Back to Journals » Vascular Health and Risk Management » Volume 16
Targeting Cardiac Metabolic Pathways: A Role in Ischemic Management
Authors Yehualashet AS ![]() , Belachew TF
, Belachew TF ![]() , Kifle ZD
, Kifle ZD ![]() , Abebe AM
, Abebe AM ![]()
Received 29 May 2020
Accepted for publication 11 August 2020
Published 14 September 2020 Volume 2020:16 Pages 353—365
DOI https://doi.org/10.2147/VHRM.S264130
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Takashi Kajiya
Awgichew Shewasinad Yehualashet,1 Teshome Fentik Belachew,2 Zemene Demelash Kifle,3 Ayele Mamo Abebe4
1Pharmacology and Toxicology Unit, Department of Pharmacy, College of Health Sciences, Debre Berhan University, Debre Berhan, Ethiopia; 2Department of Pharmacy, Debre Birhan Health Science College, Debre Birhan, Ethiopia; 3School of Pharmacy, Department of Pharmacology, University of Gondar, Gondar, Ethiopia; 4Department of Nursing, College of Health Sciences, Debre Berhan University, Debre Berhan, Ethiopia
Correspondence: Awgichew Shewasinad Yehualashet
Pharmacology and Toxicology Unit, Department of Pharmacy, College of Health Sciences, Debre Berhan University, PO Box 445, Debre Berhan, Ethiopia
Tel +251-93-545-0290
Email [email protected]
Abstract: Among the vast number of noncommunicable diseases encountered worldwide, cardiovascular diseases accounted for about 17.8 million deaths in 2017 and ischemic heart disease (IHD) remains the single-largest cause of death in countries across all income groups. Because conventional medications are not without shortcomings and patients still refractory to these medications, scientific investigation is ongoing to advance the management of IHD, and shows a great promise for better treatment modalities, but additional research can warrant improvement in terms of the quality of life of patients. Metabolic modulation is one promising strategy for the treatment of IHD, because alterations in energy metabolism are involved in progression of the disease. Therefore, the purpose of this review was to strengthen attention toward the use of metabolic modulators and to review the current level of knowledge on cardiac energy metabolic pathways.
Keywords: metabolic modulation, ischemic heart disease, cardiac energy metabolism, mitochondrial dynamics
Introduction
Among many other noncommunicable disease around the globe, cardiovascular problems accounted for about 17.8 million deaths in 2017,1 and ischemic heart disease (IHD) is the leading cause of death from cardiovascular and other diseases.2 IHD remains the single-largest cause of death in countries among all income groups, and ≥80% of deaths occur in developing countries.3,4 IHD represents diseases of related syndromes resulting from myocardial ischemia as a result of disproportionate supply and demand of the heart for perfusion of oxygenated blood.5 It includes not only inadequacy of O2 but also decreased levels of nutrients and their substrates and insufficient removal of metabolites. In the majority of cases, the cause of myocardial ischemia is reduction in coronary blood flow because of atherosclerosis- induced coronary arterial obstruction. In such scenarios, IHD manifests as coronary artery disease (CAD) or coronary HD. Coagulation disorders and functioning of endothelial and/or smooth muscles and the myocardium are actors in the disease. However, it is the myocardial consequences of the O2 supply–demand imbalance that are of pivotal significance in determining the clinical benefit of a patient with IHD.6
Primary treatment options for IHD act either by improving the supply of blood and O2 to the heart or reducing the O2 demand of the heart. Although recent advances with these approaches have been made, there are still patients ineligible or refractory to this conventional treatment.7 Metabolic modulation is among the promising strategies in the treatment of IHD, because alterations in energy metabolism are known to be involved in the progression of the disease. Insufficient energy supply to the heart impairs energy sources of the heart and contributes to the progression of the disease.8 Therefore, newer cardioprotective approaches are required to treat IHD.
All cellular function is highly reliant on O2 consumption by mitochondria in order to produce energy. In normal physiological circumstances, mitochondria are found to be in a homeostatic state of metabolic and cellular ions that contribute to normal functioning. Any change from this hemostatic point can result in abnormally high mitochondrial Ca2+ and elevated oxidative stress.8 The stress can bring about oxidative damage to mitochondrial membranes, enzymes, and components of the electron-transport chain (ETC), which impairs ATP production and thereby promotes permeability of transition pore opening, which finally tends to induce cellular apoptosis and necrosis.9
Myocardial Energy Metabolism in the Healthy Heart
The myocardium needs a huge and continuous supply of ATP from catabolism of carbohydrates and fatty acids (FAs). Glucose, lactate, and FAs are oxidized in the mitochondrion and produce a common end product, ie, acetyl-CoA, which then enters the tricarboxylic acid (TCA) cycle.10 Reducing equivalents nicotinamide adenine dinucleotide (NADH) and flavin adenine dinucleotide, responsible for moving electrons to the ETC, are generated by the TCA cycle via oxidation of FAs and glucose. The ETC accepts electrons from reducing equivalents, (Figure 1) and transforms them into ATP, which is then moved from the mitochondrial matrix to the cytoplasm by the adenine nucleotide transporter, finally allowing energy to be made available for cellular activity.11
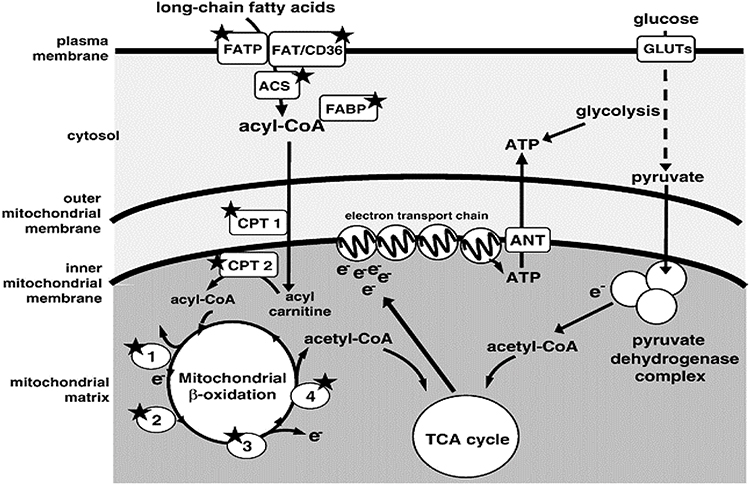 |
Figure 1 Cellular energy–metabolism pathways. Notes: Major routes for ATP production from catabolism of fatty acids and glucose in cardiomyocytes. Proteins and enzymes known to be regulated by PPARα are indicated by a star. 1) Four chain length–specific acyl-CoA dehydrogenases; 2) enoyl-CoA hydratase; 3) 3-hydroxyacyl-CoA dehydrogenase; (4) 3-ketoacyl-CoA thiolase. Reproduced from Fink BN. The PPAR regulatory system in cardiac physiology and disease. Cardiovasc Res. 2007;73(7):269–277, by permission of Oxford University Press.11Abbreviations: FATP, fatty acid–transport protein; FAT/CD36, fatty-acid translocase; FABP, fatty acid–binding protein; ACS, acyl-CoA synthetase; GLUTs, glucose transporters; CPT, carnitine palmitoyltransferase; TCA, tricarboxylic acid; ANT, adenine nucleotide translocator. |
Fatty-Acid Oxidation
About 95% of the ATP produced in the normal heart is obtained from oxidative phosphorylation in mitochondria, whereas the rest is derived from the glycolysis pathway. Although FAs produce more ATP than carbohydrates, the process is not O2-efficient. The production of ATP from FAs requires 10% more O2 than the equivalent amount of ATP produced from carbohydrates. FAs indirectly hinder the oxidation of carbohydrates because of the uncoupling of glycolysis from glucose oxidation, resulting in increased proton generation that decreases the efficiency of the heart during ischemia.12 Sources of FAs are free FAs that are bound to albumin or FAs obtained from a triacylglycerol (TAG) found in chylomicrons or very-low-density lipoprotein (VLDL). Although most FAs used by the heart coming from exogenous TAG are derived from chylomicrons, only a minor portion originate from VLDL. In addition, TAG released from LDL can also be used for FA oxidation.13 The major substrates used by the normal adult human heart under aerobic conditions are free FAs. Long chain FAs are the major components in the utilization of FAs. Entry of components of free FAs into mitochondrial cells is a complex process that is catalyzed by several enzymes. Oxidation of FA takes place in the mitochondrial matrix. The delivery of FAs to mitochondria is a determinant in the oxidation of FAs in mitochondria. Firstly, FAs should move from the plasma to the cytoplasm and then sequentially to the mitochondrial matrix. FAs join cardiomyocytes by means of passive diffusion or protein-mediated uptake. Transporters engaged in the uptake of FAs are fatty acyl translocase (FAT/CD36) and the plasma membrane isoform of FA-binding protein (FABPpm).12
