Back to Journals » OncoTargets and Therapy » Volume 10
Targeted interfering DEP domain containing 1 protein induces apoptosis in A549 lung adenocarcinoma cells through the NF-κB signaling pathway
Authors Wang Q ![]() , Li A, Jin J
, Li A, Jin J ![]() , Huang G
, Huang G ![]()
Received 20 May 2017
Accepted for publication 3 August 2017
Published 11 September 2017 Volume 2017:10 Pages 4443—4454
DOI https://doi.org/10.2147/OTT.S142244
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Samir Farghaly
Qingqing Wang,1 Aili Li,1 Junfei Jin,2,3 Guojin Huang1,2
1Laboratory of Respiratory Disease, Affiliated Hospital of Guilin Medical University, 2China-USA Lipids in Health and Disease Research Center, Guilin Medical University, 3Laboratory of Hepatobiliary and Pancreatic Surgery, Affiliated Hospital of Guilin Medical University, Guilin, Guangxi, People’s Republic of China
Abstract: Ectopic expression of DEP domain containing 1 (DEPDC1) in lung adenocarcinomas is associated with poor prognosis, but its role and the underlying mechanism remain unknown. In this study, DEPDC1 expression in lung cancer cell lines was examined with Western blot assay, and DEPDC1-positive cell A549 was selected for further experiments. DEPDC1 inhibitor miR-130a was overexpressed in A549 cells, and the proliferation and apoptosis of these cells were analyzed with cell counting and flow cytometry assay. Interfering peptide 11R-DEP:611–628 and JNK inhibitor SP600125 were used alone or in combination to treat A549 cells, and the cell proliferation and apoptosis were assessed by flow cytometry assay; caspase 3 and cleaved caspase 3, phosphor-JNK, and total JNK were detected by Western blotting; and nuclear factor kappa B (NF-κB) localization was determined by immunofluorescence staining. We found that miR-130a and 11R-DEP:611–628 peptides (5 µM) both inhibited A549 proliferation and induced apoptosis. We observed that 11R-DEP:611–628 peptide treatment resulted in elevated A20 expression, dramatically reduced nuclear NF-κB, and increased phosphor-JNK. These findings indicate that DEPDC1 inhibits apoptosis of A549 cell by suppressing A20 expression to regulate NF-κB activity, and that JNK plays a protective role upon 11R-DEP:611–628 peptide treatment. In conclusion, DEPDC1 might be a novel therapeutic target for lung cancer, and the 11R-DEP:611–628 peptide is a potent apoptosis inducer in A549 cells.
Keywords: lung cancer, A549 cells, DEPDC1, A20, NF-κB, miR-130a
Introduction
Lung cancer is one of the most common cancers in the world. The incidence and mortality of lung cancer in the United States, as estimated in 2017, are 13.0% of total new cancer cases and 26% of total cancer mortality, respectively.1 Lung cancer is the first cause of cancer death among males, and the leading cause of cancer death among females.2 Based on histological features, lung cancers are classified into four types: adenocarcinoma, squamous cell carcinoma, small-cell carcinoma, and large-cell carcinoma.3 From a genetic aspect, lung cancers occur due to chromosomal changes and alteration in gene copy numbers, DNA single-nucleotide alterations and insertions and deletions, epigenetic alterations, transcriptome alterations, or pathway alterations;4 thus, lung cancers may be categorized into different molecular subtypes, which have various genetic changes that drive and maintain lung tumorigenesis. Surgical resection is the most common treatment method for early-stage non-small cell lung cancer.5 However, most lung cancers are diagnosed at an advanced stage, and treatment is chemotherapy and/or radiation therapy.5 Current molecular targeted therapies, such as specific epidermal growth factor receptor (EGFR) kinase inhibitors and ALK inhibitors,4 are widely used in the treatment of lung cancers, and have been shown to have better outcomes than platinum-based chemotherapy.4 However, it is necessary to discover novel therapeutic targets and drugs for lung cancer to provide personalized treatment to patients who have specific molecular subtypes.
DEP domain containing 1 (DEPDC1) is an oncoprotein, and its expression is undetectable in the lung and other examined normal human tissues, except the testis.6–8 DEPDC1 was first reported in bladder cancer, where it plays an important role in carcinogenesis by forming a complex with zinc-finger protein 224 (ZNF224) to suppress the transcription of A20, an NF-κB inhibitory protein, leading to translocation of NF-κB into the nucleus, where the expression of genes related to the anti-apoptotic function is activated.9 DEPDC1 physically interacts with ZNF224 by its molecular region (598–653). The mimicking peptide of DEPDC1 611–628 region, conjugated with 11 residues of arginine (11R) at its NH2 terminus, and named the 11R-DEP:611–628 peptide, is able to efficiently disrupt interaction between DEPDC1 and ZNF224, and is capable of inducing apoptosis of UM-UC-3 bladder cancer cells both in vitro and in vivo.9 A recent study reported that, in HeLa, MCF7, or SH-SY5Y cells, DEPDC1 is involved in vincristine-induced cell death by promoting JNK-dependent degradation of MCL1.10 DEPDC1 is upregulated in a variety of tumor tissues, such as bladder, breast, and liver cancers.8,9,11,12 DEPDC1 expression is also elevated in lung adenocarcinomas and can be a prognostic marker for patients with non-small cell lung cancer.13 However, the mechanism by which DEPDC1 participates in carcinogenesis in the lung is still unknown.
In the present study, we examined the expression of DEPDC1 in several lung cancer cell lines, investigated the function of DEPDC1 in A549 cells, and tested the possibility of inducing A549 apoptosis by targeted interfering DEPDC1. Our study may provide evidence to understand the function of DEPDC1 in lung cancer and find a novel therapeutic target and drug for the treatment of lung cancer.
