Back to Journals » Journal of Asthma and Allergy » Volume 14
Targeting Airway Smooth Muscle Hypertrophy in Asthma: An Approach Whose Time Has Come
Authors Chetty A ![]() , Nielsen HC
, Nielsen HC ![]()
Received 3 September 2020
Accepted for publication 20 April 2021
Published 25 May 2021 Volume 2021:14 Pages 539—556
DOI https://doi.org/10.2147/JAA.S280247
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Amrita Dosanjh
Anne Chetty, Heber C Nielsen
Tufts Medical Center, Tufts University, Boston, MA, USA
Correspondence: Heber C Nielsen
Department of Pediatrics, Tufts Medical Center, Boston, MA, USA
Email [email protected]
Abstract: Airway smooth muscle (ASM) cell dysfunction is an important component of several obstructive pulmonary diseases, particularly asthma. External stimuli such as allergens, dust, air pollutants, and change in environmental temperatures provoke ASM cell hypertrophy, proliferation, and migration without adequate mechanistic controls. ASM cells can switch between quiescent, migratory, and proliferative phenotypes in response to extracellular matrix proteins, growth factors, and other soluble mediators. While some aspects of airway hypertrophy and remodeling could have beneficial effects, in many cases these contribute to a clinical phenotype of difficult to control asthma. In this review, we discuss the factors responsible for ASM hypertrophy and proliferation in asthma, focusing on cytokines, growth factors, and ion transporters, and discuss existing and potential approaches that specifically target ASM hypertrophy to reduce the ASM mass and improve asthma symptoms. The goal of this review is to highlight strategies that appear ready for translational investigations to improve asthma therapy.
Keywords: airway smooth muscle cells, hypertrophy, proliferation, airway remodeling
At a Glance Commentary
New approaches in asthma therapy are needed. Recently, the role of airway smooth muscle hypertrophy as a significant component of asthmatic airway obstruction has gained increased attention. There are, however, few therapeutic approaches that primarily target airway smooth muscle hypertrophy. This review brings together the current knowledge of how airway smooth muscle cell hypertrophy develops in asthma and discusses proposed and proven treatments designed specifically to reduce the airway smooth muscle cell mass.
What This Study Adds to the Field
The importance of airway smooth muscle cell hypertrophy as an important phenotypic element of clinical asthma is becoming a major area of investigation in asthma. This review focuses specifically on the mechanisms by which airway smooth muscle mass becomes increased in asthma and possible therapeutic approaches that will reverse the hypertrophic process. As such, this review presents a valuable resource for researchers and clinicians working to improve asthma therapeutics.
Introduction
The American Thoracic Society published a statement in 2017 describing therapeutic advances and challenges in asthma, in which an area of focus was the problem of airway remodeling (AR). The significant contribution of AR to clinical asthma is becoming widely recognized, accompanied by recognition of the need for identification of key biomarkers that correlate with disease phenotype and that define the induction, progression, and therapeutic responsiveness of AR.1 The identification of such biomarkers for AR requires a solid understanding of the contributing cell types and cellular mechanisms underlying AR. In this review, we discuss the airway smooth muscle (ASM) component of AR, with a particular focus on cytokines, growth factors, and ion transporters that regulate hypertrophy and proliferation of the ASM in asthma. We discuss current and proposed therapies designed specifically to reduce the increased ASM mass in asthma as a strategy to improve respiratory function. Our focus is on severe asthma, which has been defined as steroid-dependent asthma that is refractory to treatment control. This includes patients of both TH2-high and TH2-low endotypes. It has been estimated that this represents ~10% of all asthma patients.2
The ASM is an important component of normal airways. It maintains airway diameter by modulating airway tone, mediates the cough reflex, and facilitates both forced expiration and deep breathing. Besides contraction, ASM cells also are capable of secreting cytokines in response to damage or immunologic provocation. ASM cells wrap around the large and small airways in a striped circumferential pattern. The ratio of ASM thickness relative to the airway diameter increases in normal peripheral airways.3
Remodeling and dysfunction of the ASM are important components of several obstructive pulmonary diseases, particularly asthma.4 The primary physical components of AR are increased ASM mass around large and small bronchi and the extension of ASM deeper into the respiratory tree to accompany progressively smaller bronchi (Figure 1). These changes come about through increased ASM cell proliferation, hypertrophy, and migration, coupled with decreased apoptosis. It is useful to briefly review the elements of ASM hypertrophy before discussing how AR is controlled. In the normal lung, ASM cells develop and differentiate from lung mesodermal progenitor cells in response to signals derived from the airway epithelium and basement membrane components.5 The recruitment, growth, and development of ASM cells are amplified in response to inhaled allergens, resulting in increased ASM mass, a further extension of the ASM layer in the respiratory tree associated with the smallest airways, and inadequate braking of constrictor responses.6
A commonly used animal model of asthma is the adult mouse exposed to an allergen such as house dust mite (HDM) antigen. This model recapitulates many physiologic features and anatomic changes observed in clinical asthma. These include increased airway reactivity, decreased compliance, and increased ASM and submucosal thickness.7 Whereas bronchoconstriction alone, ie, without inflammation-induced swelling, increases AR,8 the combination of chronic inflammation and continuous bronchoconstriction leads to more extensive changes in the cellular and extracellular matrix (ECM). Overall, the severity of AR appears broadly correlated with the clinical severity of asthma. For example, both hypertrophy and hyperplasia of the ASM were observed in the lungs of patients who died of asthma, but only hyperplasia was found in non-fatal asthmatic lungs.9–11
The cellular immune responses to inhaled allergens are complex, involving numerous interactive pathways that create positive reinforcement through feedback mechanisms. Inhaled allergens such as dust, microbes, and gases activate plasma cells causing IL-4 production and release. Immune responses in the airway have been divided into a category of increased eosinophils and a category of increased neutrophils. This complexity led to a classification of allergic asthma according to levels of T-helper (TH2) cell subsets, as TH2-high and TH2-low. Individuals with Th2-high asthma are described as having severe, steroid-dependent disease, and TH2-low asthma includes the remainder of the allergic asthma group.2,12 Group 2 innate lymphoid cells have a critical role in type 2 immune responses.2 The Severe Asthma Research Program (SARP) and the Biomarkers for Prediction of Respiratory Disease Outcome (U-BIOPRED) leveraged these TH cell subgroups to characterize disease endotypes and clinical phenotypes according to the molecular mechanisms of allergic asthma.13 In the CD4+ T-cell subsets (TH1 and TH2 subpopulations), TH2 cells play a major role in eosinophilic airway inflammation, generating IL-4, IL-5, and IL-13 as seen in corticosteroid-dependent asthma.14
Other endotypes of non-allergic asthma continue to be grouped under the umbrella diagnosis. The treatment of asthma has become better targeted with use of drugs according to the endotype involved. It is unclear whether targeting ASM remodeling will need a similar approach by endotype, as ASM remodeling appears to be a feature of all asthma endotypes.
Origins of Increased ASM Mass in Asthma
Airway remodeling in asthma has been defined as a constellation of pathologic structural changes in the large and small airways of the lung that contribute to, and perpetuate, the clinical symptoms of the disease. Cellular and ECM changes include apoptosis of epithelial and ASM cells, expansion of ECM components such as actin subtypes and fibronectin, proliferation of ASM cells, and recruitment of additional ASM cells through mobilization of ASM precursors, activation of mesenchymal transformation, and stimulation of ASM motility.15 Remodeling involves cross-talk between ASM cells, epithelial cells, recruited inflammatory cells, and the ECM, resulting in hypertrophy, ie, increased mass, of the ASM and the ECM, and other changes (Figure 2).
The fundamental components of increased ASM mass in the asthmatic lung are stimulated by factors produced by recruited inflammatory cells, airway epithelial cells, and the ASM cells themselves (Figure 2). These basic processes establish and intensify cellular mechanisms that increase ASM mass, the thickness of the muscle layer surrounding the airways, and the volume of the ECM. Dynamic changes in the ASM cell actin cytoskeleton enhance the processes of contractile activation and mechanical adaptation of ASM cells. ASM cells adapt rapidly to local chemical and mechanical signals.16 The survival of ASM cells is regulated by several growth factors including transforming growth factor (TGF)-β and Platelet Derived Growth Factors (PDGF)s, the ECM proteins fibronectin, laminin, and collagen types I and IV, and the ECM receptors (integrins).17–19 These factors individually and together exert strong anti-apoptotic functions in airway myofibroblasts.
Myofibroblasts are the immediate precursors of ASM cells. They are primarily derived from circulating fibrocytes or fibroblasts within the airway sub-epithelial space. Myofibroblasts may also originate through injury-repair processes involving epithelial-mesenchymal transition and endothelial-mesenchymal transition in response to signals initiated by growth factors, inflammatory mediators, and mechanical force.20–22
Increased numbers of myofibroblasts in the submucosal compartment is an important factor in ASM survival. Myofibroblasts accumulate rapidly after allergen challenge. The migration of new airway myocytes from the adjacent ASM layer extends the muscle layer and creates increased airway thickness.23 Terminally differentiated ASM cells are capable of expressing and secreting cytokines, chemokines, and ECM proteins, all of which can act via paracrine, juxtacrine, and autocrine mechanisms to stimulate further ASM cell proliferation and migration. The increase in ASM cells supports new deposition of ECM proteins, collagens, laminin, and proteoglycans, which in turn promotes further increases in ASM cells. Cytokines and growth factors located within the airway ECM, including fibroblast growth factor−1 and TGF-α, stimulate expression of collagen types I and III mRNA by ASM cells.24
ASM hypertrophy and hyperplasia have significant biochemical consequences. Myofibroblasts are metabolically active not just for their contractile properties but also as a source of increased expression of a number of the important mediators of asthma responses, including matrix metalloproteinase (MMP)-9, inflammatory mediators, cytokines, and growth factors, all of which promote hyperplasia of neighboring ASM cells.25 Myofibroblasts also synthesize ECM proteins and α-smooth muscle actin (α-SMA), visible in cells as stress fibers.22 The contribution of the ASM to the endocrine milieu in asthma creates a significant amplification of the initial cellular reaction to acute and chronic stimuli in a positive feedback loop. This important feature of ASM hypertrophy and hyperplasia has not been overlooked; much of the impetus for the increasing interest in targeting ASM can be attributed to this feature.15 The appreciation of the biomechanical contribution of ASM hypertrophy to the clinical symptomatology of severe asthma has intensified this interest.
