Back to Journals » Pharmacogenomics and Personalized Medicine » Volume 7
Tailoring therapy for heart failure: the pharmacogenomics of adrenergic receptor signaling
Authors Femminella GD, Barrese V, Ferrara N, Rengo G ![]()
Received 21 May 2014
Accepted for publication 27 June 2014
Published 9 September 2014 Volume 2014:7 Pages 267—273
DOI https://doi.org/10.2147/PGPM.S49799
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Grazia Daniela Femminella,1 Vincenzo Barrese,2,3 Nicola Ferrara,1,4 Giuseppe Rengo4
1Department of Translational Medical Sciences, Federico II University, Naples, Italy; 2Department of Neuroscience, Reproductive Science and Odontostomatology, Federico II University, Naples, Italy; 3Division of Biomedical Sciences, St George’s University of London, London, UK; 4”Salvatore Maugeri” Foundation – IRCCS – Scientific Institute of Telese Terme, Telese Terme, Benevento, Italy
Abstract: Heart failure is one of the leading causes of mortality in Western countries, and β-blockers are a cornerstone of its treatment. However, the response to these drugs is variable among individuals, which might be explained, at least in part, by genetic differences. Pharmacogenomics is the study of genetic contributions to drug response variability in order to provide evidence for a tailored therapy in an individual patient. Several studies have investigated the pharmacogenomics of the adrenergic receptor system and its role in the context of the use of β-blockers in treating heart failure. In this review, we will focus on the most significant polymorphisms described in the literature involving adrenergic receptors and adrenergic receptor-related proteins, as well as genetic variations influencing β-blocker metabolism.
Keywords: adrenergic system, polymorphisms, β-blockers, functional recovery
Introduction
Heart failure (HF), the leading cause of mortality in Western countries, is a clinical syndrome that develops in response to a cardiac injury, resulting in the impairment of the contractile function of the heart.1 HF may result from disorders of the pericardium, myocardium, endocardium, or heart valves, or due to other causes, but most patients with HF have symptoms due to left ventricular dysfunction. Patients present a range of conditions ranging from normal ventricular size and preserved ejection fraction to severe dilatation and/or markedly reduced ejection fraction.2
HF incidence increases with age, rising from 20 per 1,000 individuals 65–69 years of age to >80 per 1,000 individuals among those ≥85 years of age.2 Ethnic disparities in HF prevalence have been identified; the incidence of HF is greatest among African-Americans, intermediate among whites and Hispanics, and lowest among Chinese Americans. These differences are in large part determined by the higher prevalence of hypertension and diabetes mellitus among African-Americans.3
In HF due to systolic dysfunction, in response to the reduced peripheral tissue perfusion, several compensatory mechanisms are activated, aiming to maintain the cardiovascular homeostatic balance. Among these, hyperactivation of the adrenergic nervous system (ANS) plays a crucial role.4,5 However, in the long term, excessive activation of the ANS is detrimental, inducing systemic vasoconstriction, increased sodium/water retention, and ventricular remodeling, all of which contribute to disease progression. In the 1980s, the role of neurohormonal activation in HF was documented, subsequently leading to an increasing interest in the use of β-blockers to counteract ANS hyperactivation.6 Until that time, β-blockers were contraindicated in patients with HF due to their negative inotropic effect. In the 1990s, however, large randomized controlled trials with bisoprolol, metoprolol succinate, and carvedilol provided evidence of the mortality and morbidity benefits of β-blockade.7–9 β-blockers, in fact, reduced cardiovascular mortality, sudden cardiac death, and hospitalizations, and improved New York Heart Association functional class10 compared to placebo, when added to standard background therapy. Based on these data, β-blockers are now recommended in HF as an essential therapy, unless contraindications are present.6,11 The most recent guidelines recommend the use of β-blockers in all patients with HF due to left ventricular systolic dysfunction, including among older adults and patients with peripheral vascular disease, erectile dysfunction, diabetes mellitus, interstitial pulmonary disease, and chronic obstructive pulmonary disease (COPD) without reversibility. An exception is represented by patients who have COPD with reversible obstructive pulmonary disease.2
However, not all β-blockers show favorable effects in HF patients, and variability in responses to β-blockers exists among HF patients. For example, in about 25% of HF patients, β-blockers need to be discontinued due to drug intolerance. These interindividual variations might be explained, at least in part, by genetic differences.11 Pharmacogenomics is the study of genetic contributions to differences in drug response, and the current challenge in this discipline is to investigate the array of genes whose variability contributes to variability in drug efficacy or toxicity, in order to tailor therapy in each patient, predicting who might or might not respond to a specific treatment and avoiding adverse events in susceptible individuals.12
In this review, we will focus on the pharmacogenomics of the adrenergic receptor (AR) system and its role in the context of the use of β-blockers in HF.
