Back to Journals » International Journal of Nanomedicine » Volume 13
Targeted nonviral gene therapy in prostate cancer
Authors Altwaijry N, Somani S, Dufès C ![]()
Received 16 April 2018
Accepted for publication 8 June 2018
Published 25 September 2018 Volume 2018:13 Pages 5753—5767
DOI https://doi.org/10.2147/IJN.S139080
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Thomas Webster
Najla Altwaijry, Sukrut Somani, Christine Dufès
Strathclyde Institute of Pharmacy and Biomedical Sciences, University of Strathclyde, Glasgow, UK
Abstract: Prostate cancer is the second-most widespread cancer in men worldwide. Treatment choices are limited to prostatectomy, hormonal therapy, and radiotherapy, which commonly have deleterious side effects and vary in their efficacy, depending on the stage of the disease. Among novel experimental strategies, gene therapy holds great promise for the treatment of prostate cancer. However, its use is currently limited by the lack of delivery systems able to selectively deliver the therapeutic genes to the tumors after intravenous administration without major drawbacks. To remediate this problem, a wide range of nonviral delivery approaches have been developed to specifically deliver DNA-based therapeutic agents to their site of action. This review provides an overview of the various nonviral delivery strategies and gene therapy concepts used to deliver therapeutic DNA to prostate cancer cells, and focuses on recent therapeutic advances made so far.
Keywords: prostate cancer therapy, gene delivery, tumor targeting, nanomedicine
Introduction
Prostate cancer is one of the leading causes of death in men worldwide. It is the fourth-most widespread cancer in the world and the second-most common cancer in men.1 According to the World Health Organization, prostate cancer is estimated to cause 1.1 million new diagnosed cases and 307,000 deaths annually worldwide.1 A continuous rise in the prostate cancer incidence rate has been recorded, with a threefold increase in Europe between 1975 and 2011.2 Around 190,000 new cases arise each year, with 80,000 deaths occurring annually from prostate cancer.2
Prostate cancer is usually diagnosed at the very late stages of the disease, which is one of the factors contributing to its high mortality rate, in addition to the frequent failure of localized prostate cancer therapy.3 Unfortunately, prostate cancer patients rarely seek diagnosis during the localized stages of the disease, because most notice no symptoms other than the ones that resemble those of urinary tract infection.4
In addition, prostate cancer treatment choices are limited, variable in efficiency, and can lead to side effects. The selection of the appropriate treatment plan (prostatectomy, hormone therapy, aspecific radiotherapy, and chemotherapy) depends mainly on the severity of the case, which determines the purpose of the treatment: either a total cure or prolongation of the patient’s life. Patients diagnosed in the early stages of the disease have a higher chance of total recovery, while the main goal is to prolong the patient’s life in the case of metastasis and relapse.5 However, most of the available treatment options are associated with major side effects and have limited ability to cure patients in the late stages of prostate cancer. Therefore, in light of the high and rising incidence of the disease, the search for new therapeutic approaches, including those applicable to metastasis cases, is crucial.
Gene therapy is a new approach used to treat genetic and hereditary disorders, such as cancer, Parkinson’s disease, and cystic fibrosis. The US Food and Drug Administration (FDA) defines gene therapy as any product that uses a transferred foreign genetic material to produce its effects by expressing and/or integrating the gene with the host-cell genome for the purpose of cell modification, both in vivo and ex vivo. The transferred gene is introduced to the host cells using a suitable vector, such as a viral or nonviral delivery system, in order to treat genetic mutation and regulate cellular processes.6,7
In 2003, the Chinese FDA approved the first gene therapy product to treat head-and-neck squamous-cell carcinoma. Gendicine is a nonreplicative adenovirus that carries the P53 gene. This product created much controversy, due to the lack of a Phase III clinical trial.8 Two years later, the Chinese FDA approved another gene therapy drug, Oncorine (Shanghai Sunway Biotech, Shanghai, China), which is used with chemotherapy to treat refractory nasopharyngeal cancer using a controlled replicative adenovirus. In 2012, Glybera (UniQure, Amsterdam, the Netherlands) was the first gene therapy to receive a recommendation for approval by the European Medicines Agency. It is an adenoassociated virus that carries the lipoprotein-lipase gene to the muscles for the treatment of lipoprotein-lipase deficiency.
Although gene therapy is a promising approach for treating cancer, it is nevertheless associated with certain limitations and challenges that undermine its possible efficacy, thereby slowing the speed of its progress as a prominent therapy for prostate cancer. A major obstacle to DNA-based therapy is the lack of stability of nucleic acids. Naked nucleic acids are subjected to nuclease-mediated degradation before they reach their target site. Complexing DNA with a nonviral delivery system has the potential to increase the stability of the DNA and to facilitate the delivery of the DNA to the diseased tissues. Another major challenge in the field of prostate cancer therapy is the need for systemic delivery and targeting of metastatic prostate cancer. Currently, most in vivo gene therapy strategies use intraprostatic injection of delivery systems, as this is the most efficacious route so far to treat primary prostate cancer cells. Although local administration allows a reduction in administered dose, it does not target prostate cancer metastasis in distant organs. Systemic delivery of a targeted gene delivery system could allow the treatment to reach metastases, but an intravenously administered nanomedicine would face numerous extracellular barriers (such as interaction with plasma proteins and opsonization) before reaching its target. Many gene therapy studies are under way worldwide, especially to treat cancer, which accounts for 60% of ongoing trials.6
Delivery barriers
Several factors affect the delivery of gene delivery systems to cancer cells following intravenous administration.9 These factors can be classified as extracellular and intracellular barriers (Figure 1).
 | Figure 1 Extracellular and intracellular barriers limiting the delivery of therapeutic genes to nuclei of prostate cancer cells. |
Extracellular barriers
Extracellular barriers correspond to any factors limiting the delivery of a nanomedicine, from its administration until it reaches the cancer cells. Due to their net positive charge, gene delivery systems can undergo aspecific binding to negatively charged molecules, such as serum proteins, immunoglobulin M, and complement C3, by electrostatic interactions, which can lead to macrophage uptake, dissociation of the DNA from its carrier, and then degradation of DNA by serum endonucleases.9,10 This issue can be overcome by conjugating the delivery systems with an anionic hydrophilic molecule, such as polyethylene glycol (PEG), to reduce their total positive charge and thus decrease aspecific interactions and improve transfection ability.10
Another issue that can arise during gene delivery is intake of nonviral vectors by the immune cells, which identify them as foreign entities.9 Nonviral vectors are usually less immunogenic than viral vectors. Nevertheless, a nonviral delivery system complexed with DNA can still face an immunoreaction, due to the unmethylated cytosine guanine (CpG) sites in the condensed gene. This immunoresponse can be diminished by eliminating unnecessary CpG sites from the complexed gene or reducing the administration dose.10
Intracellular barriers
Intracellular barriers can be categorized as any factors at the cellular level that lead to a reduction in the efficiency of the gene therapy. The first cellular obstacle that can face gene delivery systems when they reach the targeted cells is their uptake efficiency. Gene delivery systems are generally taken up by cells via receptor-mediated endocytosis, a cellular biological process whereby a specific molecule can enter the cell with the assistance of a specific receptor in the cell membrane. The endocytosis process, or endocytic pathway, can be considered a barrier to gene delivery, since its efficiency can be affected by several parameters, such as the size of the delivery system. It has been observed that delivery systems <50 nm show significant uptake by cells through endocytosis compared with delivery systems >50 nm, with optimum size being 25 nm.11
The second cellular barrier takes place during the cellular uptake of the delivery system, when the delivery system–receptor complex is trapped inside an endosomal sack. In the endosome, the delivery system and its carried genetic material may undergo degradation by lysosomal nucleases in a low-pH environment.9,12 In overcoming this limitation, gene delivery systems tend to escape from the endosome through the proton-sponge effect, which is initiated by the acidic pH of the endosome. The low pH increases the ionization of the delivery system (cationic polymer or dendrimer), then the ionized polymer attracts protons to the endosome, leading to an increase in osmotic pressure, endosomal swelling, and disruption, which releases the therapeutic DNA in the cytoplasm.10,12
Following endosomal escape, up to 99% of the DNA may be degraded by cytoplasmic nucleases if it stays for a long time in the cytoplasm. The last obstacle to gene delivery is effective nuclear uptake of the DNA. Nuclear transport is controlled by the nuclear pore complex, which is permeable to molecules of size <70 kDa (10 nm in diameter).10 DNA can be taken up by the nucleus by either passive or active transport. Passive transport occurs during cell division, during which the nuclear envelope lyses, allowing the genetic material to be transported to the nucleus. In active transport, nuclear localization signals bind to the nuclear pore complex to allow larger DNA molecules to pass through these pores.12
Gene delivery systems
For gene therapy to be successful, therapeutic DNA must successfully cross the cell membrane and then integrate with the nucleus genome of the targeted cell. However, naked DNA cannot achieve this without undergoing degradation during its passage through the blood circulation and biological tissue.13 Moreover, a charge repulsion can be created between the naked DNA and the cell membrane, due to their common negative charges, which can hinder the cellular uptake of DNA.7
These drawbacks can be overcome by using a suitable gene delivery system. Gene delivery can be simply defined as a process to introduce a foreign gene to a host cell using a gene carrier. It involves the complexation of the gene of interest to a suitable gene delivery vector, in order to facilitate its transfection to the targeted cells.13 The main challenge in gene delivery is to find an effective delivery system able to target the required tissue and deliver the transferred gene to the cells without secondary side effects. Gene delivery confers several advantages on the complexed DNA, such as protection from degradation in blood circulation and increased targeting specificity to the required tissue. Viral and nonviral delivery systems can be used to transfer DNA to cancer cells.
