Back to Journals » International Journal of Nanomedicine » Volume 12
Targeted delivery of ginsenoside compound K using TPGS/PEG-PCL mixed micelles for effective treatment of lung cancer
Authors Yang L, Zhang Z, Hou J, Jin X ![]() , Ke Z, Liu D, Du M, Jia X, Lv H
, Ke Z, Liu D, Du M, Jia X, Lv H ![]()
Received 19 June 2017
Accepted for publication 9 August 2017
Published 17 October 2017 Volume 2017:12 Pages 7653—7667
DOI https://doi.org/10.2147/IJN.S144305
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Linlin Sun
Lei Yang,1,2 Zhenghai Zhang,1 Jian Hou,1,2 Xin Jin,1 Zhongcheng Ke,1 Dan Liu,1 Mei Du,1 Xiaobing Jia,1,2 Huixia Lv3
1Key Laboratory of New Drug Delivery System of Chinese Materia Medica, Jiangsu Provincial Academy of Chinese Medicine, Jiangsu, Nanjing, China; 2College of Pharmacy, Jiangsu University, Jiangsu, Zhenjiang, China; 3Department of Pharmaceutics, State Key Laboratory of Natural Medicines, China Pharmaceutical University, Jiangsu Sheng, China
Abstract: Ginsenoside compound K (CK) is one of the effective ingredients in antitumor composition of ginsenoside. However, the poor water solubility and significant efflux have limited the widespread clinical use of CK. In this study, preparation of novel CK-loaded D-alpha-tocopheryl polyethylene glycol 1,000 succinate/poly(ethylene glycol)-poly(ε-caprolactone) mixed micelles (CK-M) is discussed to solve the above problems. Particle size, zeta potential, and morphology were characterized using dynamic light scattering and transmission electron microscopy. CK-M are spherical shaped with an average particle size of 53.07±1.31 nm with high drug loading of 11.19%±0.87% and entrapment efficiency of 94.60%±1.45%. Water solubility of CK was improved to 3.78±0.09 mg/mL, which was ~107.35 times higher than free CK. A549 and PC-9 cells were used to evaluate in vitro cytotoxicity and cellular uptake. IC50 values of CK-M in A549 and PC-9 cells (24 h) were 25.43±2.18 and 18.35±1.90 µg/mL, respectively. Enhanced cellular uptake of CK-M was observed in both cells. Moreover, CK-M promoted tumor cell apoptosis, inhibited tumor cell invasion, metastasis, and efflux through regulation of Bax, Bcl-2, matrix metalloproteinase-2, Caspase-3, and P-glycoprotein. In vivo imaging indicated that CK-M has excellent tumor targeting effect within 24 h, and the relative tumor inhibition rate of CK-M was 52.04%±4.62% compared with control group (P<0.01). Thus, CK-M could be an appropriate delivery agent for enhanced solubility and antitumor effect of CK.
Keywords: ginsenoside compound K, TPGS, PEG-PCL, mixed micelles, antitumor, non-small cell lung cancer
Introduction
Ginsenoside compound K (20-O-beta-D-glucopyranosyl-20(S)-protopanaxadiol, CK), one of the major metabolites of ginseng, has been identified to possess excellent biological activities such as antiaging, anti-inflammatory, antioxidant, and protecting cardiac muscle cells.1,2 Furthermore, CK was the strongest antitumor agent of the ginsenosides.3 Various tumor cells are inhibited by CK, including human colorectal cancer cells, human hepatoblastoma HepG2 cells, human breast cancer MCF-7 cells, and lung adenocarcinoma A549 cells.4–6 CK acts by various antitumor mechanisms including inhibition of tumor cell proliferation, promotion of tumor cell apoptosis, and inhibition of tumor cell invasion and metastasis.7,8
Although appreciable antitumor activity of CK has been reported, its poor water solubility and strong efflux greatly reduce the efficacy of CK.9 In recent years, various nanodelivery systems, such as CK-bearing glycol chitosan conjugates and PEG-CK, have been used to improve the limitations associated with CK.10,11 However, the lower drug loading (DL) and P-glycoprotein (P-gp) efflux are still restricting the development of CK nanopreparation.
Polymeric micelles are one of the most effective delivery systems due to their unique core–shell structure that wraps the poorly soluble drug in a hydrophobic core while the hydrophilic group is outward.12 Moreover, the size of polymeric micelles is <200 nm. These characteristics could protect the drug against in vivo degradation and from being metabolized, enhance stability and the permeability and retention (EPR) effect, avoid being swallowed up by the mononuclear phagocytic system, and improve the blood circulation time.13
Poly(ethylene glycol)-poly(ε-caprolactone) (PEG-PCL) are amphiphilic block copolymers with good biodegradability and biocompatibility that can be used for the production of polymeric micelles. The structure of PEG-PCL is different from that of membrane and condensed-single polymers and is, which is more dense, stable, and easier to degrade.14 The hydrophilic group PEG (molecular weight: 5,000) can be easily hydrated and PCL (molecular weight: 5,000) is the hydrophobic core, and thus, water cannot cross into the inner part in short times.15 Furthermore, longer the length of PCL segments, slower the degradation rate and more stable the micelles will be.16 In vivo, the flexible hydrophilic PEG shell prevents absorption of micelles by the reticuloendothelial system (RES), which increases drug stability and circulation time in vivo, and the drug in the hydrophobic nucleus diffuses slowly to achieve sustained release.17
However, there is no inhibition of P-gp by PEG-PCL, which could greatly restrict its use in the development of mixed micelles containing drugs subject to P-gp efflux. D-alpha-Tocopheryl polyethylene glycol 1,000 succinate (TPGS) has been used in various drug delivery systems, including micelles, liposomes, nanoparticles, and prodrugs,18–20 and can enhance the solubility, permeability, and stability of insoluble drugs. TPGS is also widely used to inhibit drugs being pumped out from cells by P-gp.21 P-gp is an adenosine triphosphate (ATP)-dependent drug efflux transporter. Once P-gp combines with drugs, the drug is pumped from inside the cell with energy provided by ATP, and the concentration of the drug in the cytoplasm decreases, resulting in a weak antitumor effect of the drug and removal of the drug from the cell.22 TPGS could inhibit P-gp efflux by blocking ATP that provides energy for this activity, and thus, improve the antitumor effect of a drug.
A large number of recent studies on mixed micelles show that mixed micelles have obvious advantages over single copolymer micelles.23 The loading content and stability of the drug are improved by mixed micelles. Furthermore, mixed micelles with different kinds of copolymers have some new functions that single micelles do not have. For example, the low hydrophobicity and encapsulation efficiency (EE) of TPGS can be overcome by PEG-PCL while P-gp inhibition can be contributed by TPGS in PEG-PCL self-assembly micelles.
In this study, we have used TPGS to modify PEG-PCL and then prepared CK-loaded TPGS/PEG-PCL mixed micelles (CK-M) to carry the antitumor CK. The thin-film hydration method was used to make the CK-M, and the micelles were characterized. The non-small cell lung cancer (NSCLC) A549 and PC-9 cells were used as an in vitro model to investigate the cytotoxicity, cellular uptake, pro-apoptotic effects, and inhibition of migration and invasion effects of CK-M. In vivo imaging of tumor-bearing mice evaluated the in vivo antitumor efficacy.
Materials and methods
Materials
CK of >98% purity was purchased from Nanjing Jingzhu Biotechnology Co., Ltd. (Nanjing, China). PEG-PCL (5,000:5,000) was purchased from Xi’an Ruixi Biological Technology Co., Ltd. (Xi’an, China). TPGS was purchased from Aladdin Industrial Co., Ltd. (Shanghai, China). 3-(4, 9,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazoliumbromide (MTT) and dimethyl sulfoxide (DMSO) were purchased from Nanjing Sunshine Biotechnology Co., Ltd. (Nanjing, China). Coumarin-6 and 4′,6-diamidino-2-phenylindole (DAPI) were purchased from Sigma (Shanghai, China). 1,1′-Dioctadecyl-3,3,3′,3′-tetramethylindotricarbocyanine iodide (DiR) was purchased from Nanjing KeyGen Biotech Co., Ltd. (Nanjing, China). All reagents were of analytical grade except methanol, which was of chromatographic grade.
Animals
Male athymic nude mice (22±2 g) were obtained from Changzhou Cavens Lab Animal Co., Ltd. (Changzhou, China, SCXK2011-0003). All mice were given distilled water and kept at a temperature of 25°C±0.5°C, relative humidity of 45%±5%, and 12 h/12 h light–dark cycle. All animal experiments were performed in accordance with protocols evaluated and approved by the Institutional Animal Care and Use Committee (IACUC), Jiangsu Provincial Academy of Chinese Medicine’s Experimental Animal Center.
Preparation of drug-loaded micelles
CK-M were prepared using the method of thin-film hydration. Different proportions of PEG-PCL and TPGS were co-dissolved in dichloromethane (5 mL); then, 20 mg of CK dissolved in ethanol was dropped in it and mixed via ultrasonic oscillation. The liquid was removed to form a dry drug-containing thin film under vacuum rotary evaporation. Additionally, the film was hydrated with 5 mL of water, filtered through a 0.45 μm membrane filter to remove the non-incorporated CK, and the CK-M were successfully prepared.
