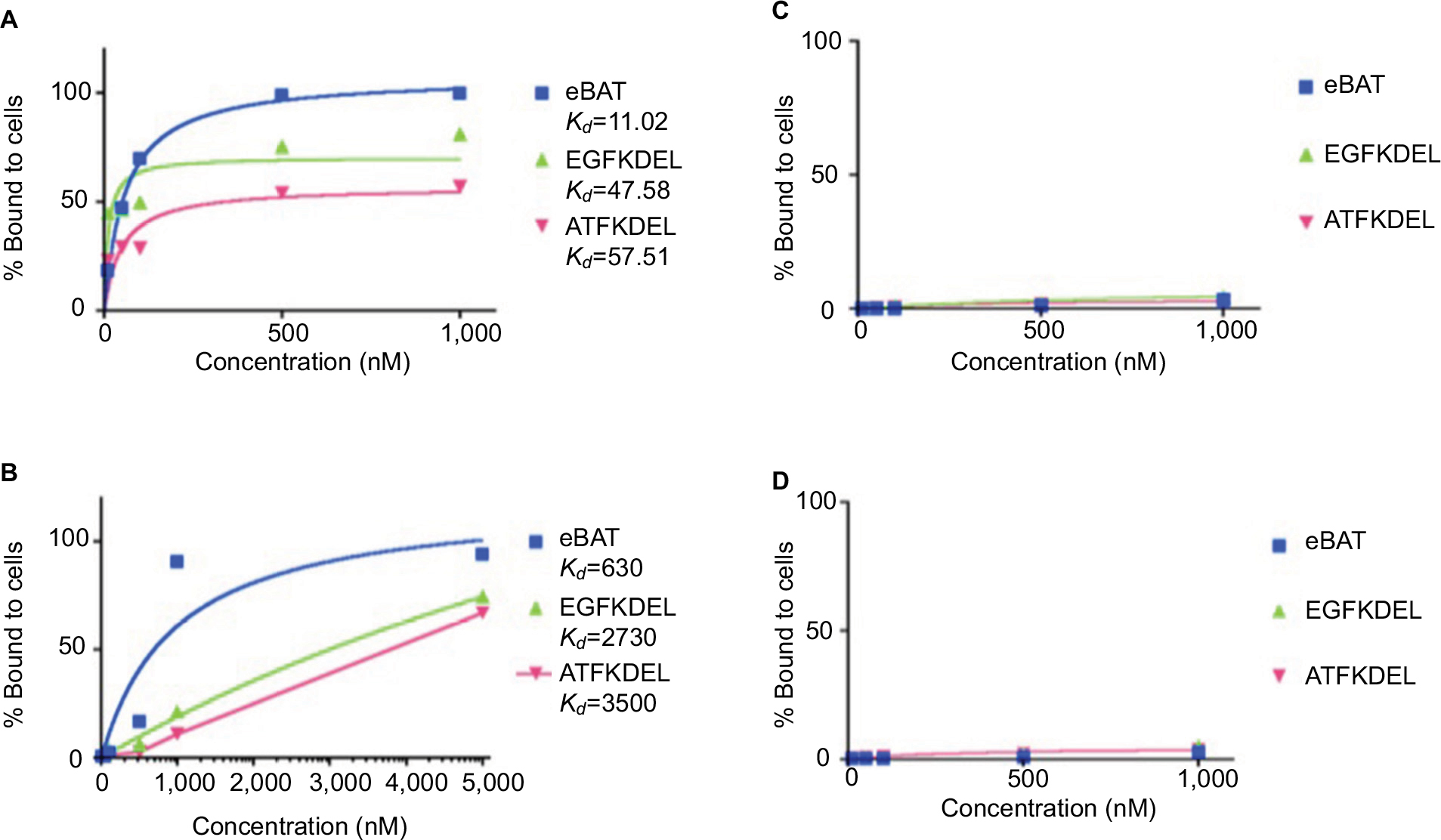 | Figure 2 Binding affinity of eBAT to sarcoma (A, B) and B-cell lymphoma (C, D) cell lines. Notes: FACS analysis was performed on the binding of fluorescently labeled eBAT to Saos2 human osteosarcoma cells and RD rhabdomyosarcoma cells. eBAT bound to EGFR- and uPAR-expressing Saos2 cells with very high affinity (>99%) at 500 and 1,000 nM. (A) In comparison, at 1,000 nM EGFKDEL binding peaked at 81.1% while ATFKDEL binding peaked at only 57.2%. eBAT showed greater binding affinity to human osteosarcoma cells than its monospecific counterparts EGFKDEL (Kd=47.58 nM) and ATFKDEL (Kd=57.51 nM) at any given concentration. RD cells exhibited similar binding patterns with the highest binding affinity to eBAT (>90%) at 5,000 nM followed by EGFKDEL and ATFKDEL. (B) The Kd value of eBAT in human rhabdomyosarcoma cells was 630 nM, indicating that it bound more effectively than EGFKDEL (Kd=2,730 nM) and ATFKDEL (Kd=3,500 nM). eBAT was not expected to bind to Raji and Daudi cells because the two cell lines negatively express EGFR and uPAR. eBAT, EGFKDEL, and ATFKDEL did not bind specifically to Raji and Daudi cells at all concentrations tested. (C, D) The graphs show the population of cancer cells relative to control cells as the concentration of each reagent increases. Kd values were not applicable as the reagents did not bind to >50% of the cells at any given concentration and no significant difference in binding was noted between the reagents. Abbreviations: ATF, amino terminal fragment; eBAT, epidermal growth factor bispecific angiotoxin; EGFR, epidermal growth factor receptor; FACS, fluorescence-activated cell sorting; uPAR, urokinase plasminogen activator receptor. |