Viral delivery systems
Viruses are highly efficient at transferring their own genome to the host cells. This unique ability has been used to transfer therapeutic genes to specific cells.14 Modified (replication-deficient) viruses are used as gene vectors to attenuate their ability to induce infection.15 The use of viruses as gene vectors has several advantages. First, viruses have a strong ability to pursue and attach to specific cells. They also exhibit high efficiency at introducing genetic material into cells. Furthermore, viral vectors are found to integrate genes into target cells for a longer period than nonviral vectors.16
On the other hand, the use of viral vectors is severely hampered by disadvantages, such as immunogenicity. The immune system can identify a virus as a foreign substance and initiate an immunoresponse against it, especially if the patient has previously been exposed to that virus. This immunoreaction thus reduces the ability of the viral vector to reach the target cells and to achieve successful gene delivery.16 In addition, viral vectors suffer from a limited ability to condense and transfer large DNA molecules.7 Using viruses as gene delivery systems can also potentially lead to toxicity and oncogenicity.17 Because of these limitations, this review thus focuses on the use of nonviral gene delivery systems for the treatment of prostate cancer.
Nonviral gene delivery systems
A variety of nonviral delivery systems, such as liposomes and polymers, have been tested for gene delivery to prostate cancer (Figure 2). The majority of nonviral vectors can condense DNA via electrostatic interactions,7 resulting in such advantages as protection of the DNA from degradation by the nuclease enzymes present in the circulation.7,9 In addition, DNA complexation reduces the charge repulsion that arises from the charge similarity between the DNA and cell membrane, which in turn facilitates the delivery of the DNA inside the cells.10 Furthermore, nonviral vectors possess higher ability to complex large DNA molecules when compared to viral vectors, due to their well-constructed chemical structure comprising cationic functional groups. In addition, the targeting ability of nonviral vectors can be improved by using targeting ligands able to recognize specific receptors in the target tissue.
 | Figure 2 Nonviral gene delivery systems and therapeutic strategies for prostate cancer therapy. |
Liposomes
Liposomes were first described in 1965 as a chemical formulation comprising a phospholipid bilayer with a hydrophilic head and hydrophobic chain, forming an overall spherical shape.18 Due to their unique chemical nature, they can be used as carriers for drugs and genes: hydrophilic compounds are encapsulated in the inner part of the liposome, while lipophilic substances can be trapped in the lipid bilayer. This gives liposomes the ability to transfer different substances through the blood circulation.13
In 1987, Felgner et al first designed a cationic liposomal system using the double-strain monovalent quaternary ammonium N-[1-(2,3-dioleyloxy)propyl]-N,N,N-trimethylammonium chloride (DOTMA) to carry genetic material. This forms a complex called a lipoplex, which is initiated by the electrostatic interactions between negative phosphate groups in the genetic material and positive amino groups in the liposome, thereby leading to the condensation of DNA.19 Several factors contribute to lipoplex characterization, such as shape, size, and surface charge. These properties can be controlled by physical methods to improve transfection. The surface-charge ratio is the most important parameter: cationic liposomes should have a higher net charge than DNA, so that the DNA complexation with the vector forms a total surface-charge ratio around one. This criterion creates full protection from degradation for the DNA and assists its penetration into cells, due to charge attraction between the lipoplex and cell membrane.7,16
Liposomes are associated with some limitations to their DNA-transfer capability, which can be overcome by various modification methods. First, liposomes tend to have a short circulation half-life in the bloodstream, due to their rapid clearance from the body. The rate of clearance is affected by the size, charge, and membrane composition of the liposome. Large liposomes tend to have a faster clearance rate, due to the high serum protein opsonization in the liposomal membrane during its circulation in bloodstream, and hence the opsonins in the liposome surface trigger its uptake via phagocytosis.20 In order to improve circulation half-life, liposomes can be chemically modified by conjugation with a ligand. For example, the conjugation of PEG with liposomes leads to the formation of long-circulating, sterically stabilized liposomes, which have a longer half-life and thus better targeting ability than regular liposomes. Circulation time is prolonged by the inability of macrophages to remove these molecules, because they are less able to detect the modified liposomes. PEG in the liposome surface impedes the coupling of opsonins with the liposome surface, thus reducing the ability of phagocytes to recognize the liposomes.18 With longer circulation time, the ability of liposomes to deliver the transferred DNA to the targeted tissue tends to increase severalfold, which improves the targeting of poorly accessible tissue, such as solid tumors.18
Secondly, the targeting proficiency of liposomes is not specific enough when compared with viral vectors, which tend to have high targeting ability, but it can be improved by conjugating an antibody to the liposome in order to target specific tissue. Antibody-coupled liposomes, also called immunoliposomes, can be removed easily from the circulation if not combined with PEG to form stabilized immunoliposomes.21 However, some disadvantages arise from the use of antibodies with liposomes, such as a possible immunoreaction, especially when using a high antibody concentration.
Finally, gene transfection efficiency varies among different types of liposomes, monovalent liposomes being the least efficacious. Neutral colipids such as dioleoylphosphatidylethanolamine (DOPE) and cholesterol can be linked to the cationic liposome to improve the liposomal transfection process through the cell membrane. Colipids promote the fusion of the endosomal membrane, facilitating endosomal escape and the release of DNA into the cytoplasm.22 They also facilitate the release of DNA from the lipoplex. Furthermore, colipid binding is critically important in improving the gene transfection of monovalent cationic liposomes, while its main effect with multivalent liposomes is to reduce the toxicity that might arise from their high charge ratio.7
Cationic polymers
Cationic polymers are macromolecules that possess positive charges due to their high density in amine groups. Among the different types of cationic polymers developed in recent years are neutral polymers (eg, chitosan), polypeptides (eg, poly-L-lysine [PLL]), and dendrimers (eg, polyamidoamine [PAMAM]). Some chemical modifications can be applied to these polymers to improve their efficiency as vectors and reduce their toxicity via the use of binding ligands. Polymers can be used as gene carriers, as their positively charged groups can form electrostatically attractive forces with the negatively charged phosphates in the DNA. The complex formed, called a polyplex, protects the complexed DNA from hydrolysis in the circulation or tissue, giving it a longer half-life and improved targeting.16
Several criteria can affect the efficiency of the polyplex. For example, a higher molecular-weight polymer provides superior condensation of the genetic material. Another important factor that can contribute to polyplex efficiency is the charge ratio, which is a parameter used to calculate the nucleotide equivalence using the ratio of nitrogen (in the polymer) to phosphate (in the DNA). The total charge should be positive in order to form a stable and effective polyplex. Although the charge ratio is important, it should be limited to a certain extent. A high total positive charge on the polyplex can have various side effects, such as increased systemic cytotoxicity of the polyplex and random interaction between the polyplex and biological membranes, thus attenuating targeting ability and causing a loss of specificity.23 In order to overcome these problems, a polypeptide, such as PEG, can be conjugated to the polyplex to reduce the overall charge ratio without affecting its gene complexation capability.23 Alternatively, the high-charge problem can be overcome by the use of newly designed cationic polymers with hydroxyl or amide groups, such as poly(vinyl alcohol) dimethylaminoacetal (PVA3), that have been shown to have effective gene transfer, less toxicity, and increased DNA release in the cytoplasm. This effect can be explained by a decrease in the total positive charges of the complex or the formation of hydrogen bonds between the DNA and hydroxyl groups.13
Examples of cationic polymers used for treatment of prostate cancer
PLL is a polypeptide cationic polymer that is one of the early polymeric vectors used in gene delivery. PLL contains a high density of primary amines that have a strong affinity with negative ions at neutral pH, since most of them are ionized at pH 7.4.24 The use of PLL as a gene carrier is associated with certain limitations. For example, its gene transfection ability is weak at biological pH, due to the strong interaction between DNA and PLL, which prevents the release of DNA to the cytoplasm. Additionally, the chemistry of PLL is associated with a certain immunogenicity and toxicity, due to its amino groups. The modification of PLL with ligands can reduce its side effects and improve its ability as a gene carrier, especially in in vivo trials, since it is biodegradable.24