External Biomechanical Pressure on Airway Epithelium
Airway inflammation that provokes the release of soluble mediators is commonly considered to be the cause of AR. Less commonly considered, but possibly no less important, is the effect of mechanical compression of the epithelial layer. Smooth muscle contraction creates stress forces on the 3-dimensional airway structure, including epithelial compression (Figure 3). The persistence of these forces are under-appreciated factors promoting ASM hypertrophy in asthma. Studies demonstrate the progression of asthmatic airway structural changes in the absence of inflammation, presumably due to airway compression.26
An acute asthma attack with enhanced ASM contraction not only causes airway constriction that reduces the airway luminal area beyond the normal contractile range, it creates mucosal folding of the airway epithelium which provokes intracellular responses that stimulate AR.27 In vitro models show that acutely compressed airway epithelial cells release factors that stimulate increased basal ASM contraction and proliferation, leading to a higher resting tone and a narrower resting airway lumen.28 For example, ASM constriction produces tension on integrin αvβ5 fibers, this in turn activates TGF-β which promotes ASM hyperreactivity and AR (Figure 3).29
Animal and human studies provide translational relevance to these findings. For example, the effect of acute bronchoconstriction on cell proliferation was studied in asthmatic patients using allergen and methacholine (MCh) challenge with or without pretreatment with albuterol. Bronchial biopsies demonstrated increased epithelial cell expression of the cell proliferation marker Ki67 after allergen- and MCh-induced bronchoconstriction. This was not found in subjects that received albuterol prior to MCh.8
ECM and ASM Cell Remodeling
At the cellular level, increased ECM proteins, particularly fibronectin and collagen type I, the presence of pro-inflammatory mediators, and increased cell adhesion receptors all influence ASM cells to undergo phenotypic alterations, manifested by increased contractility, proliferation, hypertrophy, and migration (Figure 4). There are numerous integrin subtypes in the lung, including α5β1, α7β1, and αvβ5 which are primarily in the smooth muscle compartment.30 Integrin subtypes are synthesized in response to the inflammatory factors TGF-β and PDGF. These increase the levels of the transcription factors Activator Protein-1 (AP-1) and NFKB, which in turn stimulate gene expression for integrins.31,32 The remodeled ECM, along with proinflammatory mediators, and cell adhesion receptors further promote the secretion of cytokines and growth factors, most notably TGF-β and PDGF, further promoting ASM cell growth and ECM production.33–35 As the thickness of the ASM cell layer increases, there is an accompanying increase in the ECM.36,37 Phenotypic plasticity of the ASM is a major factor underlying AR in asthma. ASM cells may alternate between contractile and proliferative phenotypes.38
Hypertrophy and hypercontractility of ASM cells indicate that the muscle is stronger, stiffer, or both. Tumor necrosis factor alpha (TNF-α) and interleukin (IL)-1β synergistically increase the stiffness of ASM cells and reduce β-adrenergic responsiveness, mediated by increased cyclooxygenase 2 and prostaglandin (PG) E2 (PGE2).39 Dystrophin-glycoprotein (DG) complexes in ASM cells anchor the cell’s actin cytoskeleton to the ECM to support contractile tension.40 Dystrophin promotes calcium responses to contractile agonists and mediates the activation (phosphorylation) of the signaling proteins Akt 1 (also known as protein kinase-B), glycogen synthase kinase 3β (GSK3β), and mammalian target of rapamycin (mTOR), all of which are required for ASM maturation, hypertrophy, and accumulation of contractile proteins.40
The levels of DG complex subunits reflect the state of ASM cell maturation. DG subunit expression in ASM is associated with phenotype maturation from a proliferative to a contractile state and is regulated by laminin-integrin binding and the induction of phosphatidylinositol 3-kinase (PI3K) signaling. Laminin-2 binds to αDG subunits, generating intracellular signaling cascades supporting myocyte maturation into a contractile phenotype.41 Laminin also retards the dedifferentiation of ASM cells. Insulin-like growth factors (IGF-I and IGF-II) and insulin prolong the differentiated (contractile) phenotype through activation of PI3K and its downstream target, Akt-1.42,43
ASM Cell Proliferation
ASM cells produce growth factors and pro-inflammatory cytokines that regulate their proliferation through autocrine and paracrine signaling (Figure 4). ASM cells from asthmatic mice have an increased Ca++ ion concentration and decreased cell apoptosis. The stimulation of proliferation in ASM cells has been described as following one or the other of two general pathways: receptor tyrosine kinase phosphorylation that is linked to the activation of an intracellular cascade of protein phosphorylation signaling events, and activation of receptors linked to heterotrimeric ATP and GTP binding proteins.44
Chemokines and prostaglandins produced by healthy ASM cells mediate muscle tone; their production is strongly increased in asthmatic ASM cells, in part due to cell hypertrophy.45 The inflammatory cytokines IL-1β, IL-4, IL-5, and IL-6 promote ASM cell proliferation and inhibit apoptosis. They also induce ASM cells to secrete more inflammatory cytokines, further affecting cell proliferation, migration, and apoptosis in a positive feedback loop. Cultured ASM cells treated with IL-1β, TNF-α, bradykinin, and IL-4 also secrete increased amounts of monocyte chemoattractant proteins (MCP), eotaxin, and granulocyte-macrophage colony-stimulating factor (GM-CSF) which act as chemoattractants for inflammatory cells and enhance the positive feedback promoting proliferation, hypertrophy, and migration.46–49 IL-33 levels are also elevated and co-expressed with TNF-α in ASM cells from humans with severe asthma. IL-6 production is increased by several mediators including TNF-α, IL-1β, TGF-β, bradykinin, and endothelin-1. Airway myocytes also directly contribute to IL-6 production.45
Many of these cytokines upregulate ASM proliferation by activation of Transient Receptor Potential channels (TRPC) types 1 and 3 on ASM cells. TRPC-1 and TRPC-3 regulate the movement of cations, particularly Ca++, across cell membranes to activate Ca++-dependent processes.50–53 They participate in the formation of store operated calcium channels (SOCC) that maintain cytoplasmic and sarcoplasmic/endoplasmic reticulum Ca++ balance. Ca++ channels control the progression of ASM cells through the cell cycle, reducing the levels of phosphorylated p38 mitogen activated protein kinase (p38 MAPK), Jun n-Terminal Kinase (JNK) and Bcl-2, and increase cell survival through decreased caspase 3 cleavage.50
TNF-α and IL-1β modulate inflammatory processes in chronic asthma and during acute exacerbations. IL-1β treatment of human ASM cells enhances the activities of other mediators such as IL-4, IL-5, IL-13, and leukotrienes.44,46 On the other hand, IL-1β desensitizes the histamine H1 receptor, leading to induction of PGE2 and reducing the responsiveness of cultured bronchial rings to histamine.54 The IL-1β receptor antagonist reduces airway hyperreactivity in mice.55
ASM Cell Hypertrophy
Separate from proliferation that increases the number of ASM cells, ASM cells become larger, gaining more contractile elements (Figure 4). Accompanying this hypertrophy are increases in ECM volume and components. Dynamic changes in the ASM cell actin cytoskeleton potentiate contractile activation and mechanical force and facilitate mechanical adaptation. ASM cells adapt rapidly to local chemical and mechanical signals at the cell surface.16 The various aforementioned survival regulators acting individually and together exert strong anti-apoptotic influences on airway myofibroblasts, whose presence and activity may arise from infiltration by myofibroblasts or circulating hematopoietic progenitor cells at the site of inflammation.
Several integrin subtypes, particularly α5β1 integrin, act as receptors for components of the ECM. These receptors promote ASM cell survival and decrease apoptosis via tyrosine kinase signaling events that activate PI3K, nuclear factor kappa B (NF-kB), p53 and Bcl-2.18 Fibronectin together with α5β1 integrin protects cells from apoptosis, whereas fibronectin with αvβ1 integrin has no effect on apoptosis. Both osteopontin and β3 integrins increase NF-kB nuclear localization and transcription activity. The αvβ3 integrin induces IK-kB phosphorylation causing it to dissociate from NF-kB; NF-kB is thus activated and undergoes nuclear translocation of its p65 and p50 sub-units.56,57 This is blocked in ASM cells by anti-β3 integrin antibody.
It is not surprising that ASM hypertrophy is an energy-dependent process, mediated by increased oxidative phosphorylation. Evidence for this includes increased numbers of mitochondria and increased mitochondrial oxygen consumption in hypertrophied ASM from asthmatics. Mitochondrial proteins are dysregulated in ASM hypertrophy, interfering with apoptosis and upregulating energy metabolism for proliferation.58–61
Related to mitochondrial oxidative energy metabolism is the process of autophagy, by which cells actively phagocytose cellular material, including other cells. Autophagy is an essential process in development, cell growth, immune protection against both invasive external organisms and internal threats such as cancer, and programmed cell death. Autophagy can be detrimental if it promotes increased tissue remodeling; lack of autophagy can be detrimental if that leads to inappropriate accumulation of cells and matrix. This is meaningful for asthma as several studies have found that the accumulation of ASM cells and ECM in airway remodeling is accompanied by evidence of dysregulated autophagy. Familial asthmatics and some childhood asthma individuals have genetic abnormalities of one or more of several autophagy pathway proteins, particularly beclin 1 and Autophagy Related 5 (ATG-5).62 There is strong enthusiasm for the concept that abnormal autophagy in asthma is a significant contributor to increased ASM mass and for targeting autophagy to reduce ASM hypertrophy. However, it is not established whether the problem is excessive autophagy or too little autophagy in the asthmatic lung (Figure 4). Studies support both upregulation and downregulation of autophagy as possible therapeutic strategies. In an attempt to resolve this, Zeki et al proposed two contrasting models of the involvement of autophagy AR. In one model, autophagy is necessary for providing substrate and energy requirements of cell and ECM repair of inflammatory damage, hence, in this model autophagy is highly active and promotes increased ASM mass. In contrast, the second model proposes that autophagy is needed to remove the dense, fibrous ECM produced by inflammatory damage, hence, in this model the fact that increased ASM mass is a prominent pathologic feature of asthma indicates that autophagy is underactive.63 While those authors favored the “autophagy is overactive” model, and proposed possible treatments to reduce autophagy, contemporary and more recent reports give strong support for the “autophagy is underactive” model.