The CK-loaded PEG-PCL micelles (CK-P), coumarin-6-loaded PEG-PCL micelles (C-P), coumarin-6-loaded mixed micelles (C-M), DiR-loaded micelles, and black micelles were prepared similarly.
Characterization of drug-loaded micelles
The determination of size, zeta potential, and morphology analysis
The size and zeta distribution of CK-M were examined using dynamic light scattering by using a Malvern system (ZEN-3600; Malvern Instruments, Worcestershire, UK). Transmission electron microscopy (TEM; JEM-200CX, JEOL, Tokyo, Japan) was used to determine the morphology of CK-M.
Drug content and entrapment efficiency
The concentration of CK contained by the micelles with core–shell structure was measured using high-performance liquid chromatography (HPLC) at 203 nm (Agilent LC1260; Agilent Technologies; Santa Clara, CA, USA). In brief, after filtration with a 0.45 μm microporous membrane, 200 μL of CK-M were dissolved in 800 μL of methanol to disrupt its structure.24 DL% and EE% are calculated using the following equations:
|
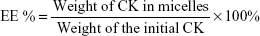
|
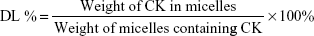
In vitro release study
The in vitro release rate of CK from CK-M was measured using dialysis and HPLC.25 Dialysis tubing (10 cm), soaked in deionized water for 10 min, was washed three times using fresh deionized water. Free CK and CK-M containing 4 mg of CK were placed into the dialysis bag (molecular weight cut-off of 3,500 Da; Green Bird Inc., Shanghai, China).26 The tubing was placed into 100 mL of dialysis medium containing 0.5% Tween 80 and stirred at 100 rpm/min at 37°C (pH =7.4). One milliliter of the release medium was removed and the same volume of fresh buffer was added at 0, 1, 2, 3, 4, 6, 8, 20, 24, and 48 h. The concentration of CK in CK-M at each time point was determined using HPLC. All measurements were performed in triplicate.
In vitro cytotoxicity analysis
MTT assay
The cell viability of CK and CK-M was measured using the MTT bioassay. A549 and PC-9 cells originate from the same NSCLC cell line; therefore, either of them can be used to determine the cell viability of CK-M. In brief, cells were seeded into 96-well plates at 5×103 cells/well with Dulbecco’s Modified Eagle’s Medium (DMEM) containing 10% fetal bovine serum (FBS) for 24 h (37°C, 5% CO2).26 The spent medium was discarded and substituted with 100 μL of fresh DMEM with CK or CK-M of different concentrations (100, 50, 25, 12.5, 6.25, and 3.125 μg/mL) while the culture medium without FBS was used as the control group. After incubation for 24 h, the drug-containing medium was replaced by 10% MTT solution (50 μg/μL). Then, 100 μL of DMSO was added to each well and oscillated for 10 min to dissolve the MTT formazan generated by live cells after incubation for 4 h. The relative cell viability (%) was measured by comparing the absorbance of each well at 490 nm with the control well by using a microplate reader (Bio-Rad Laboratories, Hercules, CA, USA). All experiments were performed in triplicate. The cell viability was calculated using the following equation:
|
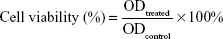
ODtreated and ODcontrol represented the absorbance of sample and control, respectively.
In vitro cellular uptake
The in vitro cellular uptake of drug-loaded micelles was measured using a fluorescence microscope. A549 and PC-9 cells were seeded into six-well plates at a concentration of 1×105 cells/chamber and incubated overnight. The spent medium was removed and replaced by C-P or C-M solution (the concentration was calculated as free coumarin-6). After culturing for 4 h, the cells were washed three times by using cool PBS (pH =7.4) to prevent the coumarin-6 entering the cells. Then, the cells were fixed with 4% formaldehyde for 20 min and washed twice with PBS buffer.27 DAPI was added for 15 min to stain the nuclei (100 μL per well), and the cells were washed again with PBS and observed under the microscope.
In vitro migration assay
The migration of A549 and PC-9 cells treated with CK and CK-M was assessed by the cell scratch test. Cells were suspended in DMEM without FBS and seeded into a six-well plate (5×105 cells/well). A wound area was drawn in the center of each well using 20 μL pipette tips and washed twice with PBS to remove floating cells. Fresh DMEM, CK, and CK-M solutions were added to the six-well plate. After incubation for 0 and 24 h, the plate was observed and photographed under an inverted microscope. The experiment was repeated three times.
In vitro invasion assay
A549 and PC-9 cells were suspended in serum-free DMEM and added to the upper chamber of the Transwell (1×105 cell/200 μL), and CK and CK-M solutions (20 μg/mL) were then added to it. Next, DMEM (600 μL) with 5% FBS was added to the lower chamber of the Transwell. Following 24 h of incubation, the upper chamber was removed, wiped with a cotton swab, and methanol was added for 3 min to fix cells. After removing the fixation liquid, 0.1% crystal violet dye was added and the cells were stained for 20 min. The microporous filtering film of the upper chamber was washed three times with distilled water, and the cells were observed and counted under the microscope.
|
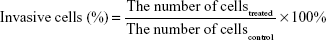
Cell cycle analysis
Cells were seeded at 2×105 cells/chamber into a six-well plate and incubated for 24 h (37°C, 5% CO2). CK or CK-M solution was added to each well. Following 24 h incubation, the cells were harvested by trypsinization and washed twice using PBS. Then, the cells were fixed with 70% ethanol overnight, washed again using PBS, and 50 μg/mL of propidium iodide (PI) was added.28 The cell cycle stage was detected using flow cytometry after cells were kept at 25°C room temperature for 30 min.
Detection of apoptosis
The flow cytometry assay was used to quantify apoptosis of A549 and PC-9 cells. Cells (1×105 cells/chamber) were seeded into a six-well plate. After 24 h at 37°C and 5% CO2, CK or CK-M solution was added to each well. Following 24 h of incubation, the cells were harvested using trypsinization and washed twice with PBS. According to kit instructions, the cells were resuspended in 250 μL of binding buffer at a concentration of 1×106 cells/mL, and then 5 μL of Annexin V-EGFP (enhanced green fluorescent protein) and PI were added, mixed, and incubated for 5–15 min at 25°C.29 Flow cytometry was used to detect the results of the assay after 1 h.
Western blot analysis
A549 and PC-9 cells were seeded into 24-well plates at a concentration of 1×105 cells/well for 24 h and treated with CK and CK-M for 24 h (concentration of CK was 20 μg/mL). Cells were collected and total protein was extracted. The protein extract was subjected to sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE), and then a PVDF (polyvinylidenedifluoride) membrane was dipped in transfer buffer at 4°C for 1.5 h. The film was blocked with 5% dried milk protein for 1 h.30 The primary antibodies (anti-Bax, anti-Bcl-2, anti-matrix metalloproteinase [MMP]-9, anti-Caspase-3, and anti-P-gp) were added at dilutions of 1:600, 1:1,000, 1:500, 1:500, and 1:200, respectively, and then incubated overnight (4°C). The samples were washed twice with triethanolamine-buffered saline (TBS) and incubated with the secondary antibody at the dilution of 1:3,000 for 1 h. The blots were visualized using electrochemiluminescence (ECL; Beyotime Biotechnology Co., Ltd., Shanghai, China) and the experiment was repeated twice.
In vivo imaging
Magnetic resonance imaging (NightOWLII LB983; Berthold, Germany) was used to investigate the targeting effect of DiR-loaded micelles. After the size of the tumors reached 60 mm3, DiR-loaded micelles were injected into the blood stream via the tail vein of the tumor-bearing mice at a dose of 5 mg/kg. Following anesthetization, the mice were imaged at 0, 1, 2, 4, 6, 8, and 24 h.
In vivo antitumor activity
A 100 μL of single-cell suspension containing 1×106 A549 cells in DMEM with 10% FBS was introduced under the right armpit of the nude mice subcutaneously. When the tumor size reached 60 mm3, the male athymic nude mice (22±2 g) were randomly divided into four groups (n=6): control group for treatment with saline solution, positive control group for treatment with cisplatin (2 mg/kg), and one each for treatment with CK (15 mg/kg) and CK-M (15 mg/kg) solutions. Mice were injected with the treatment drug through the tail vein one time every 3 days for 15 days. After the last administration, the mice were given euthanasia and picture of the tumor was taken. The tumor volume (V) is calculated as: V = a × b2/2, where a and b are length and width of the tumor, respectively. The relative tumor volume (RTV) was calculated according to the above measurement, and the relative tumor growth rate (T/C) was used to evaluate the antitumor activity.31
|
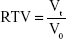
|
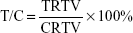
where V0 is the tumor size at 0 day, and Vt is the tumor volume at each measurement. TRTV is the RTV of the treatment groups and CRTV is the RTV of the control group.
Statistical analysis
Results are reported as mean ± SD. Graphpad Prism 6.0 (GraphPad Software, La Jolla, CA, USA) was used for drawing graphs. SPSS13.0 software (SPSS Inc., Chicago, IL, USA) was used for the statistical analysis. Differences between two groups were assessed using Student’s t-test. Statistically significant was defined as P-values <0.05.