Another cationic polymer that is commonly used as a vector in gene delivery is polyethylenimine (PEI). Its unique chemical structure, comprising a nitrogen atom to every three atoms, promotes its DNA-complexation ability to form a stable polyplex, as well as a strong ability to condense large molecules.25 At pH 7.4, only 17% of the nitrogen groups in PEI are ionized. This gives PEI several advantages. It has superior DNA condensation ability with high gene-transfection capability, since it can easily release DNA into the cytoplasm. Moreover, the low protonated PEI facilitates endosomal escape of the polyplex from the lysosome by means of the proton-sponge effect.26
Another polymer, poly(β-amino ester), has also been used in prostate cancer therapy. It is a biodegradable polymer that is able to self-assemble with DNA to form positively charged gene delivery nanoparticles. The main advantage of this polymer is its ability to hydrolyze in acidic and alkaline media, which facilitates the release of DNA inside cells. Furthermore, both the polymer and its degradation products have been shown to be nontoxic.27
Dendrimers
Dendrimers are three-dimensional branched macromolecules with a treelike structure. Their name originates from the Greek word ‘dendron’, which means “tree”. Since their discovery, dendrimers have been widely used in various applications in pharmacy, medicine, and engineering.28–31 Their chemical structure is composed of two parts: a reactant core molecule that is considered the origin of the dendrimer, and highly branched polymers that bind to the core in a specific way to form a uniformly branched spherical macromolecule.31
The use of dendrimers in gene delivery is restricted to cationic dendrimers, such as PAMAM and diaminobutyric polypropylenimine (DAB) dendrimers. Cationic dendrimers complex the genetic material by electrostatic interactions between the phosphate groups on the DNA and the amino groups on the dendrimer. Among their advantages over regular polymers, dendrimers have generally enhanced gene-transfection ability, which increases with higher generations.13 Their treelike shape also increases the surface area of the molecule and creates inner cavities, thus improving their DNA-complexation and drug-encapsulation capability. Additionally, dendrimers have been found to be nonimmunogenic molecules, which makes them suitable gene carriers for in vivo studies.31
Cancer targeting
Passive targeting
Passive targeting is a naturally occurring process that arises from the unique pathophysiology of the tumor site. Due to the nature of the blood vessels formed and disruption of lymph-node function, macromolecules and nanoparticles tend to accumulate in tumor tissue. This is called the enhanced permeability-and-retention effect or passive targeting.32 Its efficiency is affected by several factors, such as the vascular permeability of the tumor blood vessels and the size of macromolecules, which control their diffusion to cancer tissue. Most blood vessels in cancer tissue have a diffusion permeability of 200–2,000 nm, depending on the location and the type of the tumor.32
Active targeting
Active targeting is a method allowing the specific delivery of nanomedicine to an organ, tissue, or cell by chemically conjugating the delivery system with a specific ligand that has the ability to bind to a specific binding site in the targeted tissue. The ligand could be a protein, antibody, or sugar, while the targeted binding site could be a receptor, sugar, protein, or lipid that is highly expressed on the surface of the targeted cancer cells.32
Prostate cancer-targeting ligands
Iron-binding proteins
Transferrin (Tf) and lactoferrin (Lf) are iron-binding proteins that have been found to be overexpressed on various cancer cells, including prostate cancer cells. The main function of these proteins is to control the free iron level in the blood and body fluids. High iron levels increase the risk of bacterial invasions, as well as free-radical generation, which is associated with the conversion of ferrous ion (Fe2+) to ferric ion (Fe3+) in the body.33 Several studies have used Tf and Lf as targeting ligands in prostate cancer therapy.34–36
Integrins
Integrins are transmembrane glycoprotein receptors that link the cytoskeletons of cells with the extracellular matrix (ECM). They act as adhesion, migration, and signaling molecules, connecting cells to the ECM by binding with such proteins as fibronectin, vitronectin, and collagen.37,38 These adhesive molecules assist the connection between adjacent cells and surrounding ECM by creating tunnel-like junctions maintained by integrins, which facilitate the passage of ions and small molecules between cells and form and break as needed.39 Integrins also act as signaling molecules between cells in a given tissue, and between cells and the ECM.
Several integrin receptors have been found to be overexpressed in prostate cancer cells, with differentiation in their expression as the cancer progresses, whereas other receptors have been detected only in cancer tissue. The integrin heterodimer receptors ανβ6, ανβ3, αIIbβ3, α6β1, α5β1, and α6β4 have been shown to be overexpressed in prostate cancer.40,41 Among these, ανβ3 has been found to be expressed in prostate cancer cells at an intensity that varies with the metastatic and invasion tendency of the cells. PC3 cells extensively express ανβ3, while LNCaP cells do not, explaining the higher invasion capability of PC3 over LNCaP cells, as ανβ3 tends to adhere to vitronectin in the ECM.42 In addition, α5β1 has been shown to be expressed in PC3 cells, as it has a role in their adhesion and invasion behavior.43
Prostate-specific membrane antigen
Prostate-specific membrane antigen (PSMA), or glutamate carboxypeptidase II, is an integral transmembrane glycoprotein with a molecular size of 100 kDa. It was first identified on the prostate epithelial membrane in 1987, when a newly developed antibody (7E11) was able to recognize the N-terminal portion of the protein in prostate tissue, hence its common name.44,45 PSMA is expressed primarily in all forms of prostate tissue and has low expression in the nervous system, liver, kidney, and small intestine. More importantly, it has been found to be expressed in solid tumors, such as prostate, lung, breast, and colon cancer.46,47 Although PSMA has been detected in several tumor types, it is uniquely overexpressed in prostate cancer, which makes it a suitable biomarker to be investigated in therapeutic and diagnostic applications. It is strongly expressed in both primary and metastatic cancer cells, and its expression increases with the malignancy of the disease.48
PSMA expression in prostate cancer cells has been found to be correlated with androgen sensitivity. Cells of the androgen-sensitive LNCaP epithelial cancer cell line express PSMA in their membranes. Conversely, PC3 and DU145 metastatic cancer cells in bone and brain, respectively, were found not to express PSMA, although its DNA and mRNA have been detected in these cells.49 Although there is still no known natural ligand that can bind to PSMA, several researchers have succeeded in preparing synthetic binding antibodies and peptides with good ability to bind to the extracellular domain of PSMA.45,50
Prostate stem-cell antigen
Prostate stem cell antigen (PSCA) is a prostate-specific cell-surface antigen that is overexpressed in both androgen-dependent and -independent prostate cancer cells, making it a suitable marker for prostate cancer. Taylor et al51 examined the mRNA expression of different prostate cancer-targeting motifs, including PSCA, which was found to be overexpressed. Garcia-Hernandez et al52 vaccinated mice with PSCA-based vaccine to induce long-term protection against prostate cancer. The vaccination increased the expression of some cytokines (IFNγ, TNFα, and IL2), which was an indication of the successful generation of an immunoresponse against prostate cancer in the vaccinated mice.
Others
There are several other possible ligands that can target prostate cancer. HER2 is a potential target, due to its overexpression in various tumors, including prostate cancer.53 Mucin 1 is a membrane glycoprotein that has positive expression in prostate cancer tumors compared with healthy tissue, making it a suitable target for prostate cancer treatment.54
Therapeutic strategies
Cancer is considered a genetic disorder, as various genetic mutations affect several genes in cancerous cells. Those mainly affected are tumor-suppressor genes (eg, TP53 and NM23) and oncogenes (eg, RAS, c-MYC, BCL2, and c-MET).55,56 Tumor-suppressor genes are responsible for the regulation of normal cell death and cellular waste-product removal. Their inactivation by either disappearance or mutation can promote cell malignancy. Oncogenes are responsible for consistent cell growth. Their activation can boost cancer-cell growth.16
In prostate cancer, various genes are involved in genetic mutations. For example, the tumor-suppressor genes TP53 and retinoblastoma were found to be mutated in around 50% and 35% of advanced prostate cancer cases, respectively. The main functions of TP53 are to regulate the cell life cycle and repair any disruption in the DNA, and the result of a mutation of this gene is uncontrolled cell growth.55,57 In addition, the GSTP1 gene was found to be disrupted in some prostate cancer cases. The main cellular role of GSTP1 is carcinogen detoxification, and its inactivation leads to carcinogenesis.56,57
Various gene therapy strategies can be involved in prostate cancer treatment, such as gene apoptosis therapy, in which the transformed gene is replaced with one that regulates cancer cell death, corrective gene therapy, which uses a tumor-suppressor gene to restore normal cell-growth regulation, immunomodulatory gene therapy, which uses genes to stimulate the immune system, and suicide-gene therapy, which relies on introducing genes that cause changes to a chemical compound or enzyme to form toxic substances. An overview of the ongoing studies based on nonviral gene delivery systems and therapeutic approaches against prostate cancer is presented in Table 1.