Materials and methods
Antibodies, peptides, and other reagents
The information for antibodies used in this experiment is as follows. DEPDC1 antibody (#LS-C167364) was purchased from LifeSpan BioSciences Inc. (Seattle, WA, USA). Caspase 3 antibody (#9665S), JNK antibody (c-Jun N-terminal kinase; #9252S), and NF-κB antibody (#8242S) were purchased from Cell Signaling Technology (Beverly, MA, USA). p-JNK antibody (phosphorylated c-Jun N-terminal kinase; #ab76572) and cleaved caspase 3 antibody (#ab32042) were obtained from Abcam (Cambridge, MA, USA). β-actin antibody (#TA-09) was purchased from ZSGB-BIO (Beijing, People’s Republic of China). The dilutions of these antibodies in Western blotting are as followed: DEPDC1 1:500, caspase 3 1:300, cleaved caspase 3 1:500, JNK 1:500, p-JNK 1:4,000, and β-actin 1:1,000.
The 11R-DEP:611–628 peptide and its scramble peptide were synthesized by Shanghai Top-peptide Biotechnology Co. (Shanghai, People’s Republic of China) according to the sequence from a previous report.9 JNK inhibitor SP600125 was purchased from Santa Cruz Biotechnology (Dallas, TX, USA).
Cell lines and cell culture
Lung adenocarcinoma cell lines A549, H3255, HCC827, H1993, and H1995 were obtained from the Kunming cell bank of the Chinese Academy of Sciences (Kunming, Yunnan, People’s Republic of China). Cells were cultured in Roswell Park Memorial Institute (RPMI-1640) medium containing 10% fetal bovine serum at 37°C in a 5% CO2 incubator.
Western blot assay
Western blotting was conducted as described previously.14 In brief, cells were harvested and lysed with lysis buffer (#R0020; Solarbio, Beijing, People’s Republic of China) on ice for 5 min. The lysate was centrifuged at 12,000 rpm for 20 min at 4°C to obtain supernatant, in which the protein concentration was measured with a BCA Kit (Beyotime Biotechnology, Shanghai, People’s Republic of China) according the manufacturer’s instructions. Proteins were separated with 10% or 15% sodium dodecyl sulfate-polyacrylamide gels and transferred onto the membrane, which was blocked with 5% nonfat milk at room temperature (RT) for 2 h, followed by incubation with primary antibody at 4°C overnight. The membrane was then incubated with the secondary antibody at RT for 1 h. Blots were developed using ECL Kit (Beyotime Biotechnology).
Cell proliferation analysis
Cell proliferation was measured with the Cell Counting Kit-8 (CCK-8, Dojindo CK04, Kumamoto, Japan). A549 cells (7×103) were seeded into 96-well culture plates, and cultured at 37°C in a 5% CO2 incubator for 16 h. A549 cells were treated with 11R-DEP:611–628 or scramble peptide at different concentrations (0, 1, 2, 3, 4, 5, or 6 μM) in the RPMI-1640 containing 2% fetal bovine serum for the indicated time. Media with peptide in every well were replaced every 24 h to maintain peptide activity. At the end of the experiments, 10 μL CCK-8 reagent was added and incubated for 1 h at 37°C and, finally, the absorbance was read with a plate reader (Spectra, max plus 384, Sunnyvale, CA, USA) at 450 nm.
Flow cytometric analysis
The flow cytometry assay was conducted to examine the apoptotic death of cells treated with indicated reagents. A549 cells (1.5×105 cells/well) were seeded in six-well plates, and cultured for 16 h, followed by treatment with 11R-DEP:611–628 peptide or scramble peptide alone or in combination with JNK inhibitor (10 μM), as well as appropriate control for the indicated time. Then, the cells were harvested and stained with the FITC Annexin V Dead Cell Kit (Merck Millipore, Darmstadt, Germany) for 20 min at RT in a dark chamber according to the manufacturer’s instructions. Finally, the cells were analyzed with flow cytometry (Muse™ Cell Analyzer, Merck Millipore).
Light microscopic imaging
A549 cells were seeded in six-well plates, and cultured at 37°C in a 5% CO2 incubator for 16 h, then treated with indicated reagents for a certain period, and the cell morphological change was monitored with a contrast phase microscope (Olympus, Tokyo, Japan).
Quantitative real-time polymerase chain reaction
Quantitative real-time polymerase chain reaction (qRT-PCR) assay was conducted as described previously.15 In brief, total RNA from cells was extracted using TRIzol (#15596026, Thermo Fisher Scientific, Waltham, MA, USA). RNA concentration was measured with the NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific). cDNA was amplified with 2 μg of total RNA for each sample according to the protocol of the Reverse Transcription Kit (TIANGEN, Beijing, People’s Republic of China). In order to examine the expression of A20 gene, qRT-PCR was conducted using SYBR Green I (#RR820A, Takara Bio Inc., Beijing, People’s Republic of China). PCR conditions were as follows: 95°C for 30 s; 40 cycles: 95°C for 5 s, and 60°C for 30 s. All reactions were carried out in triplicate. GAPDH was used as the internal control. The primers for A20 were as followed: forward, 5′-CGTCCAGGTTCCAGAACACCATTC-3′; reverse, 5′-TGCGCTGGCTCGATCTCAGTTG-3′. The primers for GAPDH were as follows: forward, 5′-CAGGAGGCATTGCTGATGAT-3′, reverse, 5′-GAAGGCTGGGGCTCATTT-3′. For the miR-130a experiment, cDNA was amplified according to the manufacturer’s protocol using Mir-X miRNA First Strand Synthesis Kit (#638313, Takara Bio Inc., Mountain View, CA, USA). qRT-PCR was conducted according to kit instructions of Mir-X miRNA qRT-PCR SYBR kit (#638314, Takara Bio Inc). PCR conditions were as followed: 95°C for 10 s; 40 cycles: 95°C for 5 s, and 60°C for 20 s. U6 RNA was used as an internal control. The relative expression level was determined with the ΔΔCt method.
Plasmids and cell transfection
MicroRNA-130a (miR-130a)-expressing plasmids, where coding sequence has-mir-130a is inserted into XhoI/BamHI sites of vector plasmid GV514, were purchased from Shanghai Genechem Co., Ltd. Empty GV514 was used as a negative control plasmid.
A549 cells were seeded in 12-well plates 1 day prior to transfection. Cells were transfected with 1.25 μg plasmid DNA/well by using Lipofectamine 3000 (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instruction. A total of 72 h post-transfection, the cells were harvested for cell counting, imaging, and analysis for gene expression and apoptosis.