ASM Cell Migration
As already mentioned, in normal conditions the ASM population is maintained by migration from the interstitial compartment of precursor myofibroblasts and circulating fibrocytes. ASM cells migrate in response to chemotactic and chemokinetic signals in the microenvironment (Figure 4). The process of cell migration is mediated by polymerization of actin fibers that extend cell membrane protrusions (lamellipodia and filopodia). At the leading edge of migration, the actin fibers interact with adhesive structures (focal adhesions) within the ECM that stabilize the protrusions so that the body of the cell follows along. Focal adhesions of polymerized actin anchored to intracellular structures generates tension allowing the cell to propel itself through its environment. This all requires dynamic actin polymerization that is regulated by actin-related proteins and nucleation promoting factors.64,65
ASM cell migration is increased in asthmatic airways. Pro-inflammatory mediators produced by airway epithelial cells in response to allergens and inflammation, including TGF-β, PDGF, Epidermal Growth Factor (EGF), eotaxin, IL-8, and PGD2, induce ASM cell migration (Figure 2). In contrast, PGE2 inhibits migration. Cell surface integrin receptors that anchor cells to the ECM also organize the mechanical and chemical signals from matrix components. These signals in turn regulate the activities of cytoplasmic kinases, growth factor receptors, and ion channels to control the organization of the intracellular actin cytoskeleton.19
The p38 MAPK and the p21 activated kinase-1 (p21 AK-1) signaling pathways are major signaling pathways in cell migration. They are stimulated by oxidative stress and inflammatory mediators. For example, TNF-α activates p38 MAPK, which then activates TRPC-1 and TRPC-3 to upregulate Ca++ influx to stimulate cell motility and migration.50–53 The regulation of actin dynamics by p38 MAPK also involves interaction with heat shock protein 27.66
The actin polymerization and depolymerization associated with cell motility is regulated by intracellular calcium, phosphatidylinositol 4,5-bisphosphate (PIP2), G proteins, and integrins.19 Phosphorylated PI3K accumulates in the protruding edge of the cell, while phosphatase and tensin homolog (PTEN) protein accumulates in the trailing edge where they break down adhesions to allow the cell to move forward. PI3K amplifies chemotactic signals including Rac exchange factors along the leading edge, thereby promoting actin polymerization that initiates membrane protrusions.
Actin polymerization and depolymerization also occur in ASM cells during their contraction-relaxation cycle. Stimulation with a contractile agonist induces recruitment of α-actin to integrin-associated complexes within the cell. These complexes are also stimulated by phosphorylation of PI3K and p38 MAPK in response to the ECM and to signaling factors in the cellular milieu.
Immune Modulators and ASM Hypertrophy
Immune modulators activate naïve T cells, causing them to differentiate into TH2 cells that produce IL-4 and other cytokines. IL-4 stimulates allergen-specific B cells to release IgE, which binds to surface receptors on mast cells as well as basophils and eosinophils. These IgE-bound cells respond by producing more IgE, as well as more IL-4, IL-5, and histamine, which reinforce the cell-cell interactive circuit and initiate further downstream actions on airway epithelium, endothelium, ECM, and the ASM. One important consequence of damaging the airway epithelium and its barrier function is the potential of directly activating ASM hypertrophy. Serine proteases present in some allergens activate the protease-activated receptor (PAR)-2 on the airway epithelium, creating gaps in the epithelial barrier that allow allergen infiltration into sub-epithelial tissues.67 Allergens may also damage the basement membrane leading to inflammation that induces modification of ECM composition and function. Allergens can thereby penetrate more easily to make direct contact with ASM cells and initiate cellular reactions creating ASM hypertrophy.
Even individuals with mild asthma exhibit ASM cell remodeling after exposure to inhaled allergens, independent of eosinophilic infiltration. ASM cells constitutively express the low affinity (FceRII/CD23) and the high affinity (FceRI) IgE receptors. IgE linked with activating antigen stimulates ASM cell release of IL- 4, IL- 5, IL−13, and eotaxin, activate the extracellular signal regulated kinase (ERK)1/2 - MAPK pathway to promote ASM cell proliferation and hypertrophy, and deposit more collagen types I and type III.6,53,68
Leukotrienes are a large family of inflammatory mediators that are metabolites of arachidonic acid, some of which promote cell injury in response to inflammation and oxidative stress while others protect cells against these challenges. Leukotrienes that promote cell injury in asthma induce ASM proliferation and hypertrophy. Lipoxin and soluble epoxide hydrolase (sEH), which metabolize leukotrienes, are also present in the lung, where they promote cell injury (sEH) or protect against cell injury (lipoxin) and reduce inflammation in response to oxidative stress and injury. Lipoxin production is down regulated by sEH; their activity is normally balanced because of their opposite functions. However, asthmatic patients have elevated sEH levels and low lipoxin levels; this imbalance removes a valuable brake against inflammatory actions on ASM cells.69
Molecular Mediators of ASM Hypertrophy
Growth Factors and Cytokines
Intracellular signaling pathways underlying ASM hypertrophy modulate gene expression for migration, cell division, and apoptosis (Figure 4). PDGF, EGF, IGF-1, and TGF-β largely work through activation of PI3K and PTEN in stimulating ASM cell division and migration.19,70–72 Their chemokinetic, chemotactic, and cell division responses are attenuated by inhibitors of p38 MAPK and by Rho-kinase; although inhibition of PI3K affects just the chemotactic responses. Cysteinyl leukotrienes also promote ASM cell contraction, proliferation, and migration by activation of PI3K signal transduction pathways.19,70,73 Cysteinyl leukotrienes augment PDGF-stimulated ASM migration.70 TNF-α, a key modulator of cellular responses to inflammation, potentiates ASM contractile responses to serotonin and bradykinin.74
WNT1-induced secreted protein-1 (WISP-1), a member of the cysteine-rich, connective tissue growth factor and nephroblastoma overexpressed (CCN) family of growth factors, is a profibrotic protein found in several organs including the lung.75 Increased WISP1 stimulates ASM cell hypertrophy and proliferation in asthma. WISP-1 activates intracellular signaling cascades, including the PI3K, Akt, and GSK3β pathways which control additional ASM functions. For example, WISP1 mediated the deposition of collagen via activation of Akt-dependent GSK3β signaling in a rat model of asthma with AR.75 WISP-1 activity in lung fibroblasts also promotes increased type 1 collagen type 1 and fibronectin synthesis through increased phosphorylated AKT and phosphorylated GSK3β signaling.
The cellular second messenger molecules cyclic AMP and cyclic GMP have well-recognized roles in the activation of ASM contractile activity. Perhaps less frequently considered is that these molecules regulate signal transduction and ion channels involved in many of the discussed mechanisms that increase ASM hypertrophy.
Biomechanical Contributions to ASM Cell Hypertrophy
Passive Stiffness and Decreased Airway Distensibility
ASM contractility is dependent on the concentration of intracellular cytosolic calcium. Agonists that activate membrane Ca++ ion channels and the SOCCs increase free intracellular calcium to facilitate increased contractility (Figure 4). Passive airway stiffness, ie, a state of fixed airway stiffness or spasticity, can be created by a combination of increased ASM sensitivity to intracellular calcium plus increased amounts of the non-muscle components of the airway wall such as collagen, elastin, and the ECM fibrous tissue (Figure 3). In this condition, asthmatic airways are less distensible compared to normal airways, even after the force exerted by the muscle is abolished.76 Passive stiffness involves activation of actomyosin by increased free intracellular calcium. Re-sequestration of calcium away from the intracellular space restores the basal resting tension. The maintenance of passive stiffness is controlled in part by a calcium sensitive Rho kinase pathway in ASM.77
Other mechanisms that create prolonged increases in cytoplasmic calcium and thus contribute to passive stiffness have also been identified. For example, activation of the sarcoplasmic/endoplasmic reticulum calcium ATPase channel modulates passive stiffness through increases in ASM proliferation and secretory responses to IL-13 and PDGF-BB.1
ASM Contractile Responses
G-protein-coupled muscarinic (M)1, M2 and M3 receptors are present on ASM cells, in addition to mesenchymal, epithelial, and inflammatory cells. They differentially regulate contraction and proliferation of ASM cells in asthma, as well as other cellular responses including inflammation, goblet cell mucous secretion, and AR (Figure 4). Acetylcholine, released from vagal nerve endings, activates muscarinic receptors to stimulate the proliferative and pro-inflammatory responses of ASM cells. Airway inflammation increases cholinergic-mediated contractile tone as a result of exaggerated acetylcholine release in the ASM as well as increased expression of contraction-related proteins.78
Therapeutic Strategies to Target Increased ASM Mass
This section focuses on different available and potential strategies to therapeutically target ASM hypertrophy in asthma. Figure 5 summarizes cell and biomechanical elements that create increased ASM mass remodeling and categorizes potential strategies for targeting these elements. From these potential strategies, Table 1 lists drugs specifically shown to reduce ASM mass in humans or animal models of asthma or ASM cells in culture. Many of the strategies in this section involve either off-label use of existing drugs or use of drugs that are only available as research agents. Also, clinical trials described in this section generally were not powered for the desired outcome. We do not recommend using any of these therapeutic strategies except within clinical trials and with appropriate government agency approvals.
