Back to Journals » International Journal of Nanomedicine » Volume 14
Targeted acetylcholinesterase-responsive drug carriers with long duration of drug action and reduced hepatotoxicity
Authors Lin Y, Wang Y, Lv J, Wang N, Wang J, Li M ![]()
Received 12 May 2019
Accepted for publication 6 July 2019
Published 26 July 2019 Volume 2019:14 Pages 5817—5829
DOI https://doi.org/10.2147/IJN.S215404
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Linlin Sun
Yulong Lin, Yalin Wang, Jie Lv, Nannan Wang, Jing Wang, Meng Li
College of Pharmaceutical Sciences, Hebei Medical University, Shijiazhuang 050017, People’s Republic of China
Purpose: Acetylcholinesterase (AChE) plays a critical role in the transmission of nerve impulse at the cholinergic synapses. Design and synthesis of AChE inhibitors that increase the cholinergic transmission by blocking the degradation of acetylcholine can serve as a strategy for the treatment of AChE-associated disease. Herein, an operational targeted drug delivery platform based on AChE-responsive system has been presented by combining the unique properties of enzyme-controlled mesoporous silica nanoparticles (MSN) with clinical-used AChE inhibitor.
Methods: Functionalized MSNs were synthesized by liquid phase method and characterized by using different analytical methods. The biocompatibility and cytotoxicity of MSNs were determined by hemolysis experiment and MTT assay, respectively. Comparison of AChE activity between drug-loading system and inhibitor was developed with kits and by ELISA method. The efficacy of drug-loaded nanocarriers was investigated in a mouse model.
Results: Compared with AChE inhibitor itself, the inhibition efficiency of this drug delivery system was strongly dependent on the concentration of AChE. Only AChE with high concentration could cause the opening of pores in the MSN, leading to the controlled release of AChE inhibitor in disease condition. Critically, the drug delivery system can not only exhibit long duration of drug action on AChE inhibition but also reduce the hepatotoxicity in vivo.
Conclusion: In summary, AChE-responsive drug release systems have been far less explored. Our results would shed lights on the design of enzyme controlled-release multifunctional system for enzyme-associated disease treatment.
Keywords: acetylcholinesterase inhibitor, controlled drug release, mesoporous silica nanoparticles, drug action, hepatotoxicity
Introduction
Enzymes with high catalytic capability are involved in numerous important biological processes, including cellular processes and metabolic exchange.1–3 The aberrations in levels of enzyme expression have been reported to be associated with many diseases.4–6 Acetylcholinesterase (AChE), which catalyzes the hydrolysis of acetylcholine (ACh), plays a critical role in the transmission of nerve impulse at the cholinergic synapses.7,8 Design and synthesis of AChE inhibitors that increase the cholinergic transmission by blocking the degradation of ACh can serve as a strategy for the treatment of Alzheimer’s disease (AD), senile dementia, ataxia, myasthenia gravis and Parkinson’s disease.
Nowadays, small molecules such as tacrine (Tac),9,10 donepezil,11,12 and the natural product-based rivastigmine13,14 have already been used to inhibit the activity of AChE and resulted in improved cognition in clinical trials. However, these conventional small inhibitors have troubling drawbacks in their limited ability to differentiate the target enzyme associated with the disease from those normal enzymes, and exhibit adverse side effects which limit their long-term clinical use. To overcome these limitations, small-molecule drugs to covalently inhibit AChE15,16 or selective dual binding site AChE inhibitors17,18 were designed. Although promising, no clear-cut clinical evidence for these novel inhibitors on disease treatment has been demonstrated. Therefore, targeted delivery strategies that exploit disease-related biomarkers to improve treatment efficiency and reduce adverse off-target effects are an urgent need to be developed.
Recently, design and synthesis of stimuli-responsive nanocarriers have become promising approaches for targeted drug delivery and controlled release. So far, a variety of external stimuli such as temperature,19,20 pH,21,22 light23,24 and redox25,26 have been employed to realize controlled drug release. However, compared with these external stimuli, utilizing AChE as the stimuli is more suitable for AChE inhibitors delivery since the inhibitors can only be programmed to release when the enzyme is found at higher concentrations at the pathological environment. Herein, we combine the advantages of the controlled-release systems with supramolecular host–guest interaction to create a new AChE-responsive drug delivery platform (Figure 1). We adopt mesoporous silica nanoparticles (MSNs) as the carrier vehicles due to their distinctive characteristics such as thermal stability, tunable pore sizes, large loading capacity and the ease of surface functionalization.27–30 This novel system can be specific for AChE-responsive release of AChE inhibitors to realize targeted delivery and reduce side effects in disease treatment. To the best of our knowledge, AChE-responsive drug release systems have been far less explored.31 Our study will provide a general strategy to construct enzyme-inhibitors-loaded nanocarriers which are responsive to disease-associated enzymes.
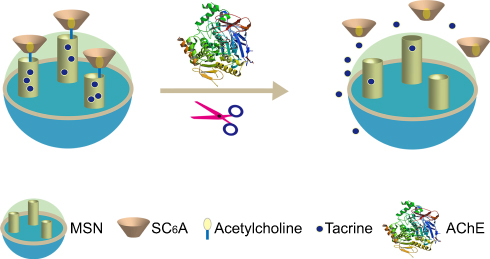 |
Figure 1 Schematic representation of AChE-fueled release of guest molecules Tac from the pores of MSN capped with SC6A. Abbreviations: Tac, tacrine; SC6A, p-sulfonatocalix[6]arene. |
As illustrated in Figure 1, our system consists of a mesoporous nanoparticle functionalized with a derivative of ACh. Biocompatible p-sulfonatocalix6arene (SC6A) and ACh are employed as the macrocyclic host and enzyme-cleavable guest, respectively.32,33 The opening protocol and delivery of the entrapped guest depend on an enzyme-catalyzed reaction in which the ACh derivative is expected to be hydrolyzed by AChE to generate choline, resulting in the breakage of the complex of SC6A and choline from the surface of MSN and the release of guest molecules after that. The capping of SC6A lids on AChE-inhibitors-loaded MSNs should therefore allow AChE-triggered drug release.
Materials and methods
Materials
Tetraethyl orthosilicate (TEOS, 99 wt.%), cetyltrimethylammonium bromide (CTAB), N,N’-dicyclohexylcarbodimide (DCC, >98%), 2-N-morpholino-ethanesulfonic acid (MES), 4-dimethylaminopyridine (DMAP) and N-hydroxysuccinimide (NHS, >98%) were purchased from Aladdin. 2-cyanoethyltriethoxysilane (CETES), 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide-HCl (EDC, >98%), epsilon-aminocaproic acid (ACA), 2-hydroxyethyl trimethylammonium chloride (ChCl), Fluorescein isothiocyanate (Flu) and acetylcholine esterase (AChE) were purchased from Sigma-Aldrich. p-Sulfonatocalix[6]arene (p-SC6A) was obtained from TCI Development Co., Ltd. (Shanghai, China). 1,2,3,4-Tetrahydro-5-aminoacridine hydrochloride (Tacrine, Tac) was purchased from Heowns Biochem. All these reagents were used as received without further purification. Deionized water (18.2 MΩ cm) used for all experiments was obtained from a Milli-Q system (Millipore, Bedford, MA).