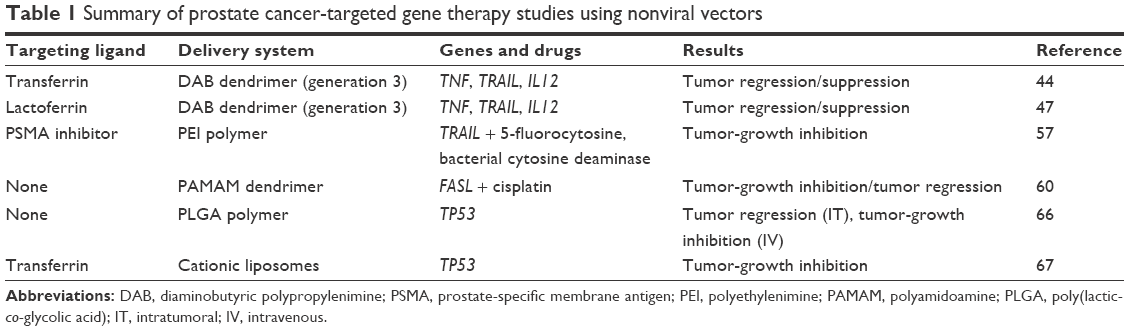 | Table 1 Summary of prostate cancer-targeted gene therapy studies using nonviral vectors |
Apoptosis-inducing gene therapy
Apoptosis is a normal physiological process that is programmed under certain circumstances to control cell death.58 In normal cells, it is usually generated to eliminate cells that have sustained genetic mutation or damage. The suppression of cellular apoptosis is one of the common problems generated by genetic mutation in cancer cells, leading to cancer development. Therefore, using gene therapy to induce cellular apoptosis in cancer cells would be an effective treatment for cancer.
TNF superfamily cytokines (TNFα, TRAIL, and FasL) are the most common cytokines to cause cellular apoptosis by binding to their specific death receptors.59 Caspase activation, which occurs externally (by binding to the death receptor) or internally (via the mitochondria and death domain), is required for stimulating cell apoptosis.58,60
Extrinsic apoptosis activation occurs through activation of the transmembrane death receptors (TNF receptors). The activation initiated by the specific binding of death ligands, such as the TNF-protein family or FasL to their death receptors TNFR1, TNFR2 and Fas receptor respectively, which creates a particular binding site specific to the adaptor protein. This causes the formation of a death-inducing signaling complex (DISC), triggering the activation of caspase 8, which in turn causes cell apoptosis through activating effector caspases such as caspase 3 and 6, which have the ability to recognize proteins’ amino-acid sequences involved in the main cellular functions and cleave them at specific motifs to form substrate residues.58,60
Alternatively, the intrinsic activation pathway can arise in the cell itself, due to genetic damage, oncogene activation, or any stress condition. It initiates from mitochondria or endoplasmic reticulum. In the mitochondrion, mitochondrial membrane permeability is controlled by a protein family called Bcl2. After apoptotic stimulation, this causes the release of cytochrome C, followed by the apoptosome, leading to the release of Apaf1 and then activating the initiator caspase 9 followed by the effector caspase 3, which initiates cell apoptosis.60 Some cytokines are well known to cause apoptosis of inflamed and mutated cells.60 Using this mechanism to suppress cancer has been found to lead to promising outcomes.58,61,62 Some common cytokines used for this purpose are TNFα, TRAIL, and FasL.
Tumor-necrosis factor-α
TNFα is an inflammatory cytokine that produces many biological activities in the immune system, at inflammation sites, and in cell death and survival.63 To generate cell apoptosis, TNFα couples with its binding receptor (TNFR1), which is expressed in most tissue and leads to activation of the death domain in the cytoplasm (TRADD) and Fas-associated death domain (FADD). This leads to the formation of DISC, which activates procaspase 8, thereby causing DNA fragmentation and cell apoptosis.64
High TNFα levels cause cell apoptosis and necrosis, a property that can be used to destroy cancer cells.64 Mauceri et al used the TNFα gene to treat epidermoid carcinoma, together with targeted radiation therapy. Their results showed highly selective tissue necrosis, with no damage to normal tissue. DNA encoding TNFα has been used before in various studies to treat different cancerous tumors, either alone or in combination with such therapies as radio- or chemotherapy, using various types of gene carrier, mostly viral ones. Delivering the TNFα gene using viral vectors to target different cancer models by intratumoral65 or intravenous injection66 results in significant inhibition in tumor growth, but without tumor suppression.
TNFα cytokine therapy was also tested against prostate cancer when Chopra et al64 investigated the effects of TNFα on normal and cancerous (LNCaP) prostate cells in vitro. The results for low doses of TNFα were promising, with significant (90%) LNCaP cell apoptosis and no effect on normal cells. In another study, Chung et al67 treated nude mice bearing xenograft prostate tumors using a combination of radiotherapy together with TNFα carried by an adenovirus injected intravenously, resulting in regression of tumor volume compared to gene therapy or radiotherapy treatment alone. In addition, studies have discussed the impact of inhibiting NFκB to improve the sensitivity of prostate cancer cells toward TNFα apoptosis by using PMS1077 and apigenin.68,69 Using a nonviral delivery system, we demonstrated that intravenous administration of Tf-bearing DAB dendriplex encoding TNFα resulted in tumor eradication of 60% of PC3 and 50% of DU145 tumors.34 This tumor-targeted TNFα gene therapy was more efficacious than TRAIL and IL12, leading to tumor regression and even some tumor disappearance. To our knowledge, it was the first time that the intravenous administration of tumor-targeted dendriplexes encoding TNFα, TRAIL, and IL12 on mice bearing prostate tumors inhibited tumor growth and even led to complete tumor suppression in some cases. In a further study, we recently replaced the tumor-targeting ligand Tf with Lf, an iron-binding member of the Tf family that has been shown to have intrinsic antitumoral activity, making it particularly attractive as part of gene medicine. Lf binds to specific receptors (LfR1, LfR2) or to Tf receptors overexpressed on most cancer cell lines.70,71 This novel intravenously administered Lf-bearing DAB dendriplex-encoding TNFα resulted in improved tumor eradication of 70% of PC3 cells and 50% of DU145 cells compared to a Tf-bearing dendriplex.36
Tumor necrosis factor-related apoptosis-inducing ligand
TRAIL or Apo2L is a transmembrane protein found in human tissue, such as the spleen, prostate, and ovaries. TRAIL binds to its specific receptors TRAILR1 (death receptor 4) and TRAILR2 (death receptor 5), causing activation of the death domain and cell apoptosis.72,73 In addition, TRAIL has also been found to bind with TRAILR3 (TRID/DcR1) and TRAILR4 (DcR2) decoy receptors, which do not contain any cytoplasmic death domain and thus cannot induce cell apoptosis. Therefore, binding TRAIL to these receptors acts as an antagonist, because it will not activate any cell-death signaling. The existence of TRAILR3 and TRAILR4 in the cells is linked to their TRAIL-apoptosis resistance.74
The mechanism by which TRAIL induces apoptosis occurs mainly through the extrinsic pathway when TRAIL binds to TRAILR1 or TRAILR2 receptors, which as a result stimulates a TRAIL-signaling cascade by forming a DISC. The DISC includes several proteins, such as FADD and procaspases 8 and 10. Therefore, after TRAIL binding and DISC activation, caspase 8 activation is initiated and so are caspases 3, 6, and 7. Caspase activation leads to the cleavage of target proteins responsible for preserving cellular function, resulting in cell apoptosis.73,74
The use of TRAIL as an agent to kill cancer cells arises from its preferential ability to induce cell apoptosis in cancer cells without harming normal healthy cells.75,76 Ashkenazi et al72 examined the apoptotic effect of TRAIL in various normal and cancerous cells. It was found to have good apoptotic activity against most cancer cells in vitro (32 of 39 cell types) and was not cytotoxic to normal cells. This important characteristic led to further investigations of the antitumor activity of this cytokine. Moreover, TRAIL caused a significant reduction in tumor progression in vivo, with an increase in survival rate. Yu et al75 also examined the effect of TRAIL cytokine in vitro in androgen-insensitive PC3 and DU145 prostate cancer cells. A TRAIL dose of 200 ng/mL resulted in 70% cell death in both cell lines. The study also investigated the mechanism behind the TRAIL effect, which was found to be the activation of caspase 8 and 3 by cross-linking and through the death receptors (DR4 and DR5).
Griffith et al77 were the first to introduce TRAIL cDNA instead of the cytokine delivered by adenovirus (Ad5-TRAIL) to examine its possible apoptosis effect against PC3 prostate cancer cells. In vitro, treating cells with Ad5-TRAIL showed an antiproliferative effect similar to that obtained using TRAIL cytokines, whereas other nontherapeutic DNA carried by the same carrier did not show any cytotoxic effect. This study also demonstrated the successful production of TRAIL protein in PC3 cells after infecting it with Ad5-TRAIL. Later, various researchers used adenovirus and adenoassociated virus as carriers for the gene encoding TRAIL to target prostate cancers, both in vitro and in vivo.78–80 These treatments resulted in the induction of cellular apoptosis in vitro and suppression of tumor growth in vivo.