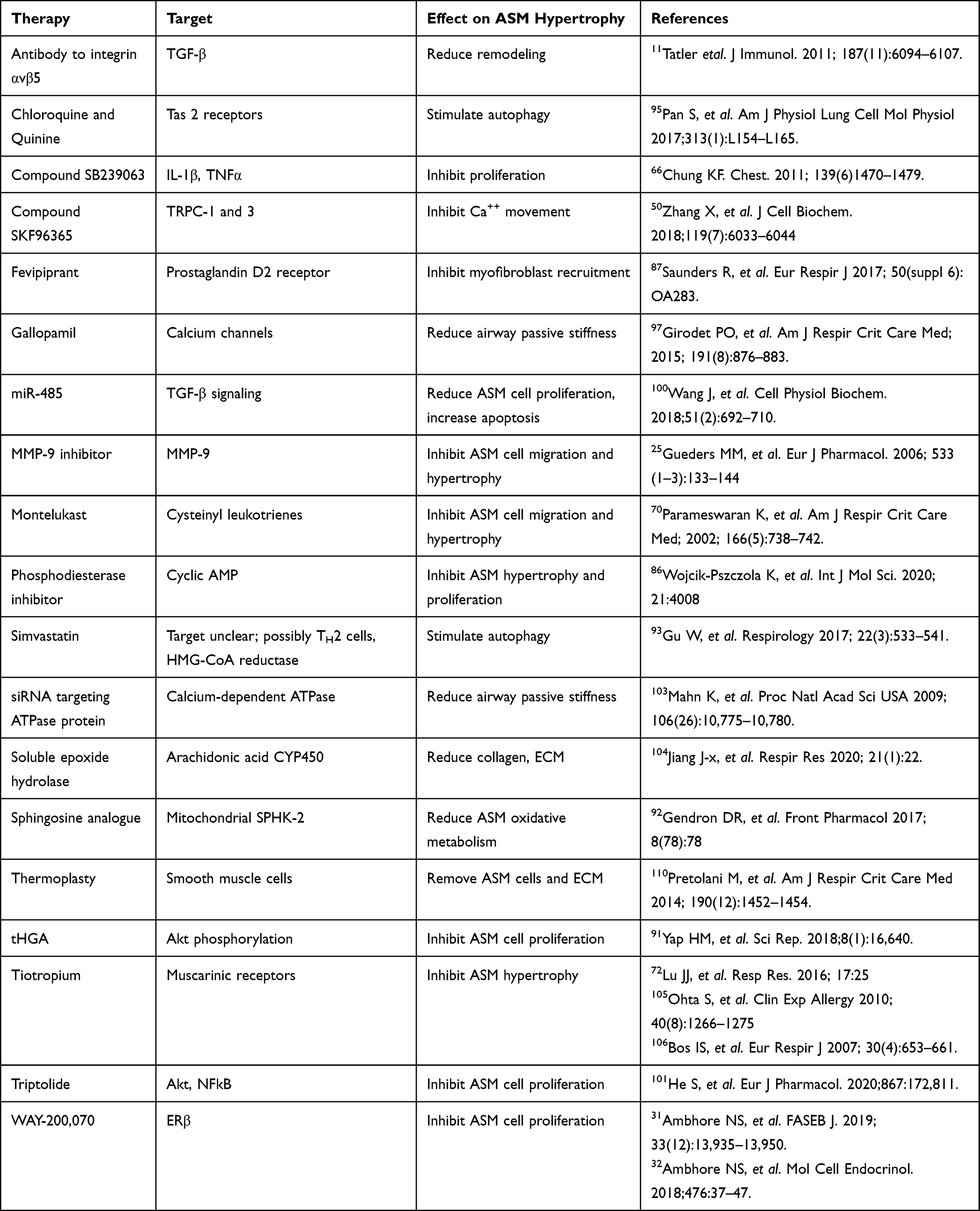 |
Table 1 Agents Shown to Reduce ASM Mass |
Pharmacologic Approaches
Therapeutic targeting of cell signaling to control bronchoconstriction underlies most pharmacologic approaches for asthma treatment79. Current pharmacologic therapy in asthma targets acute and chronic bronchoconstriction and inflammation, with little attention to potential effects on ASM mass. However, some currently available drugs might also reduce ASM mass. Phosphodiesterase (PDE) inhibitors are one such category that may deserve attention. Several PDEs actively promote ASM remodeling in asthma.80–82 PDEs mediate inflammatory responses that contribute to AR by cleaving cyclic AMP (cAMP) and cyclic GMP (cGMP). The inhibition of PDEs prevents the inactivation of cAMP and cGMP, which may result in less stimulation of ASM cell hypertrophy, proliferation, and migration if used in a chronic treatment protocol. The PDE4 inhibitor roflumilast is approved for a subset of patients with chronic obstructive pulmonary disease (COPD); studies in asthma show it may improve late bronchoconstriction in an acute asthma attack, possibly by resolving inflammation.83 A single inhaled dose of Ensifentrine, a combined PDE3/4 inhibitor, improved pulmonary function.84 Other PDE4 inhibitors have shown significant toxicity when tested in asthma.85 A recent in vitro study of a novel group of pan-PDE inhibitors found strong anti-remodeling effects mediated through cAMP and the TGF-β pathway, affecting ASM hypertrophy.86
Methylxanthines such as theophylline inhibit PDEs and were once the mainstay of asthma therapy as chronic bronchodilators. Their role was reduced from a first line approach based on concerns regarding their clinical effectiveness as bronchodilators and their therapeutic/toxicity ratio. Currently, they are employed as an alternative second therapy to improve corticosteroid effectiveness in difficult to control asthma.80 Whether methylxanthines have a significant impact on ASM cell hypertrophy is not known.
Blocking the effects of inflammatory mediators reduces ASM constriction; this approach may be appropriate for reversing ASM hypertrophy. The PGD2 receptor antagonist fevipiprant reduces ASM mass in patients with moderate to severe asthma with high eosinophil levels. This effect might reflect both reduced eosinophilic airway inflammation and a direct effect on ASM cells.87 Alternatively, reducing signals that promote TH2 maturation, and thereby reducing cytokine production that promotes ASM hypertrophy, has been considered. The factor Thymic Stromal Lymphopoietin (TSLP) is a stimulator of TH2 maturation. TSLP is produced by lung fibroblasts and airway epithelial cells; production is increased on exposure to antigens. It activates heterodimer receptors consisting of cytokine receptor-like factor 2 and the alpha chain of IL-7, leading to activation of several cytokine genes. Randomized trials testing the ability of the monoclonal antibody Tezepelumab, which targets the TSLP receptor, to treat severe asthma have reported benefit in reducing exacerbations and improving patient-reported symptoms.88,89 At this point, however, whether this is related to an effect on ASM hypertrophy is unclear.
Use of chemical compounds to specifically inhibit the activity or block the phosphorylation of signal transduction proteins regulating ASM cell proliferation is an attractive concept that has been tried. There are several known chemical inhibitors of different p38 MAPK functions invitro. The compound SB239063 blocks ASM proliferation and migration in cell culture.66 However, it is unclear from studies in an animal model if SB239063 is therapeutically useful. Dexamethasone alone reduced airway thickness in a mouse asthma model, this effect was increased by the addition of SB239063. However, in the same study SB239063 alone had no effect.90 No specific components of ASM hypertrophy were measured. Overall, there is not strong evidence that blocking p38 MAPK invivo will significantly affect ASM hypertrophy.
The synthetic non-steroidal compound 2,4,6-trihydroxy-3-geranyl acetophenone (tHGA) attenuates airway remodeling and hyperreactivity in a mouse asthma model. In cultured human ASM cells, tHGA inhibited the synthesis of TGF-β, and inhibited growth factor stimulation of cell proliferation and migration. The ASM cells were arrested in the G1 cell cycle phase, with down regulation of cyclin D1, due to direct inhibition of phosphorylation of Akt and JNK and blocking of STAT3 signal transduction. Cell apoptosis was not significantly affected.91
Mitochondrial activity sustaining cell proliferation and ASM hypertrophy can be reduced using artificial sphingosine analogs that act as a substrate for sphingosine kinase-2 (SPHK-2). The analog (R)-2-amino-4-(4-heptyloxyphenyl)-2-methylbutanol reduces ASM cell numbers by decreasing oxidative energy metabolism and reducing airway hyperresponsiveness.58,92 As artificial substrates are non-specific in their effects on energy metabolism, careful studies evaluating the safety of inhibiting mitochondrial oxidative phosphorylation to reverse ASM hypertrophy must be done.
The role of autophagy in promoting airway remodeling, and its appeal as a target for therapeutic manipulation in asthma was recently reviewed.62 However, just how autophagy should be altered is not clear at this time, based on the uncertainty of whether autophagy is increased or decreased as previously discussed (Figure 5). Pharmacologic stimulation of pathways that actively reduce ASM cell mass by promoting autophagy has been accomplished. Antibodies directed against specific integrin subtypes are permissive for ASM cell apoptosis.18 Statin drugs, such as simvastatin, induce autophagy in a variety of tissues, including the ASM.93 The pro-autophagic effects of statins appears to be potentiated by simultaneous use of Rho-kinase inhibitors. In contrast, other studies show that reducing autophagy reduces ASM mass. Unconventional but attractive targets in this regard are the bitter taste receptors (TAS2 receptors) present in the ASM. Bitter taste receptors, when activated by agonists such as chloroquine or quinine, induce ASM autophagy by altering mitochondrial function.94,95 The ideal approach, i.e. whether to stimulate or block autophagy, or even whether there is a role for both approaches in asthma therapy remains unclear.