The AR system in HF
The ARs belong to the superfamily of G-protein-coupled receptors, with seven transmembrane domains, an extracellular N-terminal region, and an intracellular C-terminus.13 Their agonists are norepinephrine and epinephrine, released by ANS nerve terminals and the adrenal gland. The ARs are divided into three types: α1AR (α1A, α1B, α1D); α2AR (α2A, α2B, α2C); and βAR (β1, β2, β3). The human heart contains all βAR subtypes, with β1AR accounting for the 80% of total βAR density in the normal myocardium and β2AR comprising 20% of cardiac βARs.14 However, under various conditions, such as during aging15 and in HF,16 the proportion of β1 subtypes decreases due to downregulation, while levels of β2-AR remain stable, resulting in a 1:1 ratio between the two AR subtypes. As for the β3ARs, their role in the myocardium has been elucidated only recently and remains incompletely explained.
In the heart, the most important role of βARs is the regulation of cardiac rate and contractility in response to norepinephrine and epinephrine. Stimulation of βARs has positive inotropic, chronotropic, and lusitropic effects in the myocardium, respectively increasing cardiac contractility, frequency, and rate of relaxation. However, the stimulation of myocardial β3ARs has opposite effects from the other two βAR subtypes, resulting in a reduced inotropic response. From a molecular point of view, stimulation of βAR pathways catalyzes the exchange of guanosine triphosphate for guanosine diphosphate on the Gα subunit of heterotrimeric G proteins, resulting in the dissociation into active Gα and free Gβγ subunits with consequent stimulation of the adenylate cyclase, an increase in intracellular cyclic adenosine monophosphate and protein kinase A-dependent phosphorylation, and modulation of the activity of important proteins involved in myocardial contractility, such as L-type Ca2+ channels, troponin I, and sarcoplasmic reticular Ca2+/adenosine triphosphatase inhibitory protein. Moreover, through the βγ subunits of the heterotrimeric G-protein, βAR stimulation activates members of the G-protein receptor kinase (GRK) family.17,18 The GRKs are a family of cytosolic serine/threonine kinases consisting of seven isoforms that share structural and functional similarities. GRK2, initially identified as βAR kinase-1, is the most abundant GRK expressed in the heart.19 Upon receptor activation, GRK2 translocates to the plasma membrane, as a consequence of its interaction with G βγ subunits released by agonist binding of the receptor, and by phosphorylating residues in the C-terminal region of the receptor, GRK2 induces the binding of protein such as β-arrestin, thus causing receptor uncoupling from the Gs protein and its transduction pathways. Moreover, GRK2-induced receptor phosphorylation results in the increase of βAR affinity for the inhibitory Gi protein, thus accelerating receptor desensitization.20
In addition to the Gs-mediated signaling, stimulation of βARs can also activate Gi proteins as well as different intracellular pathways, modulating the activity of mitogen-activated protein kinases and other proteins involved in cell cycle control and apoptosis, thus suggesting a wider role of βARs in regulating cardiac pathophysiology. Interestingly, the capability to stimulate such “alternative” signaling pathways seems to be different among βAR subtypes, arguing in favor of a unique role of specific βARs in determining cardiomyocyte death or survival.21
During HF, ANS hyperactivity is evidenced by increased plasma norepinephrine and epinephrine levels, sympathetic outflow, and norepinephrine spillover from cardiac sympathetic nerve terminals. The elevated sympathetic neurohormone levels in chronic HF lead to chronically elevated stimulation of the cardiac βAR system, which is detrimental to the failing heart. Data from both preclinical and clinical studies have indicated that the cardiac βAR system is severely affected during HF. In particular, βAR signaling and function are significantly impaired and the adrenergic cardiac reserve is reduced.17,22 In human HF, myocardial