C57BL6/J mice were obtained from the Experimental Animal Center of the Chinese Academy of Medical Sciences. Bel7402 cells were obtained from American Type Culture Collection. RPMI 1640 and fetal bovine serum (FBS) were purchased from Gibco BRL. (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) was bought from Sigma-Aldrich.
Instruments
Transmission electron microscopic (TEM) experiments were performed on a Philips Tacnai G2 20 S-TWIN microscope operating at 200 kV to characterize the size and morphologies. Small angle X-ray diffraction (SAXRD) patterns were recorded by a Rigaku SmartLab X-ray diffractometer with Cu-Kα source (λ =1.5406 Å). Nitrogen adsorption-desorption isotherms were measured using an ASAP2460 automatic surface and porosity analyzer (Micromeritics Instrument Corp., America). The BET (Brunauer-Emmett-Teller) surface area, total pore volume and average pore size were calculated from N2 adsorption-desorption isotherms. Fourier-transform infrared (FTIR) spectra was collected on a Rigaku 8400s FTIR spectrophotometer (Japan) using KBr pellets. Zeta potential was measured by a Malvern Zetasizer Nano ZS (UK). The ultraviolet-visible spectroscopy (UV-vis) of Tac and Flu were recorded by a Jasco-V550 UV-vis spectrophotometer.
Synthesis of carboxyl-functionalized MSN (MSN-COOH)
The carboxyl-modified mesoporous silica nanoparticles (MSN-COOH) were synthesized by co-condensation.34 Typically, 250 mg CTAB was dissolved in 240 mL of water. Sodium hydroxide aqueous solution (1.75 mL, 2.0 M) was introduced to the CTAB solution and the temperature of the mixture was adjusted to 80°C. After stirring for about 10 mins at 80°C, 2.5 mL of TEOS (11.2 mmol) was added into the mixture dropwise under vigorous stirring. The resulting mixture was stirred for another 2 hrs at 80°C to give rise to a white precipitate. The solid crude products were filtered, washed with water and methanol. Finally, the products were dried under high vacuum. To remove the surfactant template (CTAB), the as-synthesized MSN was refluxed at 80°C for 10 hrs in an acetonitrile solution. Then the samples were collected by centrifugation at 14,000 rpm for 5 mins, washed and re-dispersed with 50 mL toluene. 2.0 mL CETES was added to the mixture dropwise at 80°C and reacted for 4 hrs. After that, the dried product was treated with 9 mol·L−1 H2SO4 solution at 95°C for 12 hrs to produce MSN-COOH. The MSN-COOH nanoparticles were vacuum-dried for further use.
Synthesis of choline-functionalized MSN (MSN-Ch)
The purified MSN-COOH (20 mg) was dispersed in 10 mL MES buffer (10 mM, pH 6.0), and then 80 mg EDC was added to the suspension. After stirring at room temperature for 10 mins, 120 mg NHS was added into the above solution. The mixture was incubated at room temperature for another 15 mins under stirring to allow the MSN-COOH be efficiently activated. Then, 20 mL phosphate buffer saline (PBS) (10 mM, pH7.4) containing 20 mg ACA was added into the mixture, following by continuously stirring for 12 hrs at room temperature. After washed with PBS and water for three times, the ACA-functionalized MSN (MSN-COOR) was obtained. To synthesis choline-modified MSN, the obtained MSN-COOR was dispersed in 20 mL dichloromethane, and then 30 mg ChCl, 1.5 mg DMAP and 24 mg DCC were added into the above solution under continuous stirring. After 72 hrs stirring, MSN-Ch nanoparticles were collected by centrifugation, and washed with dichloromethane and water.
Synthesis of fluorescein-loaded MSN-SC6A
The obtained MSN-Ch (10.0 mg) was stirred in a solution of fluorescein (Flu) (1 mM) in PBS for 24 hrs in dark. Then, SC6A (10 mg) was added to the suspension. The mixture was stirred in dark for another 6 hrs. After that, the physisorbed Flu and uncapped SC6A were removed by centrifugation and washing with PBS. The resulting precipitate was collected and dried under high vacuum.
Synthesis of Tac-loaded MSN-SC6A (MSN-Tac-SC6A)
Similar to Flu-loaded MSN-SC6A, the purified MSN-Ch (10.0 mg) was added to the solution of Tac (1 mM) in PBS, and stirred for 24 hrs. Then, Tac-loaded MSN-Ch was harvested by centrifugation and resuspended in PBS. After SC6A (10 mg) was added, the mixture was stirred for another 6 hrs, followed by centrifugation and washing with water for three times to remove the physisorbed Tac and uncapped SC6A. The resulting precipitate was collected and dried under high vacuum.
Flu release experiments
Flu-loaded MSN-Ch (5 mg) material was dispersed in 10 mL of PBS buffer (10 mM, pH 8.0) containing different concentration of AChE. Aliquots were taken from the suspension and the delivery of Flu dye from the pore to the buffer solution was monitored via the absorbance band of the dye centered at 484 nm.
Detection of AChE activity assay
To detect the inhibition effects of Tac or MSN-Tac-SC6A on the activity of AChE, different levels of AChE were added to the PBS (10 mM, pH=7.4) composed of different concentrations of Tac or MSN-Tac-SC6A, followed by incubation at room temperature for 30 mins to make the reaction between AChE and inhibitors sufficiently. After that, acetylthiocholine (ATC) (500 μM) and dithiobisnitrobenzoic acid (DTNB) (250 μM) were added to the mixture and incubated for another 20 mins. Then the absorption value at 412 nm was monitored.
Animal treatment
Eight-week-old male C57BL6/J mice with body weights between 20 and 25 g were used to test the efficacy of drug-loading system in vivo. All the protocols and procedures for animal handling were carried out following the guidelines of the Hebei committee for care and use of laboratory animals, and were approved by the Animal Experimentation Ethics Committee of the Hebei Medical University. The mice were maintained in a germ-free environment and allowed free access to food and water. The mice were randomized divided into 3 groups: (1) PBS, (2) Tac, (3) MSN-Tac-SC6A. The mice, 6 per group, were treated subcutaneously with Tac (2.5 mg/kg body weight (B.W.)) or MSN-Tac-SC6A (the amount of Tac loaded in MSN-Tac-SC6A was 2.5 mg/kg B.W.). Subcutaneous (s.c.) application was chosen due to the availability of toxicity data and widespread use in pharmacology research. For AChE inhibition experiment, blood was collected at different time points after s.c. injection and centrifuged to obtain plasma for the detection of AChE activity. To access the hepatotoxicity, blood was collected at 24 hrs after s.c. injection and the plasma was separated for liver functions (alkaline phosphatase, ALP and aspartate aminotransferase, AST) test. For the in vivo toxicity experiment, the mice, 6 per group, were treated subcutaneously with MSN-COOH (40 mg/kg B.W.) or MSN-SC6A (40 mg/kg B.W.). After injection, the body weight of these treated mice was recorded.