Calcium channel blocking agents are commonly the first line drug for treating hypertension and other cardiovascular disorders, and their safety and therapeutic margins are well known. The fact that intracellular calcium metabolism is prominent in many of the mechanisms controlling the proliferation, hypertrophy, and motility of ASM cells suggests that use of calcium channel blockers may be a means of targeting ASM mass (Figure 5). The use of calcium channel blockers as bronchodilators has been tested in small clinical trials, both for acute and chronic asthma management, and found to have no clinical effectiveness.96 However, those trials did not propose or evaluate any possible effects on ASM hypertrophy. More recently, a randomized blinded placebo controlled clinical trial of the calcium channel blocker gallopamil found that daily treatment for a year significantly reduced ASM mass, resulting in improved airway function. This outcome was encouraging, however there has been criticism of the study design and conclusions.97,98
TRPC channels in asthma ASM cells have been blocked using the compound SKF96365 to inhibit their function or by knocking down the expression of their genes using RNA interference technology. Blocking or knocking down TRPC-1 and 3 increased the levels of phosphorylated p38 MAPK (p-p38 MAPK) and p-JNK, increased cleaved caspase 3 and decreased anti-apoptotic Bcl-2. This resulted in decreased ASM cell proliferation and ASM cell viability. Cells were arrested in G2/S phase with fewer cells in G1.50
Estrogen receptor β (ERβ) activity can reduce ECM synthesis and ASM cell proliferation. Selective activation of ERβ counteracts the stimulation of ECM production and remodeling by TGF-β and PDGF. For example, the ERβ agonist WAY-200070 downregulates the stimulation by TGF-β and PDGF of AP-1 and NFKB levels in both normal and asthmatic ASM cells.31,32 This significantly reduces the production of ECM matrix proteins in response to these inflammatory mediators. ERβ also counteracts the stimulation of ASM cell proliferation by PDGF and TNFα by suppressing their activation of intracellular signaling proteins Akt, p38 MAPK, ccaat enhancer binding protein (c/EBP), and ERK 1/2. This reduces the levels of the cell cycle proteins cyclin D1 and cyclin E1 kinase, arresting the cells in G1 phase of the cell cycle.31,32
Micro RNAs (miRs) are being developed as drugs to silence the expression of specific genes as a novel approach to medical therapy. Li et al described elevated levels of miR-378 in the serum and the airways of children with asthma. The sequence of miR-378 predicts that it will affect several signaling pathways important in AR, including Ras, calcium, MAPK, phosphatidylinositol, and estrogen signaling pathways. In cell culture, miR-378 overexpression stimulated the proliferation and migration of ASM cells and increased the production of fibronectin and collagen type 1. Use of a miR antagonist (anti-miR) against miR-378 reversed these effects, and down regulated ASM cell proliferation and migration.99
TGF-β signaling is regulated by miR-485, this has received interest in the cancer literature. In a mouse model of asthma, low levels of miR-485 correlated with high levels of IL-4, IL-5, IL-13, and IL-17, with increased ASM cell proliferation and decreased apoptosis. Overexpression of miR-485 in the mice inhibited TGF-β signaling, reduced the levels of interleukins, and inhibited ASM cell proliferation while reducing apoptosis.100 The authors cautiously suggested the possibility of using miR-453 agonists as a therapy in asthma, but also noted that much work needs to be done to best define the effects.
Triptolide, a Chinese herbal extract, has been studied for potential cell proliferation effects. He et al reported that triptolide inhibits the stimulation of ASM cell proliferation in culture. At the cellular level, there was cell cycle arrest in G0/G1, and suppression of TGF-β and Akt/NFκB/cyclin D1 signaling.101
Specifically targeting passive stiffness to modulate ASM hypertrophy is a strategy that is gaining increased attention, motivated by the need to find new approaches in asthma therapy.76 Actin polymerization mediates mechano-sensitive ASM contraction, and is inhibited by the chemotherapeutic agent latrunculin, which prevents length-sensitive tension development.102 Passive stiffness, because it is mediated by intracellular calcium, can be reduced by inhibitors of calcium channels, calcium mobilization, or downstream elements regulated by calcium. Rho-kinase pathway inhibitors reduce passive airway stiffness in animal models (Figures 4 and 5).77 Contributions of the ECM to passive stiffness can be directly targeted in vitro. Use of a specific antibody to αvβ5 reduces airway smooth muscle stiffness caused by TGF-β activation.29 The ability of the sarcoplasmic/endoplasmic reticulum calcium ATPase channel to control ASM passive stiffness has been reversed by treatment with siRNA targeting the expression of the ATPase protein.103 Loss of dystrophin suppresses the contractile response of the ASM to calcium agonists, but to date studies of dystrophin have used genetic models of dystrophin loss.40 An antibody or drug that will reduce the function of dystrophin has not been described. The arachidonic acid CYP450 pathway was targeted by oral administration of an inhibitor of sEH. This significantly reduced factors that contribute to passive stiffness, including inflammatory leukotrienes, MMP-9, collagen levels, and α-SMA. Importantly, AR and ASM cell hypertrophy were reduced.104
Muscarinic receptors are potential targets for reducing ASM hypertrophy in asthma, as they regulate mediators of ASM migration and AR.72,105 It is well-known that muscarinic receptor antagonists (anti-cholinergic agents), particularly of the M3 receptor subtype, have bronchodilating properties.72 They are sometimes used in steroid-resistant asthma, but generally their use is avoided in asthma because they also create mucous membrane dehydration. However, the use of long-acting muscarinic receptor antagonists may have value for targeting ASM hypertrophy in asthma (Figure 5). Tiotropium, a potent anticholinergic drug, is frequently used clinically to prevent worsening of COPD because of its ability to inhibit airway inflammation and remodeling. This anti-inflammatory activity has been shown in animal models of asthma to reduce ASM hypertrophy.105,106 Tiotropium also reduces the ASM migration-promoting effects of lung epithelial-derived IL-8 and TGF-β.72
Surgical Approaches
Invasive reduction of ASM using bronchial thermoplasty (BT) is currently the only therapeutic strategy approved by the US Food and Drug Administration (FDA) specifically for reducing the ASM in asthma.107 It is considered an option for patients with severe asthma that is poorly responsive to conventional treatments. BT uses controlled radio-frequency energy that is generated and delivered via a catheter into the bronchial tree through a flexible bronchoscope to thermally ablate ASM underlying the epithelial layer at the position of the bronchoscope. The goal is diminishing bronchial constriction and reducing asthma symptoms by reduction of ASM mass. BT reduced bronchial ASM cell volume and airway hyper-responsiveness in canine models.108 The clinical feasibility of BT was also demonstrated in patients without asthma who underwent lung resection for other diseases.109
Pretolani et al. reported the outcome of BT performed in patients with severe asthma who were nonresponse to steroids and bronchodilators. Three sessions of BT were given at monthly intervals, with follow-up bronchial biopsies, in which the amount of ASM was determined. This study reported a significant decrease (>45%) in ASM cell volume in both treated and nontreated areas.110,111 Other small trials reported similar results, including reduced evidence of inflammation and fewer severe asthma episodes.112,113 Donovan et al., using human lung autopsy specimens obtained from individuals who had fatal asthma, non-fatal asthma, or no asthma to model flow distributions and airway resistance changes created by thermoplasty, reported a 5% reduction in ASM after BT. However, this relatively small degree of ASM reduction in the central airways triggered a significant redistribution of flow patterns, a reopening of small airways, and a marked improvement in peripheral airway flow with improved lung function.107
However, the efficacy and long-term safety of BT continues to be a topic of controversy. A more recent trial of BT was unable to replicate the findings of Pretolani et al.110 and also raised questions over long-term complications and whether changes persisted.114,115 Conversely, a separate clinical trial supported the clinical effectiveness of BT and reported that the underlying mechanisms for success of the procedure were significant increases in airway volume and reductions in airway resistance.116 Thus, even though this therapy is FDA approved it is wise to restrict the use of this procedure to participation in carefully designed protocols that will contribute to the understanding of the risk/benefit ratio.115
Conclusions
Exposures to environmental irritants promote ASM cell hypertrophy due to inflammation and airway injury. Inflammation augments airway tension and impairs distensibility in asthma, and together with injury they induce remodeling with increased numbers of ASM cells, thickened airway walls, altered ECM, and spasticity promoting epithelial compression. Because these changes worsen mechanical obstruction and exacerbate airway hyper-reactivity, the importance of a therapeutic strategy that reduces both ASM contractile responses and also the ASM mass is becoming increasingly evident. Laboratory studies suggest many potential approaches to the problem of reducing ASM mass; some clinically available drugs are candidates for study and other effective compounds are in further development. Physical removal of hypertrophic ASM with BT is an effective but expensive strategy that has FDA approval but an unclear safety and efficacy profile. Increased attention to pharmacologic strategies has opened up a wide selection of possible strategic targets. There is strong evidence that addressing these targets with available or new drugs is likely to be effective in reducing ASM hypertrophy, and lead to better control of severe asthma.
 |
Figure 1 A cut away view of the structure of a normal and an asthmatic bronchiole, revealing the ASM cell hypertrophy with a thicker cross-sectional area and wider extension of the muscle across the outer surface of the asthmatic bronchiole. The muscle layer itself exhibits increased thickness. The airway lumen of the asthmatic bronchiole is decreased by the edematous swollen pseudo-columnar epithelium and submucosa, increased and edematous extracellular matrix, and the increased ASM thickness. The mucous secretions in the airway are also increased, contributing to the resistance to air flow. Current asthma therapies reduce the tissue swelling and edema, but not the increased thickness of the ASM and ECM. |
 |
Figure 2 The increased ASM mass in asthma is driven by cell hypertrophy, increased cell migration, and increased cell proliferation. Activation of the airway epithelium by allergens causes the release of EGF, Eotaxin, PDGF, TGF-β, IL-8, and PGD2. Part of the response of these factors is recruitment of inflammatory cells into the airway ECM that release IL-4, IL-5 and IL-13. The factors from the epithelium and the inflammatory cells stimulate myofibroblast migration, proliferation, and hypertrophy, and increased production of ECM components. The epithelial and inflammatory cell-derived factors also stimulate additional production of soluble mediators from the ASM cells that further drive increased ASM mass. |
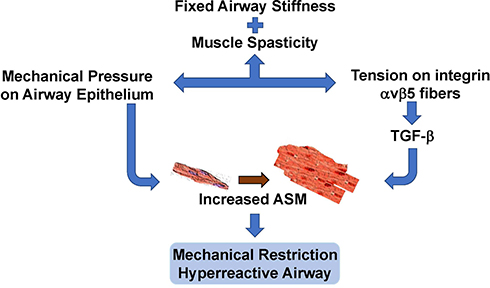 |
Figure 3 The ASM in chronic asthma exhibits fixed stiffness and muscle spasticity that maintain a baseline increased airway tone with chronic tension. This exerts mechanical compressive forces on the underlying airway epithelium, and also stimulates TGF-β activity via αvβ5 integrin. Chronic inflammation further increases the basal airway tension and epithelial compression. The constant high level of airway spasticity and airway hyperreactivity create a fixed degree of mechanical obstruction. |
 |
Figure 4 The pathways and factors that create airway stiffness, ASM proliferation, migration, and hypertrophy, and dysregulated autophagy are summarized. Each panel illustrates how different signaling elements and pathways combine to stimulate remodeling of the airway. The autophagy panel illustrates the current dilemma over whether autophagy is increased or decreased in the development of ASM hypertrophy. |
 |
Figure 5 Increased ASM cell hypertrophy and migration, ASM cell proliferation and survival, biomechanical forces, and extracellular matrix are the conditions which can be therapeutically targeted to decrease the physical impact of airway remodeling in asthma. Causal factors are listed separately for each process, along with potential strategies to inhibit these factors. The causal factors include only those for which there is good evidence that they stimulate the specific process. The listed potential remedies include only those shown to antagonize one or more of the causal factors in that column. Some causal factors and potential remedies are listed in more than one column, indicating that some factors are involved in more than one process. This figure should be examined in combination with the Table 1, which lists the treatments shown to affect the named process. |
Funding
Supported by a research grant from the Peabody Foundation, Boston MA, USA.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Prakash YS. Airway smooth muscle in airway reactivity and remodeling: what have we learned? Am J Physiol Lung Cell Mol Physiol. 2013;305(12):L912–933. doi:10.1152/ajplung.00259.2013
2. Kuruvilla ME, Lee FE, Lee GB. Understanding asthma phenotypes, endotypes, and mechanisms of disease. Clin Rev Allergy Immunol. 2019;56(2):219–233. doi:10.1007/s12016-018-8712-1
3. Ebina M, Yaegashi H, Takahashi T, Motomiya M, Tanemura M. Distribution of smooth muscles along the bronchial tree. A morphometric study of ordinary autopsy lungs. Am Rev Respir Dis. 1990;141(5 Pt 1):1322–1326.