Immunofluorescence staining
Immunofluorescence staining was conducted as described previously.16 In brief, A549 cells (3.5×104 cells/well) were seeded into 24-well plates and cultured at 37°C in a 5% CO2 incubator for 16 h, followed by treatment with 11R-DEP:611–628 peptide (5 μM) or scramble peptide (5 μM) for 3 h. Cells were fixed with 4% paraformaldehyde for 20 min at RT. The cell membrane was penetrated with 3% Triton X-100 for 15 min at RT. Nonspecific binding was blocked with 5% bovine serum albumin for 1 h at RT. The cells were incubated with anti-NF-κB antibody (1:50, CST) at 4°C overnight. The nucleus was stained with 4′,6-diamidino-2-phenylindole (DAPI) for 5 min. Finally, the cells were visualized with fluorescence microscopy (Olympus TH4-200, Tokyo, Japan), and images were obtained.
Statistical analysis
GraphPad Prism 5 (GraphPad Software Inc., San Diego, CA, USA) and the SPSS version 11.0 (SPSS Inc, Chicago, IL, USA) were used to analyze data. All experiments were conducted at least three times independently, and the results are shown as mean ± SD. Comparison between different groups was undertaken by unpaired or paired Student’s t-test, and P<0.05 was considered statistically significant.
Results
DEPDC1 is expressed in multiple lung cancer cell lines
It was reported that DEPDC1 expression was upregulated in lung tissues.13 To study the function of DEPDC1 in vitro, we need lung cell lines with DEPDC1 expression. Thus, we detected expression levels of DEPDC1 in several lung cancer cell lines with Western blot analysis. As shown in Figure 1, DEPDC1 is expressed in all of the lung cancer cell lines tested, and the expression level in H3255 and A549 cells is higher than in the rest. Because A549 is often used in lung cancer cell lines, we used it for subsequent experiments.
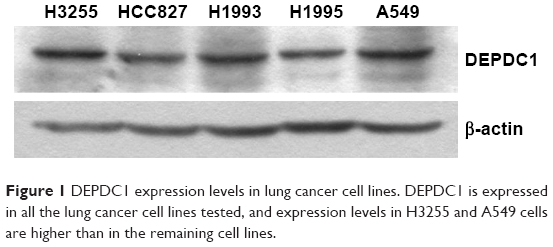 | Figure 1 DEPDC1 expression levels in lung cancer cell lines. DEPDC1 is expressed in all the lung cancer cell lines tested, and expression levels in H3255 and A549 cells are higher than in the remaining cell lines. |
Downregulating DEPDC1 by miR-130a results in inhibited proliferation and enhanced apoptosis in A549 cells
As it was reported very recently that miR-130a effectively reduced cell viability and invasion capability, while it increased apoptosis in prostate cancer cells by targeting DEPDC1 and SEC23B,17 we used miR-130a as a tool to study the function of DEPDC1 in A549 cells, which were transfected with miR-130a-expressing plasmids. We monitored morphological changes of cells, measured cell amounts, analyzed apoptosis, and detected the expression of DEPDC1 and A20 72 h after transfection. The results showed that, compared with the control group, cells appeared thinner and longer (Figure 2A) and the amounts of live cells were decreased by approximately 50% (Figure 2B) with miR-130a expression (Figure 2C); furthermore, apoptotic cells were increased by approximately 30% (Figure 2D). Compared with the cells transfected with control plasmids, cells with miR-130a expression had significantly reduced DEPDC1 level (Figure 2E) and increased A20 (Figure 2F). These results imply that DEPDC1 plays a role in promoting A549 proliferation via regulating apoptosis, and targeting interference of DEPDC1 expression might induce apoptosis of A549 cells via modulating A20 expression.