βAR dysfunction is characterized by receptor downregulation, a selective reduction of β1AR density at the plasma membrane, and by functional desensitization, the uncoupling of βARs from their cognate G proteins. On the other hand, it has been demonstrated, both in human and animal studies, that β3ARs are upregulated during HF. Along with receptor dysfunction, also postreceptor components of βAR signaling pathways are affected in HF. In particular, myocardial levels and activity of myocardial GRKs, namely GRK2 and GRK5, are elevated both in humans and in animal models of HF. Evidence suggests that ANS hyperactivity chronically stimulating cardiac βARs also triggers GRK2 upregulation in cardiomyocytes, which in turn is responsible for cardiac βAR downregulation and desensitization. Accordingly, GRK2 elevation can be considered a homeostatic mechanism aimed at protecting the heart from excessive catecholamine-induced toxicity, and several studies have confirmed the hypothesis that GRK2 has a pivotal role in the regulation of βAR-dependent cardiac contractility and function.23,24 In addition, GRK2 exerts its regulatory effects on myocardial function by modulating AR activity also in the adrenal gland.25,26 Indeed, it has been demonstrated that the inhibition of adrenal GRK2 by a small peptide (βARKct) increases cardiac contractility, possibly by reducing the uncoupling and desensitization of α2ARs, which inhibit catecholamine release from the adrenal gland.27–29 Moreover, βARKct administration normalizes neurohormonal axis activity30 and increases survival in several animal models of HF.31
Taken together, this evidence strongly suggests that changes in the activity of ANS play a pivotal and direct role in the pathogenesis of HF and offers further insights to understand the efficacy of β-blockers in the treatment of HF. Indeed, recent studies have demonstrated that β-blockers can ameliorate clinical outcomes in HF also by reducing adrenal GRK2 levels, thus normalizing catecholamine release by restoring the negative feedback mediated by α2AR (bisoprolol)32 or by increasing vascular endothelial growth factor signaling, thus stimulating neoangiogenesis (atenolol and bisoprolol).33
Pharmacogenetics of the AR system and its role in HF
The difference in response to β-blockers is influenced by interindividual genetic variations. In particular, genetic polymorphisms in the metabolic enzymes can affect the pharmacokinetics of β-blockers (ie, cytochrome P450 [CYP]2D6), while those in the receptor genes and in the other molecules involved in the signaling pathway can affect the pharmacodynamics of β-blockers.
Here, we will review the most significant polymorphisms described in the literature involving ARs and AR-related proteins, as well as genetic variations influencing β-blocker metabolism (Table 1).
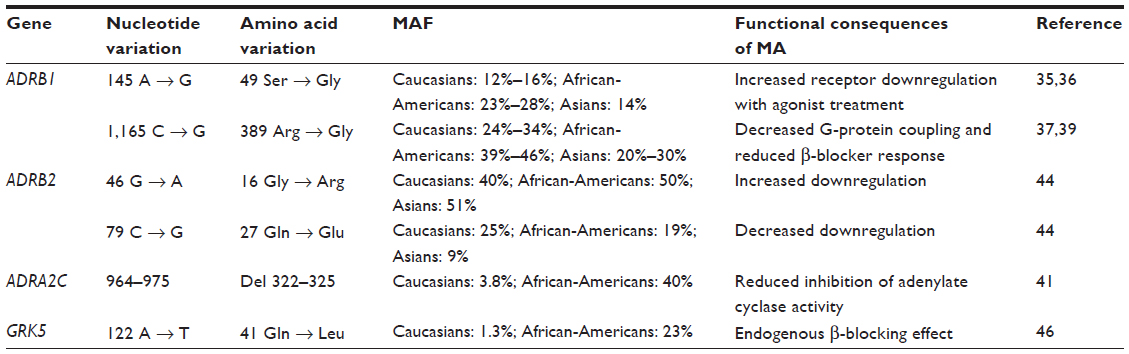 | Table 1 Common polymorphisms in adrenergic signaling genes |
β1AR genetic polymorphisms
In the human β1AR, at least 12 single nucleotide polymorphisms (SNPs) have been described, but only two of them can actually affect receptor function, being relevant from the clinical point of view. The first is Ser49Gly, which occurs in the N-terminus region where it can be involved in receptor downregulation as well as in intracellular trafficking.34 The functional consequences of this SNP have been separately studied by two different groups in recombinant cellular models. Rathz et al35 showed that both variants have similar agonist and antagonist binding affinities. However, long-term agonist-induced downregulation was greater for Gly49 compared with Ser49. Moreover, the two variants presented different patterns in N-glycosylation. Levin et al36 demonstrated that Gly49 has the features of constitutively active receptors, with both basal and agonist-stimulated adenylyl cyclase activities that are higher compared to the Ser49 variant. They also showed that after sustained catecholamine stimulation, the Gly49 variant undergoes increased desensitization and a more profound agonist-promoted downregulation than the Ser49 variant.