Collection of red blood cells
The red blood cells (RBCs) were collected by centrifugation of heparin-stabilized rat blood samples at 3500 rpm for 10 mins to remove the plasma and buffy coat. The remaining packed RBCs were washed with PBS for three times until no traces of plasma were seen.
Hemolysis
In hemolysis experiment, 400 μL of packed RBCs was diluted to 4 mL with PBS (10% hematocrit) and the diluted RBC suspension (200 μL) was mixed with 600 μL of MSN-based nanomaterial suspensions in PBS at different concentrations. PBS and water (600 μL) which incubated with 200 μL diluted RBC suspension were served as negative and positive controls, respectively. All the mixtures were gently vortexed and incubated at room temperature for 2 hrs. The mixtures were centrifuged at 3500 rpm for 10 mins. The absorbance of the supernatant was measured.
Cell toxicity assays
Bel7402 cells were cultured in RPMI 1640 medium supplemented with 10% FBS, a 5% CO2 humidified environment at 37°C. Bel7402 cells were seeded at a density of 5000 cells/cm2 for MTT assay. After 24 hrs, different concentrations of MSN-SC6A were added. 24 hrs later, the cells were treated with 10 μL MTT (5 mg·mL−1 in PBS) for 4 hrs at 37°C and then were lysed in DMSO for 10 mins at room temperature in the dark. Absorbance values of formazan were determined at 570 nm with 630 nm as the reference using a Bio-Rad model-680 microplate reader.
Statistical analysis
All the experimental data were analyzed by calculating the ± standard error of mean of three independent experiments and compared by one-way analysis of variance (ANOVA) test (using a statistical package, Origin 8.5, MA, USA) with multiple comparison and paired-samples t-tests, P<0.05 as a limit of significance.
Results and discussion
Synthesis and characterization of MSN-SC6A
To validate our design, the MSN-COOH was first synthesized by co-condensation.34 TEM images indicated that the resulting spherical MSN had a diameter of about 100 nm and an MCM-41-type channel-like mesoporous structure (BJH pore diameter =2.8 nm) (Figure 2A). The hexagonally arranged pores were further characterized by SAXRD and N2 adsorption (Figure 2C and D). Then, ACA was introduced to MSN-COOH to yield MSN-COOR. The ACA acted as not only the linker to conjugate choline on the surface of MSN, but also a spacer to avoid possible AChE activity blocking by MSN. MSN-Ch was obtained through a direct esterification process between hydroxyl groups and carboxyl groups. After that, the negatively charged SC6A macrocycles were introduced to encircle the choline stalks on the surfaces of MCM-41 nanoparticles via host–guest complexation (MSN-SC6A), leading to the pores of MSN blocked (Figure S1). Herein, we also examined the hydrodynamic radius of MSN-COOH and MSN-SC6A using dynamic light scattering (DLS) in PBS to obtain more insights into the nanoparticles. As shown in Figure S2, the MSN-SC6A was 266 nm in diameter, which was obviously smaller than MSN-COOH, indicating the possibility of MSN-SC6A used in vivo. The surface functionalization of MSN was monitored by FTIR spectroscopy (Figure S3). The emerging absorption band at around 1700 cm−1 in the MSN-COOH sample can be assigned to C=O stretching of the carboxyl groups. The successful grafting of choline onto the surface of mesoporous silica was validated by the appearance of the typical vibration peaks at 1080 cm−1 and a specific group of quaternary ammonium compounds in the range of 900–980 cm−1. After capping, the peaks appeared from 1650 cm−1 to 1446 cm−1 representing the characteristic absorption of aromatic ring and peaks at 2934 cm−1 and 2859 cm−1 correspond to the stretching band of C–H, indicating the efficient binding of SC6A onto the surface of MSN. TEM showed that after capping with SC6A, there was no clear difference in shape and average diameter of the MSN, but a layer of soft materials surrounding the MSN was observed (Figure 2B). The pore blocking or sealing effect was also indicated by the change of BET analysis (Figure 2D and Table 1) and ζ potential measurement (Table S1). Quantification of the density of SC6A anchored on MSN-Ch was accomplished by thermogravimetric analysis (TGA) (Figure S4), which corresponded to about 122.9 μg/mg MSN-Ch.
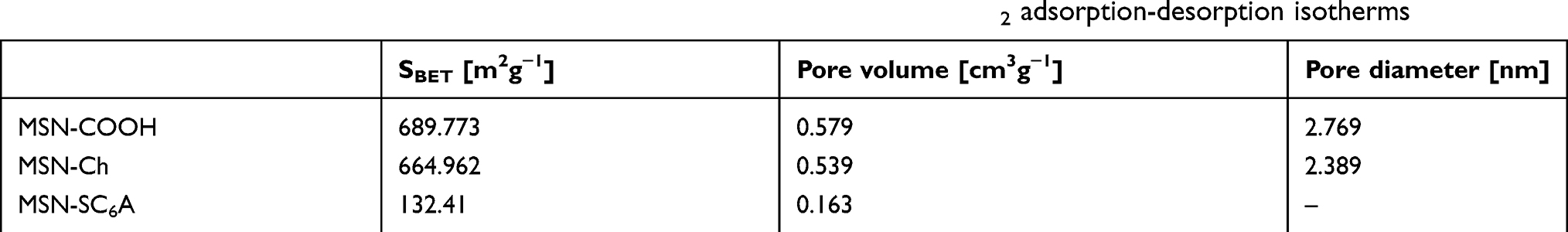 |
Table 1 BET-specific surface values, pore volumes and pore sizes calculated from N2 adsorption-desorption isotherms |
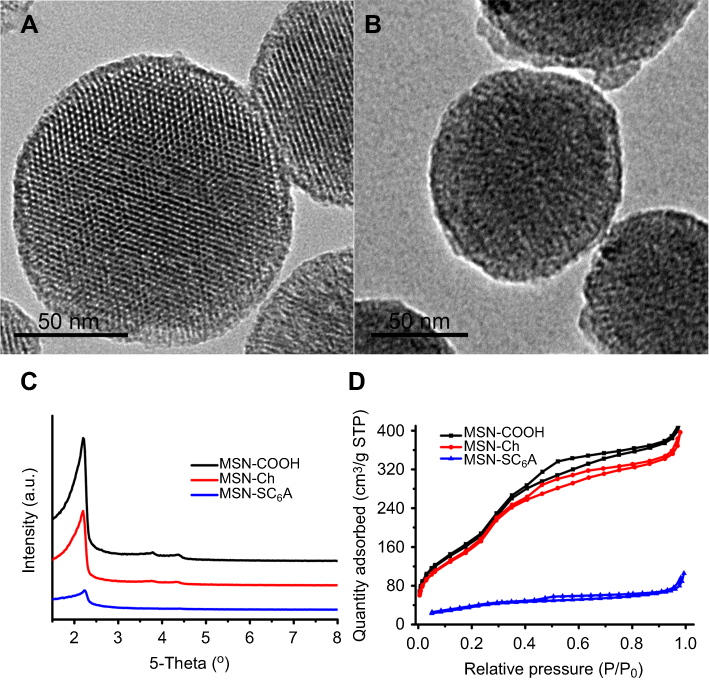 |
Figure 2 The characterization of the as-synthesized nanomaterials. (A) TEM image of MSN-COOH. (B) TEM image of MSN-SC6A. Scale bars: 50 nm. (C) X-ray diffraction pattern of MSN-COOH, MSN-Ch and Flu-loaded MSN-SC6A. Both MSN-COOH and MSN-Ch exhibited the typical diffraction patterns of MCM-41 type mesoporous silica with hexagonal symmetry. The changes in the Flu-loaded MSN-SC6A diffraction pattern might be caused by pore filling and SC6A coating effects. (D) N2 adsorption-desorption isotherms for MSN-COOH, MSN-Ch and MSN-SC6A. The MSN-SC6A exhibited relatively flat curves compared (at the same scale) to original MSN-COOH, indicating that the pores were significantly blocked by SC6A. Abbreviations: TEM, transmission electron microscopy; MSN, mesoporous silica nanoparticles; SC6A, p-sulfonatocalix[6]arene; MSN-COOH, MSN modified by carboxyl; MSN-SC6A, MSN conjugated with p-sulfonatocalix[6]arene; MSN-Ch, MSN conjugated with choline; Flu, fluorescein; MCM-41, Mobil composition of matter No.41. |