FITC-labeled eBAT functionally bound to EGFR- and uPAR-expressing Saos2 and RD cells with high affinity (>99%). At the same concentration, EGFKDEL and ATFKDEL peaked at 81.1% and 57.2% binding in Saos2 and 74.5% and 67.3% binding in RD, respectively. eBAT bound to 99.7% of Saos2 target cells at 1,000 nM with a Kd value of 11.02 nM, showing that its optimal binding to the target receptors was achieved at the lowest concentration among the reagents tested. eBAT bound to 94.1% of RD cells at 5,000 nM with a Kd value of 630 nM.
eBAT kills human sarcoma cells in vitro
A [3H] leucine incorporation assay was designed to test the ability of bispecific eBAT to kill human osteosarcoma cells in vitro. The Saos2 cells were already confirmed to overexpress both EGFR and uPAR on the cell surface and thus presented viable targets for the drug.14,15 eBAT demonstrated effective cell killing with an IC50 of <1.0 × 10–6 nM in Saos2 cells as shown in Figure 3A. As expected, the negative control, CD3CD3KDEL, had no activity. eBAT demonstrated significantly greater cytotoxicity compared with EGFKDEL at concentrations ranging from 10–6 nM to 10–3 nM (P<0.002) or ATFKDEL at concentrations ranging from 10–6 nM to 10–2 nM (P<0.02).
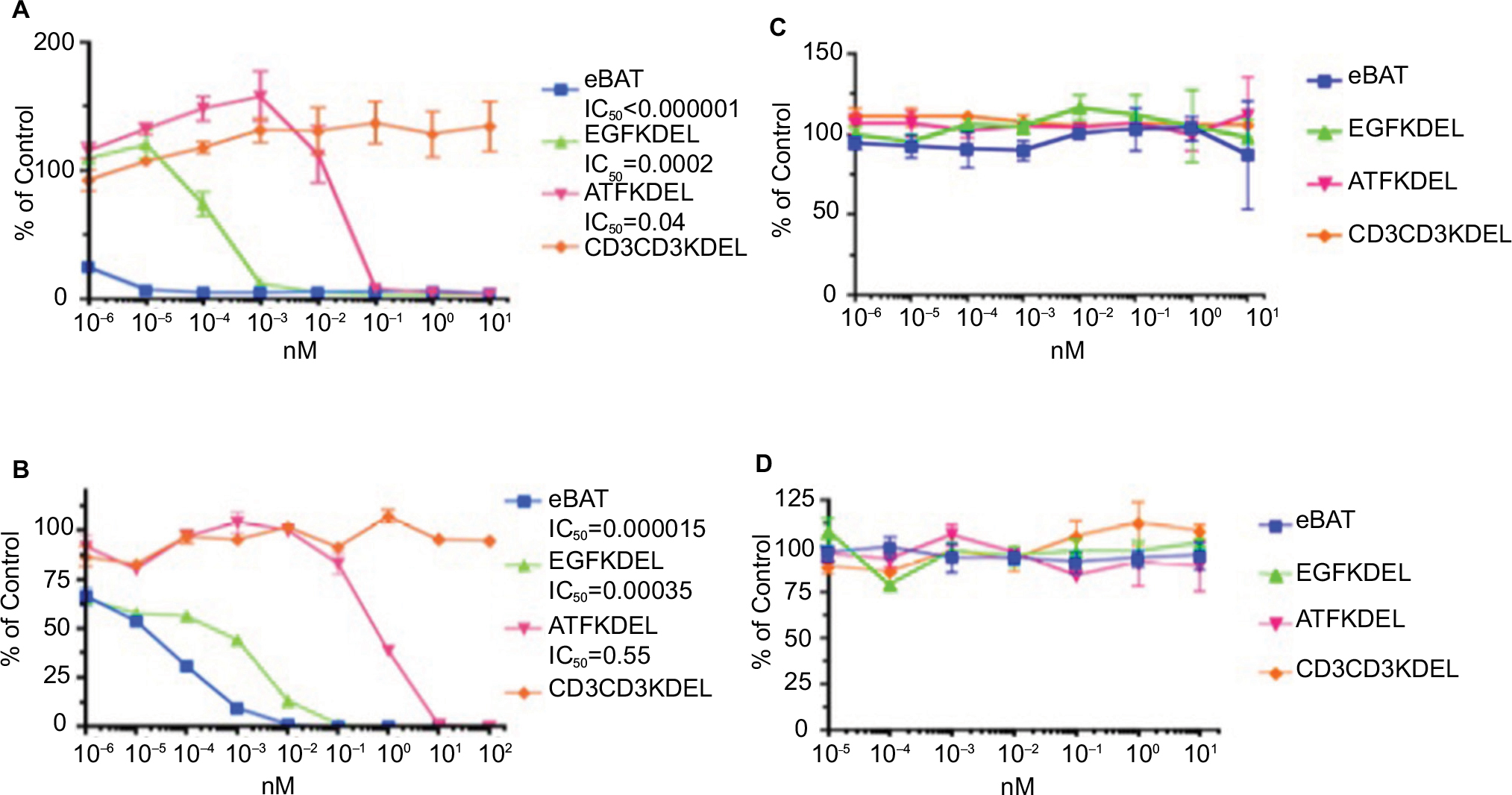 | Figure 3 Cytotoxicity of eBAT against sarcoma (A, B) and B-cell lymphoma (C, D) cell lines in vitro. Notes: Proliferation assays were performed with sarcoma cells in the presence of eBAT, EGFKDEL, ATFKDEL, and CD3CD3KDEL as the irrelevant control. The graphs show the population of cancer cells relative to control cells as the concentration of each reagent increases. In Saos2 osteosarcoma cells, eBAT demonstrated significantly increased cytotoxicity (IC50<0.000001) compared with EGFKDEL (P<0.02) and ATFKDEL (P<0.002) even at the lowest concentrations (A). In RD rhabdomyosarcoma cells, eBAT also demonstrated significantly greater cytotoxicity than both EGFKDEL (P<0.001) and ATFKDEL (P<0.0003) (B). Raji and Daudi cells were used as negative control cells due to