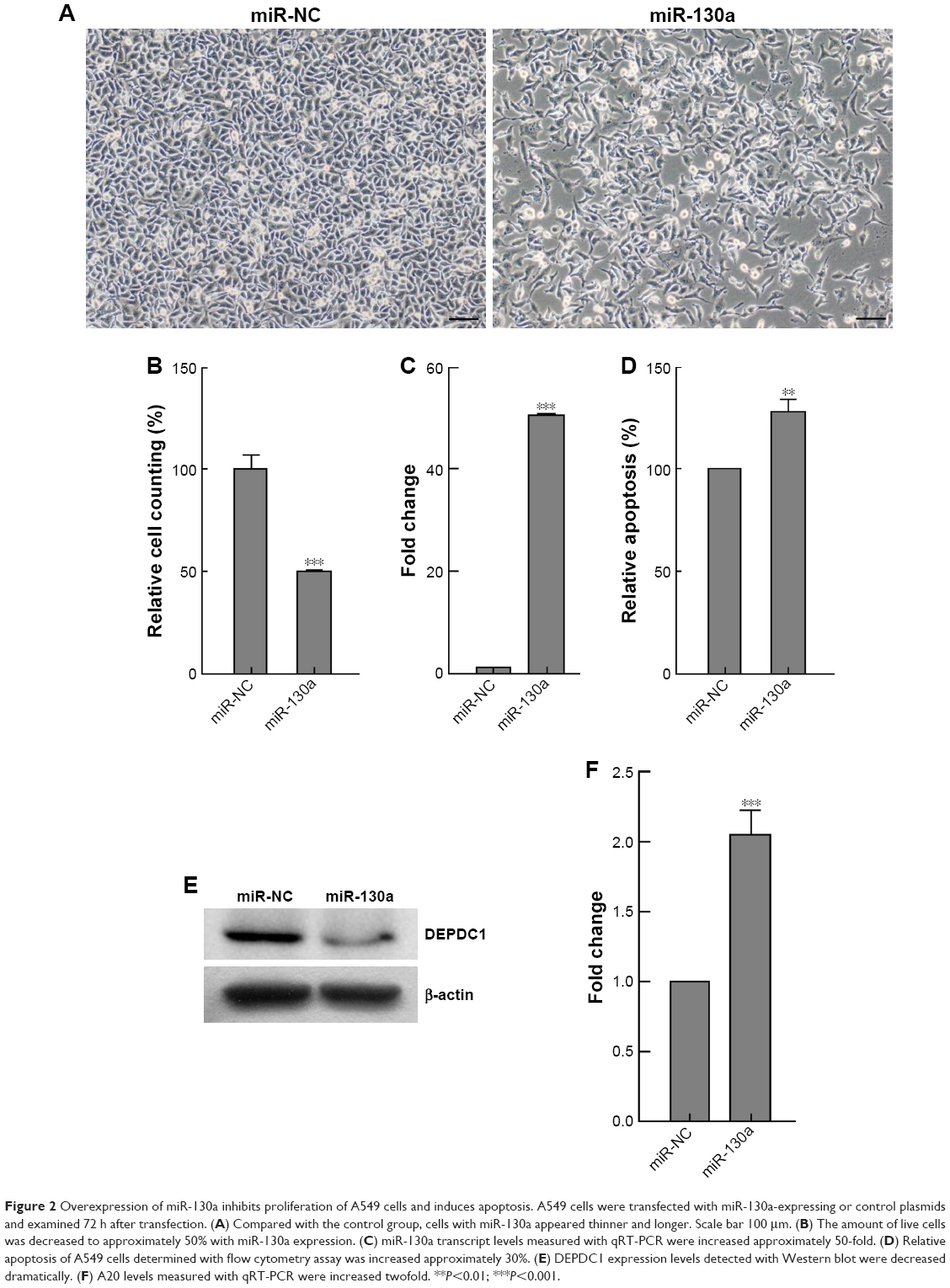 | Figure 2 Overexpression of miR-130a inhibits proliferation of A549 cells and induces apoptosis. A549 cells were transfected with miR-130a-expressing or control plasmids and examined 72 h after transfection. (A) Compared with the control group, cells with miR-130a appeared thinner and longer. Scale bar 100 μm. (B) The amount of live cells was decreased to approximately 50% with miR-130a expression. (C) miR-130a transcript levels measured with qRT-PCR were increased approximately 50-fold. (D) Relative apoptosis of A549 cells determined with flow cytometry assay was increased approximately 30%. (E) DEPDC1 expression levels detected with Western blot were decreased dramatically. (F) A20 levels measured with qRT-PCR were increased twofold. **P<0.01; ***P<0.001. |
Targeted disrupting of DEPDC1–ZNF224 complex with peptide inhibits proliferation and induces apoptosis in A549 cells
To confirm the aforementioned findings, we used interfering peptide to conduct further experiments. Because DEPDC1 physically interacts with ZNF224 to form a complex, and acts as transcription suppressor of A20 in the bladder cancer cell line UM-UC-3, a cell-permeable peptide capable of disrupting the DEPDC1–ZNF224 complex, named the 11R-DEP:611–628 peptide, is able to inhibit proliferation of UM-UC-3 cells.9 To test whether DEPDC1 functions in A549 cells through the same mechanism, we treated cells with the 11R-DEP:611–628 peptide at various concentrations, and measured cell proliferation with CCK-8 assay to determine the effect of this peptide on A549 cells and find the optimal working concentration of peptide. The results showed that the 11R-DEP:611–628 peptide at 5 μM or higher concentration inhibited proliferation of A549 cells dramatically, as compared with scramble peptide (Figure 3), and the inhibitory effect of the 11R-DEP:611–628 peptide was dose dependent. These data indicate that DEPDC1 promotes A549 cell proliferation by forming a complex with ZNF224, and that the 11R-DEP:611–628 peptide at 5 μM can inhibit the function of DEPDC1 in A549 cells efficiently. Therefore, 11R-DEP:611–628 at 5 μM was selected to conduct further experiments.
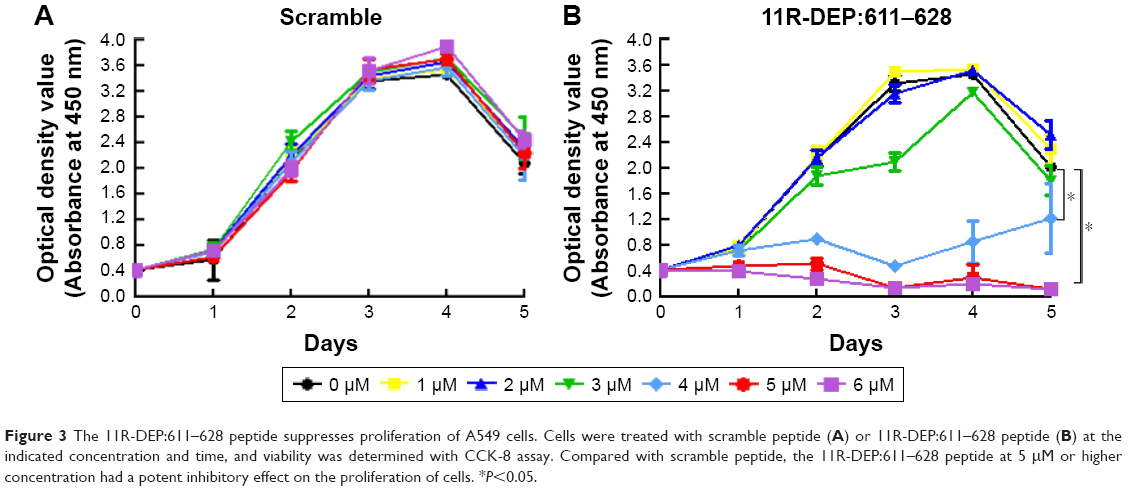 | Figure 3 The 11R-DEP:611–628 peptide suppresses proliferation of A549 cells. Cells were treated with scramble peptide (A) or 11R-DEP:611–628 peptide (B) at the indicated concentration and time, and viability was determined with CCK-8 assay. Compared with scramble peptide, the 11R-DEP:611–628 peptide at 5 μM or higher concentration had a potent inhibitory effect on the proliferation of cells. *P<0.05. |
A previous report showed that DEPDC1 promotes proliferation through suppressing apoptosis in bladder cancer cells.9 To test whether the same occurs in A549 cells, we treated A549 cells with 5 μM 11R-DEP:611–628 peptide, and monitored the morphological change of cells. The results showed that A549 cells appeared abnormal and began to shrink within 1 h after treatment with the 11R-DEP:611–628 peptide (Figure 4A). To examine the effect of the 11R-DEP:611–628 peptide on apoptosis, we first analyzed cells with flow cytometry assay. The data demonstrated that the 11R-DEP:611–628 peptide could effectively induce A549 cell apoptosis, and the effect was time dependent (Figure 4B and C). Caspase 3 is an apoptosis marker that is cleaved when cells undergo apoptosis.18 We detected the levels of caspase 3 in A549 cells treated with 5 μM 11R-DEP:611–628 peptide with Western blot analysis. The results showed that protein levels of caspase 3 in cells treated with 11R-DEP:611–628 peptide were decreased significantly; further, cleaved caspase 3 levels were increased (Figure 4D), indicating increased apoptosis.