To demonstrate the AChE-controlled actuation of the nanovalves, Flu was added as a guest molecule by soaking MSN-Ch in PBS solution. The pore was then capped via incubating with SC6A for 6 hrs in PBS. The excess amount of molecule was removed by centrifugation and repeated washing with PBS. The amount of encapsulated guest molecules in the resulting particles was spectroscopically quantitated to be 28.47 μg Flu/mg MSNs (Figure S5). Release experiments were performed in PBS with different concentrations of AChE. As shown in Figure 3, the release of Flu was almost unobvious in the absence of AChE, indicating that SC6A can act as an efficient cap for retention of guest molecules with negligible leakage. On the other hand, the amount of Flu released reached about 44.05% after incubation with 250 mU·mL−1 AChE for 60 mins, whereas about 22.09% release was obtained in the same amount of time with 50 mU·mL−1 AChE, indicating that the guest molecules' release increased as the AChE concentration increased within a certain range.
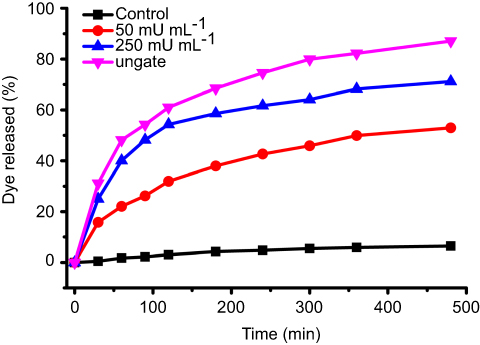 |
Figure 3 Release profiles of Flu from ungated-MSN or MSN-SC6A stimulated by different concentrations of AChE. -■-: Control, MSN-SC6A without external stimuli; -●-: MSN-SC6A triggered by 50 mU/mL AChE; -▲-: MSN-SC6A triggered by 250 mU/mL AChE; -▼-: ungated-MSN (mean ± SD, n=3 for each sample).Abbreviations: Flu, fluorescein; AChE, acetylcholine esterase; MSN-SC6A, MSN conjugated with p-sulfonatocalix[6]arene. |
The inhibition effect of MSN-Tac-SC6A on the activity of AChE in vitro
To investigate the feasibility of this drug delivery system for inhibiting the activity of AChE, Tac, a commonly used AChE inhibitor, was employed as the guest molecule. The amount of encapsulated Tac in the resulting particles was quantitated to be 27.02 μg Tac/mg MSNs. After incubation with AChE, Tac would be released from the pore of MSN, leading to the reduced activity of AChE. As shown in Figure 4A, upon treated with high concentration of AChE (250 mU·mL−1), both Tac and MSN-Tac-SC6A can inhibit the activity of AChE. The inhibitory capacity increased with increasing Tac concentration, which demonstrated that MSN could not change the activity of encapsulated Tac. However, compared with Tac, the inhibition efficiency of this drug delivery system strongly depended on the concentration of AChE. Only AChE with high level could cause the breakage of the complex of SC6A and choline from the surface of MSN, leading to the release of Tac and inhibition of AChE activity (Figure 4B). Then, the remaining AChE with reduced activity could not further break the complex of SC6A and choline on the surface of MSN. Although AChE inhibition occurs at all peripheral cholinergic synapses with the use of the inhibitors, AChE could be maintained at a certain level via the controlled release of Tac in the drug delivery system, which would significantly lower the cholinergic side effects of Tac. In addition, MSN-SC6A without Tac showed no effects on the activity of AChE, indicating that the inhibition effect of MSN-Tac-SC6A was raised from the released Tac.
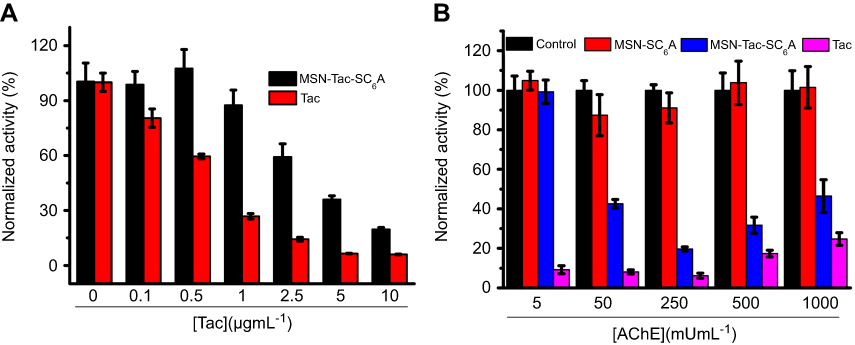 |
Figure 4 The inhibition effects of Tac or MSN-Tac-SC6A on the activity of AChE. (A) The concentration-dependent inhibition effects of Tac or MSN-Tac-SC6A on the activity of AChE. The concentration of AChE was 250 mU·mL−1. (B) The inhibition effects of Tac or MSN-Tac-SC6A on the activity of AChE with different concentrations. The concentration of Tac was 10 µg·mL−1 (mean ± SD, n=3 for each sample). Abbreviations: AChE, acetylcholine esterase; Tac, tacrine; MSN-Tac-SC6A, MSN loading with tacrine. |
The inhibition effect of MSN-Tac-SC6A on the activity of AChE in vivo
Having successfully established the excellent performance of MSN-Tac-SC6A in inhibiting the activity of AChE in vitro, we next investigated the feasibility of our drug delivery system on in vivo inhibition of AChE. We collected the blood from the mice which have been injected with different types of inhibitors within 24 hrs to determine the level of AChE. As demonstrated in Figure 5, both Tac and MSN-Tac-SC6A loaded with the same concentration of Tac showed strong inhibition effects on the activity of AChE in blood serum. It was clear that Tac itself exhibited higher inhibition effects on AChE activity at the first 2 hrs after treatment. However, the MSN-Tac-SC6A tended to stay much longer in the blood. Compared with Tac, the MSN-Tac-SC6A exhibited long duration of drug action even up to 24 hrs, while the activity of Tac became unobvious after 4 hrs. Notably, under these conditions of short-term treatment, no apparent enzyme up-regulation occurred after the administration of this drug.35 The long-acting effects of MSN-Tac-SC6A were attribute to the sustained release of Tac from the drug delivery system and the relative long blood retention time of MSN, which would decrease the dosage of AChE inhibitors used in patients and reduce the side effects of these inhibitors.