Results and discussion
Mixed micelle formulation and characterization
A range of PEG-PCL and TPGS in different ratios was used to find the suitable proportion for preparing the mixed micelles. The average size, polydispersity index (PDI), DL%, EE%, and solubility are shown in Table 1. The results show that the EE% of CK-P was much lower than that of CK-M, and the average size and PDI increased with increased proportion of TPGS, indicating that the DL% and EE% of CK improved greatly by using TPGS/PEG-PCL micelles. The most appropriate ratio of PEG-PCL to TPGS was 3:2.
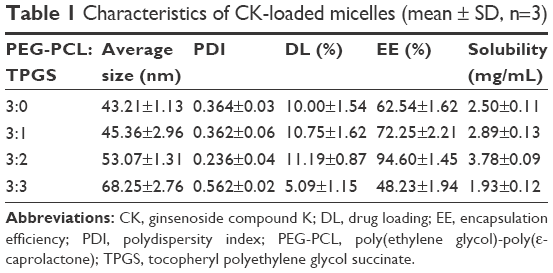 | Table 1 Characteristics of CK-loaded micelles (mean ± SD, n=3) |
The abovementioned ratio of PEG-PCL to TPGS was used to formulate CK-M. The concentration of CK in the mixed micelles was 3.78±0.09 mg/mL, which was a 107.35-fold increase than CK. The average size was 53.07±1.31 nm, the distribution profile (Figure 1A) with corresponding PDI was 0.236±0.04, and zeta potential was −8.0 mV. TEM showed that CK-M was homogeneous with spherical morphology (Figure 1B). HPLC analysis showed that EE% and DL% of CK-M were 94.60%±1.45% and 11.19%±0.87%, respectively. Moreover, it was found that the critical micelle concentration (CMC) value of the micelles formed by two excipients is lower than that of the single-excipient micelles. Micelles with lower CMC could slow down the release of the drug in vivo and achieve a long cycle.32 Moreover, when amphiphilic polymers are dissolved in aqueous solution, the hydrophilic groups orient on the outside, while the hydrophobic groups are facing inside. The stable system was formed. This stable system can effectively improve system stability and drug solubility. This might be relative to the appropriate proportion of hydrophobic PEG and hydrophilic PCL.33 The hydrophobic interaction would be in favor of increasing solubility of CK, but when the length of the hydrophobic PCL exceeds a limit, it would lead to decreasing the solubility of CK. Thus, CK-M was successfully prepared, and the small size of the micelles could enhance CK concentration in tumor tissue.
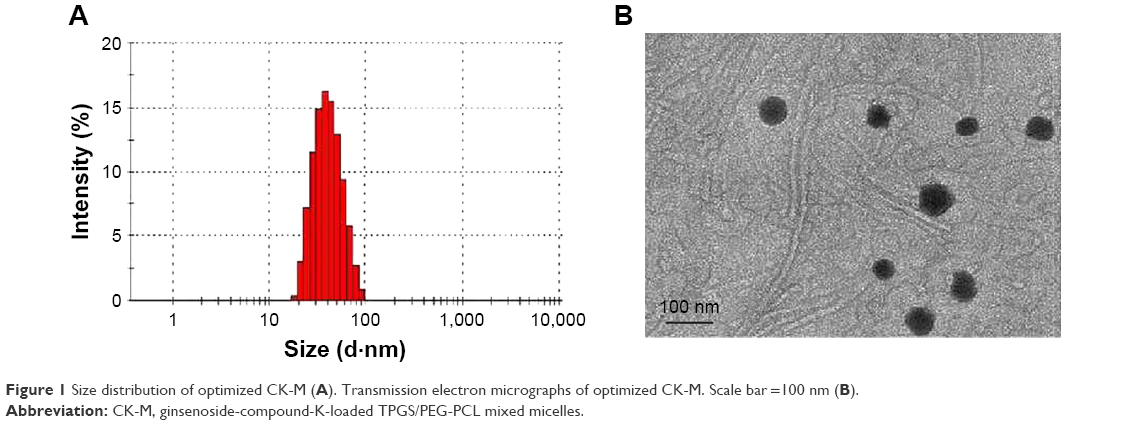 | Figure 1 Size distribution of optimized CK-M (A). Transmission electron micrographs of optimized CK-M. Scale bar =100 nm (B). |
In vitro release of CK-loaded mixed micelles
The in vitro release curves of free CK and CK-M are shown in Figure 2. To improve the solubility of CK, 0.5% Tween 80 was added to the dialysis medium. In the first 8 h, >50% of CK from free CK was burst released. CK from CK-M was released slowly within 48 h without a burst release, and the drug release percentage of CK-M was >42.13%±3.23% after 48 h. Moreover, the release percentage of CK was 84.48%±4.26%. The different release rates show that the TPGS/PEG-PCL mixed micelles could improve the solubility and delay release of CK, and hence, may increase its blood circulation time. The slow release of drug from CK-M was attributed to dense micelles structure, which retains the wrapped CK in the hydrophobic core, while water is difficult to enter inside at a short time.
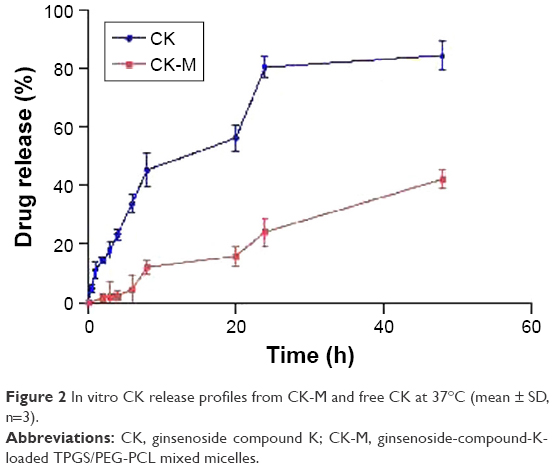 | Figure 2 In vitro CK release profiles from CK-M and free CK at 37°C (mean ± SD, n=3). |
In vitro cytotoxicity of micelles
MTT assay
The MTT assay was performed to investigate the in vitro cytotoxic activity of CK-M against A549 and PC-9 cells in 24 h. The IC50 values of CK and CK-M in A549 cells were 21.97±1.50 and 25.43±2.18 μg/mL, respectively (Figure 3A). The IC50 values of CK and CK-M in PC-9 cells were 14.46±1.24 and 18.35±1.90 μg/mL, respectively (Figure 3B). These results show that CK-M has an obvious inhibitory effect on A549 and PC-9 cells. However, the viability (%) of both cell types with CK-M was lower than that with CK; this may be because of the slower release from CK-M which resulted in a lower number of cells.33 Free CK could possibly pass through the cells quickly by a passive-diffusion mechanism, while CK-M is transported into cells through endocytosis and shows a delayed release in cells. In vitro release results also support the above results. Additionally, blank TPGS/PEG-PCL are noncytotoxic to A549 and PC-9 cells.
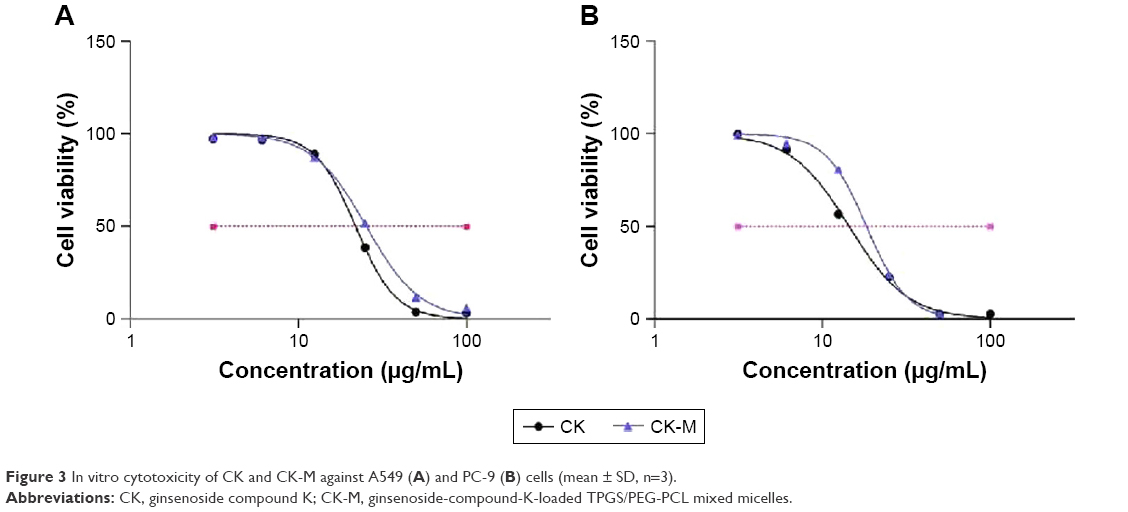 | Figure 3 In vitro cytotoxicity of CK and CK-M against A549 (A) and PC-9 (B) cells (mean ± SD, n=3). |
In vitro cellular uptake
The in vitro cellular uptake of coumarin-6 control and C-M in A549 and PC-9 cells are shown in Figure 4A and B. After excitation, the coumarin-6 dye in the cells has green fluorescence while the DAPI in the cell nuclei has blue fluorescence. It can be directly observed that the green fluorescence intensities of C-P in both cells were similar, while that of the coumarin-6 control and C-M were stronger than that of C-P. It has been reported that coumarin-6 can be used as a hydrophobic drug model to replace CK to investigate the cellular uptake of CK-M.34 The results show that the uptake of coumarin-6 by A549 and PC-9 cells was enhanced by coumarin-6 mixed micelles modified with TPGS, which may be due to the P-gp inhibition of TPGS in the mixed micelles.