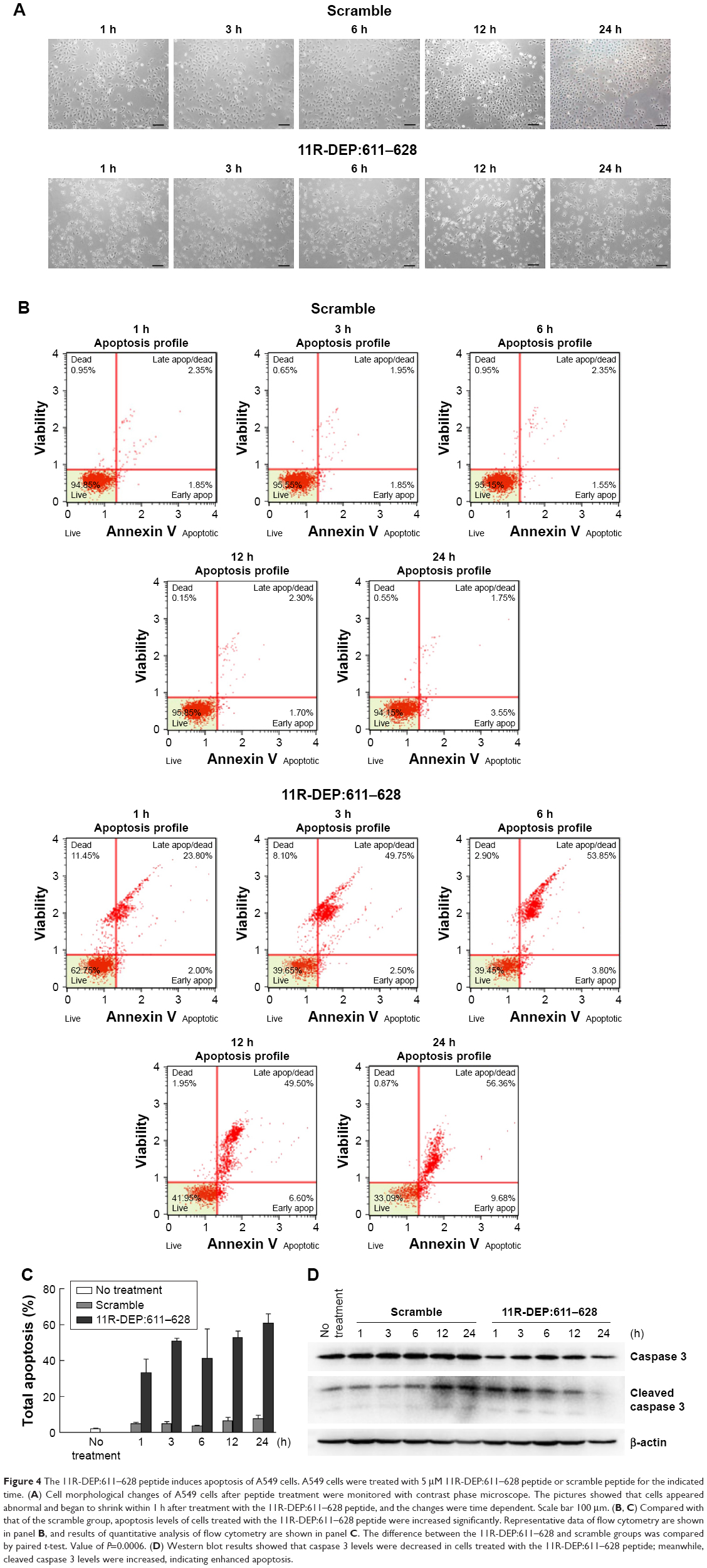 | Figure 4 The 11R-DEP:611–628 peptide induces apoptosis of A549 cells. A549 cells were treated with 5 μM 11R-DEP:611–628 peptide or scramble peptide for the indicated time. (A) Cell morphological changes of A549 cells after peptide treatment were monitored with contrast phase microscope. The pictures showed that cells appeared abnormal and began to shrink within 1 h after treatment with the 11R-DEP:611–628 peptide, and the changes were time dependent. Scale bar 100 μm. (B, C) Compared with that of the scramble group, apoptosis levels of cells treated with the 11R-DEP:611–628 peptide were increased significantly. Representative data of flow cytometry are shown in panel B, and results of quantitative analysis of flow cytometry are shown in panel C. The difference between the 11R-DEP:611–628 and scramble groups was compared by paired t-test. Value of P=0.0006. (D) Western blot results showed that caspase 3 levels were decreased in cells treated with the 11R-DEP:611–628 peptide; meanwhile, cleaved caspase 3 levels were increased, indicating enhanced apoptosis. |
These findings suggest that DEPDC1 plays a vital role in the regulation of proliferation of A549 cells by modulating apoptosis via forming a complex with ZNF224.