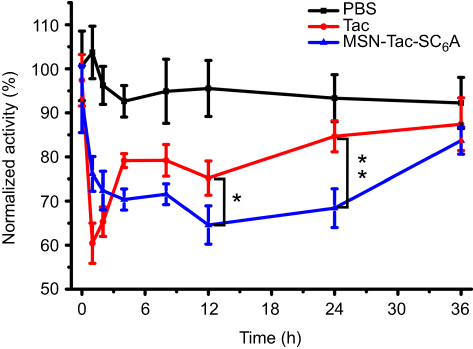 |
Figure 5 The blood AChE inhibition levels in the mice after s.c. injection of different kinds of inhibitors. The male C57BL6/J mice were randomized divided into 3 groups: (1) PBS, (2) Tac, (3) MSN-Tac-SC6A. The mice, 6 per group, were treated subcutaneously with Tac (2.5 mg/kg B.W.) or MSN-Tac-SC6A (the amount of Tac loaded in MSN-Tac-SC6A was 2.5 mg/kg B.W.). The blood was collected at different time points after s.c. injection and centrifuged to obtain plasma (mean ± SD, n=6. *P<0.05, **P<0.01, significantly different from the Tac group). Abbreviations: AChE, acetylcholine esterase; PBS, phosphate buffer saline; Tac, tacrine; MSN-Tac-SC6A, MSN loading with tacrine; B.W., body weight; s.c., subcutaneous. |
The hepatotoxicity of MSN-Tac-SC6A in vivo
Although Tac is effective in the treatment of AChE-associated disease, a major adverse effect of this drug is hepatotoxicity, which affects almost one-half of the treated patients.36–38 Based on this, we also monitored the hepatotoxicity induced by our drug delivery system. After treating mice with highest tolerated dose of Tac or MSN-Tac-SC6A loaded with the equal dose of Tac for 24 hrs, the activities of ALP and AST in serum were measured. As illustrated in Figure 6, Tac caused significant hepatotoxicity with the increased activities of both ALP and AST. Compared with the control group, the activities of ALP and AST increased up to 1.49 times and 1.57 times, respectively, while MSN-Tac-SC6A only slightly altered these parameters at 24 hrs. These results strongly demonstrated that introduction of the controlled-release system could reduce the hepatotoxicity of Tac. Due to the controlled release of Tac only in disease condition and lowered toxicity of the drugs, our strategy can be used in AChE-associated diseases, such as AD, senile dementia, ataxia, myasthenia gravis, Parkinson’s disease in elder adults.
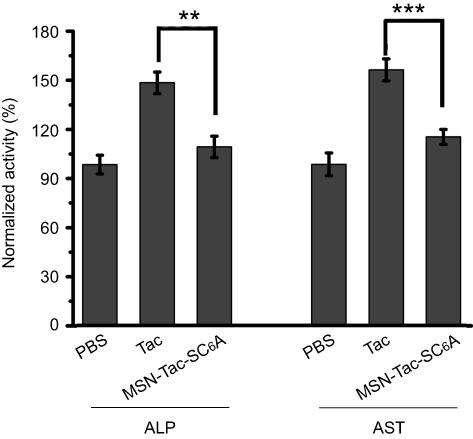 |
Figure 6 Levels of plasma ALP and AST. Plasma was collected from the mice treated with different kinds of inhibitors, and liver function was tested. The male C57BL6/J mice were randomized divided into 3 groups: (1) PBS, (2) Tac, (3) MSN-Tac-SC6A. The mice, 6 per group, were treated subcutaneously with Tac (2.5 mg/kg B.W.) or MSN-Tac-SC6A (the amount of Tac loaded in MSN-Tac-SC6A was 2.5 mg/kg B.W.). The blood was collected at 24 hrs after s.c. injection. Two groups were compared by paired-samples t-test (mean ± SD, n=6. **P<0.01, ***P<0.001, significantly different from the Tac group). Abbreviations: ALP, alkaline phosphatase; AST, aspartate aminotransferase; PBS, phosphate buffer saline; Tac, tacrine; MSN-Tac-SC6A, MSN loading with tacrine; B.W., body weight; s.c., subcutaneous. |
The toxicity of MSN-SC6A materials
As AChE inhibitor carriers, these MSN-based materials must be highly biocompatible. To demonstrate this, the toxicity of these materials was detected. As shown in Figure S6, both the carboxyl-group-modified MSN and MSN-SC6A exhibited no obvious influence on the body weight change of the mice, compared with the control group. As is known to all, the hemoglobin can be released from the damaged RBCs, leading to a red solution and an enhanced absorbance of the supernatant at 570 nm.39,40 Therefore, the hemolytic effect can be a suitable model to study the cell damage effect of these AChE inhibitor carriers. For the hemolysis experiment, a wide range of concentrations of these MSN-based materials in PBS solution were incubated with RBCs. As shown in Figure 7, both MSN-COOH and MSN-SC6A revealed no significant hemolysis effects on RBCs with the concentration varyingd from 10 μg·mL−1 to 1000 μg·mL−1, which were quite similar with the negative sample. Furthermore, MSN-SC6A showed no obvious cytotoxicity on Bel7402 cell even with the concentration up to 1000 μg·mL−1 (Figure 8). All these results illustrated that modification with SC6A on the surface of the MSN could not induce significant toxicity. As a widely used drug carrier, the toxicity of MSN has been comprehensively investigated both in vitro and in vivo.41–43 It has been reported that the spherical MSNs were distributed in the liver, spleen, brain, kidney and bladder, and excreted through urine.44 The MSNs were not toxic to tissues even after 1 month in vivo.45 Together with our results, the MSNs can be suitable to construct AChE-responsive drug release systems.