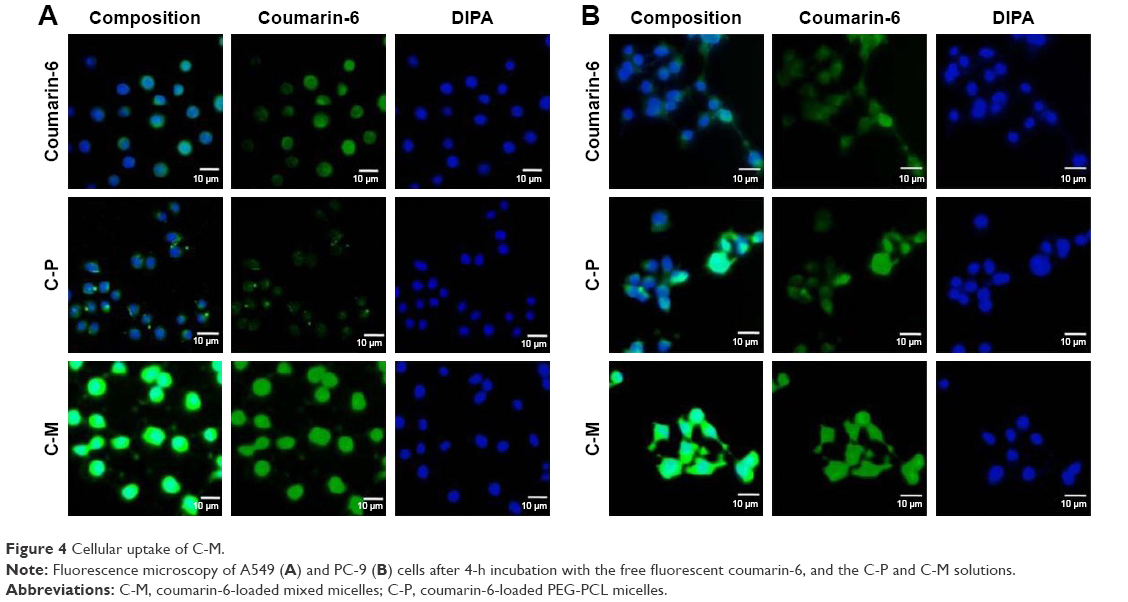 | Figure 4 Cellular uptake of C-M. |
Inhibitory effect of CK-M on cell invasion and migration
The inhibitory effect of CK and CK-M on cell migration was investigated by comparing the number of A549 and PC-9 cells at the scratch wound for 0 and 24 h (Figure 5). In the following experiments, 21.97 and 14.46 μg/mL of free CK were applied to A549 and PC-9 cells, respectively. After 24 h, the cells of both control groups migrated to the center of the scratch on the plate, while in the CK and CK-M treatment groups only a few cells migrated to the scratch, indicating that both CK and CK-M inhibit cell migration and the effect of CK can be enhanced by CK-M.
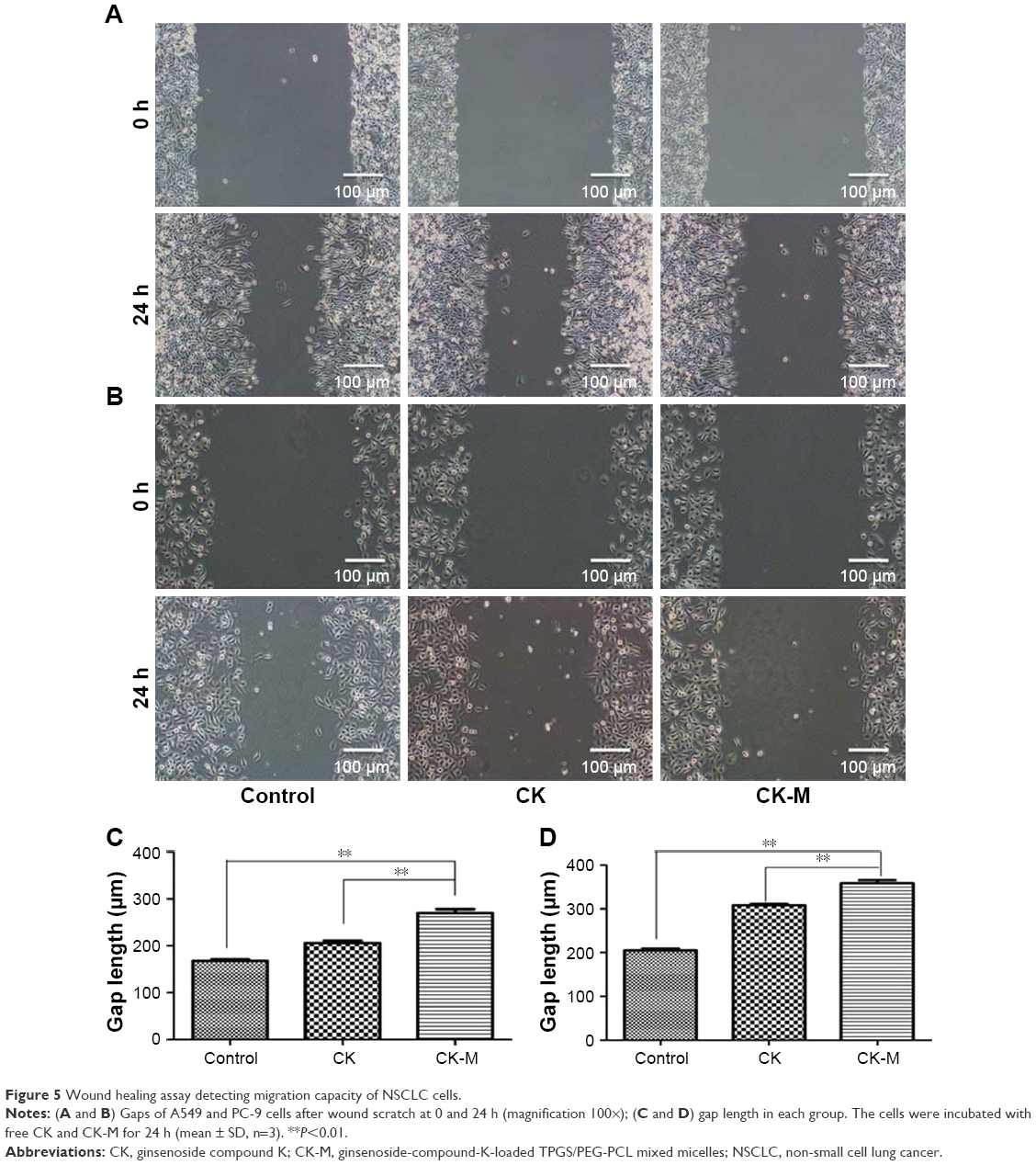 | Figure 5 Wound healing assay detecting migration capacity of NSCLC cells. |
The number of A549 and PC-9 cells through the matrix glue was representative of the inhibition of cell invasion ability after CK and CK-M treatment. After 24 h of treatment, cells were stained with crystal violet and the numbers of A549 and PC-9 cells were counted. The numbers of cells after CK-M treatment were 28.33±2.52 and 29.33±2.08 for A549 and PC-9 cells, respectively. As shown in Figure 6, free CK has a certain inhibitory effect on cell invasion, compared with control group. Furthermore, after CK-M treatment, the number of both cell types through the matrix glue significantly reduced compared with free CK (P<0.05). These results indicate that CK-M is effective in enhancing the cell invasion inhibition of CK.