the lack of EGFR and uPAR expression on the cell surface. (C, D). eBAT was expected to demonstrate minimal cytotoxicity against both Raji and Daudi cells for this reason. eBAT, EGFKDEL, and ATFKDEL had no effect on the proliferation of Raji and Daudi cells and there was no significant difference in cytotoxicity between the reagents. All significance was determined using Student’s t-test and calculated alongside the SD. The error bars indicate standard deviation. Abbreviations: ATF, amino terminal fragment; eBAT, epidermal growth factor bispecific angiotoxin; EGFR, epidermal growth factor receptor; uPAR, urokinase plasminogen activator receptor. |
RD rhabdomyosarcoma cells, which positively express EGFR and uPAR, were also effectively killed by eBAT with an IC50 of 1.5× 10–5 nM, while CD3CD3KDEL had no activity at all tested concentrations as shown in Figure 3B.14,15 eBAT had a greater cytotoxicity on RD cells than both EGFKDEL, at concentrations from 10–4 to 10–2 nM (P<0.001), and ATFKDEL, at concentrations from 10–6 to 102 nM (P<0.0003). Specifically, eBAT cytotoxicity was 23-fold greater than EGFKDEL and 36,666-fold greater than ATFKDEL.
eBAT binds to and kills human adenocarcinoma cells in vitro
SKOV3 ovarian adenocarcinoma cells also express EGFR and uPAR on the cell surface, and we hypothesized they could be an effective target for eBAT.18,19 FACS was performed to determine the functional binding ability of eBAT to the EGFR and uPAR receptors on SKOV3 cells as shown in Figure 4. FITC-labeled eBAT functionally bound to EGFR- and uPAR-expressing SKOV3 cells with a high affinity (93.8%) at 5,000 nM. At the same concentration, EGFKDEL and ATFKDEL peaked at 90.2% and 70.9% binding. eBAT had a Kd value of 680 nM against SKOV3, showing that its optimal binding to the target receptors was achieved at a lower concentration than its monospecific counterparts.