Esterification of FAs is the next process, and is mediated by a group of fatty acyl-CoA synthase enzymes. Carnitine palmitoyltransferase (CPT; I and II) and acyl translocase (CAT) are the enzymes responsible for the uptake of fatty acyl-CoA by mitochondria. CPTI, located on the outer mitochondrial membrane, catalyzes the synthesis of fatty acyl carnitines after binding to fatty acyl-CoAs. Fatty acyl carnitine then moves to the matrix by CAT for the sake of exchange with carnitine. Just after exchange, acyl carnitines will be re-esterified by CPTII to acyl-CoAs, which are then metabolized by FA oxidation.12 Fatty acyl-CoAs are broken down to synthesize acetyl-CoA, which enters the TCA cycle for the formation of ATP. Acyl-CoA dehydrogenase, 2-enoyl CoA hydratase, 3-hydroxyacyl CoA dehydrogenase, and 3-ketoacyl CoA thiolase (3-KAT) are the major enzymes in mitochondrial FA oxidation.14
During high rates of glucose oxidation, NADH levels become high, and the redox state of mitochondria triggers inhibition of FA oxidation. The regulation of FA oxidation is initiated at the level of 3-KAT, which is sensitive to the acetyl-CoA:CoA ratio, and with the existence of increased rates of glucose oxidation, accumulation of acetyl-CoA & inhibition of 3-KAT happens.15 In consideration of the enzymes responsible in oxidation of FA, there are many drug targets that are promising to optimize and balance myocardial energy metabolism so as to prevent unexpected and undesirable effects of ischemia, and these are included in the following discussion.
Carbohydrate Metabolism
Carbohydrate metabolism is another source of energy, assumed to contribute about 10%–40% of energy produced in the normal adult heart. Glucose taken up by mitochondrial cells is either stored as glycogen or undergoes glycolysis to yield pyruvate, which is then oxidized by pyruvate dehydrogenase (PDH) into acetyl-CoA within mitochondria. Glucose employed for ATP production is derived either from the bloodstream or endogenous stores. Glucose can access cardiomyocytes using glucose transporters (GLUTs), of which GLUT4 is the major transporter that is highly sensitive to insulin stimulation in the myocardium, although some portion of glucose transport takes place with the insulin-insensitive transporter GLUT1.8
By glycolysis, glucose is changed to pyruvate with net production of 2ATP and 2NADH. With use of O2, pyruvate is oxidized by the PDH complex to generate acetyl-CoA, which then enters the TCA cycle. In contrary, in the absence of adequate O2, pyruvate will be converted to lactate by lactate dehydrogenase (LDH) to regenerate the NADP required to maintain glycolysis. The PDH complex is the rate determinant in the process and highly sensitive to inhibition by acetyl-CoA. At increased rates of FA oxidation, there exists an increment in the level of acetyl CoA, which then inhibits glucose oxidation.15
Energy Metabolism in the Ischemic Heart
Because of the lack of O2supply to the heart during myocardial ischemia, both FAs and carbohydrate oxidation declineand production of ATP is disrupted. So called-glycolysis, which is a minor source of ATP in the aerobic heart, can be a more predominant source of energy during myocardial ischemia.16 The relative contribution of glucose and FA oxidation to myocardial energy production is a predictor of both cardiac function and its efficiency. Therefore, any disturbance to homeostasis of the metabolic process can negatively influence the heart and consequently expose it to many other cardiac diseases. During ischemic conditions, FA oxidation dominates to be the remaining source of oxidative phosphorylation, since an increase in the level of FA in the coronary circulation and of subcellular changes occurs that ends in dysregulation of FA oxidation. This increased reliance on FAs is both inefficient and undesirable during O2 shortages. Increased rates of FA oxidation can inhibit glucose oxidation, which then promotes uncoupling of glucose oxidation from glycolysis. This uncoupling finally tends to provoke proton overload and intracellular acidosis, which results in decreases in cardiac efficiency and increases the risk of ischemic injury while compromising cardiac contractility.17
Myocardial ischemia triggers the production of H+ from hydrolysis of ATP derived from glycolysis and tends to form acidosis.18 Lactic acid accumulation increases levels of intracellular Na+ and Ca2+. Accumulation of FAs and their intermediates during myocardial ischemia results in metabolic and ionic disturbance that can dysregulate myocardial function to cause myocardial abnormalities.16 As such, the strategy for modulation of cardiac energy metabolism is to stimulate glucose oxidation or inhibit the oxidation of FAs. Because the oxidation of one molecule of glucose consumes less O2 than that of FA, it makes the heart efficient in the production of energy. Direct stimulation of glucose oxidation or secondary inhibition of FA oxidation can result in improved coupling between glycolysis and glucose oxidation and consequently decrease proton production and alleviate myocardial acidosis ultimately to improve cardiac efficiency.12
Agents Targeting Energy Metabolic Pathways
Major pharmacological targets in the metabolic pathway includes FA transport into cardiomyocytes, FA uptake into mitochondria, the enzymatic machinery of the β-oxidative pathway itself, targeting availability of circulating energy substrates, and reducing oxidative stress during myocardial energy metabolism. Predominantly, the myocardium employs FAs as an energy source, as the chemical bonds of FA molecules have higher energy content and the potential to generate more ATP molecules per molecule of FA consumed. Inhibition of oxidation can allow the heart to produce ATP with lower O2 need. Therefore, it is predictable that an inhibitor of oxidation prevents damage to the myocardium during ischemic injury.19
The major aim of optimization of cardiac energy metabolism is to reduce the rate of FA oxidation by the heart and enhance the oxidation of pyruvate derived from glucose, glycogen, and lactate by switching the source of acetyl-CoA from FAs to pyruvate. This results in greater amounts of ATP, reduces undesirable effects of harmful FA metabolites, and decreases lactate and H+ production under low-flow conditions and during postischemic reperfusion.20 In general, metabolic modulators do not affect blood pressure, pulse rate, or left-ventricle systolic function, giving them advantages over conventional drugs, which may induce symptomatic hypotension, inappropriate bradycardia, or worsening cardiac symptoms.8
Targeting Fatty-Acid and Glucose Supplies
CPTI Inhibition
Mitochondrial entry of FAs is one of the direct strategies in the optimization of FA metabolism. The CPT1 enzyme has become the target of several pharmacological agents, as it catalyzes the rate-limiting step in the uptake of long-chain FAs to mitochondria, as depicted in Figure 2. CPTI inhibitors, (etomoxir, oxfenicine, and perhexiline) reduce CPTI activity and hence impede FA oxidation while favoring glucose oxidation. An alternative approach to inhibit the activity of CPTI and decrease uptake of FAs is through inhibition of malonyl cCoA decarboxylase (MCD), an enzyme used in degradation of malonyl CoA that is a potent endogenous reversible inhibitor of CPTI.17,21
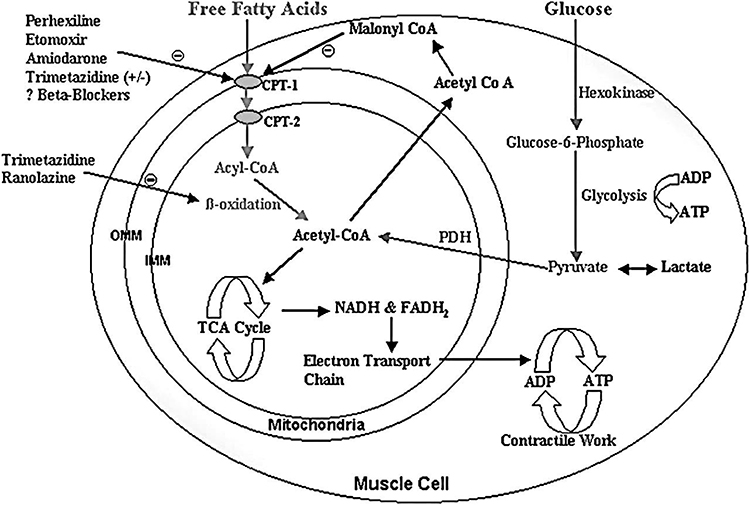 |