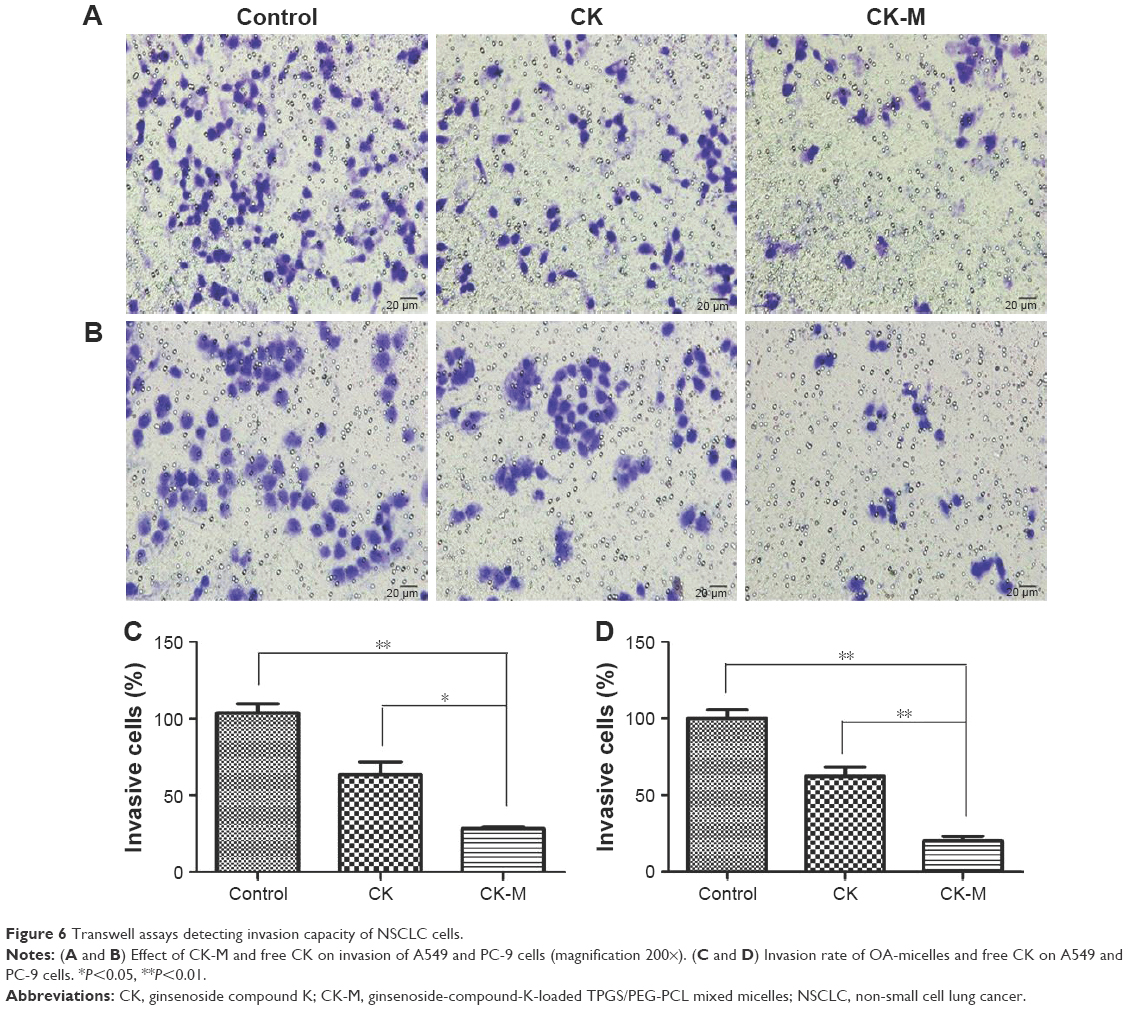 | Figure 6 Transwell assays detecting invasion capacity of NSCLC cells. |
CK-M induces cell cycle arrest and apoptosis of A549 and PC-9 cells
The cell cycle refers to the whole process from the beginning of cell division to the end of the next cell division. Here, a choice was made to use the IC50 of free CK for A549 and PC-9 cells (21.97 and 14.46 μg/mL, respectively) in the following experiments. The cell cycle is divided into two phases: the interphase (G1, S, G2) and mitosis (M). The entire cell cycle process is hindered if there is a delay in one of the phases. The A549 and PC-9 cell cycle distributions after treatment with CK and CK-M were evaluated to show the effects of CK on early cellular events. The percentage of cells at different cell cycle phases is shown in Figure 7A. The percentage of G1-phase cells of control group, CK group, and CK-M group for A549 cell were 53.50%, 60.52%, and 68.18%, respectively. The results for PC-9 cells were 43.47%, 50.95%, and 56.40%, respectively. Furthermore, there was an obvious apoptotic peak in the distribution after CK and CK-M treatment. Significant G1 arrest in both cell types was observed upon CK-M treatment.
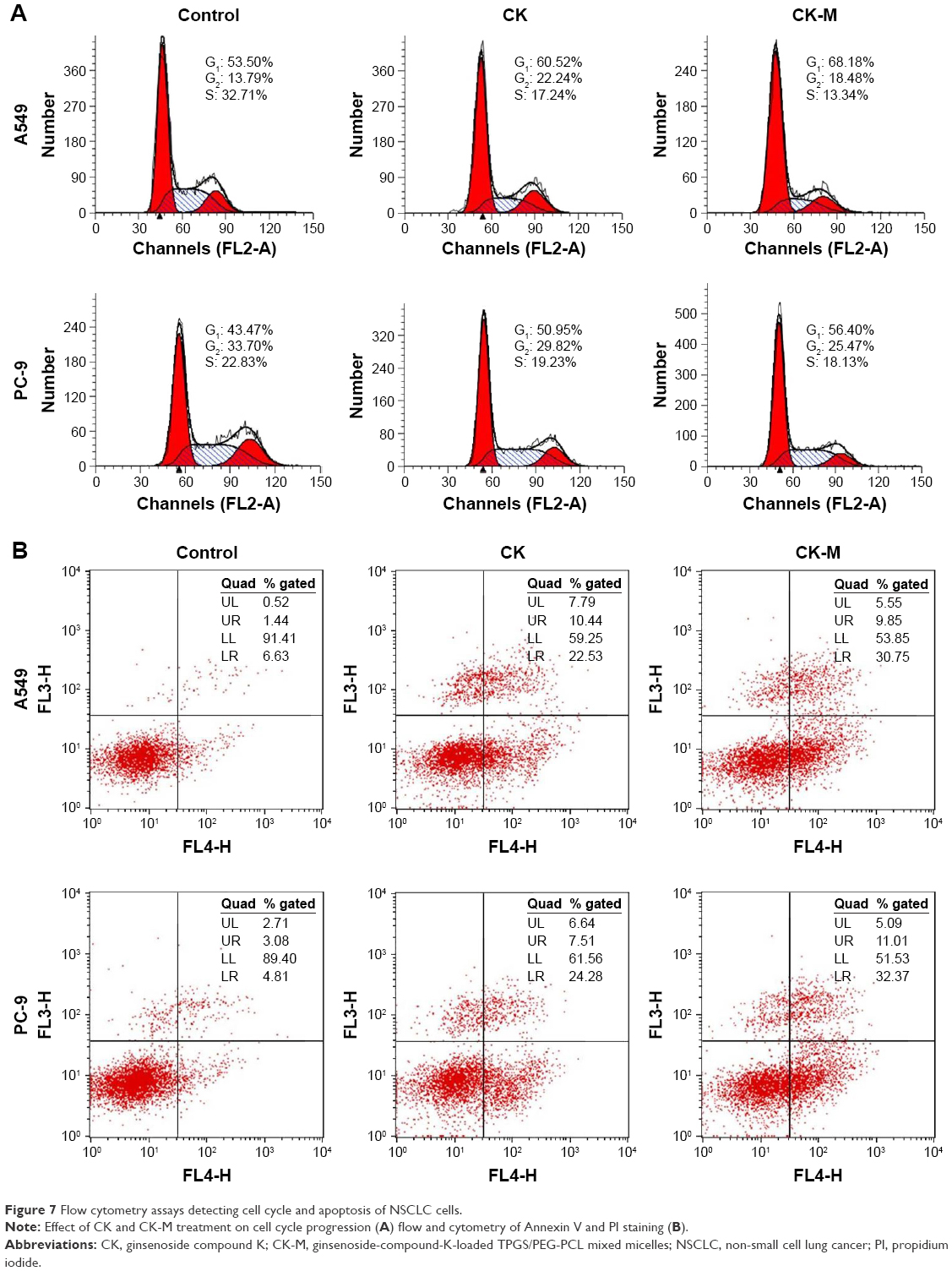 | Figure 7 Flow cytometry assays detecting cell cycle and apoptosis of NSCLC cells. |
In the process of apoptosis, phosphatidylserine (PS) would be turned over. Hence, V-EGFP/PI Annex in double staining was used to detect apoptosis of A549 and PC-9 cells. The apoptosis rates for CK and CK-M are presented in Figure 7B. The pro-apoptotic effect of CK-M (40.6% in A549 cells and 43.38% in PC-9 cells) was significantly stronger than that of control group (8.07% in A549 and 7.89% in PC-9 cells). A higher percentage of apoptotic cells were observed in wells treated with CK-M than those treated with free CK. Taken together, these observations suggest that the growth inhibitory effect of CK on A549 and PC-9 cells could be attributable to suppression of cell proliferation, induction of G1 cell cycle arrest, and apoptosis. In addition, the formation of CK-M could enhance the cytotoxicity of the free CK.
Western blot analysis
A549 and PC-9 cells were treated with CK, CK-M, or the DMEM (control) for 24 h, and the total cellular protein was extracted. The western blot results are shown in Figure 8. After CK-M treatment, expression levels of MMP-9 and Bcl-2 declined, while Bax and caspase-3 levels were significantly elevated compared with that of control. These results further confirm that CK can inhibit the proliferation of tumor cells by inhibiting apoptosis, invasion, and metastasis of tumor cells. Moreover, after CK-M treatment, the effect of CK on the proteins evaluated also enhanced significantly compared with control (P<0.05). P-gp level of CK-M was declined compared with CK, indicating CK-M could enhance cellular uptake through inhibition of P-gp expression.