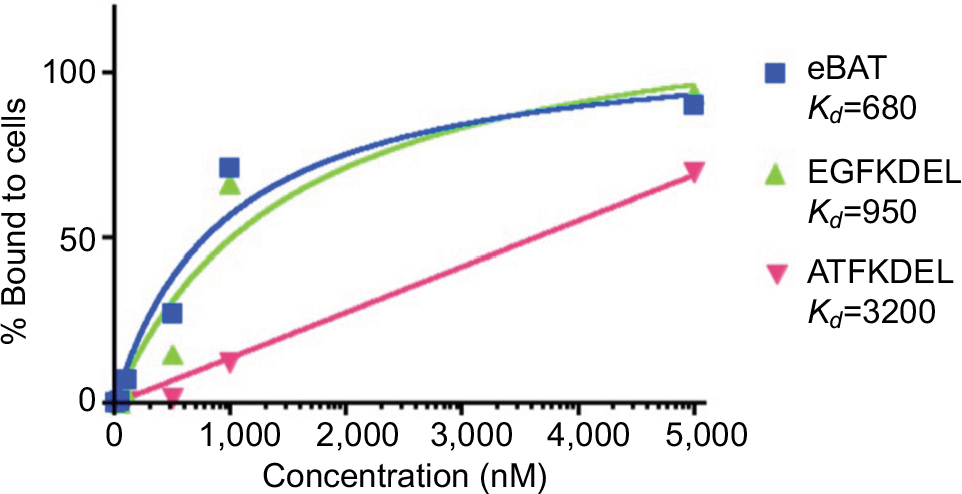 | Figure 4 Binding affinity of eBAT to SKOV3 cells. Notes: FACS analysis was performed on the binding of fluorescently labeled eBAT to SKOV3 ovarian adenocarcinoma cells. eBAT bound to EGFR- and uPAR-expressing SKOV3 cells with 93.8% affinity at 5,000 nM. At the same concentration, EGFKDEL binding peaked at 90.2% while ATFKDEL binding peaked at 70.9%. eBAT had a Kd value of 680 nM, showing greater binding affinity to human carcinoma cells than its monospecific counterparts EGFKDEL (Kd=950 nM) and ATFKDEL (Kd=3,200 nM). Abbreviations: ATF, amino terminal fragment; eBAT, epidermal growth factor bispecific angiotoxin; EGFR, epidermal growth factor receptor; FACS, fluorescence-activated cell sorting; uPAR, urokinase plasminogen activator receptor. |
eBAT demonstrated potent cell killing of SKOV3 cells with an IC50 of 5.0× 10–6 nM as shown in Figure 5. Cytotoxicity of eBAT was greater than EGFKDEL, at concentrations from 10–5 to 10–4 nM (P<0.02), and ATFKDEL at concentrations from 10–5 to 10–1 nM (P<0.001). eBAT was 1.6-fold and 50,000-fold more cytotoxic than EGFKDEL and ATFKDEL against this cell line.
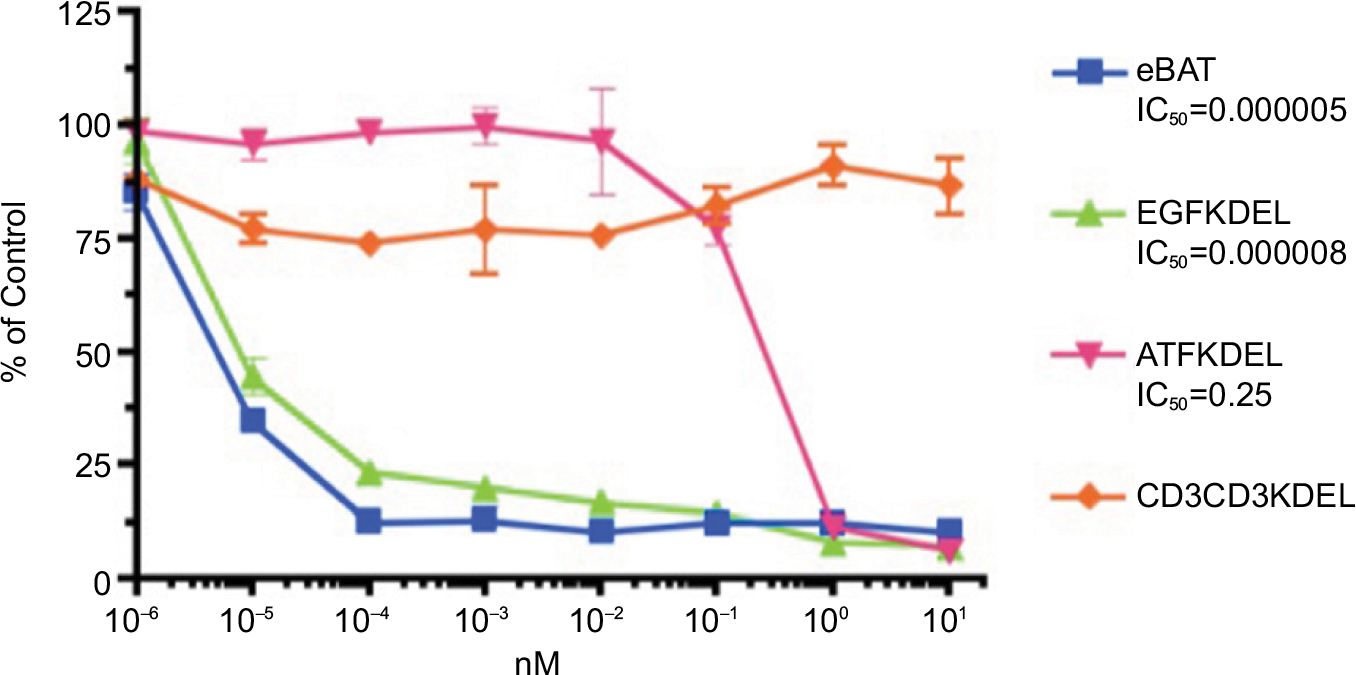 | Figure 5 Cytotoxicity of eBAT against SKOV3 cells in vitro. Notes: Proliferation assays were performed with SKOV3 ovarian adenocarcinoma cells in the presence of eBAT, EGFKDEL, ATFKDEL, and CD3CD3KDEL as the irrelevant control. The graphs show the population of cancer cells relative to control cells as the concentration of each reagent increases. Despite not being a sarcoma, SKOV3 is demonstrative of the specific cytotoxicity of eBAT against all EGFR- and uPAR-expressing tumors. In SKOV3 cells, eBAT consistently demonstrated equal or greater cytotoxicity than EGFKDEL and significantly greater cytotoxicity than ATFKDEL (P<0.001). The error bars indicate standard deviation. Abbreviations: ATF, amino terminal fragment; eBAT, epidermal growth factor bispecific angiotoxin; EGFR, epidermal growth factor receptor; uPAR, urokinase plasminogen activator receptor. |
eBAT demonstrates specificity for EGFR- and uPAR-expressing cells in vitro