The other relevant SNP in β1AR is Arg389Gly in the intracellular C-terminus, which is an important site for G-protein binding. Functional studies have demonstrated that Arg389 receptors have higher basal and isoproterenol-stimulated levels of adenylyl cyclase activity compared to the Gly389 variant. Moreover, the Arg389 receptor also showed increased agonist-induced guanosine triphosphate binding with enhanced Gs coupling.37 Initially, this behavior led to the credence that an Arg389 variant could provide an enhanced response to β-blockers, but further studies indicated that this was not the case.38,39 In fact, Arg389 showed a higher degree of agonist-induced desensitization compared to Gly389, indicating that increased G-protein coupling also resulted in enhanced susceptibility to GRK-mediated desensitization.38 Transgenic mice expressing Arg389 have enhanced cardiac receptor function and contractility compared with those expressing Gly389.39 However, with aging, Arg389 mice have decreased β-agonist signaling to adenylyl cyclase and decreased cardiac contractility compared with those with Gly389 hearts. Moreover, Arg389 hearts showed increased fibrosis, but had a greater response to β-blocker treatment, thus indicating that the human Arg389 variant can predispose individuals to HF, leading to reduced receptor coupling and ventricular dysfunction, and influencing the therapeutic response to β-blockers.
Similar findings were also obtained in clinical studies. Arg389 homozygous HF patients treated with bucindolol had reduced mortality and hospitalization compared to those treated with placebo. In contrast, Gly389 carriers had no clinical response to bucindolol compared with placebo, suggesting that the β1AR389 variation alters signaling and affects the therapeutic response to β-blockers. Thus, this variation should be taken into account to individualize treatment of the syndrome.40
Polymorphisms of the α2CARs
As mentioned in the section on the AR system and its role in HF, dysregulation of ANS also involves alterations of the signaling pathways mediated by presynaptic ARs (in particular, the α2C subtype), which decrease catecholamine release from the sympathetic nerve endings. Therefore, polymorphisms of these receptors might influence adrenergic signaling and, consequently, the clinical history of HF as well as the response to the treatment. In particular, an in-frame deletion of 12 nucleotides, leading to the loss of four amino acids (Gly-Ala-Gly-Pro) within the α2CAR protein has been identified and associated with reduced stimulation of the Gi pathway.41 In vitro, the α2C-Del322–325 mutation causes a reduced inhibition of adenylate cyclase activity when compared to the wild-type receptor. Moreover, the inositol triphosphate pathway, as well as extracellular signal-regulated kinase signaling, is severely impaired. Because the changes in α2C-AR activity prompted by a Del322–325 mutation increase (the response to catecholamine stimulation), blockade of the adrenergic system might be particularly effective in HF treatment in patients carrying this mutation, thus highlighting Del322–325 as a possible pharmacogenomic locus to predict response to therapy with β-blockers. Indeed, recent studies have suggested that patients carrying α2C-AR Del322–325 mutations together with the β1AR-Arg389 variant have a more pronounced increase in left ventricular ejection fraction (LVEF) with respect to other genotypes.42
Polymorphisms of the β2ARs
Although β1ARs are the main cardiac ARs, it has been demonstrated that β2AR expression is increased in the failing heart, balancing the reduced expression of β1ARs. Moreover, many β-blockers are nonselective and might also affect β2AR-mediated signaling. Therefore, β2AR polymorphisms might contribute to the modulation of the HF phenotype, as well as the response to β-blocker treatment. Three variants affecting amino acids in positions 16, 27, and 164 have been identified in the β2AR protein: Arg or Gly16; Gln or Glu27; and Ile164, respectively.43 Among them, polymorphisms in positions 16 and 27 are more common. Although β2ARs incorporating an Ile residue show reduced adenylate cyclase activity and decreased cardiac contractility after agonist stimulation, the Ile164 variant is very rare and its clinical relevance is restricted to a small number of patients. By contrast, Arg/Gly16 and Gln/Glu27 polymorphisms affect receptor downregulation after agonist stimulation.44 Among the possible haplotypes generated by different combinations of amino acids in positions 16 and 27, β2ARs incorporating a Gly16 residue seem to display a higher agonist-promoted downregulation. Also, SNPs have been found in noncoding regions of the β2AR gene, namely the promoter or the 3′-untranslated region: -20 T-C; -47 T-C; -367 T-C; -468 C-G; -654 G-A; -1343 A-G; and -1429 T-A.45 Some of these variants are in linkage disequilibrium with the Arg16Gly and Gln27Glu polymorphisms and seem to be associated with different basal expression levels and agonist-induced downregulation of the receptors. Therefore, characterization of SNPs affecting amino acids in positions 16 and 27, as well as noncoding regions, might help to predict response to therapy and HF progression.