11R-DEP:611–628 peptide blocks NF-κB nuclear translocation via activation of A20 expression in A549 cells
Previous reports proposed the mechanism that, in bladder cancer cells, DEPDC1 suppresses A20 expression by forming a complex with ZNF224, leading to NF-κB nuclear translocation. To investigate whether DEPDC1 has an effect on A20 and NF-κB in A549 cells, we treated cells with 11R-DEP:611–628 peptide (5 μM for 1, 3, 6, 12, and 24 h) and analyzed A20 expression and NF-κB localization. The qRT-PCR results demonstrated that the expression of A20 was elevated to 1.5-fold at 1 h, peaked around 3.5-fold at 3 h, and was maintained around twofold during the rest period (Figure 5A). Immunofluorescence staining images showed that the amount of NF-κB in the nuclei was significantly decreased 3 h after 11R-DEP:611–628 peptide treatment (Figure 5B and C). The data demonstrated that the 11R-DEP:611–628 peptide is capable of effectively inducing A20 expression, and blocking NF-κB nuclear translocation in A549 cells. These findings imply that DEPDC1 serves as a regulatory factor of apoptosis in A549 cells by suppressing A20 expression to regulate NF-κB cellular localization.
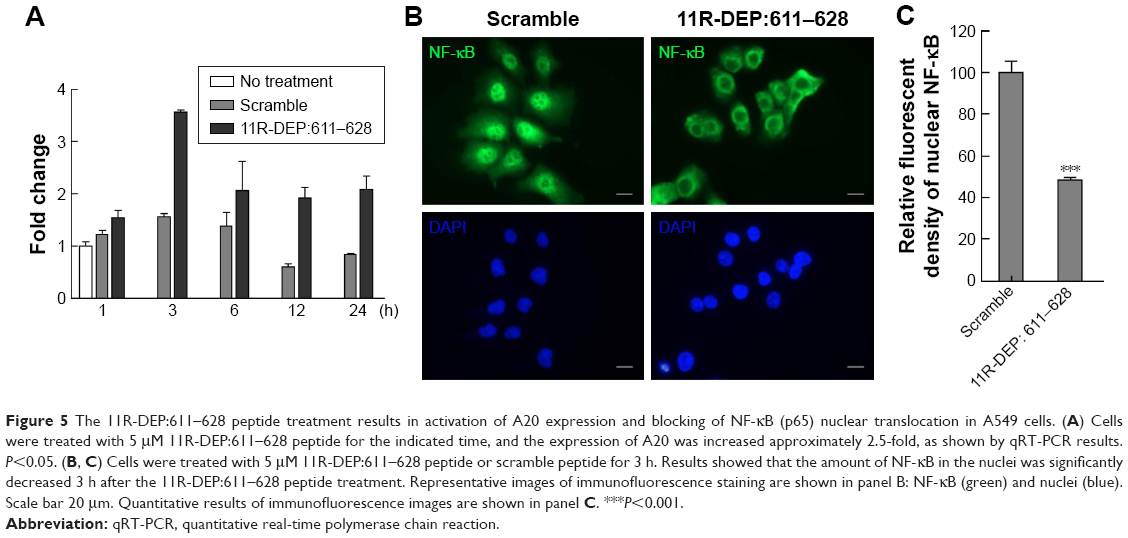 | Figure 5 The 11R-DEP:611–628 peptide treatment results in activation of A20 expression and blocking of NF-κB (p65) nuclear translocation in A549 cells. (A) Cells were treated with 5 μM 11R-DEP:611–628 peptide for the indicated time, and the expression of A20 was increased approximately 2.5-fold, as shown by qRT-PCR results. P<0.05. (B, C) Cells were treated with 5 μM 11R-DEP:611–628 peptide or scramble peptide for 3 h. Results showed that the amount of NF-κB in the nuclei was significantly decreased 3 h after the 11R-DEP:611–628 peptide treatment. Representative images of immunofluorescence staining are shown in panel B: NF-κB (green) and nuclei (blue). Scale bar 20 μm. Quantitative results of immunofluorescence images are shown in panel C. ***P<0.001. |
11R-DEP:611–628 peptide treatment results in activation of JNK in A549 cells
A study reported that, in HeLa and MCF7 cells, DEPDC1 mediates vincristine-induced apoptosis via promoting JNK-dependent MCL1 degradation.10 Therefore, we reasoned that the 11R-DEP:611–628 peptide might induce apoptosis of A549 cells via regulating JNK activity. To test this idea, we treated A549 cells with the 11R-DEP:611–628 peptide and examined JNK activity by detecting the levels of p-JNK. The results showed that cells treated with the 11R-DEP:611–628 peptide had obviously increased protein levels of p-JNK (Figure 6A), indicating that JNK is activated upon treatment and JNK is inhibited by DEPDC1 in A549 cells, which is the opposite of previous observations in HeLa and MCF-7 cells.10 To further confirm these findings and reveal the role of JNK in apoptosis of A549 cells, we treated A549 cells by combining JNK inhibitor (SP600125; 10 μM) and the 11R-DEP:611–628 peptide (5 μM), and then examined apoptosis with flow cytometry assay. The results showed that SP600125 could induce apoptosis in A549 cells. Moreover, SP600125 enhanced 11R-DEP:611–628 peptide-induced apoptosis (Figure 6B and C). These findings imply that JNK plays an antiapoptotic role in A549 cells.