The Raji and Daudi B-cell lymphoma cell lines that do not express EGFR and uPAR were used as negative controls to determine if eBAT would only specifically kill cells that overexpressed the target receptors.20 As predicted, eBAT, EGFKDEL, and ATFKDEL did not have any specific binding affinity to the cells as shown in Figure 2C and D and there was no effect on cell proliferation in Figure 3C and D. CD3CD3KDEL also exhibited negligible cytotoxic effects on cell proliferation as a non EGFR- and uPAR-targeting negative control.
Discussion
We showed for the first time that eBAT has significantly greater binding ability and cytotoxicity against human sarcoma cells than its monospecific counterparts in vitro. Despite the relatively low level of binding of ATFKDEL-FITC to uPAR (57.2% at 1,000 nM), the 18.6% difference between the peak binding ability of eBAT-FITC and that of EGFKDEL-FITC at 1,000 nM suggests that the inclusion of the ATF portion may play an important role in increasing binding affinity. This is relevant because it suggests that, despite the lower individual binding of EGFKDEL and ATFKDEL, the combined targeting of both receptors significantly improves the affinity of eBAT to EGFR- and uPAR-expressing sarcoma cells, possibly increasing killing ability. This is in line with prior studies which have suggested that the bispecificity of eBAT may enhance cytotoxicity as well as the ability to kill resistant cancer cell outliers.22 Further investigation is needed to determine exactly the role of uPAR targeting in improving the binding ability of eBAT in vitro.
The descending cytotoxicity of eBAT, EGFKDEL, and ATFKDEL, illustrated by the proliferation assays in Figure 3, mirrors the binding ability of the respective reagents shown in Figure 2. Interestingly, negligible binding activity was seen at concentrations below 10 nM even though eBAT, EGFKDEL, and ATFKDEL all demonstrated cell killing activity in the proliferation assays at concentrations as low as 10–4 nM in Figure 3. This may be a result of the off-target toxicity of Pseudomonas exotoxin or the substantially cytotoxic effect of miniscule EGFR and uPAR binding; however, the negligible cell death of Raji cells and the negligible cytotoxicity of an irrelevant control toxin suggest that eBAT may demonstrate greater cytotoxicity than what would be predicted based on its binding ability.