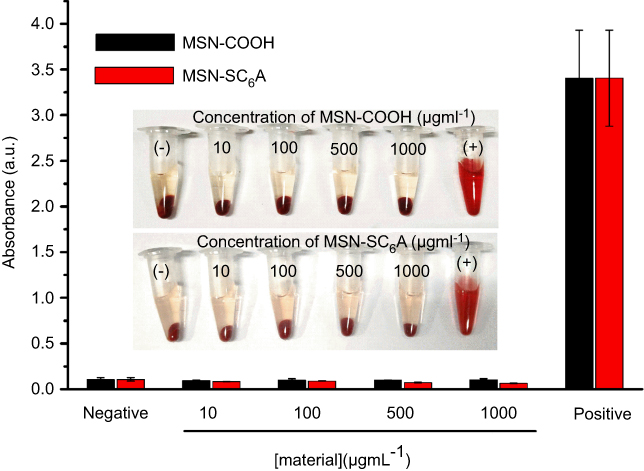 |
Figure 7 Hemolytic assays for MSN-COOH and MSN-SC6A. The RBCs were collected by centrifugation of heparin-stabilized rat blood samples. The concentration of MSN-COOH or MSN-SC6A varied from 10 μg·mL−1 to 1000 μg·mL−1. PBS and water which incubated with the diluted RBC suspension were served as negative and positive controls, respectively (mean ± SD, n=3 for each sample). Abbreviations: MSN-COOH, MSN modified by carboxyl; MSN-SC6A, MSN conjugated with p-sulfonatocalix[6]arene; RBCs, red blood cells; PBS, phosphate buffer saline. |
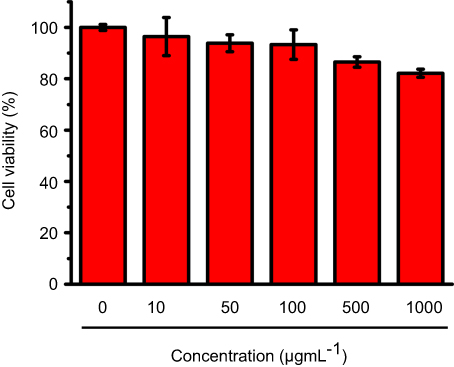 |
Figure 8 The effect of MSN-SC6A on cell viability. Cell viability was determined using the MTT method. The concentration of MSN-SC6A was in the range from 0 μg· mL−1 to 1000 μg· mL−1 (mean ± SD, n=3 for each concentration). Abbreviations: MSN-SC6A, MSN conjugated with p-sulfonatocalix[6]arene; MTT, (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide. |
Conclusion
In summary, an operational targeted drug delivery platform based on AChE-responsive system has been constructed for AChE-associated disease treatment. Our design combines the unique properties of enzyme-controlled MSN with clinical-used AChE inhibitors. By taking the advantage of host–guest chemistry, the drug can be controlled-released by the action of the elevated enzymes. The zero premature release characteristic is of importance for delivery of toxic inhibitors in AChE-associated disease therapy. Compared with AChE inhibitors themselves, the drug delivery system can not only exhibit long duration of drug action on AChE inhibition but also reduce the hepatotoxicity. Our results would shed lights on the design of enzyme-controlled-release multifunctional system for enzyme-associated disease treatment.
Acknowledgments
Financial support was provided by the National Natural Science Foundation of China (Grant No. 21807024), the Youth Top-Notch Talents Supporting Plan of Hebei Province (BJ2018007), the Hundred Persons Plan of Hebei Province (E2018050012) and the Natural Science Foundation of Hebei Province (Grant Nos. H2016206280 and H2017206281).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Allen RK. A review of angiotensin converting enzyme in health and disease. Sarcoidosis. 1991;8(2):95–100.
2. Darveau CA, Suarez RK, Andrews RD, Hochachka PW. Allometric cascade as a unifying principle of body mass effects on metabolism. Nature. 2002;417(6885):166–170. doi:10.1038/417166a
3. Holmström KM, Finkel T. Cellular mechanisms and physiological consequences of redox-dependent signaling. Nat Rev Mol Cell Biol. 2014;15(6):411–421. doi:10.1038/nrm3801
4. Weiss KM, Terwilliger JD. How many diseases does it take to map a gene with SNPs? Nat Genet. 2000;26(2):151–157. doi:10.1038/79866
5. Riley DP. Functional mimics of superoxide dismutase enzymes as therapeutic agents. Chem Rev. 1999;99(9):2573–2588.
6. López-Otín C, Bond JS. Proteases: multifunctional enzymes in life and disease. J Biol Chem. 2008;283(45):30433–30437. doi:10.1074/jbc.R800035200
7. Zimmerman G, Soreq H. Termination and beyond: acetylcholinesterase as a modulator of synaptic transmission. Cell Tissue Res. 2006;326(2):655–669. doi:10.1007/s00441-006-0239-8
8. Soreq H, Seidman S. Acetylcholinesterase – new roles for an old actor. Nat Rev Neurosci. 2001;2(4):294–302. doi:10.1038/35067589
9. Dawson RM. Reversibility of the inhibition of acetylcholinesterase by tacrine. Neurosci Lett. 1990;118(1):85–87. doi:10.1016/0304-3940(90)90254-7
10. Wlodek ST, Clark TW, Scott LR, McCammon JA. Molecular dynamics of acetylcholinesterase dimer complexed with tacrine. J Am Chem Soc. 1997;119(40):9513–9522. doi:10.1021/ja971226d
11. Rogers SL, Friedhoff LT. Long-term efficacy and safety of donepezil in the treatment of Alzheimer’s disease: an interim analysis of the results of a US multicentre open label extension study. Eur Neuropsychopharmacol. 1998;8(1):67–75.
12. Geerts H, Guillaumat PO, Grantham C, Bode W, Anciaux K, Sachak S. Brain levels and acetylcholinesterase inhibition with galantamine and donepezil in rats, mice, and rabbits. Brain Res. 2005;1033(2):186–193. doi:10.1016/j.brainres.2004.11.042
13. Polinsky RJ. Clinical pharmacology of rivastigmine: a new-generation acetylcholinesterase inhibitor for the treatment of Alzheimer’s disease. Clin Ther. 1998;20(4):634–647.
14. Bar-On P, Millard CB, Harel M, et al. Kinetic and structural studies on the interaction of cholinesterases with the anti-Alzheimer drug rivastigmine. Biochemistry. 2002;41(11):3555–3564. doi:10.1021/bi020016x
15. Lewis WG, Green LG, Grynszpan F, et al. Click chemistry in situ: acetylcholinesterase as a reaction vessel for the selective assembly of a femtomolar inhibitor from an array of building blocks. Angew Chem Int Ed Engl. 2002;41(6):1053–1057.