Figure 2 Myocardial metabolism.Notes: Reproduced from Lee L, Horowitz J, Frenneaux M. Metabolic manipulation in ischaemic heart disease, a novel approach to treatment. Eur Heart J. 2004;25(80):634–641, by permission of Oxford University Press.17 Abbreviations: CPT, carnitine palmitoyltransferase; IMM, inner mitochondrial membrane; OMM, outer mitochondrial membrane; PDH, pyruvate dehydrogenase. |
Inhibition of Malonyl CoA
Under normal and pathological conditions, malonyl CoA is important in the control of cardiac energy metabolism, and cardiac malonyl CoA levels, which are reduced in the ischemic heart, may be responsible for increased FA-oxidation rates.22 Alterations in cardiac energy metabolism impair production of ATP, important in maintain energy requirements of the heart. There is evidence implying that a decrease in cardiac efficiency because of cardiac FA oxidation and an increase in the uncoupling of glycolysis from glucose oxidation impairs cardiac function and leads to cardiac disease. In order to address this, certain therapeutic strategies are identified, among which is increasing cardiac malonyl CoA levels.23 Inhibition of MCD or increased cardiac malonyl CoA levels decreases cardiac FA-oxidation rates and further improves cardiac efficiency.7
Inhibition of Enzymatic Machinery of β-Oxidation Pathway
Considering the enzymes employed during FA oxidation, there are a number of therapeutic targets present in modulating myocardial energy metabolism so as to limit the undesirable effects of ischemia. Pharmacological targets to optimize cardiac energy metabolism to keep the balance between metabolism of FA and glucose are presented in Figure 2.
3-Ketoacyl CoA Thiolase (3-KAT) Inhibitors
3-KAT inhibitors, a new group of metabolic drugs, are able to trigger an increase in glucose and lactate oxidation with partial inhibition of oxidation of FA, producing consistent clinical advantage in patients with IHD.21 Trimetazidine is from this class of agents, and is available for clinical use through selective inhibition of 3-KAT. According to many studies, it has been shown that trimetazidine is effective in the management of IHD.24 Glucose uptake is regulated inversely by FFA. Inhibition of free FA oxidation shifts metabolism toward glucose oxidation.8 This effect may be achieved by targeting the β-oxidative pathway using 3-KAT inhibitors.
Ranolazine
Ranolazine, an active piperazine analogue, is a more recent anti-ischemic drug that inhibits FA oxidation and promotes partial oxidation of glucose. The more probable molecular mechanism is linked to the inhibition of the late inward Na+ channel that pathologically remains open during adverse stimuli to the myocardium, including myocardial ischemia, hypertrophy, or oxidative stress. Overload of Na+ inside the cell brings about major metabolic, contractile, and electrophysiological problems.25
PDH kinase and PDH phosphatase regulate activity of PDH: PDH kinase phosphorylates the active form of PDH to its inactive form, and PDH phosphatase dephosphorylates the inactive form of PDH to its active form (Figure 2). Ranolazine has no direct action on PDH kinase or phosphatase, because of inhibition of β-oxidation of FAs. When β-oxidation of FAs is inhibited, the amount of acetyl-CoA reduces and triggers an increase in the active form of PDH, which facilitates carbohydrate oxidation. Eventually, it switches myocardial substrate utilization from FAs to carbohydrates,26 and O2 requirements of the heart are reduced while cardiac work is intact. A further benefit is that glycolysis continues to be coupled to pyruvate oxidation, and thus lactate accumulation is minimized.27 The glucose pathway requires less O2 for a given level of myocardial work, and this increased O2 efficiency may be an important component of the anti-ischemic action.24 In the immediate-release formulation, it significantly decreases episodes of angina and use of nitroglycerin and shows remarkable improvement in exercise duration and time to exerciseelicited myocardial ischemia in patients with stable coronary disease.28
β-Blockers and Myocardial Energy-Substrate Metabolism
Besides their action on cardiac contractility, β-blockers also exhibit anti-ischemic properties due to the O2-saving potential of the myocardial energy metabolism induced, because of negative inotropic and chronotropic effects. Preventing neurohormonal activation by β-blockers can attenuate lipolysis induced by catecholamine, with secondary reduction in the level of circulating plasma FA (Figure 2). According to several clinical studies, β-blockers decrease the uptake of FA and increase left-ventricle function, independently decreasing O2 consumption, whichcan be reflective of increased cardiac efficiency.15 These effects may be due to the capacity of β-blockers to hinder CPTI activity (Figure 2), and promote a shift of energy metabolism from FA to glucose oxidation.29
Stimulation of Glucose Oxidation During Myocardial Ischemi
Dichloroacetate
At high levels of free-FA concentration during myocardial ischemia, dichloroacetate inhibits oxidation of FA and enhances glucose metabolism. It activates the PDH complex, a group of enzymes at the membrane of the inner mitochondria and is a rate-determining step of glucose oxidation. PDH is inactivated when phosphorylated by PDH kinase. This agent increases the activity of the PDH complex via inhibition of PDH kinase.21 A study revealed that when it was given to patients with CAD by IV infusion, left-ventrice stroke volume was improved. This response was observed without effects on heart rate, left-ventricle end-diastolic pressure, or myocardialO2consumption, and hence it is plausible that more efficient carbohydrate metabolism will be of paramount importance in stroke volume.24
Ribose
A pentose sugar, ribose has been shown in many animal experiments to improve generation of ATP and improve cardiac function21 by enhancing metabolism via the pentose phosphate pathway and bypassing the rate-determining enzymes G6PDH and 6-phosphogluconate dehydrogenase, and hence facilitate glucose metabolism. In addition, ribose has been shown to have no effect on coronary blood flow, myocardial O2consumption, or hemodynamics in the healthy heart.30
In general, D-ribose may give rise to important metabolic end products to compensate for energy deficits of the heart. As no unwanted or undesirable effects have been observed with ribose and this nutraceutical does not affect hemodynamics, it is becoming a promising adjunctive treatment for patients who are on polytherapy. However, its exact mechanism of action equires further investigation.24
Targeting Availability of Circulating Energy Substrates
Glucose–Insulin–Potassium (GIK)
It has been suggested that GIK therapy can support metabolism during myocardial ischemia. During an episode of cardiac ischemia, there is a shift from aerobic carbohydrate metabolism to anaerobic free-FA metabolism to promote production of substrates and free radicals, which are harmful to the myocardium. Contrarily, it has been indicated that infusion of insulin and glucose in GIK therapy renders a transition from harmful free-FA metabolism to glucose metabolism during myocardial ischemia, such that reduction of plasma free FA-levels by insulin would further promote this effect.31
GIK was initially considered an antiarrhythmic solution, improving incidence of cardiac rhythm disorders in the case of ischemic hearts, and a metabolic cocktail providing the heart with an energy-saving substrate and improving myocardial O2 efficiency.32 Suppressing lipolysis, reducing the amount of free FA, and increasing glucose entry into the myocardium give rise to more efficient cardiac metabolism, which specifically is relevant in ischemic myocardial tissue.33 Insulin in the GIK exerts its effect through the PI3K–Akt–endothelial nitric oxide synthase–signaling pathway, and the nitric oxide generated from this signaling protects the myocardium. Different animal models have shown that insulin given during myocardial reperfusion to reduce myocardial ischemia or reperfusion injury, partly by suppression of apoptosis.32 Because apoptosis may contribute to reperfusion injury, GIK infusion may prevent additional myocardial consequences after restoring adequate perfusion to the heart.34