To avoid the immunogenicity problem associated with viral vectors, the use of nonviral gene carriers to carry DNA encoding TRAIL for prostate cancer therapy has been reported. Chen et al81 developed a theranostic nanoplex allowing combined TRAIL pDNA gene therapy and prodrug enzyme treatment. PEI was used as the delivery system. In addition to the TRAIL pDNA, the nanoplex carried the prodrug enzyme bacterial cytosine deaminase, which is able to convert the nontoxic prodrug 5-fluorocytosine to the cytotoxic anticancer drug 5-fluorouracil. Furthermore, a urea-based small-molecule inhibitor of PSMA, was conjugated to the nanoplex for prostate cancer targeting. The combination strategy of TRAIL gene therapy and 5-fluorocytosine/bacterial cytosine deaminase treatment demonstrated significant inhibition of the growth of PC3-PIP prostate tumors: following intravenous administration of the nanoplex, tumor volumes increased to only 300% compared to the 700% increase observed following treatment with saline solution as a control at day 10 after injection.
In another study using TRAIL gene therapy as sole therapeutic strategy, we demonstrated that treatment with intravenously injected DAB-Tf dendriplex encoding TRAIL led to tumor eradication of 10% of PC3 tumors in mice bearing subcutaneous tumors.34 The replacement of Tf by Lf as the tumor-targeting moiety further increased the therapeutic efficacy of the system, leading to tumor eradication of 40% of PC3 tumors and 20% of DU145 tumors. The enhanced therapeutic efficacy observed with DAB-Lf dendriplex treatment extended mouse survival by up to 21 days compared to untreated animals.36
Fas ligand
FasL (Apo1 or CD95) is a 40 kDa membrane protein that belongs to the TNFα family. Since its discovery in 1989, the FasL cellular apoptotic effect has become the focus of many studies. FasL binds to its specific receptors in the cell membrane Fas, thereby initiating a number of events leading to cellular apoptosis. The mechanism by which FasL induces cell death is instigated by Fas binding, which causes the clustering and trimerization of Fas that trigger the activation of DISC in the cytoplasm and in turn activate FADD and procaspase 8. Caspase 8 activation triggers the release of caspases 3 and 7, which causes the cleavage of vital cellular proteins through protease activation.82,83
A key criterion that makes FasL a desirable apoptosis inducer is the bystander effect of its transfected gene. Therefore, successful transfection of the FASL gene to some cancer cells would be therapeutically sufficient, as it would infect adjacent cells such that cells in neighboring tissues will hold the same effect.84 This feature can help overcome weak targeting obstacle in gene delivery. Hyer et al85 and Xiong et al86 examined the bystander effect of the FASL gene carried by an adenovirus in prostate cancer cells. Flow cytometry analysis of prostate cancer cells treated for 6 hours with FASL carried by an adenovirus demonstrated that there was roughly a 25% increase in apoptosis of labeled cells compared with control cells. The mechanism believed to cause this bystander effect to surrounding cells emanates from apoptotic cellular debris and apoptotic vesicles of infected cells, regardless of FasL-Fas binding status in these cells.
FASL gene delivery and its apoptosis effect have been examined against different cancer types, including prostate cancer, both in vitro and in vivo. An adenovirus was the carrier of choice for most of these studies. In prostate cancer, significant tumor growth inhibition, with tumor regression in some cases, was detected after intratumoral injections.86,87 Xiong et al86 treated prostate xenograft tumors with intratumoral injections of FASL carried by an adenovirus, and recorded significant tumor growth suppression, with 25% of tumors completely disappearing.
Studies have also reported the use of nonviral delivery systems for FAS gene therapy in prostate cancer. Nakanishi et al84 used the PAMAM dendrimer to condense plasmid DNA encoding FasL. Intratumoral injection of FASL DNA (10 μg/injection) complexed with PAMAM (ratio 1:10) led to a decrease in growth rate of PC3 tumors and regression of LNCaP tumors in mice bearing subcutaneous tumors. In order to obtain a more pronounced therapeutic outcome, a PAMAM dendriplex encoding Fas was combined with the chemotherapeutic drug cisplatin. Intratumoral administration of FAS gene therapy combined with intravenously administered cisplatin limited the growth of PC3 tumors, thus extending the survival of tumor-bearing mice. The therapeutic effect resulting from the combination therapy was enhanced compared to gene therapy or chemotherapy alone.
Corrective gene therapy
In corrective gene therapy, a defective or absent gene is replaced by a therapeutic one. The most common tumor-suppressor gene found mutated or absent in prostate cancer is TP53, which has been selected as a target in this treatment approach with nonviral delivery systems.88 TP53 is a tumor-suppressor gene that plays an important role in genome stability through different cellular mechanisms. It has been found to be mutated in around 50%–75% of prostate cancer cases,89 resulting in an increase in cancer cell resistance to chemotherapy and radiotherapy.
Nonviral gene carriers, such as polymers and liposomes, have been used to deliver p53 to prostate cancer cells. Sharma et al90 developed biodegradable nanoparticles based on poly(lactic-co-glycolic acid) polymer for sustained delivery of the TP53 gene and examined their therapeutic efficacy against PC3 xenograft tumors following intratumoral and systemic administration. Intratumoral injections of this formulation resulted in significant tumor regression after only one dose, whereas intravenous injection showed a decrease in tumor growth, but without regression. In another study, Seki et al91 used Tf-bearing cationic liposomes to target prostate cancer. This intravenously injected formulation led to significant tumor-growth suppression compared to controls.
Immunomodulatory gene therapy
Cancer cells are well known to have a weak ability to elicit an immunoresponse, which reduces the immune system’s ability to identify and destroy cancer cells. The poor immunogenicity of cancer cells arises from the weak expression of major histocompatibility complex antigens in their cell membranes. These antigens are responsible for T-cell activation, which is the initial step in generating an immunoreaction. Furthermore, in several tumor types, cancer cells release certain immunosuppressive molecules, such as IL10 and TGFβ, which reduce the possibility of generating an immunoresponse against these.88,92,93
Several methods are used to produce an immunoresponse against cancer cells. For example, clinical trials have used the systemic administration of such cytokines as IFNs and interleukins (ILs) to improve immunity to cancer cells, resulting in tumor regression, but with some drawbacks that reduced their chances of success. Cytokines are proteins that undergo rapid degradation in the circulation after systemic administration, which reduces their ability to stimulate an immunoresponse against tumor cells. In order to counter this systemic degradation, they must be administered in high doses to ensure that sufficient cytokines will survive to reach the tumor site. However, this method is not practicable, since cytokines have been found to be cytotoxic in high doses.92
In order to overcome the limitations of systemic administration of cytokines, local or targeted administration has been tried, with improved results.95 However, cytokine/protein injections have to be given repeatedly to maintain their effect in increasing immunity. One way to eliminate the need for repeated injections is to use immunomodulation gene therapy, where instead of the proteins, genes encoding for cytokines are injected into the patient to promote the continuous release of cytokines from the cells.
Two approaches are commonly used for delivery of immunomodulatory genes. First, a vector (usually viral) is used to deliver a suitable gene to the cancer site, where it promotes the continuous release of the cytokine encoded by the gene. It thus activates T cells against the tumor antigens, resulting in the development of an immunoreaction against these tumor cells and their elimination by the immune system. The second approach, called cancer vaccine, involves introducing the gene encoding the cytokines of interest to extracted cancer cells (ex vivo), then reinjecting them into the patient, where they elicit an immunoreaction against the cancer cells due to the effect of the antigens expressed by the injected cells.90–92
Many studies have used genes encoding specific prostate antigens to induce an immunoresponse directed against prostate cancer cells only. Xiao et al94 used lentivirus to deliver PSCA to prostate cancer. They reported a significant increase in CD8+ and CD4+ T cells after injecting the drug into mice. This procedure also showed a possible prophylactic effect against tumor growth, as well as a preventive effect against metastasis. Similarly, Maurer et al95 investigated the immunostimulatory effect of adding the cytosine–phosphorothioate–guanine gene to bone-marrow dendritic cells. This produced a significant release of IL6 and IL12, making it a potential adjuvant for ex vivo vaccination.
Other researchers have upregulated immunoresponse by engineering ex vivo prostate cancer cells to hold cytokines and/or cytokine-receptor genes so that they respond better to cancer cells and provide immunity against cancer. Morris et al96 reconstructed murine prostate cancer cells (TRAMP-C2) to express IL15 and its specific receptor IL15Rα, then used them to vaccinate mice. Their results showed significant expression of CD8+ T cells and natural killer cells. Hull et al97 prepared a vaccine to be used in situ in mice with the RM9 model of prostate cancer. The vaccine contained a combination of two genes encoding IL12 and B71 carried by an adenovirus. In vivo results after in situ injections showed significant suppression of tumor growth.