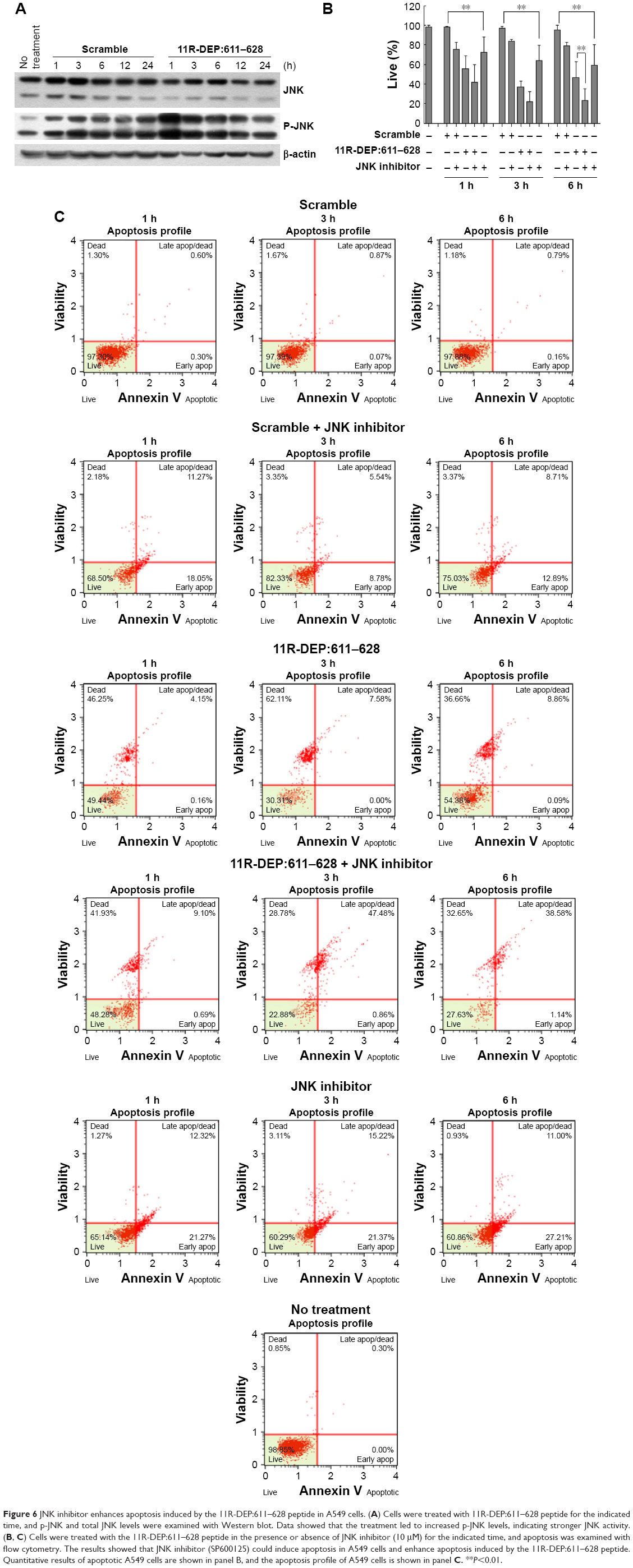 | Figure 6 JNK inhibitor enhances apoptosis induced by the 11R-DEP:611–628 peptide in A549 cells. (A) Cells were treated with 11R-DEP:611–628 peptide for the indicated time, and p-JNK and total JNK levels were examined with Western blot. Data showed that the treatment led to increased p-JNK levels, indicating stronger JNK activity. (B, C) Cells were treated with the 11R-DEP:611–628 peptide in the presence or absence of JNK inhibitor (10 μM) for the indicated time, and apoptosis was examined with flow cytometry. The results showed that JNK inhibitor (SP600125) could induce apoptosis in A549 cells and enhance apoptosis induced by the 11R-DEP:611–628 peptide. Quantitative results of apoptotic A549 cells are shown in panel B, and the apoptosis profile of A549 cells is shown in panel C. **P<0.01. |
Discussion
Lung cancer is a serious health threat for people worldwide, and discovery of novel therapeutic targets and drugs for lung cancer is imperative. In this study, we investigated the role of DEPDC1 and the mechanism by which DEPDC1 participates in carcinogenesis in A549 by using miR-130a and the 11R-DEP:611–628 peptide as interfering tools. Our results demonstrated that DEPDC1 is expressed in all of the examined lung cancer cell lines, and that miR-130a and 11R-DEP:611–628 peptide both are able to inhibit A549 cell proliferation and promote apoptosis. Our data showed that, in A549 cells, the 11R-DEP:611–628 peptide could induce A20 expression and block nuclear translocation of NF-κB. The results also showed that the 11R-DEP:611–628 peptide treatment led to increased JNK phosphorylation, and JNK inhibitor could enhance apoptosis induced by the 11R-DEP:611–628 peptide.
It has been a decade since DEPDC1 was first reported to be involved in bladder carcinogenesis, but the role of DEPDC1 in lung cancer remains unknown although a study revealed that the DEPDC1 level is elevated in EGFR/KRAS/ALK-negative lung adenocarcinomas.13 In bladder cancer cells, DEPDC1 regulates cell proliferation via forming a functional complex to suppress the expression of A20, an NF-κB inhibitory and antiapoptotic signaling protein, leading to abnormal NF-κB activation and translocation into the nucleus, where expression of antiapoptotic proteins is activated by NF-κB.9 To reveal the role of DEPDC1 and the underlying mechanism, we used the 11R-DEP:611–628 peptide, which is capable of specifically disrupting physical interaction between DEPDC1 and ZNF224, to treat cells and observed elevated A20 and decreased nuclear NF-κB expression. These findings not only indicate that, in A549 cells, DEPDC1 functions by forming a complex with ZNF224 to suppress A20 expression and ultimately regulates the function of NF-κB, but also provides evidence to support the previously proposed mechanism by which DEPDC1 regulates apoptosis. More importantly, the data suggest that the DEPDC1–ZNF224 complex is a potential therapeutic target for lung cancer where DEPDC1 is expressed, and the 11R-DEP:611–628 peptide is a promising therapeutic candidate. Because DEPDC1 expression is increased in multiple tumor tissues, it is worth investigating whether DEPDC1 functions by the same mechanism in other types of cancer and whether the 11R-DEP:611–628 peptide works effectively in other cancer cells. Because A20 is the only defined target gene regulated by the DEPDC1–ZNF224 complex, it is worth exploring whether any other genes are regulated by the DEPDC1–ZNF224 complex, and whether and how they are involved in carcinogenesis.