Peroxisome Proliferator–Activated Receptor Agonists
As nuclear receptor–transcription factors, peroxisome proliferator–activated receptors (PPAR; α, β, δ, γ) are important regulators of cardiac metabolism, and has been viewed as a potential target for pharmacologic therapy in the optimization of cardiac metabolism.35 This receptor family has a vital role in regulating myocardial lipid and energy metabolism by stimulating the transcription of genes involved in the process of cardiomyocyte-FA utilization from uptake to mitochondrial FA oxidation leading to ATP production. In common myocardial disorders, the PPAR gene–regulatory pathway is altered. For instance, in the hypertrophied heart, the expression of PPAR and its activity reduced, resulting diminished capacity for FA oxidation and enhancing rates of glucose utilization.36
Identification of PPARα is based on its ability to regulate genes encoding peroxisomal FA-oxidation enzymes in response to peroxisome proliferators, such as fibric acid derivatives. PPARα regulates the transcription of genes encoding peroxisomal, mitochondrial, and certain CYP450 enzymes that are used in the oxidation of long-chain FAs.37 PPARα is appears to be expressed at relatively high levels in the heart and vasculature and plays a significant role in maintaining cardiac metabolic homeostasis. Although the role of PPARs in the pathogenesis of heart disorders remains unclear, in PPARα-null mice altered expression of PPARα-modulated FA-oxidizing enzymes leads to age-dependent cardiac damage. In addition, metabolic stress due to suppression of the flux of cellular FAs ends with massive cardiac and hepatic lipid accumulation and death.38
PPAR activation has a positive correlation with adipocyte function, sensitivity of insulin, metabolism of lipoproteins, and function and structure of the vasculature. Although these effects have been reported, a reduction in cardiovascular mortality and morbidity of thiazolidinedione (PPARα activator) has not been supported by clinical trials to obtain conclusive evidence. Basically, the significant difference among effects on laboratory measurements and their clinical outcomes could be explained by limitations of clinical trials, possible adverse effects of activation of PPARα, or effects of thiazolidinedione agents away from the target.39
Although there is a strong biological rationale for PPARα activation to attenuate cardiovascular risk, clinical evidence needs to prove this hypothesis. Supporting evidence will be of paramount importance to establish a clinical advantage for pioglitazone. To come up with advances in the cardiovascular therapeutics of pioglitazone, alternatively other selective PPARα agonists, or dual agonists, continued clinical investigation needs to happen beyond the limitations of previous studies.
Nicotinic Acid
Niacin/nicotinic acid and nicotinamide is synthesized in humans from the essential amino acid tryptophan. In vivo nicotinic acid is converted to nicotinamide, a precursor for NAD and NADP, which are indispensable to cells and involved in vast biochemical processes. Niacin exists in the plasma in the form of nicotinamide and nicotinic acid, which are transported to cells and tissue. To perform the intracellular activities of niacin, they enter by means of diffusion, and niacin is trapped within the cell as NAD or NADP.40
Therapeutically, it is an agent to treat dyslipidemias, specifically by inhibiting lipolysis and the production of VLDL while increasing high-density lipoprotein. Additionally, it has also been shown to reduce events of ischemia in patients with dyslipidemias. Although these findings are probably attributable majorly to the systemic effect of niacin on metabolism of lipids, there is evidence on the cardiac effects of niacin in limiting ischemic injury, regardless of systemic lipids.41 Studies have shown the direct effects of niacin on myocardial metabolism, wherein many researchers observed that high amounts of niacin can restrict mobilization and accumulation of free FAs from myocardial TG stores during prolonged ischemia.42
Reducing Oxidative Stress
Role of Antioxidants
In normal circumstances, low levels of O2and oxidants are produced in cells and play an important role in cellular homeostasis, mitosis, differentiation, and signaling. After ischemia and reperfusion, the formation of radicals is highly increased and elicits cellular injury. Cardiomyocytes, like other mammalian cells, can express endogenous antioxidants or free radical–scavenging enzymes such as SOD, catalase, and glutathione peroxidase; however, these antioxidative defense mechanisms are overwhelmed in conditions of ischemia and reperfusion.43
Hyperoxic radicals, including superoxide anions (O2), hydroxyl radicals (OH), and hydrogen peroxide (H2O2), are obtained from cells after removal by systems of enzymes with free radical–scavenging activity, and they are generally found in myocardial physiology. During the normal metabolic process, these mechanisms of scavenging free radicals by endogenous antioxidants are important in limiting the intracellular accumulation of O2– and H2O2 and reducing oxidative damage to proteins and lipids.44 To date, more emphasis has been given to investigating drugs with cytoprotective potential on elements of cellular metabolism that are used separately or as an adjuvant to reduce cellular injury in myocardial cells.45
Lipoic Acid and CoQ
Lipoic acid and CoQ are essential for protection from mitochondria to defend themselves against harmful effects of the O2atmosphere. Lipoic acid and its reduced form are excellent metabolic antioxidants, since they are involved in the antioxidant–redox cycle. Lipoic acid, a cofactor of PDH, is synthesized in the human body and this FA has an important role for the function of mitochondrial PDH, thus enhancing glucose metabolism. CoQ, also called ubiquinone or ubidecarenone, is a mitochondrial coenzyme that is essential for production of ATP. The term “ubiquinone” refers to its occurrence in various places. CQ is important because of its inhibitory effect on lipid peroxidation and the oxidation of endogenous CoQ9. In addition, it can improve mitochondrial respiration. CoQ10 improves blood flow to cardiac muscle by reducing the viscosity of blood in patients with IHD.46
In IHD, CoQ10 has been shown to decrease the spillover of inflammatory cytokines and prevent hyperglycemia-induced endothelial cell injury, monocyte adhesion, and evolution of atherosclerotic lesions in human umbilical vein endothelial cells from diabetic patients. In line with this, CoQ10 was recently shown to be effective in enhancing the function of endothelial cells in patients with coronary disease by counteracting nitric oxide oxidation. Several controlled trials in patients with IHD showed significant improvement in exercise tolerance and reductions in ST-segment depression and angina, with no alteration in heart rate or blood pressure.21,47 Lack of supply of O2and nutrients to cardiomyocytes during acute myocardial ischemia in patients with acute myocardial infarction (AMI) triggers a series of severe biochemical and metabolic perturbations in cardiomyocytes that adversely influence the function of mitochondria and ATP production.48 The role of mitochondrial dynamics in cardiac ischemia is in included in the following discussion.