In 2010, the first ex vivo cancer vaccine, Provenge (Dendreon Corporation, Seal Beach, CA) (sipuleucel-T) was approved by the US FDA to treat prostate cancer. It targets the prostatic acid phosphatase antigen, which is released by prostate cancer cells. When the vaccine was used in clinical trials to treat patients at the metastatic stage of the disease, a significant increase was found in survival time between control and treated groups by up to 4.5 months.98
Nonviral gene delivery systems have also been used to upregulate immunoresponse. We demonstrated that treatment with a DAB-Lf dendriplex encoding IL12 resulted in tumor disappearance of 20% of both PC3 and DU145 prostate tumors and tumor regression of 10% on both tumor types.36 In our experiments, we had to use immunodeficient BALB/c mice able to produce B cells, but not T cells, in order for them to grow subcutaneous tumors of human origin. We thus hypothesize that the therapeutic effect could be further enhanced by using a fully immunocompetent mouse bearing a murine prostate-tumor model. Other studies have also demonstrated the ability of IL12-encoding DNA to induce a therapeutic effect on prostate tumors, but using different modalities of treatment: intratumoral injection, use of a virus as a delivery system or cotreatment with mifepristone,99 radiotherapy,100 oncolytic herpes simplex viruses,101 adenoviral vector-mediated herpes simplex virus–thymidine kinase and ganciclovir (Gcv).102 These studies mainly showed a slowdown in prostate tumor growth, rather than the tumor regression observed in some instances in our experiments.
Suicide gene therapy
One of the most important approaches in gene therapy is suicide gene therapy, the purpose of which is to destroy cancer cells selectively without harming normal ones. It is based on the principle of delivering a specific gene that encodes an enzyme able to convert prodrugs in cancer cells into cytotoxins that destroy the host cells.103,104 Two main genes encoding enzymes have been examined extensively in prostate cancer suicide therapy: Escherichia coli cytosine deaminase (CD), which has the ability to convert the prodrug 5-fluorocytosine (5FC) to 5-fluorouracil (5FU) and the herpes simplex virus-thymidine kinase gene (HSV TK), which converts the prodrug Gcv to Gcv mono- or triphosphate.102,105
The main advantage of this therapeutic approach is the bystander effect, which can be defined as the ability of the activated prodrug to induce a cytotoxic effect in the targeted cells, as well as other surrounding cancer cells, thus creating a killing zone around the cells transfected with the suicide gene.106 This effect may be due to passive transport, gap junctions, and stimulation in the microenvironment of the tumor site.105 The HSV TK-GCV suicide gene therapy system has been found to have this bystander effect, which is probably due to the intracellular diffusion of the activated prodrug Gcv triphosphate through the gap junctions between adjacent cells. However, the bystander effect of the HSV TK-GCV system is considerably weaker than that of CD-5FU.105,106 The CD-5FU suicide-gene therapy system has been found to have a significant bystander effect. It diffuses readily through the tumor site without the need of cell–cell adhesion, which has been attributed to the ability of this system to stimulate the immune system against cancer cells by activating the natural killer cells CD8 and CD4, making it more efficacious to treat metastasized tumors.104,105
The combination of the two suicide-therapy systems has been found to improve therapeutic results on prostate cancer. Yoshimura et al107 measured the therapeutic effects of HSV TK-Gcv and CD-5FC alone and in combination using a cationic lipid as vector in the LNCaP prostate cancer cell line. Each system alone gave around a 60% decrease in cell viability, while in combination they killed 77% of LNCaP cells. Similarly, HSV TK and CD genes were constructed together in pIRES plasmid, the Cytomegalovirus promoter of which was replaced by PSMA to achieve more specific targeting to prostate cancer cells. In vitro results in LNCaP and PC3 cell lines showed 70% and 60% growth inhibition, respectively. These results were improved compared to those observed with either therapy alone.108
Conclusion
Prostate cancer remains one of the most significant therapeutic challenges to address in the coming years. Although several standard therapies, such as prostatectomy, hormone therapy, chemotherapy, and radiotherapy, are currently available in the clinic, there is currently no effective treatment for advanced prostate cancer. In order to overcome this issue, novel gene-based therapeutic approaches are rapidly evolving and represent promising strategies for the treatment of this cancer. The use of highly efficient, safe, tumor-targeted delivery systems able to carry therapeutic pDNA exploiting various therapeutic strategies has been proven to be a novel and effective approach to treat prostate cancer. These nanomedicines may thus represent a new promising avenue for the treatment of prostate cancer and should be further investigated.
Disclosure
This study was financially supported by a grant from WorldCancer Research (grant 16 – 1303) to CD. NA is in receipt of a PhD studentship from the Saudi Cultural Bureau and Princess Nourah bint Abdulrahman University (Kingdom of Saudi Arabia). SS is funded by a research grant from the Dunhill Medical Trust (grant R463/0216). The authors report no other conflicts of interest in this work.
References
International Agency for Research on Cancer. All cancers (excluding non-melanoma skin cancer): estimated incidence, mortality and prevalence worldwide in 2012. Available from: http://globocan.iarc.fr/Pages/fact_sheets_cancer.aspx. Accessed November 30, 2017. | ||
Cancer Research UK. Prostate cancer incidence statistics. 2015. Available from: http://www.cancerresearchuk.org/cancer-info/cancerstats/types/prostate/incidence/uk-prostate-cancer-incidence-statistics. Accessed November 19, 2017. | ||
Freytag SO, Stricker H, Movsas B, Kim JH. Prostate cancer gene therapy clinical trials. Mol Ther. 2007;15:1042–1052. | ||
Philippou Y, Dev H, Sooriakumaran P. Diagnosis and screening. In: Tewari AK, Whelan P, Graham JD, editors. Prostate Cancer: Diagnosis and Clinical Management. Chichester: John Wiley & Sons; 2014:16–33. | ||
Bagnall P. Diagnosis and treatment of prostate cancer. Nurs Times. 2014;110:12–15. | ||
Wirth T, Parker N, Ylä-Herttuala S. History of gene therapy. Gene. 2013;525:162–169. | ||
Wang WW, Li W, Ma N, Steinhoff G. Non-viral gene delivery methods. Curr Pharm Biotechnol. 2013;14:46–60. | ||
Ma G, Shimada H, Hiroshima K, Tada Y, Suzuki N, Tagawa M. Gene medicine for cancer treatment: commercially available medicine and accumulated clinical data in China. Drug Des Devel Ther. 2009;2:115–122. | ||
Ogris M, Wagner E. Targeting tumors with non-viral gene delivery systems. Drug Discov Today. 2002;7:479–485. | ||
McCrudden CM, McCarthy HO. Cancer gene therapy: key biological concepts in the design of multifunctional non-viral delivery systems. In: Molina F, editor. Gene Therapy: Tools and Potential Applications. London: InTech; 2013:213–248. | ||
Gao HJ, Shi WD, Freund LB. Mechanics of receptor-mediated endocytosis. Proc Natl Acad Sci U S A. 2005;102:9469–9474. | ||
Pérez-Martínez FC, Guerra J, Posadas I, Ceña V. Barriers to non-viral vector-mediated gene delivery in the nervous system. Pharm Res. 2011;28:1843–1858. | ||
Taira K, Kataoka K, Niidome T. Non-Viral Gene Therapy: Gene Design and Delivery. Heidelberg: Springer; 2005. | ||
Mctaggart S, Al-Rubeai M. Retroviral vectors for human gene delivery. Biotechnol Adv. 2002;20:1–31. | ||
Kamimura K, Suda T, Zhang G, Liu D. Advances in gene delivery systems. Pharmaceut Med. 2011;25:293–306. | ||
Curiel DT, Douglas JT. Cancer Gene Therapy. Totowa (NJ): Humana Press; 2005. | ||