A previous study observed that DEPDC1 is involved in vincristine-induced apoptosis of HeLa and MCF-7 cells.10 The cellular response to vincristine was reduced if treated with DEPDC1 siRNA, but increased if DEPDC1 was overexpressed. The underlying mechanism is that DEPDC1 promotes JNK-dependent degradation of MCL1. We, therefore, examined JNK activity in A549 cells by detecting JNK phosphorylation upon treatment with the 11R-DEP:611–628 peptide. To our surprise, the results showed that JNK phosphorylation increased, indicating that disrupting the DEPDC1–ZNF224 complex leads to JNK activation. Moreover, JNK inhibitor SP600125 could enhance apoptosis induced by 11R-DEP:611–628 peptide, implying that DEPDC1 inhibits JNK activity and JNK prevents cells from apoptosis under this circumstance. These data raise an argument against the previous study as to the relationship between DEPDC1 and JNK in HeLa and MCF-7 cells,10 but are consistent with another report with regard to the protective role of JNK in A549 cells.19
A recent study reported that overexpression of miR-130a reduces cell viability and increases apoptosis by downregulating the expression of DEPDC1 and SEC23B in prostate cancer cells.17 Our data suggest miR-130a also plays a key role in regulating proliferation of A549 cells. Given that miR-130a has multiple targets in mammalian cells, we may draw a conclusion cautiously that miR-130a fulfills its function in A549 partially via regulating the DEPDC1 because we are unable to rule out whether miR-130a modulates cellular function through other targets. In addition, our findings suggest miR-130a might be a potential therapeutic factor for lung cancers with DEPDC1 expression.
In summary, our current study for the first time reveals the role of DEPDC1 and the mechanism by which DEPDC1 participates in carcinogenesis of lung cancer. Moreover, the study demonstrates in vitro that DEPDC1 is a therapeutic target for lung cancer, and the 11R-DEP:611–628 peptide and miR-130a are potential therapeutic agents for lung cancer with DEPDC1 expression, although further in vivo evidence is needed to confirm our findings.
Acknowledgments
The present study was supported in part by the National Natural Science Foundation of China (grant no 81560453), the Natural Science Foundation of Guangxi (grant no 2015GXNSFAA139178), the Guangxi Health and Family Planning Commission (grant no S2015-34), the Lijiang Scholar Award (to JJ), the Innovation Project of Guangxi Graduate Education (grant no YCSW 2017209), and the ‘Sphingolipids and Related Diseases’ Program for Innovative Research Team of Guilin Medical University (to JJ). GH was supported by the Hundred Talents Program “The Introduction of Overseas High-Level Talents in Colleges and Universities in Guangxi”.
Disclosure
The authors report no conflicts of interests in this work.
References
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2017. CA Cancer J Clin. 2017;67(1):7–30. | ||
Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65(2):87–108. | ||
Brambilla E, Travis WD, Brennan P, et al. Lung cancer. In: Stewart B, Wild CP, editors. World Cancer Report 2014. The International Agency for Research on Cancer; 2014:489–508; Lyon, France. | ||
Swanton C, Govindan R. Clinical implications of genomic discoveries in lung cancer. N Engl J Med. 2016;374(19):1864–1873. | ||
Siegel R, DeSantis C, Virgo K, et al. Cancer treatment and survivorship statistics, 2012. CA Cancer J Clin. 2012;62(4):220–241. | ||
Ponting CP, Bork P. Pleckstrin’s repeat performance: a novel domain in G-protein signaling? Trends Biochem Sci. 1996;21(7):245–246. | ||
Kharrat A, Millevoi S, Baraldi E, Ponting CP, Bork P, Pastore A. Conformational stability studies of the pleckstrin DEP domain: definition of the domain boundaries. Biochim Biophys Acta. 1998;1385(1):157–164. | ||
Kanehira M, Harada Y, Takata R, et al. Involvement of upregulation of DEPDC1 (DEP domain containing 1) in bladder carcinogenesis. Oncogene. 2007;26(44):6448–6455. | ||
Harada Y, Kanehira M, Fujisawa Y, et al. Cell-permeable peptide DEPDC1-ZNF224 interferes with transcriptional repression and oncogenicity in bladder cancer cells. Cancer Res. 2010;70(14):5829–5839. | ||
Sendoel A, Maida S, Zheng X, et al. DEPDC1/LET-99 participates in an evolutionarily conserved pathway for anti-tubulin drug-induced apoptosis. Nat Cell Biol. 2014;16(8):812–820. | ||
Kretschmer C, Sterner-Kock A, Siedentopf F, Schoenegg W, Schlag PM, Kemmner W. Identification of early molecular markers for breast cancer. Mol Cancer. 2011;10(1):15. | ||
Yuan SG, Liao WJ, Yang JJ, Huang GJ, Huang ZQ. DEP domain containing 1 is a novel diagnostic marker and prognostic predictor for hepatocellular carcinoma. Asian Pac J Cancer Prev. 2014;15(24):10917–10922. | ||
Okayama H, Kohno T, Ishii Y, et al. Identification of genes upregulated in ALK-positive and EGFR/KRAS/ALK-negative lung adenocarcinomas. Cancer Res. 2012;72(1):100–111. | ||
Huang G, Kim JY, Dehoff M, et al. Ca2+ signaling in microdomains: Homer1 mediates the interaction between RyR2 and Cav1.2 to regulate excitation-contraction coupling. J Biol Chem. 2007;282(19):14283–14290. | ||
Liao W, Huang G, Liao Y, et al. High KIF18A expression correlates with unfavorable prognosis in primary hepatocellular carcinoma. Oncotarget. 2014;5(21):10271–10279. | ||
Huang G, Yao J, Zeng W, et al. ER stress disrupts Ca2+-signaling complexes and Ca2+ regulation in secretory and muscle cells from PERK-knockout mice. J Cell Sci. 2006;119(Pt 1):153–161. | ||
Ramalho-Carvalho J, Martins JB, Cekaite L, et al. Epigenetic disruption of miR-130a promotes prostate cancer by targeting SEC23B and DEPDC1. Cancer Lett. 2017;385:150–159. | ||
Nicholson DW, Ali A, Thornberry NA, et al. Identification and inhibition of the ICE/CED-3 protease necessary for mammalian apoptosis. Nature. 1995;376(6535):37–43. | ||
Okada M, Shibuya K, Sato A, et al. Specific role of JNK in the maintenance of the tumor-initiating capacity of A549 human non-small cell lung cancer cells. Oncol Rep. 2013;30(4):1957–1964. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.