Mitochondrial Dynamics in Cardiac Ischemia
Mitochondria are the main energy-producing organelles involved in oxidative metabolism, exhibit crucial roles in both physiological and pathological processes of cardiomyocytes, and have been well established to be important regulators of apoptosis in cardiomyocytes in response to hypoxia and oxidative stress.49 During oxidative stress, mitochondria can activate mitochondrial quality control (MQC) to maintain the structure and function of mitochondria. MQC is an adaptive response that regulates mitochondrial biogenesis, fusion, fission, and mitophagy. It is important for the rapid removal of defective mitochondrial debris and timely restoration of the mitochondrial network to further protect mitochondria from damage, and thus reduces exposure of cardiomyocytes to ischemic injury.50,51 Maintaining homeostasis of cardiomyocytes requires a dynamic balance between mitochondrial fission (allocates mitochondrial contents during cell division, generates heterogeneity, and aids in eradicating damaged mitochondria) and fusion (enables mitochondrial content exchange and calcium and ROS buffering, promoting overall mitochondrial function). Coordinated biogenesis and mitophagy (removing unhealthy mitochondrial fragments) ensure sustainable mitochondrial functions. Impairment of the balance in mitochondrial dynamics contributes significantly to pathogenesis of the heart.49,52,53
Two opposing processes employ mitochondrial fission proteins (Drp1, hFis1, and Mff) andmitochondrial fusion proteins (OPA1, Mfn1, andMfn2). Imbalance in mitochondrial fusion and fission affects mitochondrial respiratory function, MQC, and susceptibility to cell death in acute myocardial ischemia–reperfusion (IR) injury, indicating that mitochondrial fusion and fission proteins are important targets in cardioprotection.54
Roles of Mitochondrial Dynamic Modulators in Cardiac Ischemia–Reperfusion Injury
Various in vitro, ex vivo, and in vivo investigations have reported that hindering morphological changes of mitochondria in myocardial IR-injury models can be achieved through inhibition of Drp1.53 Ex vivo studies in mice55 have revealed that Drp1 inhibition can reduce cardiac damage by decreasing myocardial infarct size, and cTnI/cardiac troponin/and improving left-ventricule function. Similarly, ex vivo animal experiment56 showed that Drp1 inhibitors can restore mitochondrial dynamics and functions to reduce infarct size. Another study57 showed that Drp1 inhibition improved function of mitochondria following IR injury by enhancing rates of O2 consumption, decreasing mitochondrial fission and improving cardiac function.
According to an in vivo study,58 Drp1 inhibitors saved the heart from IR injury by reducing excessive mitochondrial fission and minimizing oxidative stress and related cell death through SUMOylation in C57BL/6J mice and DJ1-deficient (knockout) mice. In male C57BL/6 mice,59 inhibition of mitochondrial fission reduces infract size and can protect the heart from further IR injury. In FVB mice,60 inhibition of Drp1 prevents the transport of Drp1 tomitochondria, maintains mitochondrial morphology, and minimizes the size of the infarct due to IR injury.
Currently, there is insufficient evidence showing the effect of mitochondrial fusion promoters during cardiac IR injury. Even the very few existing reports on the roles of mitochondrial fusion in cardiac IR injury are inconsistent. Overexpression of mitochondrial fusion proteins through transfection with Mfn1, Mfn2, or Drp1K38A into HL1 cells enhances resistance to IR injury, resulting in reduced cell death and delaying induction of mitochondrial permeability-transition pore (mPTP) opening.61 On the contrary, there has been a single conflicting study revealing that both Mfn1- and Mfn2-deficient hearts were protected from AMI, and it was found that the abnormal mitochondrial morphology, decreased mitochondrial respiration, and impaired myocardial contractile function caused by IR injury were abolished in Mfn1- and Mfn2-deficient mice. Furthermore, acute cardioprotection during Mfn1/2 deficiency was correlated with enhanced mitochondrial function, measured by resistance to mPTP opening, reduced mitochondrial Ca2+ overload, and oxidative stress. Long-term consequences of ablating mitochondrial fusion proteins are not beneficial, since cardiomyopathy and cardiac death have been reported.53,62 As result of these observed inconsistencies, investigations focusing on mitochondrial fusion promoters in relation to cardiac IR injury are urgently needed from the scientific community.
Mitophagy is a selective process of autophagy by which cells eliminate damaged or dysfunctional mitochondria, and is essential for regulating mitochondrial homeostasis. Impaired mitophagy may lead to accumulation of damaged organelles within cells, underlying the pathogenesis of a number of chronic conditions, including cardiovascular disease.63,64 When cellular energy demand is low, excess mitochondria are not important, since they can produce excessive ROS and can also consume ATP if they are left uncoupled. Besides, damaged or unstable mitochondria may trigger the release of cytochrome C, apoptosis-inducing factor (AIF), and other apoptosis-promoting factors that cause damage to the adjacent mitochondria and the entire cell. Mitophagy is stimulated by the same factors mentioned for autophagy, the most important triggeris being Bnip3, Nix,PINK1, Parkin, protein p62/SQSTM1, mitochondrial depolarization, and mPTP.48,65 Under stressful conditions, mitophagy can also recycle metabolic substrates involved in cardiomyocyte metabolism. Since mitophagy is a self-engulfing process, excessive mitophagy is maladaptive and results in cell death. Blocking the process of mitophagy genetically or pharmacologically can reduce cell death, and thus is regarded as a therapeutic target in regulating myocardial dynamics.50
Damaged mitochondria are compensated for by mitochondrial biogenesis that can provide the cell with functional mitochondria and thereby resolves the energy demands of the heart. Biogenesis of mitochondrial mass is regulated by the PPARγ coactivator (PGC) family of transcriptional coactivators, most importantly PGC1α, PGC1β, and the PGC-related coactivator PRC. PGC1α controls NRF1 and NRF2, thereby controlling regulatory factors that are important for mitochondrial DNA transcription and translation. Overexpression of PGC1α can elicit mitochondrial biogenesis, whereas acetylation of PGC1α limits the action of the transcriptional coactivator, hindering mitochondrial biogenesis. Therefore, deacetylation of PGC1α by Sirt1 enhances its activity and promotes mitochondrial biogenesis.48,65 Sirtuins control the process of autophagy by employing FOXO1 and O3, and AMPK also provokes Sirt1-dependent deacetylation of PGC1α. As such, the regulatory activity of autophagy and mitochondrial biogenesis is continued and moves coordinately. Overexpression of PGC1α in the myocardium raises mitochondrial numbers and cardiomyopathy, leading to failure, implying that maintaining the balance in mitochondrial numbers is highly important in regulating cardiac health.48,65
Role of PI3K–Akt–mTOR Signaling Pathway in Cardioprotection
The PI3K–Akt signaling pathway, one of the most important reperfusion–injury salvage kinase (pathways, can limit cardiomyocyte apoptosis and promote cell survival.66 Akt is a target of PI3K in downstream cascades: during Akt activation by the upstream signaling factor PI3K, it stimulates GSK3β to phosphorylated GSK3β, which is assumed to be the integrated point of many pathways and has a tremendous contribution in cardioprotection. Caspase 3 is an important enzyme in the caspase family and employed in the process of apoptosis after triggering by other members of the caspase family. Caspase 3 is the main enzyme of cell apoptosis. Therefore, when Akt is stimulated, myocardial infarction–induced myocardial injury can be attenuated by GSK3β and caspase 3. It has been found that short-term pretreatment with hesperidin decreases inflammatory response and oxidative stress by activating the PI3K–Akt pathway, reduces myocardial cell apoptosis in myocardial infarction IR injury, and plays a role in myocardial protection.67