4. Rydell-Tormanen K, Risse PA, Kanabar V, Bagchi R, Czubryt MP, Johnson JR. Smooth muscle in tissue remodeling and hyper-reactivity: airways and arteries. Pulm Pharmacol Ther. 2013;26(1):13–23. doi:10.1016/j.pupt.2012.04.003
5. Weaver M, Dunn NR, Hogan BLM. Bmp4 and Fgf10 play opposing roles during lung bud morphogenesis. Development. 2000;127:2695–2704. doi:10.1242/dev.127.12.2695
6. Redhu NS, Gounni AS. The high affinity IgE receptor (FcepsilonRI) expression and function in airway smooth muscle. Pulm Pharmacol Ther. 2013;26(1):86–94. doi:10.1016/j.pupt.2012.04.004
7. Chetty A, Sharda A, Warburton R, et al. A purinergic P2Y6 receptor agonist prodrug modulates airway inflammation, remodeling, and hyperreactivity in a mouse model of asthma. J Asthma Allergy. 2018;11:159–171. doi:10.2147/JAA.S151849
8. Grainge CL, Lau LC, Ward JA, et al. Effect of bronchoconstriction on airway remodeling in asthma. N Engl J Med. 2011;364(21):2006–2015. doi:10.1056/NEJMoa1014350
9. James AL, Maxwell PS, Pearce-Pinto G, Elliot JG, Carroll NG. The relationship of reticular basement membrane thickness to airway wall remodeling in asthma. Am J Respir Crit Care Med. 2002;166(12 Pt 1):1590–1595. doi:10.1164/rccm.2108069
10. James AL, Elliot JG, Jones RL, et al. Airway smooth muscle hypertrophy and hyperplasia in asthma. Am J Respir Crit Care Med. 2012;185(10):1058–1064. doi:10.1164/rccm.201110-1849OC
11. McParland BE, Macklem PT, Pare PD. Airway wall remodeling: friend or foe? J Appl Physiol. 2003;95(1):426–434. doi:10.1152/japplphysiol.00159.2003
12. Wenzel SE. Severe adult asthmas: integrating clinical features, biology and therapeutics to improve outcomes. Am J Respir Crit Care Med. 2021;203(7):809–821.
13. Moore WC, Meyers DA, Wenzel SE, et al. Identification of asthma phenotypes using cluster analysis in the Severe Asthma Research Program. Am J Respir Crit Care Med. 2010;181(4):315–323. doi:10.1164/rccm.200906-0896OC
14. Wenzel SE, Schwartz LB, Langmack EL, et al. Evidence that severe asthma can be divided pathologically into two inflammatory subtypes with distinct physiologic and clinical characteristics. Am J Respir Crit Care Med. 1999;160(3):1001–1008. doi:10.1164/ajrccm.160.3.9812110
15. Hough KP, Curtiss ML, Blain TJ, et al. Airway remodeling in asthma. Front Med. 2020;7:191. doi:10.3389/fmed.2020.00191
16. Gerthoffer WT, Gunst SJ. Invited review: focal adhesion and small heat shock proteins in the regulation of actin remodeling and contractility in smooth muscle. J Appl Physiol. 2001;91(2):963–972. doi:10.1152/jappl.2001.91.2.963
17. Yap HM, Israf DA, Harith HH, Tham CL, Sulaiman MR. Crosstalk between signaling pathways involved in the regulation of airway smooth muscle cell hyperplasia. Front Pharmacol. 2019;10(1148):1148. doi:10.3389/fphar.2019.01148
18. Freyer AM, Johnson SR, Hall IP. Effects of growth factors and extracellular matrix on survival of human airway smooth muscle cells. Am J Respir Cell Mol Biol. 2001;25(5):569–576. doi:10.1165/ajrcmb.25.5.4605
19. Salter B, Pray C, Radford K, Martin JG, Nair P. Regulation of human airway smooth muscle cell migration and relevance to asthma. Respir Res. 2017;18(1):156. doi:10.1186/s12931-017-0640-8
20. Schmidt M, Sun G, Stacey MA, Mori L, Mattoli S. Identification of circulating fibrocytes as precursors of bronchial myofibroblasts in asthma. J Immunol. 2003;171(1):380–389. doi:10.4049/jimmunol.171.1.380
21. Paw M, Wnuk D, Kadziolka D, et al. Fenofibrate reduces the asthma-related fibroblast-to-myofibroblast transition by TGF-Beta/Smad2/3 signaling attenuation and connexin 43-dependent phenotype destabilization. Int J Mol Sci. 2018;19:9. doi:10.3390/ijms19092571
22. Michalik M, Wojcik-Pszczola K, Paw M, et al. Fibroblast-to-myofibroblast transition in bronchial asthma. Cell Mol Life Sci. 2018;75(21):3943–3961.
23. Gerthoffer WT, Schaafsma D, Sharma P, Ghavami S, Halayko AJ. Motility, survival, and proliferation. Compr Physiol. 2012;2(1):255–281. doi:10.1002/cphy.c110018
24. Parameswaran K, Willems-Widyastuti A, Alagappan VK, Radford K, Kranenburg AR, Sharma HS. Role of extracellular matrix and its regulators in human airway smooth muscle biology. Cell Biochem Biophys. 2006;44(1):139–146. doi:10.1385/CBB:44:1:139
25. Gueders MM, Foidart JM, Noel A, Cataldo DD. Matrix metalloproteinases (MMPs) and tissue inhibitors of MMPs in the respiratory tract: potential implications in asthma and other lung diseases. Eur J Pharmacol. 2006;533(1–3):133–144. doi:10.1016/j.ejphar.2005.12.082
26. Tschumperlin DJ, Drazen JM. Mechanical stimuli to airway remodeling. Am J Respir Crit Care Med. 2001;164(10 Pt 2):S90–S94. doi:10.1164/ajrccm.164.supplement_2.2106060
27. Wiggs BR, Hrousis CA, Drazen JM, Kamm RD. On the mechanism of mucosal folding in normal and asthmatic airways. J Appl Physiol. 1997;83(6):1814–1821. doi:10.1152/jappl.1997.83.6.1814
28. Lan B, Mitchel JA, O’Sullivan MJ, et al. Airway epithelial compression promotes airway smooth muscle proliferation and contraction. Am J Physiol Lung Cell Mol Physiol. 2018;315(5):L645–L652. doi:10.1152/ajplung.00261.2018
29. Tatler AL, John AE, Jolly L, et al. Integrin alphavbeta5-mediated TGF-beta activation by airway smooth muscle cells in asthma. J Immunol. 2011;187(11):6094–6107. doi:10.4049/jimmunol.1003507
30. Teoh CM, Tan SS, Tran T. Integrins as therapeutic targets for respiratory diseases. Curr Mol Med. 2015;15(8):714–734. doi:10.2174/1566524015666150921105339
31. Ambhore NS, Kalidhindi RSR, Pabelick CM, Hawse JR, Prakash YS, Sathish V. Differential estrogen-receptor activation regulates extracellular matrix deposition in human airway smooth muscle remodeling via NF-kappaB pathway. FASEB J. 2019;33(12):13935–13950. doi:10.1096/fj.201901340R
32. Ambhore NS, Katragadda R, Raju Kalidhindi RS, et al. Estrogen receptor beta signaling inhibits PDGF induced human airway smooth muscle proliferation. Mol Cell Endocrinol. 2018;476:37–47. doi:10.1016/j.mce.2018.04.007
33. Panettieri RA
34. Tran T, Halayko AJ. Extracellular matrix and airway smooth muscle interactions: a target for modulating airway wall remodelling and hyperresponsiveness? Can J Physiol Pharmacol. 2007;85(7):666–671. doi:10.1139/Y07-050
35. Halayko AJ, Tran T, Ji SY, Yamasaki A, Gosens R. Airway smooth muscle phenotype and function: interactions with current asthma therapies. Curr Drug Targets. 2006;7(5):525–540. doi:10.2174/138945006776818728
36. Ebina M, Takahashi T, Chiba T, Motomiya M. Cellular hypertrophy and hyperplasia of airway smooth muscles underlying bronchial asthma. A 3-D morphometric study. Am Rev Respir Dis. 1993;148(3):720–726.