16. Krasiński A, Radić Z, Manetsch R, et al. In situ selection of lead compounds by click chemistry: target-guided optimization of acetylcholinesterase inhibitors. J Am Chem Soc. 2005;127(18):6686–6692. doi:10.1021/ja043031t
17. Bolognesi ML, Cavalli A, Valgimigli L, et al. Multi-target-directed drug design strategy: from a dual binding site acetylcholinesterase inhibitor to a trifunctional compound against Alzheimer’s disease. J Med Chem. 2007;50(26):6446–6449. doi:10.1021/jm701225u
18. Catto M, Pisani L, Leonetti F, et al. Design, synthesis and biological evaluation of coumarin alkylamines as potent and selective dual binding site inhibitors of acetylcholinesterase. Bioorg Med Chem. 2013;21(1):146–152. doi:10.1016/j.bmc.2012.10.045
19. Chen C, Geng J, Pu F, Yang XJ, Ren JS, Qu XG. Polyvalent nucleic acid/mesoporous silica nanoparticle conjugates: dual stimuli-responsive vehicles for intracellular drug delivery. Angew Chem Int Ed Engl. 2011;50(4):882–886. doi:10.1002/anie.201005471
20. Yang XJ, Liu X, Liu Z, Pu F, Ren JS, Qu XG. Near-infrared light-triggered, targeted drug delivery to cancer cells by aptamer gated nanovehicles. Adv Mater. 2012;24(21):2890–2895. doi:10.1002/adma.201104797
21. Chen ZW, Li ZH, Lin YH, Yin ML, Ren JS, Qu XG. Biomineralization inspired surface engineering of nanocarriers for pH-responsive, targeted drug delivery. Biomaterials. 2013;34(4):1364–1371. doi:10.1016/j.biomaterials.2012.10.060
22. Shi P, Qu KG, Wang JS, Li M, Ren JS, Qu XG. pH-responsive NIR enhanced drug release from gold nanocages possesses high potency against cancer cells. Chem Commun. 2012;48(61):7640–7642. doi:10.1039/c2cc33543c
23. Chen ZW, Zhou L, Bing W, et al. Light controlled reversible inversion of nanophosphor-stabilized pickering emulsions for biphasic enantioselective biocatalysis. J Am Chem Soc. 2014;136(20):7498–7504. doi:10.1021/ja503123m
24. Li M, Liu Z, Ren JS, Qu XG. Inhibition of metal-induced amyloid aggregation using light-responsive magnetic nanoparticle prochelator conjugates. Chem Sci. 2012;3:868–873. doi:10.1039/C1SC00631B
25. Luo Z, Cai KY, Hu Y, et al. Mesoporous silica nanoparticles end-capped with collagen: redox-responsive nanoreservoirs for targeted drug delivery. Angew Chem Int Ed Engl. 2011;50(3):640–643. doi:10.1002/anie.201005061
26. Cho H, Bae J, Garripelli VK, Anderson JM, Jun HW, Jo S. Redox-sensitive polymeric nanoparticles for drug delivery. Chem Commun. 2012;48(48):6043–6045. doi:10.1039/c2cc31463k
27. Lai CY, Trewyn BG, Jeftinija DM, et al. A mesoporous silica nanosphere-based carrier system with chemically removable CdS nanoparticle caps for stimuli-responsive controlled release of neurotransmitters and drug molecules. J Am Chem Soc. 2003;125(15):4451–4459. doi:10.1021/ja028650l
28. Singh N, Karambelkar A, Gu L, et al. Bioresponsive mesoporous silica nanoparticles for triggered drug release. J Am Chem Soc. 2011;133(49):19582–19585. doi:10.1021/ja206998x
29. Tang FQ, Li LL, Chen D. Mesoporous silica nanoparticles: synthesis, biocompatibility and drug delivery. Adv Mater. 2012;24(12):1504–1534. doi:10.1002/adma.201104763
30. Trewyn BG, Slowing II, Giri S, Chen HT, Lin VS. Synthesis and functionalization of a mesoporous silica nanoparticle based on the sol-gel process and applications in controlled release. Acc Chem Res. 2007;40(9):846–853. doi:10.1021/ar600032u
31. Guo DS, Wang K, Wang YX, Liu Y. Cholinesterase-responsive supramolecular vesicle. J Am Chem Soc. 2012;134(24):10244–10250. doi:10.1021/ja303280r
32. Lv J, He BN, Wang N, Li M, Lin YL. A gold nanoparticle based colorimetric and fluorescent dual-channel probe for acetylcholinesterase detection and inhibitor screening. RSC Adv. 2018;8(57):32893–32898. doi:10.1039/C8RA06165C
33. Guo DS, Liu Y. Supramolecular chemistry of p-sulfonatocalixarenes and its biological applications. Acc Chem Res. 2014;47(7):1925–1934. doi:10.1021/ar500009g
34. Chen M, He XX, Wang KM, et al. A pH-responsive polymer/mesoporous silica nano-container linked through an acid cleavable linker for intracellular controlled release and tumor therapy in vivo. J Mater Chem B. 2014;2(4):428–436. doi:10.1039/C3TB21268H
35. Scali C, Casamenti F, Bellucci A, Costagli C, Schmidt B, Pepeu G. Effect of subchronic administration of metrifonate, rivastigmine and donepezil on brain acetylcholine in aged F344 rats. J Neural Transm. 2002;109(7–8):1067–1068. doi:10.1007/s007020200090
36. Eagger SA, Levy R, Sahakian BJ. Tacrine in Alzheimer’s disease. Lancet. 1991;338(11):50–51. doi:10.1016/0140-6736(91)90035-n
37. Watkins PB, Zimmerman HJ, Knapp MJ, Gracon SI, Lewis KW. Hepatotoxic effects of tacrine administration in patients with Alzheimer’s disease. JAMA. 1994;271(13):992–998.
38. Anand P, Singh B. A review on cholinesterase inhibitors for Alzheimer’s disease. Arch Pharm Res. 2013;36(4):375–399. doi:10.1007/s12272-013-0036-3
39. Lin YS, Haynes CL. Impacts of mesoporous silica nanoparticle size, pore ordering, and pore integrity on hemolytic activity. J Am Chem Soc. 2010;132(13):4834–4842. doi:10.1021/ja910846q
40. Yildirim A, Ozgur E, Bayindir M. Impact of mesoporous silica nanoparticle surface functionality on hemolytic activity, thrombogenicity and non-specific protein adsorption. J Mater Chem B. 2013;1(14):1909–1920. doi:10.1039/c3tb20139b
41. Park JH, Gu L, Von Maltzahn G, Ruoslahti E, Bhatia SN, Sailor MJ. Biodegradable luminescent porous silicon nanoparticles for in vivo applications. Nat Mater. 2009;8(4):331–336. doi:10.1038/nmat2398
42. Croissant JG, Fatieiev Y, Khashab NM. Degradability and clearance of silicon, organosilica, silsesquioxane, silica mixed oxide, and mesoporous silica nanoparticles. Adv Mater. 2017;29(9):1604634. doi:10.1002/adma.201700681
43. Chen G, Teng Z, Su X, Liu Y, Lu G. Unique biological degradation behavior of Stöber mesoporous silica nanoparticles from their interiors to their exteriors. J Biomed Nanotechnol. 2015;11(4):722–729.