The prevalence of EGFR on the cell membrane and the angiogenic properties of uPAR render them attractive targets for targeted therapies. The high expression rate of EGFR on glioma and glioblastoma tumor cells in the brain has led to the development of EGF-targeting bacterial toxins, molecularly analogous to the monospecific counterparts of eBAT, with potent specific cytotoxicity.23,24 Several anticancer therapeutics designed to target EGFR and uPAR individually are available commercially.25–27 Unfortunately, EGFR inhibitors including cetuximab and gefitinib have been associated with undesirable side effects, often requiring therapy discontinuation.25,26 Furthermore, though numerous angiogenic inhibitors such as thalidomide and bevacizumab are readily accessible, their effectiveness is limited by the high adaptability of tumors to hypoxic environments and historically poor tumor regression rates.27
By combining the two targeting mechanisms of EGF and ATF, eBAT retains the versatile cell killing ability of EGFR toxins while simultaneously acquiring the antiangiogenic properties of uPAR toxins. Increased targeting specificity should presumably reduce the off-target effects of standalone EGFR-targeting therapeutics. Interestingly, a recent study of eBAT combined with standard of care surgery and chemotherapy for treatment of naturally occurring canine hemangiosarcoma, a highly aggressive, incurable sarcoma affecting the spleen of dogs, showed that eBAT was remarkably safe and potentially effective. None of the treated dogs experienced the adverse effects that are typically associated with EGFR-targeted treatments, suggesting that bispecificity likely abated toxicity.28 This could also be true in human cancer patients, and additional studies will be needed to specifically evaluate the safety and efficacy of eBAT in humans.
We showed that carcinomas may represent a targetable cancer for eBAT, in addition to sarcomas. Both sarcomas and carcinomas require angiogenesis, the production of nutrient-rich vasculature for tumor proliferation, and show similarities in their expression pattern of surface receptors, namely EGFR and uPAR.14–19 Carcinomas are the most prevalent type of cancer and they include a wide variety of subtypes potentially targetable by eBAT, including the ovarian adenocarcinoma we tested.2 Our results showed that eBAT induced specific cytotoxicity in ovarian adenocarcinoma at a lower concentration than its monospecific counterparts, especially ATFKDEL. While statistically significant, the difference in IC50 between eBAT and EGFKDEL was not as pronounced as the difference between eBAT and ATFKDEL. This could have been due to the relatively low expression of uPAR compared with the expression of EGFR, which was the primary determinant of cytotoxicity. Nevertheless, eBAT effectively targeted and killed carcinoma cells. The potential efficacy of eBAT in the treatment of carcinomas is further supported by the specific cytotoxicity of the reagent against glioblastoma tumors in mouse xenograft models.22 Testing the cytotoxicity of eBAT as well as analyzing the specific binding ability of the reagent against different cell lines will continue to provide a better understanding of what cancer subtypes eBAT may be effective against in the clinical setting.
Before eBAT can undergo further clinical development, several practical application issues must be addressed. The nature of sarcomas in humans requires repeated and sustained treatment in order to penetrate solid tumors, and thus eBAT faces several obstacles including delivery method and immunogenicity.9 To circumvent antibody response to the reagent, Kreitman et al designed a modified Pseudomonas exotoxin in which seven distinct immunogenic regions were mutated to avoid the development of antitoxin antibodies and reduce the likelihood of treatment resistance.29 This truncated toxin has previously been shown to induce almost no immunogenic change in mice after eight injections when compared with the unmutated parental strand which incited antibody production in mice after only four injections.30 eBAT has demonstrated similar success in overcoming antibody resistance in canines as well. Canine neutralizing antibody (NA) levels were measured before and after the administration of eBAT, and less than half of the dog population developed antibodies against the reagent. Even then, no significant association was found between the development of NAs and survival.28 Though there is no animal experimentation in our data, the viability of murine and canine models for human immunogenic responses to the Pseudomonas exotoxin in eBAT warrants further study.
Even though sarcomas make up <2% of diagnosed cancers in humans, they typically demonstrate highly aggressive behavior and often require extensive treatment including invasive surgery and chemotherapy.4,31 The results of the canine study are encouraging. In fact, despite the typically dismal prognosis of dogs diagnosed with hemangiosarcoma, 6-month survival rates increased from approximately 40% in a comparison group of dogs treated with standard of care therapy to almost 70% in the group of dogs treated with eBAT at the biologically active dose. Additionally, six dogs lived longer than 450 days and were considered long-term survivors.28 Canine sarcomas are physiologically analogous to human sarcomas in the expression of target receptors but occur at a much higher frequency in dogs and therefore offer a larger patient population for the study of the disease.32 Further development of eBAT presents an opportunity to potentially supplement conventional therapies to hopefully overcome drug resistance mechanisms that lead to tumor recurrence and progression in dogs as well as in people.