37. Thomson RJ, Bramley AM, Schellenberg RR. Airway muscle stereology: implications for increased shortening in asthma. Am J Respir Crit Care Med. 1996;154(3 Pt 1):749–757. doi:10.1164/ajrccm.154.3.8810615
38. Wright DB, Trian T, Siddiqui S, et al. Functional phenotype of airway myocytes from asthmatic airways. Pulm Pharmacol Ther. 2013;26(1):95–104. doi:10.1016/j.pupt.2012.08.003
39. Moore PE, Lahiri T, Laporte JD, Church T, Panettieri RA
40. Sharma P, Basu S, Mitchell RW, Stelmack GL, Anderson JE, Halayko AJ. Role of dystrophin in airway smooth muscle phenotype, contraction and lung function. PLoS One. 2014;9(7):e102737. doi:10.1371/journal.pone.0102737
41. Sharma P, Tran T, Stelmack GL, et al. Expression of the dystrophin-glycoprotein complex is a marker for human airway smooth muscle phenotype maturation. Am J Physiol Lung Cell Mol Physiol. 2008;294:L57–L68. doi:10.1152/ajplung.00378.2007
42. Hayashi K, Saga H, Chimori Y, Kimura K, Yamanaka Y, Sobue K. Differentiated phenotype of smooth muscle cells depends on signaling pathways through insulin-like growth factors and phosphatidylinositol 3-kinase. J Biol Chem. 1998;273(44):28860–28867. doi:10.1074/jbc.273.44.28860
43. Chetty A, Cao GJ, Nielsen HC. Insulin-like growth factor-I signaling mechanisms, type I collagen and alpha smooth muscle actin in human fetal lung fibroblasts. Pediatr Res. 2006;60(4):389–394. doi:10.1203/01.pdr.0000238257.15502.f4
44. Hirst SJ, Martin JG, Bonacci JV, et al. Proliferative aspects of airway smooth muscle. J Allergy Clin Immunol. 2004;114(2 Suppl):S2–S17. doi:10.1016/j.jaci.2004.04.039
45. Damera G, Panettieri RA
46. Khan MA. Inflammation signals airway smooth muscle cell proliferation in asthma pathogenesis. Multidiscip Respir Med. 2013;8(1):11. doi:10.1186/2049-6958-8-11
47. Bonini M, Usmani OS. The role of the small airways in the pathophysiology of asthma and chronic obstructive pulmonary disease. Ther Adv Respir Dis. 2015;9(6):281–293. doi:10.1177/1753465815588064
48. Carroll N, Cooke C, James A. The distribution of eosinophils and lymphocytes in the large and small airways of asthmatics. Eur Respir J. 1997;10(2):292–300. doi:10.1183/09031936.97.10020292
49. Hamada K, Goldsmith CA, Goldman A, Kobzik L. Resistance of very young mice to inhaled allergen sensitization is overcome by coexposure to an air-pollutant aerosol. Am J Respir Crit Care Med. 2000;161(4 Pt 1):1285–1293. doi:10.1164/ajrccm.161.4.9906137
50. Zhang X, Zhao Z, Ma L, et al. The effects of transient receptor potential channel (TRPC) on airway smooth muscle cell isolated from asthma model mice. J Cell Biochem. 2018;119(7):6033–6044. doi:10.1002/jcb.26801
51. Pelaia C, Vatrella A, Gallelli L, et al. Role of p38 mitogen-activated protein kinase in asthma and COPD: pathogenic aspects and potential targeted therapies. Drug Des Devel Ther. 2021;15:1275–1284. doi:10.2147/DDDT.S300988
52. El Karim I, McCrudden MT, Linden GJ, et al. TNF-alpha-induced p38MAPK activation regulates TRPA1 and TRPV4 activity in odontoblast-like cells. Am J Pathol. 2015;185(11):2994–3002. doi:10.1016/j.ajpath.2015.07.020
53. Xiao J-H, Zheng Y-M, Liao B, Wang Y-X. Functional role of canonical transient receptor potential 1 and canonical transient receptor potential 3 in normal and asthmatic airway smooth muscle cells. Am J Respir Cell Mol Biol. 2010;43(1):17–25. doi:10.1165/rcmb.2009-0091OC
54. Pype JL, Xu H, Schuermans M, et al. Mechanisms of interleukin 1beta-induced human airway smooth muscle hyporesponsiveness to histamine. Involvement of p38 MAPK NF-kappaB. Am J Respir Crit Care Med. 2001;163(4):1010–1017. doi:10.1164/ajrccm.163.4.9911091
55. Barnes PJ. The cytokine network in asthma and chronic obstructive pulmonary disease. J Clin Invest. 2008;118(11):3546–3556. doi:10.1172/JCI36130
56. Zhang Z, Vuori K, Reed JC, Ruoslahti E. The alpha 5 beta 1 integrin supports survival of cells on fibronectin and up-regulates Bcl-2 expression. Proc Natl Acad Sci U S A. 1995;92(13):6161–6165. doi:10.1073/pnas.92.13.6161
57. Scatena M, Almeida M, Chaisson ML, Fausto N, Nicosia RF, Giachelli CM. NF-κB mediates αvβ3 integrin-induced endothelial cell survival. J Cell Biol. 1998;141(4):1083–1093. doi:10.1083/jcb.141.4.1083
58. Blais-Lecours P, Laouafa S, Arias-Reyes C, et al. Metabolic adaptation of airway smooth muscle cells to a SPHK2 substrate precedes cytostasis. Am J Respir Cell Mol Biol. 2019;62(1):35–42. doi:10.1165/rcmb.2018-0397OC
59. Pan S, Shah SD, Panettieri RA
60. Beaufils F, Marthan R, Berger P. Bnip3 as a potential target to treat airway smooth muscle remodeling in asthma? Am J Physiol Lung Cell Mol Physiol. 2020;318(1):L212. doi:10.1152/ajplung.00431.2019
61. Pan S, Deshpande DA. Reply to letter to the editor: “Bnip3 as a potential target to treat airway smooth muscle remodeling in asthma?”. Am J Physiol Lung Cell Mol Physiol. 2020;318(1):L213–L214. doi:10.1152/ajplung.00470.2019
62. Yadav SK, Shah SD, Penn RB. Give me a fork: can autophagy research solve the riddle of airway remodeling in asthma? Am J Respir Cell Mol Biol. 2019;60(5):494–496. doi:10.1165/rcmb.2018-0353ED
63. Zeki AA, Yeganeh B, Kenyon NJ, Post M, Ghavami S. Autophagy in airway diseases: a new frontier in human asthma? Allergy. 2016;71(1):5–14. doi:10.1111/all.12761
64. Tang DD. Critical role of actin-associated proteins in smooth muscle contraction, cell proliferation, airway hyperresponsiveness and airway remodeling. Respir Res. 2015;16:134. doi:10.1186/s12931-015-0296-1
65. Gerlach BD, Tubbesing K, Liao G, et al. Phosphorylation of GMFgamma by c-Abl coordinates lamellipodial and focal adhesion dynamics to regulate airway smooth muscle cell migration. Am J Respir Cell Mol Biol. 2019;61(2):219–231. doi:10.1165/rcmb.2018-0352OC
66. Chung KF. p38 mitogen-activated protein kinase pathways in asthma and COPD. Chest. 2011;139(6):1470–1479. doi:10.1378/chest.10-1914
67. Post S, Heijink IH, Petersen AH, de Bruin HG, van Oosterhout AJ, Nawijn MC. Protease-activated receptor-2 activation contributes to house dust mite-induced IgE responses in mice. PLoS One. 2014;9(3):e91206. doi:10.1371/journal.pone.0091206
68. Grunstein MM, Hakonarson H, Leiter J, et al. IL-13-dependent autocrine signaling mediates altered responsiveness of IgE-sensitized airway smooth muscle. Am J Physiol Lung Cell Mol Physiol. 2002;282(3):L520–528. doi:10.1152/ajplung.00343.2001
69. Ono E, Dutile S, Kazani S, et al. Lipoxin generation is related to soluble epoxide hydrolase activity in severe asthma. Am J Respir Crit Care Med. 2014;190(8):886–897. doi:10.1164/rccm.201403-0544OC
70. Parameswaran K, Cox G, Radford K, Janssen LJ, Sehmi R, O’Byrne PM. Cysteinyl leukotrienes promote human airway smooth muscle migration. Am J Respir Crit Care Med. 2002;166(5):738–742. doi:10.1164/rccm.200204-291OC
71. Lee H, Kim SR, Oh Y, Cho SH, Schleimer RP, Lee YC. Targeting insulin-like growth factor-I and insulin-like growth factor-binding protein-3 signaling pathways. A novel therapeutic approach for asthma. Am J Respir Cell Mol Biol. 2014;50(4):667–677. doi:10.1165/rcmb.2013-0397TR
72. Lu JJ, Xu GN, Yu P, et al. The activation of M3 mAChR in airway epithelial cells promotes IL-8 and TGF-beta1 secretion and airway smooth muscle cell migration. Respir Res. 2016;17:25. doi:10.1186/s12931-016-0344-5
73. Parameswaran K, Radford K, Fanat A, et al. Modulation of human airway smooth muscle migration by lipid mediators and Th-2 cytokines. Am J Respir Cell Mol Biol. 2007;37(2):240–247. doi:10.1165/rcmb.2006-0172OC
74. Brightling C, Berry M, Amrani Y. Targeting TNF-alpha: a novel therapeutic approach for asthma. J Allergy Clin Immunol. 2008;121(1):5–10. doi:10.1016/j.jaci.2007.10.028
75. Yang M, Zhao X, Liu Y, Tian Y, Ran X, Jiang Y. A role for WNT1-inducible signaling protein-1 in airway remodeling in a rat asthma model. Int Immunopharmacol. 2013;17(2):350–357. doi:10.1016/j.intimp.2013.06.011
76. Seow CY. Passive stiffness of airway smooth muscle: the next target for improving airway distensibility and treatment for asthma? Pulm Pharmacol Ther. 2013;26(1):37–41. doi:10.1016/j.pupt.2012.06.012
77. Raqeeb A, Jiao Y, Syyong HT, Pare PD, Seow CY. Regulatable stiffness in relaxed airway smooth muscle: a target for asthma treatment? J Appl Physiol. 2012;112(3):337–346. doi:10.1152/japplphysiol.01036.2011
78. Meurs H, Dekkers BG, Maarsingh H, Halayko AJ, Zaagsma J, Gosens R. Muscarinic receptors on airway mesenchymal cells: novel findings for an ancient target. Pulm Pharmacol Ther. 2013;26(1):145–155. doi:10.1016/j.pupt.2012.07.003
79. Athari SS. Targeting cell signaling in allergic asthma. Signal Transduct Target Ther. 2019;4(1):45. doi.org/10.1038/s41392-019-0079-0
80. Barnes PJ. Theophylline. Am J Respir Crit Care Med. 2013;188(8):901–906. doi:10.1164/rccm.201302-0388PP
81. Page CP. Phosphodiesterase inhibitors for the treatment of asthma and chronic obstructive pulmonary disease. Int Arch Allergy Immunol. 2014;165(3):152–164. doi:10.1159/000368800
82. Johnstone TB, Smith KH, Koziol-White CJ, et al. PDE8 is expressed in human airway smooth muscle and selectively regulates cAMP signaling by beta2-adrenergic receptors and adenylyl cyclase 6. Am J Respir Cell Mol Biol. 2018;58(4):530–541. doi:10.1165/rcmb.2017-0294OC
83. Bardin P, Kanniess F, Gauvreau G, Bredenbroker D, Rabe KF. Roflumilast for asthma: efficacy findings in mechanism of action studies. Pulm Pharmacol Ther. 2015;35(Suppl):S4–10. doi:10.1016/j.pupt.2015.08.006
84. Bjermer L, Abbott-Banner K, Newman K. Efficacy and safety of a first-in-class inhaled PDE3/4 inhibitor (ensifentrine) vs salbutamol in asthma. Pulm Pharmacol Ther. 2019;58:101814. doi:10.1016/j.pupt.2019.101814
85. Phillips JE. Inhaled Phosphodiesterase 4 (PDE4) inhibitors for inflammatory respiratory diseases. Front Pharmacol. 2020;11(259):259. doi:10.3389/fphar.2020.00259
86. Wojcik-Pszczola K, Chlon-Rzepa G, Jankowska A, et al. A novel, Pan-PDE inhibitor exerts anti-fibrotic effects in human lung fibroblasts via inhibition of TGF-beta signaling and activation of cAMP/PKA signaling. Int J Mol Sci. 2020;21(11):4008. doi:10.3390/ijms21114008
87. Saunders R, Kaul H, Berair R, et al. Fevipiprant reduces airway smooth muscle mass in asthmatics via PGD2 receptor antagonism. Eur Respir J. 2017;50(suppl61):OA283.