Polymorphisms of the GRKs
As stated before, the most abundant GRK isoform in the heart is GRK2, but GRK5 is also expressed in the heart. Levels of both GRK2 and GRK5 are upregulated during HF, possibly as a compensatory mechanism to reduce catecholamine stimulation. A variant affecting the residue in position 41 (Leu in place of the more common Glu) has been identified in the GRK5 sequence.46 The molecular and pathophysiological effects of Leu41 substitution have been investigated in in vitro studies, demonstrating that this polymorphism confers a higher capability to uncouple the adrenoreceptor, thus acting as a genetic β-blocker. Indeed, transgenic mice expressing GRK5-Leu41 in which HF was induced by infusion of isoproterenol showed a better LVEF compared to controls (GRK5-Glu41). Moreover, while β-blocker administration ameliorated cardiac function in animals carrying the Glu41 variant, only little increases in LVEF were observed in GRK5-Leu41 mice, a result reflecting the endogenous β-blocking effect prompted by this polymorphism.
A recent study by Lobmeyer et al47 characterized SNPs in the GRK2 gene (ADRBK1), finding that one of them (rs1894111 G>A) shows a signal for association with the systolic and diastolic blood pressure response to hydrochlorothiazide in white patients, while a novel SNP in the promoter region of ADRBK1 was not associated with differential GRK2 expression.
Polymorphisms in metabolic enzymes
The main liver CYP enzyme isoform involved in the metabolism of most β-blockers is 2D6. As a consequence, polymorphisms affecting CYP2D6 activity might potentially influence response to therapy with β-blockers. Based on the different allelic variants of CYP2D6, which can reduce (CYP2D6*10, CYP2D6*17, and CYP2D6*41) or completely abolish (CYP2D6*4 and CYP2D6*5) enzymatic activity, patients can be divided in groups ranging from poor metabolizers (PMs) to extensive metabolizers. CYP2D6 alleles show interethnic/geographic differences, with PMs mainly localized in Europe, and the extensive metabolizer haplotype found diffusely in North Africa and Oceania.48 Pharmacogenomic characterization of patients might have beneficial effects in choosing an adequate dose of β-blockers to be administered, thus achieving a “tailored” and patient-based therapy.
Potential clinical implications of β-blocker pharmacogenetics
Previous evidence demonstrated that polymorphisms in genes involved in catecholamine signaling might modulate HF progression and response to therapy. To better address this issue, several meta-analyses and studies have been conducted, and the results obtained are often controversial and do not fully clarify the real impact of a given gene variant in modifying clinical outcomes. For example, an increased improvement of cardiac function (measured by evaluation of LVEF) has been found in patients carrying β1AR-Arg389 treated with metoprolol succinate or carvedilol.39,49 By contrast, no association was found in other studies.40,50 Similar results also arise from studies evaluating possible correlations between β-blockers and polymorphisms in β2AR and α2CAR.11 However, the results could be influenced by differences in many variables such as the β-blocker used, the population, and the duration of treatment. For this reason, large dedicated, randomized clinical trials should be carried out to clarify these issues. In addition, it might be possible that a stronger link between pharmacogenomic variability and clinical outcomes could be found by considering two or more polymorphisms influencing adrenoreceptor pathophysiology, as well as β-blocker metabolism. As an example, it has been demonstrated that α2CARdel322–325 causes a more pronounced increase in LVEF compared to other genotypes when associated with the β1AR-Arg389. Similarly, patients carrying the Gln41 variant in GRK5 in a β1AR-Arg389 background show a decreased mortality rate after therapy with β-blockers.42
Moreover, one of the most important aspects of tailored therapy and pharmacogenetics is ethnic variability in responses to certain treatment strategies. As for β-blockers, a general hypothesis is that the African-American population is less responsive to β-blockers than the Caucasian population because genotypes with poor β-blocker response are more common in African-American patients.34 The β-blocker Evaluation of Survival Trial (BEST), a study enrolling a higher percentage of African-American patients compared to similar studies, has shown a trend towards higher mortality for African-American patients receiving bucindolol compared to those receiving placebo, despite improved cardiac function.51 This effect could be partly explained by the fact that bucindolol has an intrinsic sympathomimetic activity, which could be detrimental in the African-American HF population, which has lower baseline neurohumoral activation. In another study evaluating the effects of atenolol administration before and after exercise, the reduction in heart rate was significantly more evident in Caucasian patients than in African-American patients.52
Another possible application of pharmacogenetic characterization might be the possibility of finding the correct dose of β-blocker to be administered. For instance, it has been demonstrated that, although not influencing the response to therapy, patients classified as PMs for CYP2D6 require a higher dose of carvedilol to obtain clinical improvement. Similar results were obtained in patients homozygous for the β1ARArg389 variant.53 Therefore, patients carrying both variants (in β1AR and CYP2D6) might require a higher dose of carvedilol to reach beneficial effects on HF.54 Finally, genetic information could be used to identify patients at high risk for developing serious adverse events. This strategy might be very useful also in the enrollment of patients in clinical trials, thus avoiding the selection of people at high risk.11 A possible polymorphism to be screened might be the β2ARArg16Gln27 haplotype, because two studies have demonstrated that patients carrying this genetic configuration show an increased risk of adverse events.55,56
Disclosure
The authors report no conflicts of interest in this work.