Shan Y, Luo T, Peng C, et al. Gene delivery using dendrimer-entrapped gold nanoparticles as nonviral vectors. Biomaterials. 2012;33:3025–3035. | ||
Allen TM. Liposomes: opportunities in drug delivery. Drugs. 1997;54:8–14. | ||
Felgner PL, Gadek TR, Holm M, et al. Lipofection: a highly efficient, lipid-mediated DNA-transfection procedure. Proc Natl Acad Sci U S A. 1987;84:7413–7417. | ||
Longmire M, Choyke PL, Kobayashi H. Clearance properties of nano-sized particles and molecules as imaging agents: considerations and caveats. Nanomedicine (Lond). 2008;3:703–717. | ||
Bendas G. Immunoliposomes: a promising approach to targeting cancer therapy. Biodrugs. 2001;15:215–224. | ||
Fasbender A, Marshall J, Moninger TO, Grunst T, Cheng S, Welsh MJ. Effect of co-lipids in enhancing cationic lipid-mediated gene transfer in vitro and in vivo. Gene Ther. 1997;4:716–725. | ||
Mastrobattista E, Hennink WE. Polymers for gene delivery: charged for success. Nat Mater. 2012;11:10–12. | ||
Männistö M, Vanderkerken S, Toncheva V, et al. Structure-activity relationships of poly(L-lysines): effects of pegylation and molecular shape on physicochemical and biological properties in gene delivery. J Control Release. 2002;83:169–182. | ||
Lemkine GF, Demeneix BA. Polyethylenimines for in vivo gene delivery. Curr Opin Mol Ther. 2001;3:178–182. | ||
Benjaminsen RV, Mattebjerg MA, Henriksen JR, Moghimi SM, Andresen TL. The possible “proton sponge” effect of polyethylenimine (PEI) does not include change in lysosomal pH. Mol Ther. 2013;21:149–157. | ||
Lynn DM, Langer R. Degradable poly(β-amino esters): synthesis, characterization, and self-assembly with plasmid DNA. J Am Chem Soc. 2000;122:10761–10768. | ||
Buhleier E, Wehner W, Vögtle F. “Cascade”- and “nonskid-chain-like” syntheses of molecular cavity topologies. Synthesis. 1978;2:155–158. | ||
Tomalia DA, Baker H, Dewald J, et al. A new class of polymers: starburst-dendritic macromolecules. Polym J. 1985;17:117–132. | ||
Klajnert B, Bryszewska M. Dendrimers: properties and applications. Acta Biochim Pol. 2001;48:199–208. | ||
Dufès C, Uchegbu IF, Schätzlein AG. Dendrimers in gene delivery. Adv Drug Deliv Rev. 2005;57:2177–2202. | ||
Bertrand N, Wu J, Xu X, Kamaly N, Farokhzad OC. Cancer nanotechnology: the impact of passive and active targeting in the era of modern cancer biology. Adv Drug Deliv Rev. 2014;66:2–25. | ||
Wally J, Buchanan SK. A structural comparison of human serum transferrin and human lactoferrin. Biometals. 2007;20:249–262. | ||
Al Robaian M, Chiam KY, Blatchford DR, Dufès C. Therapeutic efficacy of intravenously administered transferrin-conjugated dendriplexes on prostate carcinomas. Nanomedicine (Lond). 2014;9:421–434. | ||
Shankaranarayanan JS, Kanwar JR, Al-Juhaishi AJ, Kanwar RK. Doxorubicin conjugated to immunomodulatory anticancer lactoferrin displays improved cytotoxicity overcoming prostate cancer chemo resistance and inhibits tumour development in TRAMP mice. Sci Rep. 2016;6:32062. | ||
Altwaijry N, Somani S, Parkinson JA, et al. Regression of prostate tumors after intravenous administration of lactoferrin-bearing polypropylenimine dendriplexes encoding TNF-α, TRAIL, and interleukin-12. Drug Deliv. 2018;25:679–689. | ||
Liu Z, Wang F, Chen X, Integrin αvβ3-targeted cancer therapy. Drug Dev Res. 2008;69:329–339. | ||
Marelli UK, Rechenmacher F, Sobahi TR, Mas-Moruno C, Kessler H. Tumor targeting via integrin ligands. Front Oncol. 2013;3:222. | ||
Bonkhoff H, Stein U, Remberger K. Differential expression of α6 and α2 very late antigen integrins in the normal, hyperplastic, and neoplastic prostate: simultaneous demonstration of cell surface receptors and their extracellular ligands. Hum Pathol. 1993;24:243–248. | ||
Fornaro M, Manes T, Languino LR. Integrins and prostate cancer metastases. Cancer Metastasis Rev. 2001;20:321–331. | ||
Suyin PC, Dickinson JL, Holloway AF. Integrins in prostate cancer invasion and metastasis. In: Hamilton G, editor. Advances in Prostate Cancer. Rijeka, Croatia: InTech; 2013:621–639. | ||
Zheng DQ, Woodard AS, Fornaro M, Tallini G, Languino LR. Prostatic carcinoma cell migration via αvβ3 integrin is modulated by a focal adhesion kinase pathway. Cancer Res. 1999;59:1655–1664. | ||
Stachurska A, Elbanowski J, Kowalczyńska HM. Role of α5β1 and αvβ3 integrins in relation to adhesion and spreading dynamics of prostate cancer cells interacting with fibronectin under in vitro conditions. Cell Biol Int. 2012;36:883–892. | ||
Horoszewicz JS, Kawinski E, Murphy GP. Monoclonal-antibodies to a new antigenic marker in epithelial prostatic cells and serum of prostatic-cancer patients. Anticancer Res. 1987;7:927–936. | ||
Kinoshita Y, Kuratsukuri K, Landas S, et al. Expression of prostate-specific membrane antigen in normal and malignant human tissues. World J Surg. 2006;30:628–636. | ||
Shen D, Xie F, Edwards WB. Evaluation of phage display discovered peptides as ligands for prostate-specific membrane antigen (PSMA). PLoS One. 2013;8:68339. | ||
Dassie JP, Hernandez LI, Thomas GS, et al. Targeted inhibition of prostate cancer metastases with an RNA aptamer to prostate-specific membrane antigen. Mol Ther. 2014;22:1910–1922. | ||
Goodman OB, Barwe SP, Ritter B, et al. Interaction of prostate specific membrane antigen with clathrin and the adaptor protein complex-2. Int J Oncol. 2007;31:1199–1203. | ||
Laidler P, Dulińska J, Lekka M, Lekki J. Expression of prostate specific membrane antigen in androgen-independent prostate cancer cell line PC-3. Arch Biochem Biophys. 2005;435:1–14. | ||
Aggarwal S, Singh P, Topaloglu O, Isaacs JT, Denmeade SR. A dimeric peptide that binds selectively to prostate-specific membrane antigen and inhibits its enzymatic activity. Cancer Res. 2006;66:9171–9177. | ||
Taylor RM, Severns V, Brown DC, Bisoffi M, Sillerud LO. Prostate cancer targeting motifs: expression of ανβ3, neurotensin receptor 1, prostate specific membrane antigen, and prostate stem cell antigen in human prostate cancer cell lines and xenografts. Prostate. 2012;72:523–532. | ||
Garcia-Hernandez ML, Gray A, Hubby B, Klinger OJ, Kast WM. Prostate stem cell antigen vaccination induces a long-term protective immune response against prostate cancer in the absence of autoimmunity. Cancer Res. 2008;68:861–869. | ||
Sharifi N, Salmaninejad A, Ferdosi S, et al. HER2 gene amplification in patients with prostate cancer: evaluating a CISH-based method. Oncol Lett. 2016;12:4651–4658. | ||
Rabiau N, Dechelotte P, Guy L, et al. Immunohistochemical staining of mucin 1 in prostate tissues. In Vivo. 2009;23:203–207. | ||
Bookstein R, Rio P, Madreperla SA, et al. Promoter deletion and loss of retinoblastoma gene expression in human prostate carcinoma. Proc Natl Acad Sci U S A. 1990;87:7762–7766. | ||
Mazhar D, Waxman J. Gene therapy for prostate cancer. BJU Int. 2004;93:465–469. | ||
Shalev M, Thompson TC, Kadmon D, Ayala G, Kernen K, Miles BJ. Gene therapy for prostate cancer. Urology. 2001;57:8–16. | ||
Jia LT, Chen SY, Yang AG. Cancer gene therapy targeting cellular apoptosis machinery. Cancer Treat Rev. 2012;38:868–876. | ||
Walczak H. Death receptor-ligand systems in cancer, cell death, and inflammation. Cold Spring Harb Perspect Biol. 2013;5:a008698. | ||
Wong RS. Apoptosis in cancer: from pathogenesis to treatment. J Exp Clin Cancer Res. 2011;30:87. | ||
Tüting T, Gambotto A, Baar J, et al. Interferon-α gene therapy for cancer: retroviral transduction of fibroblasts and particle-mediated transfection of tumor cells are both effective strategies for gene delivery in murine tumor models. Gene Ther. 1997;4:1053–1060. | ||
Wang CY, Mayo MW, Baldwin AS. TNF- and cancer therapy-induced apoptosis: potentiation by inhibition of NF-κB. Science. 1996;274:784–787. | ||
van Horssen R, ten Hagen TL, Eggermont AM. TNF-α in cancer treatment: molecular insights, antitumor effects, and clinical utility. Oncologist. 2006;11:397–408. | ||
Chopra DP, Menard RE, Januszewski J, Mattingly RR. TNF-α-mediated apoptosis, in normal human prostate epithelial cells and tumor cell lines. Cancer Lett. 2004;203:145–154. | ||