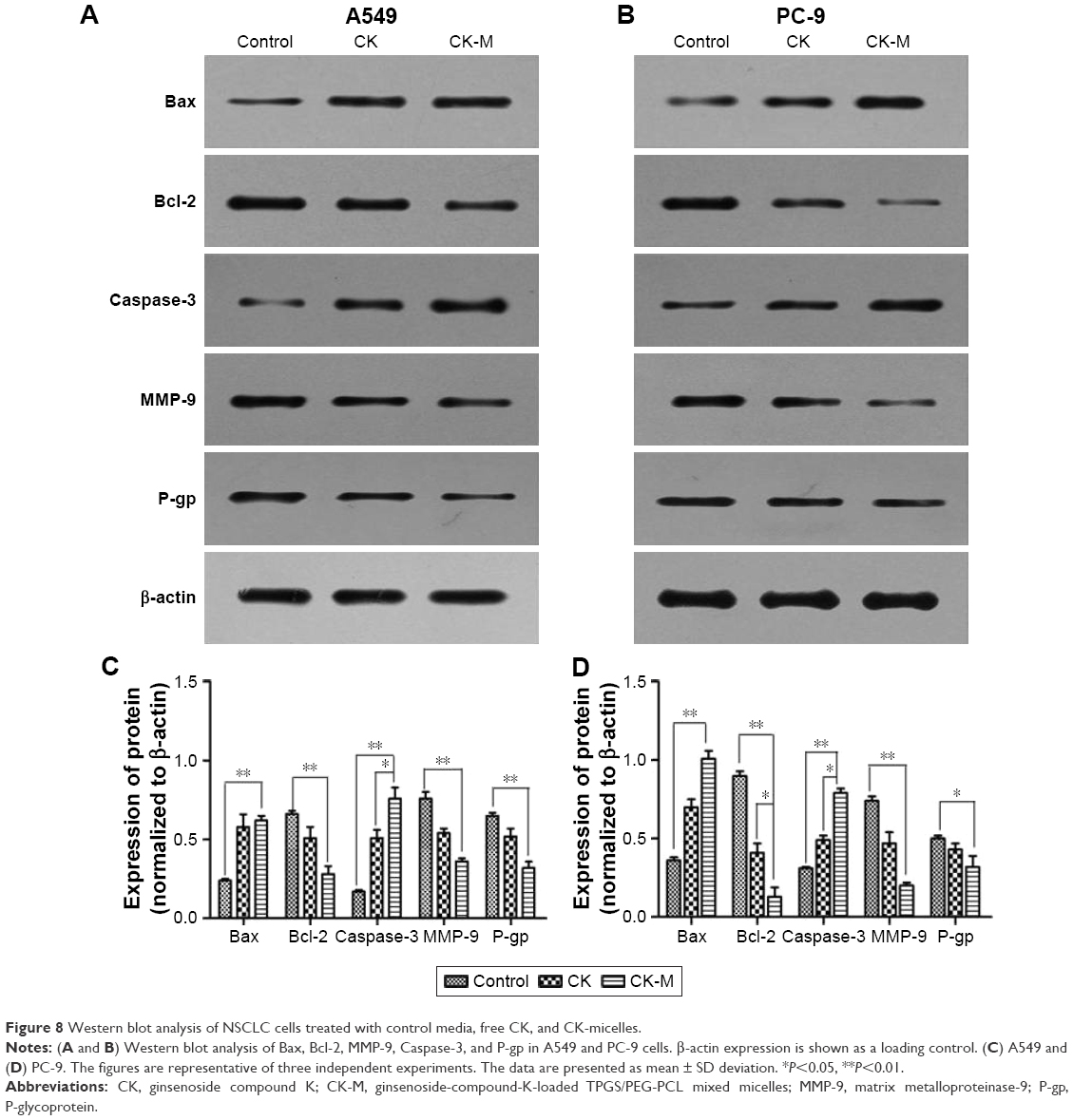 | Figure 8 Western blot analysis of NSCLC cells treated with control media, free CK, and CK-micelles. |
In vivo imaging
In vivo imaging demonstrated the targeting ability of the mixed micelles. As shown in Figure 9, the DiR mixed micelles were concentrated in the liver at 1 h. The fluorescence intensity in tumor tissue steadily enhanced at 2 h and continued to increase without any reduction until 24 h. The heart, liver, spleen, lung, kidney, and tumor were taken out and imaged. Stronger fluorescence absorption was observed in tumor tissue after 24 h while other visceral organs, except liver and spleen, did not show fluorescence. After this time point, fluorescence at the tumor site was noticeably observed and was maintained for more than 24 h. Therefore, CK-M has excellent targeting to the tumor site, and it enhances the permeability and retention time in tumor cells. Furthermore, the longer maintenance time in the tumor tissue also confirms that CK-M have a better sustained-release effect in vivo.
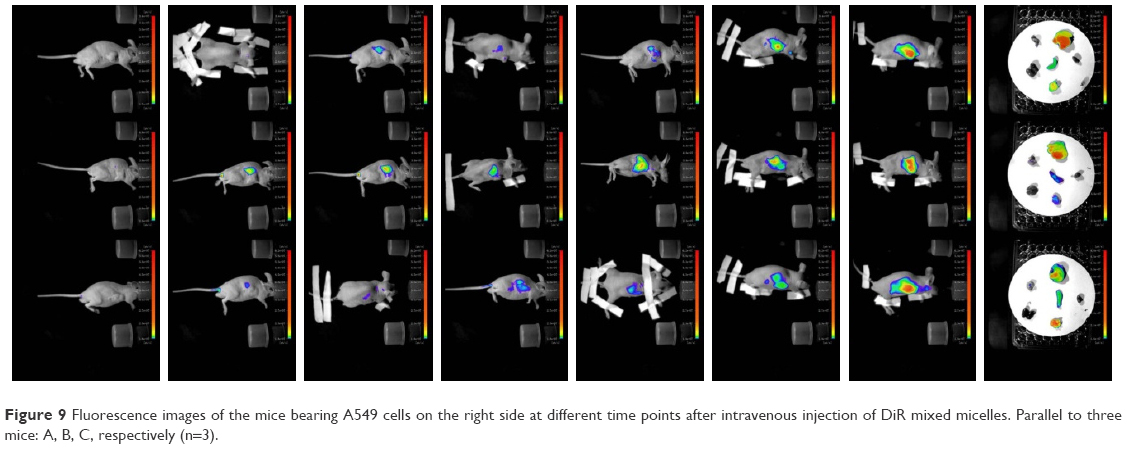 | Figure 9 Fluorescence images of the mice bearing A549 cells on the right side at different time points after intravenous injection of DiR mixed micelles. Parallel to three mice: A, B, C, respectively (n=3). |
In vivo antitumor activity
The in vivo antitumor effect of CK-M has been shown in tumor-bearing athymic nude mice. The tumor volume time curve and weight analysis are presented in Figure 10. The tumors of free CK group grew slowly compared with that of control group, indicating CK was effective in inhibiting tumor growth. Moreover, the relative tumor size of CK-M group was 2.67±0.88 at the end of the study, and the increase in tumor volume was much lower than the CK group (4.27±0.35; Figure 10A and B). The relative tumor growth rate of CK-M gradually decreased from 79.12%±0.60% to 52.04%±4.62% with the increase of number of drug administrations while that of CK was not significantly reduced (Figure 10C). This indicated that CK-M were significantly effective in the inhibition of tumor growth compared to the control group (P<0.01).