References
Rengo F, Leosco D, Iacovoni A, et al. [Epidemiology and risk factors for heart failure in the elderly]. Ital Heart J. 2004;5(Suppl 10):9S–16S. Italian. | |
Yancy CW, Jessup M, Bozkurt B, et al; Writing Committee Members; American College of Cardiology Foundation/American Heart Association Task force on Practice Guidelines. 2013 ACCF/AHA guideline for the management of heart failure: a report of the American College of Cardiology Foundation/American Heart Association Task Force on practice guidelines. Circulation. 2013;128(16):e240–e327. | |
Bahrami H, Kronmal R, Bluemke DA, et al. Differences in the incidence of congestive heart failure by ethnicity: the multi-ethnic study of atherosclerosis. Arch Intern Med. 2008;168(19):2138–2145. | |
Lymperopoulos A, Rengo G, Koch WJ. Adrenal adrenoceptors in heart failure: fine-tuning cardiac stimulation. Trends Mol Med. 2007;13(12):503–511. | |
Lymperopoulos A, Rengo G, Koch WJ. Adrenergic nervous system in heart failure: pathophysiology and therapy. Circ Res. 2013;113(6):739–753. | |
Azuma J, Nonen S. Chronic heart failure: beta-blockers and pharmacogenetics. Eur J Clin Pharmacol. 2009;65(1):3–17. | |
CIBIS II Investigators and Committees. The Cardiac Insufficiency Bisoprolol Study II (CIBIS-II): a randomised trial. Lancet. 1999;353:9–13. | |
MERIT-HF Study Group. Effect of metoprolol CR/XL in chronic heart failure: Metoprolol CR/XL Randomized Intervention Trial in Congestive Heart Failure (MERIT-HF). Lancet. 1999;353:2001–2009. | |
Packer M, Coats AJ, Fowler MB, et al. Effect of carvedilol on survival in severe chronic heart failure. N Engl J Med. 2001;344:1651–1658. | |
Fleg JL, Piña IL, Balady GJ, et al. Assessment of functional capacity in clinical and research applications: An advisory from the Committee on Exercise, Rehabilitation, and Prevention, Council on Clinical Cardiology, American Heart Association. Circulation. 2000;102(13):1591–1597. | |
Shin J, Johnson JA. Beta-blocker pharmacogenetics in heart failure. Heart Fail Rev. 2010;15(3):187–196. | |
Johnson JA, Liggett SB. Cardiovascular pharmacogenomics of adrenergic receptor signaling: clinical implications and future directions. Clin Pharmacol Ther. 2011;89(3):366–378. | |
Rengo G, Lymperopoulos A, Koch WJ. Future g protein-coupled receptor targets for treatment of heart failure. Curr Treat Options Cardiovasc Med. 2009;11(4):328–338. | |
Rengo G, Lymperopoulos A, Leosco D, Koch WJ. GRK2 as a novel gene therapy target in heart failure. J Mol Cell Cardiol. 2011;50(5):785–792. | |
Leosco D, Rengo G, Iaccarino G, et al. Exercise training and beta-blocker treatment ameliorate age-dependent impairment of beta-adrenergic receptor signaling and enhance cardiac responsiveness to adrenergic stimulation. Am J Physiol Heart Circ Physiol. 2007;293(3):H1596–H1603. | |
Ciccarelli M, Chuprun JK, Rengo G, et al. G protein-coupled receptor kinase 2 activity impairs cardiac glucose uptake and promotes insulin resistance after myocardial ischemia. Circulation. 2011;123(18):1953–1962. | |
Rengo G, Perrone-Filardi P, Femminella GD, et al. Targeting the β-adrenergic receptor system through G-protein-coupled receptor kinase 2: a new paradigm for therapy and prognostic evaluation in heart failure: from bench to bedside. Circ Heart Fail. 2012;5(3):385–391. | |
Lymperopoulos A, Rengo G, Koch WJ. GRK2 inhibition in heart failure: something old, something new. Curr Pharm Des. 2012;18(2):186–191. | |
Petrofski JA, Koch WJ. The beta-adrenergic receptor kinase in heart failure. J Mol Cell Cardiol. 2003;35:1167–1174. | |
Rengo G, Zincarelli C, Femminella GD, et al. Myocardial β(2) -adrenoceptor gene delivery promotes coordinated cardiac adaptive remodelling and angiogenesis in heart failure. Br J Pharmacol. 2012;166(8):2348–2361. | |