mTOR is one of the most extensively studied complexes, because of its established role in cell biology and cell-signaling mechanisms. Dysregulation of mTOR signaling is assumed to cause several diseases, including cardiovascular diseases. mTOR is a serine/threonine protein kinase found in two complexes: mTORC1 mTORC2. The rproteins RAPTOR and RICTOR represent mTORC1 and mTORC2, respectively. Removal of mTORC1 or RAPTOR in cardiac-specific knockout mice produces reduced cardiomyocyte mitochondrial mass, apoptosis, and finally death. Any ablation of proteins responsible for mTOR signaling in genetic knockout mice shows a cardiac phenotype similar to mTOR-knockout mice.68
Several scientific studies have shown that cellular hypoxia disrupts homeostasis of the microenvironment, resulting in cardiomyocyte necrosis and apoptosis. Apoptosis of cardiomyocytes is part of the pathological cascade in hypoxia/reoxygenation (H/R)–induced myocardial injury and highly related to cardiac dysfunction. It is also strongly thought that oxidative stress is a major factor in cardiomyocyte apoptosis in resulting H/R-induced myocardial injury. As such, inhibition of oxidative stress and cardiomyocyte apoptosis could be regarded as one treatment option in the prevention of H/R-induced myocardial injury in AMI. Studies have shown that activation of H/R can attenuate cellular viability, raise the release of LDH, and elicit cellular apoptosis and oxidative stress in H9c2 cardiomyocytes, suggesting H/R might provoke myocardial injury.69
Aggregated evidence has revealed that Akt–mTOR signaling is employed in cardioprotection against hypoxia-induced injury. It has also been illustrated that ginsenoside Rg1 protects the heart from hypoxia-induced cell injury by activating the PI3K–Akt–mTOR pathway. In addition, other studies have indicated that miR21 inhibits H/R-induced autophagy and apoptosis in clonal embryonic rat-heart tissue (H9c2) cells by stimulating the Akt–mTOR pathway, and it has also been shown that dragon’s blood extracts exhibit cardioprotection against myocardial injury in AMI mice by activating the PI3K–Akt–mTOR signaling pathway. I was demonstrated that H/R exposure suppresses activation of the Akt–mTOR pathway in H9c2 cardiomyocytes. PK2 activated the Akt–mTOR pathway in H/R-exposed H9c2 cardiomyocytes, and PK2 treatment recuperated H/R-induced enhancement of malondialdehyde content and inhibition of SOD, CAT, and GSH-Px activities by activation of the Akt–mTOR pathway in H9c2 cardiomyocytes.Nrf2 is linked with the expression of cytoprotective genes in response to oxidative stress. Knockdown of Nrf2 inhibits the expression of Nrf2 downstream target genes like SOD, CAT, and GSHPX at mRNA and protein levels in H9c2 cells. Because the Akt–mTOR pathway mediates expression of Nrf2, it was deduced that PK2 might enhance protein levels of SOD, CAT, and GSH-Px in H/R-exposed H9c2 cardiomyocytes.69
Bioenergetic–Metabolite Interactome for Novel Future Potential Metabolotherapies
The influence of metabolism in modulating tissue health and remodeling is a recent technological advance in the process of bioenergetic measurements and metabolomics. These advances pave the way to a new paradigm highlighting deregulation in the plasticity of the metabolic network to be regarded as an etiology for other severe disorders, and great emphasis are given to the need to understand and elaborate on metabolic networks in healthy individuals. The deregulation might be because of problems in the metabolic system or toxicological challenge exposure, and might even further provoke metabolic error.71,72
In addition to energy generation and synthesis of building blocks, metabolism is also important for controlling the cellular redox state.73 Metabolism helps in the process of cellular proliferation, tissue development, and adaptation through metabolite signaling, since metabolites are employed in extracellular and intracellular signaling cascades. For instance, such intracellular metabolites as G6P, 5-amino-4-imidazole-carboxamide ribonucleotide and AMP regulate mTOR and AMPK. Likewise, circulating metabolites are used as robust signaling molecules. FAs, such as palmitoleate, produced during exercise enhance cardiac growth through stimulation of GPCRs, Akt, or nuclear receptors. It has recently been recognized that gut microbiota–derived metabolites, bile acids, and several metabolites from intermediary metabolism like succinate, lactate, and kynurenic acid function as extracellular signaling molecules by binding to cognate GPCRs.
Therapeutics targeting mitochondria encompass newer pharmacological agents that are entering clinical practice and also the repurposing of drugs that work through metabolic mechanisms. Recent studies have supported the idea that mitochondrial metabolites are emerging as important modulators of health.70 As far as mitochondrial metabolites are assumed to have several cellular targets, their effect on the bioenergetic–metabolite interactome is a multifeatured idea. Although expertise regarding metabolomics continues to advance, there are huge expectations for the field to overcome challenges and show deeper insights into the role of metabolism in health and disease. A better understanding of the bioenergetic–metabolite interactome will also aid in the development of future metabolotherapies.
Summary
Energy metabolism is a process of production of ATP, which is a determinant of cardiac health. In a healthy heart, the major proportion of ATP is obtained through oxidation of FAs and a balance between the use of FAs and other energy sources maintained. Optimization of cardiac energy metabolism, specifically shifting utilization of energy from FAs to glucose, is a promising therapeutic strategy for various IHDs. This transition in energy-substrate preference can be achieved by employing agents that act on FAs and glucose-metabolism pathways, regulating balance and engendering these pathways to contribute in overall cardiac energetics and hence enhance the efficiency of both the generation and utilization of ATP.
Provided that cardiac mitochondria exhibit an established role in providing energy requirements, it is an important therapeutic strategy for cardioprotection. Aggregated evidence shows that mitochondrial dynamics can be altered by modulating MQC mechanisms. Becauseconventional medications are not without their shortcomings and patients still refractory to these medications, the scientific community is trying to scrutinize potential drug targets and other management modalities to advance the treatment of IHD. As an emerging field, there are many areas to be answered and elaborated in modulating cardiac energy metabolism targeting mitochondrial dynamics. The area requiri in depth understanding of metabolic paths in mitochondrial dynamics, and we believe that further scrutiny and deep insight is highly needed. Moreover, the search for new and better strategies should take continue along with employing ever-evolving therapeutic interventions to manage IHD.
Abbreviations
ADP, adenine diphosphate; ATP, adenine triphosphate; CAD, coronary artery disease; CPT, carnitine palmitoyltransferase; ETC, electron-transport chain; FADH, flavin adenine dinucleotide; FAT, fatty-acid translocase; FATP, fatty acid–binding protein; GIK, glucose insulin potassium; GLUTs glucose transporters; IHD, ischemic heart disease; KAT, ketoacyl thiolase; LDH, lactate dehydrogenase; MCD, malonyl-CoA decarboxylase; NAD, nicotinamide adenine dinucleotide; NADP, nicotinamide adenine dinucleotide phosphate; PDH, pyruvate dehydrogenase; PPAR, peroxisome proliferator–activated receptor; TAG, triacylglycerol; TCA, tricarboxylic acid; VLDL, very-low-density lipoprotein.
Author Contributions
All authors made a significant contribution to the work reported, whether in conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas, took part in drafting, revising, or critically reviewing the article, gave final approval to the version to be published, agreed on the journal to which the article has been submitted, and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Kaptoge S, Pennells L, De Bacquer D; The WHO CVD Risk Chart Working Group. World Health Organization cardiovascular disease risk charts: revised models to estimate risk in 21 global regions. Lancet Glob Health. 2019;7:e1332–45. doi:10.1016/S2214-109X(19)30318-3
2. Sakboonyarat B, Rangsin R. Prevalence and associated factors of ischemic heart disease (IHD) among patients with diabetes mellitus: a nationwide, cross-sectional survey. BMC Cardiovasc Disord. 2018;18(151):1–7.