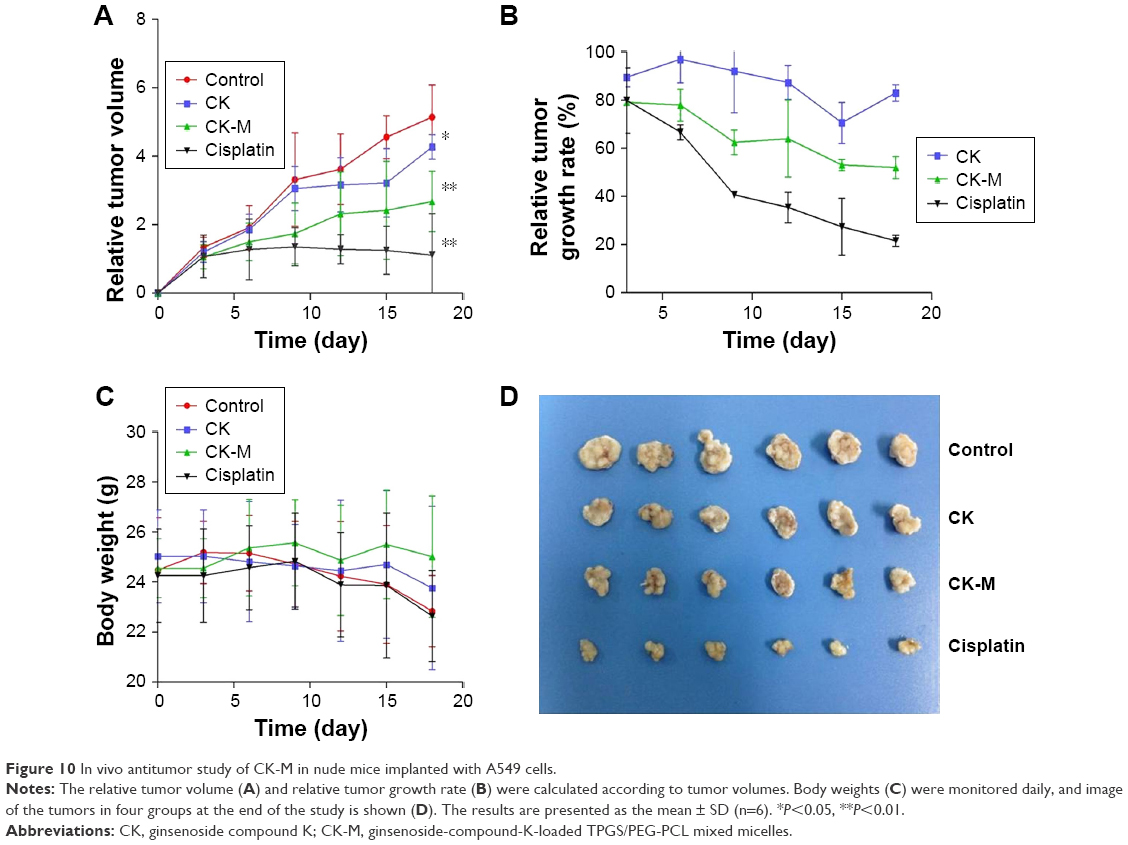 | Figure 10 In vivo antitumor study of CK-M in nude mice implanted with A549 cells. |
During the whole experiment, no nude mice were killed. The body weight of CK-M group was 25.02±2.42 while that of control group was 22.83±1.83 after 19 days, indicating CK-M had low toxicity to the mice (Figure 10D). Although the positive cisplatin group was effective in tumor inhibition (with a relative tumor growth rate of 21.59%±2.33%), the serious side effect caused was the significant decrease of body weight. On the contrary, the role of CK in enhancing immune function can increase the ability of mice to resist tumor and do not damage the organs. This formulation is thus safe and can provide support for further preparation.
Discussion
Since 1998, the antitumor mechanisms of CK in vivo have been studied.35 Unfortunately, the clinical use of CK is limited by its low water solubility and significant efflux. Polymeric micelles have been widely used as carriers for the solubilization of drugs with poor water solubility. Moreover, block copolymers with biocompatible hydrophilic groups and biodegradable lipophilic groups can spontaneously form micelles with a core–shell structure, wrapping the drug and increasing solubility. This stable system, consisting of TPGS and PEG-PCL, involves a smaller particle size (53.07±1.31 nm) and a complete ellipsoidal structure. Similarly, the release percentage of CK from CK-M was slower than that of free CK.
Additionally, P-gp efflux is a major factor limiting the antitumor efficacy of CK. It has been reported that TPGS can effectively reduce the basolateral transport of paclitaxel in a Caco-2 cell culture model, thereby reducing the efflux and increasing the antitumor efficacy of paclitaxel.36 The green fluorescence intensity of C-M was significantly higher than that of C-P in vitro cellular uptake, which was associated with the role of TPGS in suppressing efflux. P-gp levels were detected via western blot analysis; the protein expression of P-gp declined following treatment with CK-M compared with CK, indicating that reduced efflux greatly improved the antitumor efficiency of CK-M. Meanwhile, increased drug accumulation and extended maintenance time in tumors may be another way to improve the antitumor effect of CK-M. The in vivo imaging of DiR mixed micelles demonstrated that the drug contained in TPGS/PEG-PCL mixed micelles could quickly reach the tumors and be maintained for more than 24 h. Therefore, the inherent properties of amphiphilic block copolymers, the P-gp inhibition of TPGS, and the passive targeting of drug-resistant tumors by TPGS/PEG-PCL mixed micelles may underlie tumor growth inhibition in the CK-M treatment groups.
In vivo antitumor experiments showed that the tumor growth rate of the CK-M group was significantly lower than that of the CK group, indicating that CK-M could significantly increase the antitumor effect of CK. Cell cytotoxicity assays also demonstrated that the inhibitory effect of CK-M on tumor cell proliferation was more potent than that of CK.
The inhibition of tumor cell proliferation is a crucial indicator of antitumor drug efficacy. The CK-M-mediated inhibition of lung cancer cell proliferation was studied by examining the cell cycle distribution in tumors administered free CK and CK-M. The significant G1 phase arrest in A549 and PC-9 cells following treatment with CK-M showed that cell growth was successfully inhibited by CK-M. The growth rate of tumor cells is faster than that of normal cells, not only due to abnormal cell proliferation and differentiation, but also due to abnormal cell apoptosis. It was previously reported that apoptosis was significantly inhibited in malignant cancer cells. Therefore, the effect of drugs on apoptosis is critical in antitumor mechanism studies. The flow cytometry results demonstrated that CK-M inhibits tumor growth by inducing apoptosis. Furthermore, it has been reported that CK significantly induces apoptosis in tumor cells through the caspase-dependent pathway.37 High protein levels of Bc1-2, which is currently recognized as an anti-apoptotic protein, can inhibit cell death to prolong cell life. Bax induces cytochrome c release to activate caspase-3, causing vesicle formation, nuclear fragmentation, and cell death. Conversely, Bcl-2 antagonizes Bax, so the high expression levels of Bcl-2 inhibitor block caspase-3-induced apoptosis.38 These results indicate that CK promotes the downregulation of the Bcl-2/Bax ratio and the upregulation of caspase-3, inferring that CK-M can affect the apoptosis of NSCLC cells.
The migration and invasion abilities of tumor cells are the major mechanisms by which they invade adjacent tissues or organs and transfer via the blood. Wound healing and transwell migration assays were used to investigate the anti-metastasis effects of CK-M on A549 and PC-9 cells. The results showed that both CK and CK-M can significantly inhibit cancer cell invasion and migration. It has been revealed that the CK-mediated inhibition of cell invasion and migration is associated with MMP-9 expression.39 MMPs are a class of proteases that primarily degrade extracellular matrix components. MMP-9 is the most abundant enzyme in the MMP class. MMP-9 degrades and destroys the extracellular matrix and basement membrane of the tumor surface; consequently, the tumor cells can infiltrate the surrounding tissue along the absent basement membrane and promote tumor invasion and metastasis.40 Additionally, MMP-9 can encourage tumor growth by promoting capillary angiogenesis. The western blot results indicate that the MMP-9 expression levels declined following treatment, indicating the significant anti-migration and anti-invasion abilities of CK-M on NSCLC cells.
Conclusion
In this study, the CK-M were uniform in size and had a complete ellipsoidal shape with a sustained-release effect. The solubility of CK was enhanced, and efflux was inhibited by CK-M with TPGS/PEG-PCL. In vivo images indicated the passive accumulation of micelles in tumor tissues. The CK-M can achieve an improved growth inhibitory effect on tumor in vivo. Cytotoxicity, cell migration, and invasion assays showed CK-M has a stronger in vitro antitumor effect than free CK and induces an antitumor effect through the induction of apoptosis. Western blot analysis demonstrated that the apoptotic degree of CK-M was higher than that of free CK. Significantly, the construction of CK-M with TPGS/PEG-PCL can enhance the CK sensitivity of A549 and PC-9 cells and provide an effective method for the treatment of lung cancer. Thus, this system could serve as a promising carrier of CK for cancer therapy.
Acknowledgments
The study was financially supported by Program for Innovative Research Team of Six Talent Peaks Project in Jiangsu Province (SWYY-CXTD-004), Innovative Research Team of Health Development Project with Science and Education in Jiangsu Province, National Nature Science Foundation of China (Grant No 81403119, 81573620, 81503265), and Anhui Universities Provincial Natural Science Key Research project (No KJ2017A394).
Disclosure
The authors report no conflicts of interest in this work.