Barrese V, Taglialatela M. New advances in beta-blocker therapy in heart failure. Front Physiol. 2013;4:323. | |
Rengo G, Galasso G, Femminella GD, et al. Reduction of lymphocyte G protein-coupled receptor kinase-2 (GRK2) after exercise training predicts survival in patients with heart failure. Eur J Prev Cardiol. 2014;21(1):4–11. | |
Rengo G, Leosco D, Zincarelli C, et al. Adrenal GRK2 lowering is an underlying mechanism for the beneficial sympathetic effects of exercise training in heart failure. Am J Physiol Heart Circ Physiol. 2010;298(6):H2032–H2038. | |
Raake PW, Vinge LE, Gao E, et al. G protein-coupled receptor kinase 2 ablation in cardiac myocytes before or after myocardial infarction prevents heart failure. Circ Res. 2008;103(4):413–422. | |
Lymperopoulos A, Rengo G, Zincarelli C, Kim J, Koch WJ. Adrenal beta-arrestin 1 inhibition in vivo attenuates post-myocardial infarction progression to heart failure and adverse remodeling via reduction of circulating aldosterone levels. J Am Coll Cardiol. 2011;57(3):356–365. | |
Lymperopoulos A, Rengo G, Zincarelli C, Kim J, Soltys S, Koch WJ. An adrenal beta-arrestin 1-mediated signaling pathway underlies angiotensin II-induced aldosterone production in vitro and in vivo. Proc Natl Acad Sci U S A. 2009;106(14):5825–5830. | |
Lymperopoulos A, Rengo G, Zincarelli C, Soltys S, Koch WJ. Modulation of adrenal catecholamine secretion by in vivo gene transfer and manipulation of G protein-coupled receptor kinase-2 activity. Mol Ther. 2008;16(2):302–307. | |
Lymperopoulos A, Rengo G, Funakoshi H, Eckhart AD, Koch WJ. Adrenal GRK2 upregulation mediates sympathetic overdrive in heart failure. Nat Med. 2007;13(3):315–323. | |
Lymperopoulos A, Rengo G, Gao E, Ebert SN, Dorn GW 2nd, Koch WJ. Reduction of sympathetic activity via adrenal-targeted GRK2 gene deletion attenuates heart failure progression and improves cardiac function after myocardial infarction. J Biol Chem. 2010;285(21):16378–16386. | |
Rengo G, Lymperopoulos A, Zincarelli C, et al. Myocardial adeno-associated virus serotype 6-betaARKct gene therapy improves cardiac function and normalizes the neurohormonal axis in chronic heart failure. Circulation. 2009;119(1):89–98. | |
Rockman HA, Chien KR, Choi DJ, et al. Expression of a beta-adrenergic receptor kinase 1 inhibitor prevents the development of myocardial failure in gene-targeted mice. Proc Natl Acad Sci U S A. 1998;95(12):7000–7005. | |
Rengo G, Lymperopoulos A, Zincarelli C, et al. Blockade of β-adrenoceptors restores the GRK2-mediated adrenal α(2)-adrenoceptor-catecholamine production axis in heart failure. Br J Pharmacol. 2012;166(8):2430–2440. | |
Rengo G, Cannavo A, Liccardo D, et al. Vascular endothelial growth factor blockade prevents the beneficial effects of β-blocker therapy on cardiac function, angiogenesis and remodeling in heart failure. Circ Heart Fail. 2013;6(6):1259–1267. | |
von Homeyer P, Schwinn DA. Pharmacogenomics of β-adrenergic receptor physiology and response to beta-blockade. Anesth Analg. 2011;113(6):1305–1318. | |
Rathz DA, Brown KM, Kramer LA, Liggett SB. Amino acid 49 polymorphisms of the human beta1-adrenergic receptor affect agonist-promoted trafficking. J Cardiovasc Pharmacol. 2002;39(2):155–160. | |
Levin MC, Marullo S, Muntaner O, Andersson B, Magnusson Y. The myocardium-protective Gly-49 variant of the beta 1-adrenergic receptor exhibits constitutive activity and increased desensitization and down-regulation. J Biol Chem. 2002;277(34):30429–30435. | |
Mason DA, Moore JD, Green SA, Liggett SB. A gain-of-function polymorphism in a G-protein coupling domain of the human beta1-adrenergic receptor. J Biol Chem. 1999;274(18):12670–12674. | |
Rathz DA, Gregory KN, Fang Y, Brown KM, Liggett SB. Hierarchy of polymorphic variation and desensitization permutations relative to beta 1- and beta 2-adrenergic receptor signaling. J Biol Chem. 2003;278(12):10784–10789. | |