3. Alexandra N, et al. Mortality from Ischemic Heart Disease. Circ Cardiovasc Qual Outcomes. 2019;12(6).
4. Roth GA, Johnson C, Abajobir A, et al. Global, regional, and national burden of cardiovascular, Diseases for 10 Causes, 1990 to 2015. J Am Coll Cardiol. 2017;70(1):1–25. doi:10.1016/j.jacc.2017.04.052
5. Kaski JC, FCrea F, Gersh BJ, Paolo G. Reappraisal of Ischemic Heart Disease. Circulation. 2018;138(14):1463–1480. doi:10.1161/CIRCULATIONAHA.118.031373
6. Pepine J, Wilmer W, Nichols. The pathophysiology of chronic ischemic heart disease. Clin Cardiol. 2007;30(Suppl.I):
7. Balla C, Pavasini R, Ferrari R. Treatment of angina: where are we? Cardiology. 2018;140(1):52–67. doi:10.1159/000487936
8. Leea L, John H, Michael F.Metabolic manipulation in ischemic heart disease, a novel approach to treatment. Eur Heart J. 2004;25:634–641.
9. Panel M, Ghaleh B, Morin D. Mitochondria and aging: a role for the mitochondrial transition pore? Aging Cell. 2018;1–15.
10. Pietrocola F, Lorenzo L, Manuel Bravo-San Pedro J, Madeo F, Kroemer G. Acetyl Coenzyme A: a central metabolite and second messenger. Cell Metab. 2015;21:805–821. doi:10.1016/j.cmet.2015.05.014
11. Fink BN. The PPAR regulatory system in cardiac physiology and disease. Cardiovasc Res. 2007;73(7):269–277. doi:10.1016/j.cardiores.2006.08.023
12. Ussher J, Lopaschuk G. The Malonyl CoA axis as a potential target for treating ischemic heart disease. Cardiovasc Res Advan. 2008;79(2):259–268. doi:10.1093/cvr/cvn130
13. Lopaschuk G, et al. Fatty acid metabolism in health and disease. Physiol Rev. 2010;90:207–258. doi:10.1152/physrev.00015.2009
14. Sharpe AJ, McKenzie M. Mitochondrial fatty acid oxidation disorders associated with short-chain Enoyl-CoA Hydratase (ECHS1) deficiency. Cells. 2018;7(46):1–13. doi:10.3390/cells7060046
15. Agdip S, et al. Optimizing cardiac energy substrate metabolism: a novel therapeutic intervention for ischemic heart disease. Heart Metab. 2008;38:5–14.
16. Nagoshi T, Yoshimura M, Rosano G, et al. Optimization of cardiac metabolism in heart failure. Curr Pharm Des. 2011;17(35):3846–3853. doi:10.2174/138161211798357773
17. Lee L, Horowitz J, Frenneaux M. Metabolic manipulation in ischaemic heart disease, a novel approach to treatment. Eur Heart J. 2004;25(80):634–641.
18. Gao Q, Deng H, Li H, et al. Glycolysis and fatty acid β-oxidation, which one is the culprit of ischemic reperfusion injury? Int J Clin Exp Med. 2018;11(1):59–68.
19. Karwi OG, Uddin GM, Ho KL, Lopaschuk GD. Loss of metabolic flexibility in the failing heart. Front Cardiovasc Med. 2018;5(68):1–13. doi:10.3389/fcvm.2018.00068
20. Lopaschuk GD. Metabolic changes in the acutely ischemic heart. Heart Metab. 2016;70:32–35.
21. Lionetti V, Stanley WC, Recchia FA. Modulating fatty acid oxidation in heart failure. Cardiovasc Res. 2011;90:202–209. doi:10.1093/cvr/cvr038
22. Folmes C, Sowah D, Clanachan A, Lopaschuk G. High rates of residual fatty acid oxidation during mild ischemia decrease cardiac work and efficiency. J Mol Cell Cardiol. 2009;47:142–148. doi:10.1016/j.yjmcc.2009.03.005
23. Natasha Fillmore N, Lopaschuk GD. Malonyl CoA: a promising target for the treatment of cardiac disease. 2014. Int Union Biochem Mol Biol. 2014;66(3):139–146.
24. Pauly D, Pipen J. Ischemic heart disease: metabolic approaches to management. Clin Cardiol. 2004;27:439–444. doi:10.1002/clc.4960270802
25. Horváth, Horváth B, Hézső T, et al. Late sodium current inhibitors as potential antiarrhythmic agents. Front Pharmacol. 2020;11(413):1–17. doi:10.3389/fphar.2020.00413
26. Kuntz MJ, Harris RA. Pyruvate Dehydrogenase Kinase. In: Choi S, editor. Encyclopedia of Signaling Molecules. New York, NY: Springer; 2018. https://doi.org/10.1007/978-1-4614-6438-9_101636-2.
27. Richard C. Partial Fatty Acid Oxidation (pFOX) inhibition: a new therapy for chronic stable angina. Clin Cardiol. 2003;26:161–162. doi:10.1002/clc.4960260402
28. Antzelevitch C, Belardinelli L, Zygmunt AC, et al. Electrophysiological effects of ranolazine, a novel antianginal agent with antiarrhythmic properties. Circulation. 2004;110:904–910. doi:10.1161/01.CIR.0000139333.83620.5D
29. Podbregar M, Voga G. Effect of selective and nonselective beta-blockers on resting energy production rate and total body substrate utilization in chronic heart failure. J Card Fail. 2002;8:369–378. doi:10.1054/jcaf.2002.130238
30. Teitelbaum J. Enhancing Mitochondrial Function with D-Ribose. Integrative Med. 2008;7:2.
31. Mamas MA, Neyses L, Fath-Ordoubadi F, et al. A meta-analysis of glucose-insulin-potassium therapy for treatment of acute myocardial infarction. Exp Clin Cardiol. 2010;15:2.
32. Vlasselaers D. Gluco-insulin-potassium: much more than enriched myocardial fuel. Circulation. 2011;123(2):129–130. doi:10.1161/CIRCULATIONAHA.110.002709
33. Vander Horst J, Zijlstra F, Van’t Hof AWJ. Glucose-insulin-potassium infusion in patients treated with primary angioplasty for acute myocardial infarction (GIPS): a randomized trial. J Am Coll Cardiol. 2003;42:784–791. doi:10.1016/S0735-1097(03)00830-1
34. Legtenberg R, Houston R, Oeseburg B, Smits P. Physiological insulin concentrations protect against ischemia-induced loss of cardiac function in rats. Comp Biochem Physiol Mol Integr Physiol. 2002;132:161–167. doi:10.1016/S1095-6433(01)00543-8
35. Ahmed W, et al. PPARs and their metabolic modulation: new mechanisms for transcriptional regulation. J Int Med. 2007;262(2):184–198. doi:10.1111/j.1365-2796.2007.01825.x
36. Abdelrahman M, Sivarajah A, Thiemermann C, et al. Beneficial effects of PPAR-g ligands in ischemia–reperfusion injury, inflammation and shock. Cardiovasc Res. 2005;65:772–778. doi:10.1016/j.cardiores.2004.12.008
37. Desvergne B, Michalik L, Wahli W. Transcriptional regulation of metabolism. Physiol Rev. 2006;86:465–514. doi:10.1152/physrev.00025.2005
38. Yue, W Bao, BM Jucker, et al. Activation of peroxisome proliferator–activated receptor-protects the heart from ischemia/reperfusion injury. Circulation. 2003;11.
39. Huang JV, Greyson CR, Schwartz GG. PPAR-as a therapeutic target in cardiovascular disease: evidence and uncertainty. J Lipid Res. 2012;53.