In the future, the methodology of this study could be extended to additional EGFR- and uPAR-expressing cancers including other carcinoma and sarcoma subtypes that have been historically treatment resistant.2,3 Annexin V and propidium iodide assays may be used in the future to affirm the cytotoxic effect of eBAT on cancers in vitro by measuring the populations of apoptotic and live cells after binding. Further testing of eBAT will provide greater insight into the cytotoxic ability as well as the clinical applicability of the toxin against tumors of various origins that share physiological characteristics.
Conclusion
In conclusion, the results of our study suggest that eBAT should be further investigated as a potential therapeutic approach for sarcomas and carcinomas as well as possibly other refractory malignancies that express EGFR and uPAR. We showed that eBAT binds with high affinity to and effectively kills osteosarcoma, rhabdomyosarcoma, and ovarian adenocarcinoma cells in vitro. Specifically, eBAT bound with the highest affinity to EGFR- and uPAR-expressing cell lines and achieved the greatest specific cytotoxicity at clinically relevant concentrations. In the future, eBAT will be a promising treatment option for patients with human sarcoma and carcinoma, and further study will expand upon its applicability to a wide variety of EGFR- and uPAR-expressing tumors.
Acknowledgments
This work was supported in part by the US Public Health Service Grant R01-CA36725, R01-CA72669, P01-CA65493, P01-CA111412, and R35 CA197292 awarded by the NCI and the NIAID, DHHS. It was also supported by an NIH Research Evaluation and Commercialization Hub (REACH) Award (U01), the Killebrew-Thompson Memorial Golf Tournament, the Lion Fund, William Lawrence and Blanche Hughes Fund, the Randy Shaver Research and Community Foundation, the Atwater Cancer Drug Development Award, the Deutsche Krebshilfe (J.U.S., 111548), and a CETI translational award from the University of Minnesota Masonic Cancer Center.
Disclosure
Dr Vallera is a member of the Oxis Biotech Scientific Advisory Board and holds equity in the company. This relationship has been reviewed and managed by the University of Minnesota in accordance with its conflict of interest policies. A Borgatti and D Vallera have ownership interest (including patents) in a patent entitled “Reduction of EGFR therapeutic toxicity” filed by the University of Minnesota Office of Technology Commercialization. The other authors report no conflicts of interest in this work.
References
World Health Organization. Classification of tumours. In: Fletcher CDM, Unni KK, Mertens F, editors. Pathology and Genetics of Tumors of Soft Tissue. Lyon, France: IARC Press; 2002:12–224. | ||
American Cancer Society. Cancer Facts & Figures 2018. Atlanta, Ga: American Cancer Society; 2018. | ||
Rosenberg SA, Tepper J, Glatstein E, et al. The treatment of soft-tissue sarcomas of the extremities: prospective randomized evaluations of (1) limb-sparing surgery plus radiation therapy compared with amputation and (2) the role of adjuvant chemotherapy. Ann Surg. 1982;196(3):305–315. | ||
Survival rates for ovarian cancer, by stage. 2018. Available from: https://www.cancer.org/cancer/ovarian-cancer/detection-diagnosis-staging/survival-rates. Accessed April 17, 2018. | ||
Dorsam RT, Gutkind JS. G-protein-coupled receptors and cancer. Nat Rev Cancer. 2007;7(2):79–94. | ||
Li S, Huang S, Peng SB. Overexpression of G protein-coupled receptors in cancer cells: involvement in tumor progression. Int J Oncol. 2005;27(5):1329–1339. | ||
Broad-Novartis Cancer Cell Line Encyclopedia. 2018. https://portals.broadinstitute.org/ccle_legacy/home. Accessed May 3, 2018. | ||
Vallera DA, Todhunter DA, Kuroki DW, Shu Y, Sicheneder A, Chen H. A bispecific recombinant immunotoxin, DT2219, targeting human CD19 and CD22 receptors in a mouse xenograft model of B-cell leukemia/lymphoma. Clin Cancer Res. 2005;11(10):3879–3888. | ||
Alewine C, Hassan R, Pastan I. Advances in anticancer immunotoxin therapy. Oncologist. 2015;20(2):176–185. | ||
Pastan I, Hassan R, Fitzgerald DJ, Kreitman RJ. Immunotoxin treatment of cancer. Annu Rev Med. 2007;58:221–237. | ||