88. Corren J, Parnes JR, Wang L, et al. Tezepelumab in adults with uncontrolled asthma. N Engl J Med. 2017;377(10):936–946. doi:10.1056/NEJMoa1704064
89. Corren J, Garcia Gil E, Griffiths JM, et al. Tezepelumab improves patient-reported outcomes in patients with severe, uncontrolled asthma in PATHWAY. Ann Allergy Asthma Immunol. 2021;126(2):187–193. doi:10.1016/j.anai.2020.10.008
90. Liang L, Li F, Bao A, Zhang M, Chung KF, Zhou X. Activation of p38 mitogen-activated protein kinase in ovalbumin and ozone-induced mouse model of asthma. Respirology. 2013;18(S3):20–29. doi:10.1111/resp.12189
91. Yap HM, Lee YZ, Harith HH, et al. The geranyl acetophenone tHGA attenuates human bronchial smooth muscle proliferation via inhibition of AKT phosphorylation. Sci Rep. 2018;8(1):16640. doi:10.1038/s41598-018-34847-0
92. Gendron DR, Lecours PB, Lemay AM, et al. A phosphorylatable sphingosine analog induces airway smooth muscle cytostasis and reverses airway hyperresponsiveness in experimental asthma. Front Pharmacol. 2017;8(78):78. doi:10.3389/fphar.2017.00078
93. Gu W, Cui R, Ding T, et al. Simvastatin alleviates airway inflammation and remodelling through up-regulation of autophagy in mouse models of asthma. Respirology. 2017;22(3):533–541. doi:10.1111/resp.12926
94. McAlinden KD, Deshpande DA, Ghavami S, et al. Autophagy activation in asthma airways remodeling. Am J Respir Cell Mol Biol. 2019;60(5):541–553. doi:10.1165/rcmb.2018-0169OC
95. Pan S, Sharma P, Shah SD, Deshpande DA. Bitter taste receptor agonists alter mitochondrial function and induce autophagy in airway smooth muscle cells. Am J Physiol Lung Cell Mol Physiol. 2017;313(1):L154–L165. doi:10.1152/ajplung.00106.2017
96. Twiss MA, Harman E, Chesrown S, Hendeles L. Efficacy of calcium channel blockers as maintenance therapy for asthma. Br J Clin Pharmacol. 2002;53:243–249. doi:10.1046/j.0306-5251.2001.01560.x
97. Girodet PO, Dournes G, Thumerel M, et al. Calcium channel blocker reduces airway remodeling in severe asthma. A proof-of-concept study. Am J Respir Crit Care Med. 2015;191(8):876–883. doi:10.1164/rccm.201410-1874OC
98. Sumino K, Sheshadri A, Castro M. Calcium channel blocker reduces airway remodeling - or does it? Am J Respir Crit Care Med. 2015;191:863–864.
99. Li P, Lang X, Xia S. Elevated expression of microRNA-378 in children with asthma aggravates airway remodeling by promoting the proliferation and apoptosis resistance of airway smooth muscle cells. Exp Ther Med. 2019;17(3):1529–1536. doi:10.3892/etm.2018.7141
100. Wang J, Li HY, Wang HS, Su ZB. MicroRNA-485 modulates the TGF-beta/Smads signaling pathway in chronic asthmatic mice by targeting Smurf2. Cell Physiol Biochem. 2018;51(2):692–710. doi:10.1159/000495327
101. He S, Chen M, Lin X, Lv Z, Liang R, Huang L. Triptolide inhibits PDGF-induced proliferation of ASMCs through G0/G1 cell cycle arrest and suppression of the AKT/NF-kappaB/cyclinD1 signaling pathway. Eur J Pharmacol. 2020;867:172811. doi:10.1016/j.ejphar.2019.172811
102. Mehta D, Gunst SJ. Actin polymerization stimulated by contractile activation regulates force development in canine tracheal smooth muscle. J Physiol. 1999;519(Pt 3):829–840. doi:10.1111/j.1469-7793.1999.0829n.x
103. Mahn K, Hirst SJ, Ying S, et al. Diminished sarco/endoplasmic reticulum Ca2+ ATPase (SERCA) expression contributes to airway remodelling in bronchial asthma. Proc Natl Acad Sci U S A. 2009;106(26):10775–10780. doi:10.1073/pnas.0902295106
104. Jiang J-X, Shen H-J, Guan Y, et al. ZDHXB-101 (3′,5-Diallyl-2, 4′-dihydroxy-[1,1′-biphen-yl]-3,5′-dicarbaldehyde) protects against airway remodeling and hyperresponsiveness via inhibiting both the activation of the mitogen-activated protein kinase and the signal transducer and activator of transcription-3 signaling pathways. Respir Res. 2020;21(1):22.
105. Ohta S, Oda N, Yokoe T, et al. Effect of tiotropium bromide on airway inflammation and remodelling in a mouse model of asthma. Clin Exp Allergy. 2010;40(8):1266–1275. doi:10.1111/j.1365-2222.2010.03478.x
106. Bos IS, Gosens R, Zuidhof AB, et al. Inhibition of allergen-induced airway remodelling by tiotropium and budesonide: a comparison. Eur Respir J. 2007;30(4):653–661. doi:10.1183/09031936.00004907
107. Donovan GM, Elliot JG, Green FHY, James AL, Noble PB. Unraveling a clinical paradox: why does bronchial thermoplasty work in asthma? Am J Respir Cell Mol Biol. 2018;59(3):355–362. doi:10.1165/rcmb.2018-0011OC
108. Danek CJ, Lombard CM, Dungworth DL, et al. Reduction in airway hyperresponsiveness to methacholine by the application of RF energy in dogs. J Appl Physiol. 2004;97(5):1946–1953. doi:10.1152/japplphysiol.01282.2003
109. Miller JD, Cox G, Vincic L, Lombard CM, Loomas BE, Danek CJ. A prospective feasibility study of bronchial thermoplasty in the human airway. Chest. 2005;127(6):1999–2006. doi:10.1378/chest.127.6.1999
110. Pretolani M, Dombret MC, Thabut G, et al. Reduction of airway smooth muscle mass by bronchial thermoplasty in patients with severe asthma. Am J Respir Crit Care Med. 2014;190(12):1452–1454. doi:10.1164/rccm.201407-1374LE
111. Bonta PI, d’Hooghe J, Sterk PJ, Bel EH, Annema JT. Reduction of airway smooth muscle mass after bronchial thermoplasty: are we there yet? Am J Respir Crit Care Med. 2015;191(10):1207–1208. doi:10.1164/rccm.201502-0334LE
112. Chakir J, Haj-Salem I, Gras D, et al. Effects of bronchial thermoplasty on airway smooth muscle and collagen deposition in asthma. Ann Am Thorac Soc. 2015;12(11):1612–1618. doi:10.1513/AnnalsATS.201504-208OC
113. Denner DR, Doeing DC, Hogarth DK, Dugan K, Naureckas ET, White SR. Airway inflammation after bronchial thermoplasty for severe asthma. Ann Am Thorac Soc. 2015;12(9):1302–1309. doi:10.1513/AnnalsATS.201502-082OC
114. Goorsenberg AWM, d’Hooghe JNS, Srikanthan K, et al. Bronchial thermoplasty induced airway smooth muscle reduction and clinical response in severe asthma. The TASMA randomized trial. Am J Respir Crit Care Med. 2021;203(2):175–184. doi:10.1164/rccm.201911-2298OC
115. Krishnan JA, Husain AN. One step forward, two steps back: bronchial thermoplasty for asthma. Am J Respir Crit Care Med. 2021;203(2):153–154. doi:10.1164/rccm.202008-3173ED
116. Langton D, Bennetts K, Noble P, Plummer V, Thien F. Bronchial thermoplasty reduces airway resistance. Respir Res. 2020;21(1):76. doi:10.1186/s12931-020-1330-5
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.