40. EFSA Panel on Dietetic Products. Scientific opinion on dietary reference values for niacin. EFSA J. 2014;12(7):3759. doi:10.2903/j.efsa.2014.3759
41. Markel A, et al. The resurgence of niacin: from nicotinic acid to niaspan/laropiprant. IMAJ. 2011;13.
42. Carlso L. Nicotinic acid: the broad-spectrum lipid drug. A 50th anniversary review. J Intern Med. 2005;258:94–114. doi:10.1111/j.1365-2796.2005.01528.x
43. Sang J, Weon K. Mechanism of ischemia and reperfusion injury to the heart: from the viewpoint of nitric oxide and mitochondria. Chonnam Med J. 2010;46(3):129–139. doi:10.4068/cmj.2010.46.3.129
44. Patel BP, Rawal UM, Dave TK, et al. Lipid peroxidation, total antioxidant status and total thiol levels predict overall survival in patients with oral squamous cell carcinoma. Invest Cancer Ther. 2007;6(4):365–372. doi:10.1177/1534735407309760
45. Soares, Soares ROS, Losada DM, et al. Ischemia/reperfusion injury revisited: an overview of the latest pharmacological strategies. Int J Mol Sci. 2019;20(5034):1–45. doi:10.3390/ijms20205034
46. Nojiri S, Daida H, Mokuno H. Association of serum antioxidant capacity with coronary artery disease in middle-aged men. Heart J. 2001;42:677–690.
47. Wimmer NJ, Stone PH. Anti-anginal and anti-ischemic effects of late sodium current inhibition. Cardiovasc Drugs Ther. 2013;27(1):69–77. doi:10.1007/s10557-012-6431-z
48. Ramachandra CJA, Hernandez-Resendiz S, Crespo-Avilan GE, et al. Mitochondria in acute myocardial infarction and cardioprotection. EBioMedicine. 2020;57(2020):102884. doi:10.1016/j.ebiom.2020.102884
49. Nan J, Zhu W, Rahman MS, et al. Molecular regulation of mitochondrial dynamics in cardiac disease. Biochim Biophys Acta. 2017;1864:1260–1273. doi:10.1016/j.bbamcr.2017.03.006
50. Boyman L, Karbowski M, Lederer WJ. Regulation of mitochondrial ATP production: ca2+ signaling and quality control. Trends Mol Med. 2020;2020(26):21–39. doi:10.1016/j.molmed.2019.10.007
51. Wang J, Zhou H. Mitochondrial quality control mechanisms as molecular targets in cardiac ischemia-reperfusion injury. Acta Pharm Sin B. 2020. doi:10.1016/j.apsb.2020.03.004
52. Fu W, Liu Y, Yin H. Mitochondrial dynamics: biogenesis, fission, fusion, and mitophagy in the regulation of stem cell behaviors. Hindawi. 2019;2019(9757201):1–15.
53. Maneechote C, Siripong Palee S, Chattipakorn SC, Chattipakorn N. Roles of mitochondrial dynamics modulators in cardiac ischaemia/reperfusion injury. J Cell Mol Med. 2017;21(11):2643–2653. doi:10.1111/jcmm.13330
54. Carreira RS, Lee P, Gottlieb RA. Mitochondrial therapeutics for cardioprotection. Curr Pharm Des. 2011;17(20):2017–2035. doi:10.2174/138161211796904777
55. Gao D, Zhang L, Dhillon R, et al. Dynasore protects mitochondria and improves cardiac lusitropy in Langendorff perfused mouse heart. PLoS One. 2013;8:e60967. doi:10.1371/journal.pone.0060967
56. Disatnik MH, Ferreira JC, Campos JC, et al. Acute inhibition of excessive mitochondrial fission after myocardial infarction prevents long-term cardiac dysfunction. J Am Heart Assoc. 2013;2:e000461. doi:10.1161/JAHA.113.000461
57. Sharp WW, Fang YH, Han M, et al. Dynamin-related protein 1 (Drp1)-mediated diastolic dysfunction in myocardial ischemia-reperfusion injury: therapeutic benefits of Drp1 inhibition to reduce mitochondrial fission. FASEB J. 2014;28:316–326. doi:10.1096/fj.12-226225
58. Shimizu Y, Lambert JP, Nicholson CK, et al. DJ-1 protects the heart against ischemia-reperfusion injury by regulating mitochondrial fission. J Mol Cell Cardiol. 2016;97:56–66. doi:10.1016/j.yjmcc.2016.04.008
59. Ong SB, Subrayan S, Lim SY, et al. Inhibiting mitochondrial fission protects the heart against ischemia/reperfusion injury. Circulation. 2010;121:2012–2022. doi:10.1161/CIRCULATIONAHA.109.906610
60. Din S, Mason M, Volkers M, et al. Pim-1 preserves mitochondrial morphology by inhibiting dynamin-related protein 1 translocation. Proc Natl Acad Sci USA. 2013;110:5969–5974. doi:10.1073/pnas.1213294110
61. Hall AR, Burke N, Dongworth RK, et al. Hearts deficient in both Mfn1 and Mfn2 are protected against acute myocardial infarction. Cell Death Dis. 2016;7:e2238. doi:10.1038/cddis.2016.139
62. Yang Y, Li T, Li Z, Liu N, Yan Y, Liu B. Role of mitophagy in cardiovascular disease. Aging Dis. 2020;11(2):419–437. doi:10.14336/AD.2019.0518
63. Redmann M, Dodson M, Boyer-Guittaut M, Darley-Usmar V, Zhang J. Mitophagy mechanisms and role in human diseases. Int J Biochem Cell Biol. 2014;53:127–133. doi:10.1016/j.biocel.2014.05.010
64. Guzy RD, Schumacker PT. O_2 sensing by mitochondria at complex III: the paradox of increased reactive O_2 species during hypoxia. Exp Physiol. 2006;91:807–819. doi:10.1113/expphysiol.2006.033506
65. Chu D, Zhang Z. Trichosanthis pericarpium aqueous extract protects H9c2 Cardiomyocytes from Hypoxia/ReO_2ation Injury by Regulating PI3K/Akt/NO pathway. Molecules. 2018;23:2409. doi:10.3390/molecules23102409
66. Yuan, Yuan X, Juan Z, et al. Clemastine fumarate protects against myocardial ischemia reperfusion injury by Activating the TLR4/PI3K/Akt signaling pathway. Front Pharmacol. 2020;11:28. doi:10.3389/fphar.2020.00028
67. Zhang, Zhang L, Li Y, et al. The PI3K subunits, P110α and P110β are potential targets for overcoming P-gp and BCRP-mediated MDR in cancer. Mol Cancer. 2020;19(1):10. doi:10.1186/s12943-019-1112-1
68. Gang S, Sun G, Liu H, Shu L, Zhang W, Liang Z. Prokineticin 2 relieves hypoxia/reO_2ation-induced injury through activation of Akt/mTOR pathway in H9c2 cardiomyocytes. Artif Cells Nanomed Biotechnol. 2020;48:345–352. doi:10.1080/21691401.2019.1709850
69. Hill BG, et al. Bioenergetics and translational metabolism: implications for genetics. Physiol Prec Med Biol Chem. 2020;401(1):3–29.
70. Nadtochiy SM, Schafer X, Fu D, Nehrke K, Munger J, Brookes PS. Acidic pH is a metabolic switch for 2-hydroxyglutarate generation and signaling. J Biol Chem. 2016;291:20188–20197. doi:10.1074/jbc.M116.738799
71. Rzem R, Achouri Y, Marbaix E, et al. A mouse model of L-2-hydroxyglutaric aciduria, a disorder of metabolite repair. PLoS One. 2015;10:e0119540. doi:10.1371/journal.pone.0119540
72. Fessel JP, Oldham WM. Pyridine dinucleotides from molecules to man. Antioxid Redox Signal. 2018;28:180–212. doi:10.1089/ars.2017.7120
73. Husted AS, Trauelsen M, Rudenko O, Hjorth SA, Schwartz TW. GPCR-mediated signaling of metabolites. Cell Metab. 2017;25:777–796. doi:10.1016/j.cmet.2017.03.008
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.