Normanno N, de Luca A, Bianco C, et al. Epidermal growth factor receptor (EGFR) signaling in cancer. Gene. 2006;366(1):2–16. | ||
Breuss JM, Uhrin P. VEGF-initiated angiogenesis and the uPA/uPAR system. Cell Adh Migr. 2012;6:535–540. | ||
Tsai AK, Oh S, Chen H, Shu Y, Ohlfest JR, Vallera DA. A novel bispecific ligand-directed toxin designed to simultaneously target EGFR on human glioblastoma cells and uPAR on tumor neovasculature. J Neurooncol. 2011;103(2):255–266. | ||
Tschoep K, Kohlmann A, Schlemmer M, Haferlach T, Issels RD. Gene expression profiling in sarcomas. Crit Rev Oncol Hematol. 2007;63(2):111–124. | ||
Yang JL, Hannan MT, Russell PJ, Crowe PJ. Expression of HER1/EGFR protein in human soft tissue sarcomas. Eur J Surg Oncol. 2006;32(4):466–468. | ||
Albritton KH, Randall RL. Prospects for targeted therapy of synovial sarcoma. J Pediatr Hematol Oncol. 2005;27(4):219–222. | ||
Benassi MS, Ponticelli F, Azzoni E, et al. Altered expression of urokinase-type plasminogen activator and plasminogen activator inhibitor in high-risk soft tissue sarcomas. Histol Histopathol. 2007;22(9):1017–1024. | ||
Bifulco K, Votta G, Ingangi V, et al. Urokinase receptor promotes ovarian cancer cell dissemination through its 84-95 sequence. Oncotarget. 2014;5(12):4154–4169. | ||
Li AR, Chitale D, Riely GJ, et al. EGFR mutations in lung adenocarcinomas: clinical testing experience and relationship to EGFR gene copy number and immunohistochemical expression. J Mol Diagn. 2008;10:242–248. | ||
Clevers H, Alarcon B, Wileman T, Terhorst C. The T cell receptor/CD3 complex: a dynamic protein ensemble. Annu Rev Immunol. 1988;6:629–662. | ||
Denton C. Leucine incorporation and thymidine incorporation. Methods Mol Biol. 1997;79(1):169–178. | ||
Waldron NN, Oh S, Vallera DA. Bispecific targeting of EGFR and uPAR in a mouse model of head and neck squamous cell carcinoma. Oral Oncol. 2012;48(12):1202–1207. | ||
Liu TF, Cohen KA, Ramage JG, Willingham MC, Thorburn AM, Frankel AE. A diphtheria toxin-epidermal growth factor fusion protein is cytotoxic to human glioblastoma multiforme cells. Cancer Res. 2003;63(8):1834–1837. | ||
Liu TF, Tatter SB, Willingham MC, Yang M, Hu JJ, Frankel AE. Growth factor receptor expression varies among high-grade gliomas and normal brain: epidermal growth factor receptor has excellent properties for interstitial fusion protein therapy. Mol Cancer Ther. 2003;2(8):783–787. | ||
Herrmann D, Seitz G, Warmann SW, Bonin M, Fuchs J, Armeanu-Ebinger S. Cetuximab promotes immunotoxicity against rhabdomyosarcoma in vitro. J Immunother. 2010;33(3):279–286. | ||
Wykosky J, Fenton T, Furnari F, Cavenee WK. Therapeutic targeting of epidermal growth factor receptor in human cancer: successes and limitations. Chin J Cancer. 2011;30(1):5–12. | ||
Elaraj DM, White DE, Steinberg SM, Haworth L, Rosenberg SA, Yang JC. A pilot study of antiangiogenic therapy with bevacizumab and thalidomide in patients with metastatic renal cell carcinoma. J Immunother. 2004;27(4):259–264. | ||
Borgatti A, Koopmeiners JS, Sarver AL, et al. Safe and effective sarcoma therapy through bispecific targeting of EGFR and uPAR. Mol Cancer Ther. 2017;16(5):956–965. | ||
Kreitman RJ, Stetler-Stevenson M, Margulies I, et al. Phase II trial of recombinant immunotoxin RFB4(dsFv)-PE38 (BL22) in patients with hairy cell leukemia. J Clin Oncol. 2009;27(18):2983–2990. | ||
Onda M, Beers R, Xiang L, Nagata S, Wang Q-C, Pastan I. An immunotoxin with greatly reduced immunogenicity by identification and removal of B cell epitopes. Proc Natl Acad Sci U S A. 2008;105(32):11311–11316. | ||
Eilber FR, Mirra JJ, Grant TT, Weisenburger T, Morton DL. Is amputation necessary for sarcomas? A seven-year experience with limb salvage. Ann Surg. 1980;192(4):431–438. | ||
Schappa JT, Frantz AM, Gorden BH, Dickerson EB, Vallera DA, Modiano JF. Hemangiosarcoma and its cancer stem cell subpopulation are effectively killed by a toxin targeted through epidermal growth factor and urokinase receptors. Int J Cancer. 2013;133(8):1936–1944. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.