References
Yang XD, Yang YY, Ouyang DS, Yang GP. A review of biotransformation and pharmacology of ginsenoside compound K. Fitoterapia. 2015;100:208–220. | ||
Kim S, Kang BY, Cho SY, et al. Compound K induces expression of hyaluronan synthase 2 gene in transformed human keratinocytes and increases hyaluronan in hairless mouse skin. Biochem Biophys Res. 2004;316(2):348–355. | ||
Han Y, Sun B, Hu X, et al. Transformation of bioactive compounds by Fusarium sacchari fungus isolated from the soil-cultivated ginseng. J Agric Food Chem. 2007;55(23):9373–9379. | ||
Kim AD, Kang KA, Kim HS, et al. A ginseng metabolite, compound K, induces autophagy and apoptosis via generation of reactive oxygen species and activation of JNK in human colon cancer cells. Cell Death Dis. 2013;4:e750. | ||
Wei S, Li W, Yu Y, et al. Ginsenoside Compound K suppresses the hepatic gluconeogenesis via activating adenosine-5′monophosphate kinase: a study in vitro and in vivo. Life Sic. 2015;139:8–15. | ||
Li Y, Zhou T, Ma C, Song W, Zhang J, Yu Z. Ginsenoside metabolite compound K enhances the efficacy of cisplatin in lung cancer cells. J Thorac Dis. 2015;7(3):400–406. | ||
Kang KA, Piao MJ, Kim KC, et al. Compound K, a metabolite of ginseng saponin, inhibits colorectal cancer cell growth and induces apoptosis through inhibition of histone deacetylase activity. Int J Oncol. 2013;43(6):1907–1914. | ||
Choo MK, Sakurai H, Kim DH, Saiki I. A ginseng saponin metabolite suppresses tumor necrosis factor-alpha-promoted metastasis by suppressing nuclear factor-kappaB signaling in murine colon cancer cells. Oncol Rep. 2008;19(3):595–600. | ||
Zhang B, Zhu XM, Hu JN, et al. Absorption mechanism of ginsenoside compound K and its butyl and octyl ester prodrugs in Caco-2 cells. J Agric Food Chem. 2012;60(41):10278–10284. | ||
Mathiyalagan R, Subramaniyam S, Kim YJ, Kim YC, Yang DC. Ginsenoside compound K-bearing glycol chitosan conjugates: synthesis, physicochemical characterization, and in vitro biological studies. Carbohyd Polym. 2014;112:359–366. | ||
Mathiyalagan R, Subramaniyam S, Kim YJ, et al. Synthesis and pharmacokinetic characterization of a pH-sensitive polyethylene glycol ginsenoside CK (PEG-CK) conjugate. Biosci Biotechnol Biochem. 2014;78(3):466–468. | ||
Kumari P, Muddineti OS, Rompicharla SV, et al. Cholesterol-conjugated poly(D, L-lactide)-based micelles as a nanocarrier system for effective delivery of curcumin in cancer therapy. Drug Deliv. 2017;24(1):209–223. | ||
Lee GY, Park K, Kim SY, Byun Y. MMPs-specific PEGylated peptide-DOX conjugate micelles that can contain free doxorubicin. Eur J Pharm Biopharm. 2007;67(3):646–654. | ||
Zhong Y, Wang C, Meng F, Liu Z, Zhong Z. cRGD-Functionalized AuNR-cored PEG-PCL nanoparticles for efficacious glioma chemotherapy. J Control Release. 2015;213:e135. | ||
Xu S, Wang W, Li X, Liu J, Dong A, Deng L. Sustained release of PTX-incorporated nanoparticles synergized by burst release of DOXHCl from thermosensitive modified PEG/PCL hydrogel to improve anti-tumor efficiency. Eur J Pharm Sci. 2014;62:267–273. | ||
Zhao Y, Duan S, Zeng X, et al. Prodrug strategy for PSMA-targeted delivery of TGX-221 to prostate cancer cells. Mol Pharm. 2012;9(6):1705–1716. | ||
Yin T, Cai H, Liu J, et al. Biological evaluation of PEG modified nanosuspensions based on human serum albumin for tumor targeted delivery of paclitaxel. Eur J Pharm Biopharm. 2016;83:79–87. | ||
Li J, Cheng X, Chen Y, et al. Vitamin E TPGS modified liposomes enhance cellular uptake and targeted delivery of luteolin: an in vivo/in vitro evaluation. Int J Pharm. 2016;512(1):262–272. | ||
Duarte MD, Gaspar VM, Costa EC, et al. Combinatorial delivery of Crizotinib-Palbociclib-Sildenafil using TPGS-PLA micelles for improved cancer treatment. Eur J Pharm Biopharm. 2014;88(3):718–729. | ||
Ji H, Tang J, Li M, Ren J, Zheng N, Wu L. Curcumin-loaded solid lipid nanoparticles with Brij78 and TPGS improved in vivo oral bioavailability and in situ intestinal absorption of curcumin. Drug Deliv. 2016;23(2):459–470. | ||
Song IS, Cha JS, Choi MK. Characterization, in vivo and in vitro evaluation of solid dispersion of curcumin containing D-alpha-tocopheryl polyethylene glycol 1000 succinate and mannitol. Molecules. 2016;21(10):pii:E1386. | ||
Esser L, Zhou F, Pluchino KM, et al. Structures of the multidrug transporter P-glycoprotein reveal asymmetric ATP binding and the mechanism of polyspecificity. J Biol Chem. 2017;292(2):446–461. | ||
Yang L, El Ghzaoui A, Li S. In vitro degradation behavior of poly(lactide)-poly(ethylene glycol) block copolymer micelles in aqueous solution. Int J Pharm. 2010;400(1–2):96–103. | ||
Yang L, Xin J, Zhang Z, et al. TPGS-modified liposomes for the delivery of ginsenoside compound K against non-small cell lung cancer: formulation design and its evaluation in vitro and in vivo. J Pharm Pharmacol. 2016;68(9):1109–1118. | ||
Yan H, Wei P, Song J, Jia X, Zhang Z. Enhanced anticancer activity in vitro and in vivo of luteolin incorporated into long-circulating micelles based on DSPE-PEG2000 and TPGS. J Pharm Pharmacol. 2016;68(10):1290–1298. | ||
Hou J, Sun E, Sun C, et al. Improved oral bioavailability and anticancer efficacy on breast cancer of paclitaxel via Novel Soluplus(®)-Solutol(®) HS15 binary mixed micelles system. Int J Pharm. 2016;512(1):186–193. | ||
Gu F, Hu C, Tai Z, et al. Tumour microenvironment-responsive lipoic acid nanoparticles for targeted delivery of docetaxel to lung cancer. Sci Rep. 2016;6:36281. | ||
Obchoei S, Saeeng R, Wongkham C, Wongkham S. Novel synthetic mono-triazole glycosides induce G0/G1 cell-cycle arrest and apoptosis in cholangiocarcinoma cells. Anticancer Res. 2016;36(11):5965–5973. | ||
Zhang Y, Zhang H, Wu W, et al. Folate-targeted paclitaxel-conjugated polymeric micelles inhibits pulmonary metastatic hepatoma in experimental murine H22 metastasis models. Int J Nanomed. 2014;9:2019–2030. | ||
Firempong CK, Zhang HY, Wang Y, et al. Segetoside I, a plant-derived bisdesmosidic saponin, induces apoptosis in human hepatoma cells in vitro and inhibits tumor growth in vivo. Pharmacol Res. 2016;110:101–110. | ||
Zhang C, Qu G, Sun Y, et al. Pharmacokinetics, biodistribution, efficacy and safety of N-octyl-O-sulfate chitosan micelles loaded with paclitaxel. Biomaterials. 2008;29(9):1233–1241. | ||
Wang C, Zhang G, Liu G, Hu J, Liu S. Photo- and thermo-responsive multicompartment hydrogels for synergistic delivery of gemcitabine and doxorubicin. J Control Release. 2017;259:149–159. | ||
Song ZM, Zhu WX, Liu N, Yang FY, Feng RL. Linolenic acid-modified PEG-PCL micelles for curcumin delivery. Int J Pharm. 2014;417(1–2):312–321. | ||
Tariq M, Alam MA, Singh AT, Panda AK, Talegaonkar S. Surface decorated nanoparticles as surrogate carriers for improved transport and absorption of epirubicin across the gastrointestinal tract: pharmacokinetic and pharmacodynamic investigations. Int J Pharm. 2016;501(1–2):18–31. | ||
Akao T, Kida H, Kanaoka M, Hattori M, Kobashi K. Intestinal bacterial hydrolysis is required for the appearance of compound K in rat plasma after oral administration of ginsenoside Rb1 from Panax ginseng. J Pharm Pharmacol. 1998;50(10):1155–1160. | ||
Hou J, Sun E, Zhang ZH, et al. Improved oral absorption and anti-lung cancer activity of paclitaxel-loaded mixed micelles. Drug Deliv. 2017;24(1):261–269. | ||
Zheng ZZ, Ming YL, Chen LH, Zheng GH, Liu SS, Chen QX. Compound K-induced apoptosis of human hepatocellular carcinoma MHCC97-H cells in vitro. Oncol Rep. 2014;32(1):325–331. | ||
Zhao Y, Zhang S, Wang P, Fu S, Wu D, Liu A. Seleno-short-chain chitosan induces apoptosis in human non-small-cell lung cancer A549 cells through ROS-mediated mitochondrial pathway. Cytotechnology. Epub 2017 Apr 18. | ||
Ming Y, Chen Z, Chen L, et al. Ginsenoside compound K attenuates metastatic growth of hepatocellular carcinoma, which is associated with the translocation of nuclear factor-kB p65 and reduction of matrix metalloproteinase-2/9. Planta Med. 2011;77(5):428–433. | ||
Liu X, Lv Z, Zou J, et al. Elevated AEG-1 expression in macrophages promotes hypopharyngeal cancer invasion through the STAT3-MMP-9 signaling pathway. Oncotarget. 2016;7(47):77244–77256. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.