Mialet Perez J, Rathz DA, Petrashevskaya NN, et al. Beta 1-adrenergic receptor polymorphisms confer differential function and predisposition to heart failure. Nat Med. 2003;9(10):1300–1305. | |
Liggett SB, Mialet-Perez J, Thaneemit-Chen S, et al. A polymorphism within a conserved beta(1)-adrenergic receptor motif alters cardiac function and beta-blocker response in human heart failure. Proc Natl Acad Sci U S A. 2006;103(30):11288–11293. | |
Small KM, Forbes SL, Rahman FF, Bridges KM, Liggett SB. A four amino acid deletion polymorphism in the third intracellular loop of the human alpha 2C-adrenergic receptor confers impaired coupling to multiple effectors. J Biol Chem. 2000;275(30):23059–23064. | |
Talameh JA, Lanfear DE. Pharmacogenetics in chronic heart failure: new developments and current challenges. Curr Heart Fail Rep. 2012;9(1):23–32. | |
Small KM, McGraw DW, Liggett SB. Pharmacology and physiology of human adrenergic receptor polymorphisms. Annu Rev Pharmacol Toxicol. 2003;43:381–411. | |
Green SA, Turki J, Innis M, Liggett SB. Amino-terminal polymorphisms of the human beta 2-adrenergic receptor impart distinct agonist-promoted regulatory properties. Biochemistry. 1994;33(32):9414–9419. | |
Panebra A, Wang WC, Malone MM, et al. Common ADRB2 haplotypes derived from 26 polymorphic sites direct beta2-adrenergic receptor expression and regulation phenotypes. PloS One. 2010;5(7):e11819. | |
Liggett SB, Cresci S, Kelly RJ, et al. A GRK5 polymorphism that inhibits beta-adrenergic receptor signaling is protective in heart failure. Nat Med. 2008;14(5):510–517. | |
Lobmeyer MT, Wang L, Zineh I, et al. Polymorphisms in genes coding for GRK2 and GRK5 and response differences in antihypertensive-treated patients. Pharmacogenet Genomics. 2011;21(1):42–49. | |
Chan SW, Hu M, Tomlinson B. The pharmacogenetics of β-adrenergic receptor antagonists in the treatment of hypertension and heart failure. Expert Opin Drug Metab Toxicol. 2012;8(7):767–790. | |
Terra SG, Hamilton KK, Pauly DF, et al. Beta1-adrenergic receptor polymorphisms and left ventricular remodeling changes in response to beta-blocker therapy. Pharmacogenet Genomics. 2005;15(4):227–234. | |
de Groote P, Helbecque N, Lamblin N, et al. Association between beta-1 and beta-2 adrenergic receptor gene polymorphisms and the response to beta-blockade in patients with stable congestive heart failure. Pharmacogenet Genomics. 2005;15(3):137–142. | |
Beta-Blocker Evaluation of Survival Trial Investigators. A trial of the beta-blocker bucindolol in patients with advanced chronic heart failure. N Engl J Med. 2001;344(22):1659–1667. | |
Kurnik D, Li C, Sofowora GG, et al. Beta-1-adrenoceptor genetic variants and ethnicity independently affect response to beta-blockade. Pharmacogene Genomics. 2008;18(10):895–902. | |
Fiuzat M, Neely ML, Starr AZ, et al. Association between adrenergic receptor genotypes and beta-blocker dose in heart failure patients: analysis from the HF-ACTION DNA substudy. Eur J Heart Fail. 2013;15(3):258–266. | |
Baudhuin LM, Miller WL, Train L, et al. Relation of ADRB1, CYP2D6, and UGT1A1 polymorphisms with dose of, and response to, carvedilol or metoprolol therapy in patients with chronic heart failure. Am J Cardiol. 2010;106(3):402–408. | |
Shin J, Lobmeyer MT, Gong Y, et al. Relation of beta(2)-adrenoceptor haplotype to risk of death and heart transplantation in patients with heart failure. Am J Cardiol. 2007;99(2):250–255. | |
Lanfear DE, Jones PG, Marsh S, Cresci S, McLeod HL, Spertus JA. Beta2-adrenergic receptor genotype and survival among patients receiving beta-blocker therapy after an acute coronary syndrome. JAMA. 2005;294(12):1526–1533. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.