Mauceri HJ, Hanna NN, Wayne JD. Tumor necrosis factor α (TNF-α) gene therapy targeted by ionizing radiation selectively damages tumor vasculature. Cancer Res. 1996;56:4311–4314. | ||
Tandle A, Hanna E, Lorang D, et al. Tumor vasculature-targeted delivery of tumor necrosis factor-α. Cancer. 2009;115:128–139. | ||
Chung TD, Mauceri HJ, Hallahan DE, et al. Tumor necrosis factor-alpha-based gene therapy enhances radiation cytotoxicity in human prostate cancer. Cancer Gene Ther. 1998;5:344–349. | ||
Shukla S, Gupta S. Suppression of constitutive and tumor necrosis factor-induced nuclear factor (NF)-κB activation and induction of apoptosis by apigenin in human prostate carcinoma PC-3 cells: correlation with down-regulation of NF-κB-responsive genes. Clin Cancer Res. 2004;10:3169–3178. | ||
Shi J, Chen J, Serradji N, et al. PMS1077 sensitizes TNF-α induced apoptosis in human prostate cancer cells by blocking NF-κB signaling pathway. PLoS One. 2013;8:e61132. | ||
Barresi G, Tuccari G. Lactoferrin in benign hypertrophy and carcinomas of the prostatic gland. Virchows Arch A Pathol Anat Histopathol. 1984;403:59–66. | ||
Tuccari G, Barresi G. Lactoferrin in human tumours: immunohistochemical investigations during more than 25 years. Biometals. 2011;24:775–784. | ||
Ashkenazi A, Pai RC, Fong S, et al. Safety and antitumor activity of recombinant soluble Apo2 ligand. J Clin Invest. 1999;104:155–162. | ||
Norian L, James B, Griffith T. Advances in viral vector-based TRAIL gene therapy for cancer. Cancers. 2011;3:603–620. | ||
Griffith T, Stokes B, Kucaba T, et al. TRAIL gene therapy: from preclinical development to clinical application. Curr Gene Ther. 2009;9:9–19. | ||
Yu R, Mandlekar S, Ruben S, Ni J, Kong AN. Tumor necrosis factor-related apoptosis-inducing ligand-mediated apoptosis in androgen-independent prostate cancer cells. Cancer Res. 2000;60:2384–2389. | ||
Farooqi AA, de Rosa G. TRAIL and microRNAs in the treatment of prostate cancer: therapeutic potential and role of nanotechnology. Appl Microbiol Biotechnol. 2013;97:8849–8857. | ||
Griffith TS, Anderson RD, Davidson BL, Williams RD, Ratliff TL. Adenoviral-mediated transfer of the TNF-related apoptosis-inducing ligand/Apo-2 ligand gene induces tumor cell apoptosis. J Immunol. 2000;165:2886–2894. | ||
Lin T, Zhang L, Davis J, et al. Combination of TRAIL gene therapy and chemotherapy enhances antitumor and antimetastasis effects in chemosensitive and chemoresistant breast cancers. Mol Ther. 2003;8:441–448. | ||
Seol JY, Park KH, Hwang CI, et al. Adenovirus-TRAIL can overcome TRAIL resistance and induce a bystander effect. Cancer Gene Ther. 2003;10:540–548. | ||
Mohr A, Henderson G, Dudus L, et al. AAV-encoded expression of TRAIL in experimental human colorectal cancer leads to tumor regression. Gene Ther. 2004;11:534–543. | ||
Chen Z, Penet MF, Krishnamachary B, et al. PSMA-specific theranostic nanoplex for combination of TRAIL gene and 5-FC prodrug therapy of prostate cancer. Biomaterials. 2016;80:57–67. | ||
Elojeimy S, Mckillop JC, el-Zawahry AM, et al. FasL gene therapy: a new therapeutic modality for head and neck cancer. Cancer Gene Ther. 2006;13:739–745. | ||
Xiong L, Liu YH, Zhang YP, et al. Fas ligand delivery by a prostate-restricted replicative adenovirus enhances safety and antitumor efficacy. Clin Cancer Res. 2007;13:5463–5473. | ||
Nakanishi H, Mazda O, Satoh E, et al. Nonviral genetic transfer of Fas ligand induced significant growth suppression and apoptotic tumor cell death in prostate cancer in vivo. Gene Ther. 2003;10:434–442. | ||
Hyer ML, Sudarshan S, Schwartz DA, et al. Quantification and characterization of the bystander effect in prostate cancer cells following adenovirus-mediated FasL expression. Cancer Gene Ther. 2003;10:330–339. | ||
Xiong L, Liu YH, Zhang YP, et al. Fas ligand delivery by a prostate-restricted replicative adenovirus enhances safety and antitumor efficacy. Clin Cancer Res. 2007;13:5463–5473. | ||
Hedlund TE, Meech SJ, Srikanth S, et al. Adenovirus-mediated expression of Fas ligand induces apoptosis of human prostate cancer cells. Cell Death Differ. 1999;6:175–182. | ||
Agha-Mohammadi S, Lotze MT. Immunomodulation of cancer: potential use of selectively replicating agents. J Clin Invest. 2000;105:1173–1176. | ||
Mabjeesh N, Zhong H, Simons JW. Gene therapy of prostate cancer: current and future directions. Endocr Relat Cancer. 2002;9:115–139. | ||
Sharma B, Ma W, Adjei IM, et al. Nanoparticle-mediated p53 gene therapy for tumor inhibition. Drug Deliv Transl Res. 2011;1:43–52. | ||
Seki M, Iwakawa J, Cheng H, Cheng PW. p53 and PTEN/MMAC1/TEP1 gene therapy of human prostate PC-3 carcinoma xenograft, using transferrin-facilitated lipofection gene delivery strategy. Hum Gene Ther. 2002;13:761–773. | ||
Burdelski C, Dieckmann T, Heumann A, et al. p16 upregulation is linked to poor prognosis in ERG negative prostate cancer. Tumor Biology. 2016;37:12655–12663. | ||
Shalev M, Thompson TC, Kadmon D, et al. Gene therapy for prostate cancer. Urology. 2001;57:8–16. | ||
Xiao L, Joo KI, Lim M, Wang P. Dendritic cell-directed vaccination with a lentivector encoding PSCA for prostate cancer in mice. PLoS One. 2012;7:e48866. | ||
Maurer T, Pournaras C, Aguilar-Pimentel JA, et al. Immunostimulatory CpG-DNA and PSA-peptide vaccination elicits profound cytotoxic T cell responses. Urol Oncol. 2013;31:1395–1401. | ||
Morris JC, Ramlogan-Steel CA, Yu P, et al. Vaccination with tumor cells expressing IL-15 and IL-15Rα inhibits murine breast and prostate cancer. Gene Ther. 2014;21:393–401. | ||
Hull GW, Mccurdy MA, Nasu Y, et al. Prostate cancer gene therapy: comparison of adenovirus-mediated expression of interleukin 12 with interleukin 12 plus B7-1 for in situ gene therapy and gene-modified, cell-based vaccines. Clin Cancer Res. 2000;6:4101–4109. | ||
Cheever MA, Higano CS. Provenge (sipuleucel-T) in prostate cancer: the first FDA-approved therapeutic cancer vaccine. Clin Cancer Res. 2011;17:3520–3526. | ||
Gabaglia C, Delaney A, Gee J, et al. Treatment combining RU486 and Ad5IL-12 vector attenuates the growth of experimentally formed prostate tumors and induces changes in the sentinel lymph nodes of mice. J Transl Med. 2010;8:98. | ||
Fujita T, Timme TL, Tabata K, et al. Cooperative effects of adenoviral vector-mediated interleukin 12 gene therapy with radiotherapy in a preclinical model of metastatic prostate cancer. Gene Ther. 2007;14:227–236. | ||
Varghese S, Rabkin SD, Liu R, et al. Enhanced therapeutic efficacy of IL-12, but not GM-CSF, expressing oncolytic herpes simplex virus for transgenic mouse derived prostate cancers. Cancer Gene Ther. 2006;13:253–265. | ||
Nasu Y, Bangma CH, Hull GW, et al. Combination gene therapy with adenoviral vector-mediated HSV-tk + GCV and IL-12 in an orthotopic mouse model for prostate cancer. Prostate Cancer Prostatic Dis. 2001;4:44–55. | ||
Lu Y. Transcriptionally regulated, prostate-targeted gene therapy for prostate cancer. Adv Drug Deliv Rev. 2009;61:572–588. | ||
Kim KM, Won YW, Adhikary PP, Hwang YM, Kim YH. Suicidal gene therapy against tumor using reducible poly (oligo-d-arginine). J Control Release. 2011;152:e148–e149. | ||
Zarogoulidis P, Darwiche K, Sakkas A, et al. Suicide gene therapy for cancer: current strategies. J Genet Syndr Gene Ther. 2013;4:16849. | ||
Sato T, Neschadim A, Lavie A, Yanagisawa T, Medin JA. The engineered thymidylate kinase (TMPK)/AZT enzyme-prodrug axis offers efficient bystander cell killing for suicide gene therapy of cancer. PLoS One. 2013;8:e78711. | ||
Yoshimura I, Ikegami S, Suzuki S, Tadakuma T, Hayakawa M. Adenovirus mediated prostate specific enzyme prodrug gene therapy using prostate specific antigen promoter enhanced by the Cre-loxP system. J Urol. 2002;168:2659–2664. | ||
Yue QH, Hu XB, Yin Y, et al. Inhibition of prostate cancer by suicide gene targeting the FCY1 and HSV-TK genes. Oncol Rep. 2009;22:1341–1347. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.