44. Huang X, Li L, Liu T, et al. The shape effect of mesoporous silica nanoparticles on biodistribution, clearance, and biocompatibility in vivo. ACS Nano. 2011;5(7):5390–5399. doi:10.1021/nn200365a
45. He Q, Zhang Z, Gao F, Li Y, Shi J. In vivo biodistribution and urinary excretion of mesoporous silica nanoparticles: effects of particle size and PEGylation. Small. 2011;7(2):271–280. doi:10.1002/smll.201001459
Supplementary materials
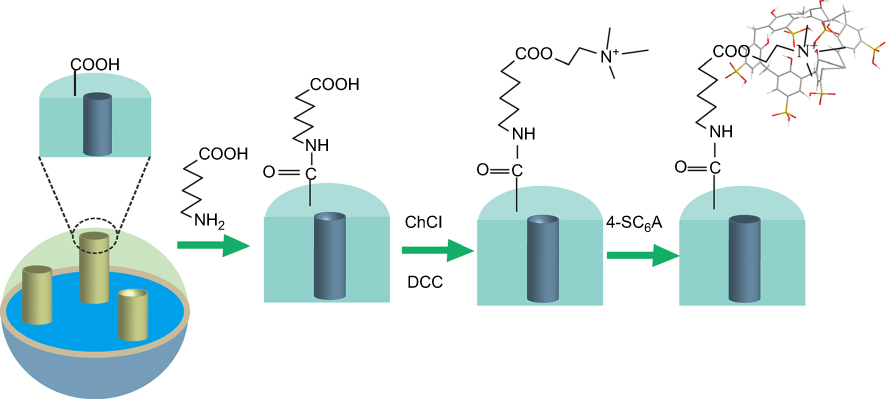 |
Figure S1 The chemistry for the preparation of MSN-SC6A. Abbreviations: DCC, N,N’-Dicyclohexylcarbodimide; ChCl, 2-Hydroxyethyl trimethylammonium chloride; 4-SC6A, p-sulfonatocalix[6]arene. |
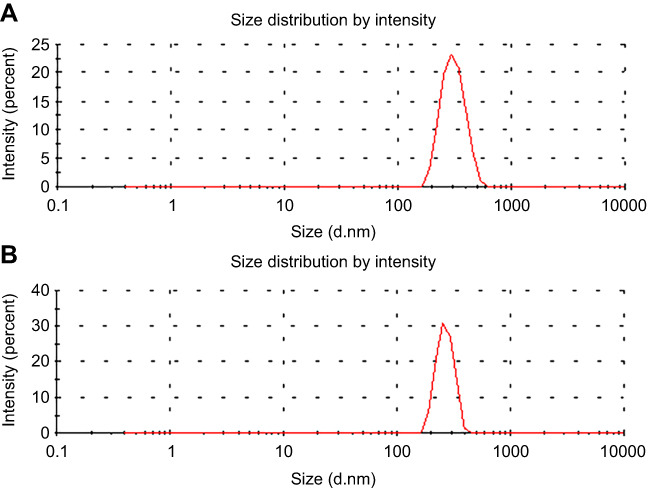 |
Figure S2 The hydrodynamic radius of (A) MSN-COOH and (B) MSN-SC6A. Abbreviations: MSN, mesoporous silica nanoparticles; MSN-COOH, MSN modified by carboxyl; MSN-SC6A, MSN conjugated with p-sulfonatocalix[6]arene. |
 |
Figure S3 FTIR spectra of MSN-CN (A), MSN-COOH (B), MSN-COOR (C), MSN-Ch (D) and MSN-SC6A (E). Abbreviations: FTIR, Fourier-transform infrared; MSN, mesoporous silica nanoparticles; SC6A, p-sulfonatocalix[6]arene; MSN-CN, MSN modified by cyano group; MSN-COOH, MSN modified by carboxyl; MSN-COOR, MSN modified by epsilon-aminocaproic acid; MSN-Ch, MSN conjugated with choline; MSN-SC6A, MSN conjugated with p-sulfonatocalix[6]arene. |
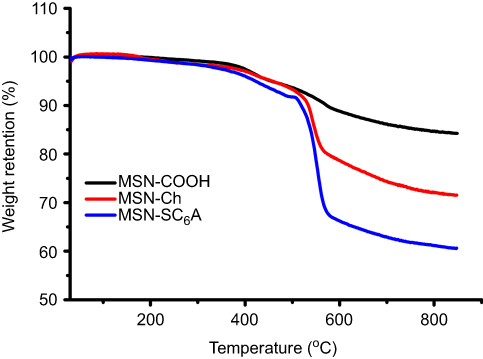 |
Figure S4 Thermogravimetric analysis of the samples: MSN-COOH, MSN-Ch, MSN-SC6A. Abbreviations: MSN, mesoporous silica nanoparticles; MSN-COOH, MSN modified by carboxyl; MSN-Ch, MSN conjugated with choline; MSN-SC6A, MSN conjugated with p-sulfonatocalix[6]arene. |
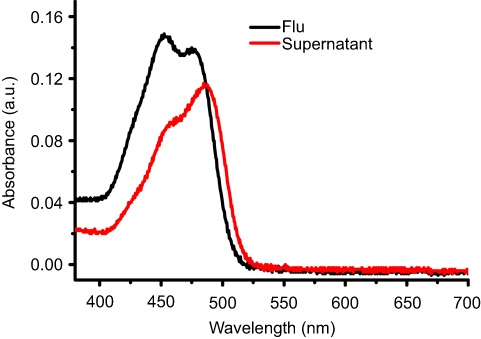 |
Figure S5 The UV-Vis absorbance of Flu before or after loaded into the MSN. Abbreviations: UV-Vis, Ultraviolet-Visible; Flu, fluorescein; MSN, mesoporous silica nanoparticles. |
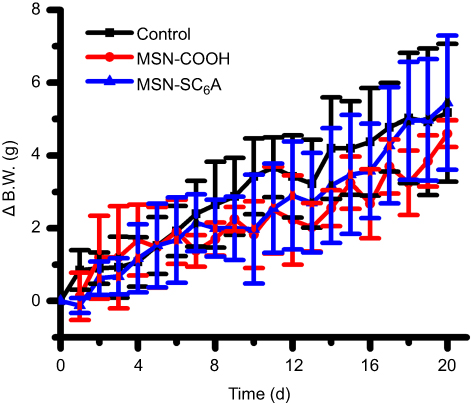 |
Figure S6 The effects of MSN-COOH and MSN-SC6A on body weight change of the mice. Abbreviations: B.W., body weight; MSN, mesoporous silica nanoparticles; MSN-COOH, MSN modified by carboxyl; MSN-SC6A, MSN conjugated with p-sulfonatocalix[6]arene (mean ± SD, n=6 for each group). |
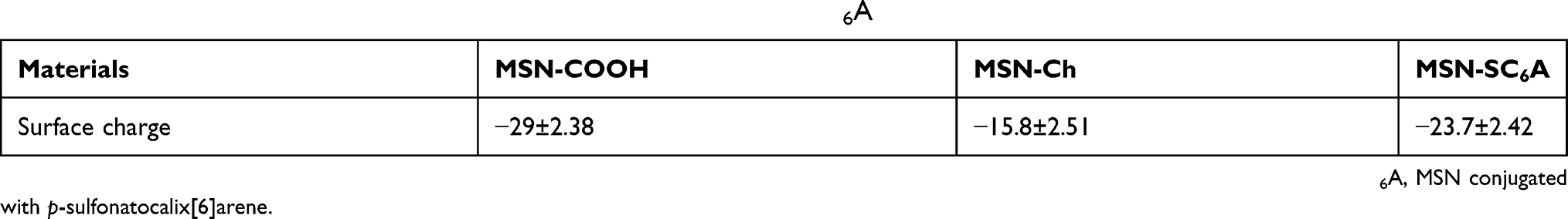 |
Table S1 The surface charges of MSN-COOH, MSN-Ch and MSN-SC6A |
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.