Back to Journals » Research and Reports in Urology » Volume 9
Systematic genetic screening in a prospective group of Danish patients with pheochromocytoma
Authors Hansen MSS, Jacobsen N, Frederiksen AL ![]() , Lund L
, Lund L ![]() , Andersen MS, Glintborg D
, Andersen MS, Glintborg D
Received 9 February 2017
Accepted for publication 11 May 2017
Published 27 June 2017 Volume 2017:9 Pages 113—119
DOI https://doi.org/10.2147/RRU.S134385
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Jan Colli
Morten Steen Svarer Hansen,1 Niels Jacobsen,1 Anja Lisbeth Frederiksen,2 Lars Lund,3 Marianne Skovsager Andersen,1 Dorte Glintborg1
1Department of Endocrinology and Metabolism, 2Department of Clinical Genetics, 3Department of Urology, Odense University Hospital, Odense, Denmark
Abstract: Recent guidelines recommend consideration of genetic screening in all newly diagnosed patients with pheochromocytoma. Patients diagnosed with pheochromocytoma in the Region of Southern Denmark during 2006–2013 without previously recognized monogenetic etiology were offered genetic screening for mutations in the VHL, RET, SDHB, SDHC, and SDHD genes. A total of 41 patients were included, and genetic data were available in 35. In four of the 35 patients, a pathogenic variant was identified prior to the diagnosis of pheochromocytoma (von Hippel–Lindau disease, n=2; neurofibromatosis type 1, n=2). The patients carrying a genetic mutation were all younger than 45 years at time of diagnosis of pheochromocytoma, two patients presented with bilateral tumors, and one patient had a positive family history of pheochromocytoma. Genetic screening of the remaining 31 patients did not identify any mutations. The sporadic cases had a median age of 58 years (range 33–80 years). Three of 31 sporadic cases (ages 60, 69, and 76 years at time of diagnosis) presented with bilateral adrenal tumors, one patient had multiple adrenal tumors in both adrenal glands, and no patients had a positive family history of pheochromocytoma. Of the 31 patients, 24 (68.6%) were diagnosed with pheochromocytoma due to evaluation of an adrenal incidentaloma. In conclusion, monogenetic etiology was identified in four of 35 (11.4%) patients diagnosed with pheochromocytoma.
Keywords: pheochromocytoma, genetic screening, guidelines, adrenal incidentaloma
Introduction
Pheochromocytomas are rare neuroendocrine catecholamine-secreting tumors arising from chromaffin cells in the adrenal medulla with an incidence of approximately two to ten per 1,000,000 per year.1 Diagnosis is based on elevated levels of plasma-fractionated metanephrines or urinary catecholamines.2 Pheochromocytomas are often sporadic, but 8%–24% are caused by germ-line mutations and most commonly associated with the NF1, VHL, RET, SDHA, SDHB, SDHC, and SDHD genes.3,4 The diagnosis of NF1 and von Hippel–Lindau (VHL) disease is established in probands fulfilling the clinical criteria,5,6 and the diagnosis can be genetically verified in approximately 95% of cases.6,7
The prevalence of SDHC-mutation carriers is low, but they may develop all the stigmata of the disease.4 Additionally, patients with mutations of the SDHB gene demand special attention, because they have a high risk of malignant disease that reflects both the typically large sizes and extra-adrenal location of associated tumors.4
The main criteria for focused genetic evaluation include young age at diagnosis, bilateral/multiple tumors, or a positive family history with pheochromocytoma or tumor syndrome.8 Different algorithms have been applied to identify patients eligible for genetic screening.8 However, recent guidelines for pheochromocytomas and paragangliomas recommend that genetic screening should be considered in all patients with pheochromocytoma, prioritizing young patients with positive family history or presence of multifocal or bilateral tumors.4 Identification of pathogenic genetic mutation in patients with pheochromocytoma confirms the clinical diagnosis and allows for genetic counseling and predictive testing of family members. Subsequently, mutation-positive carriers may enter individual surveillance programs.
Pheochromocytoma may be diagnosed during the evaluation of an adrenal incidentaloma, which is defined as an adrenal mass discovered incidentally during imaging performed due to problems unrelated to adrenal disease.9 The prevalence of adrenal incidentalomas is approximately 4% and increases with age, and pheochromocytomas are reported to account for 8% of all adrenal incidentalomas.9,10 To our knowledge, no studies have evaluated the impact of systematic genetic screening of pheochromocytomas diagnosed during the evaluation of adrenal incidentalomas. In the present retrospective cohort study, we evaluated the results of systematic genetic screening in patients newly diagnosed with pheochromocytoma and compared characteristics in patients with monogenetic etiology and patients with sporadic pheochromocytoma.
Materials and methods
Study eligibility
All patients referred to the Department of Endocrinology and Metabolism at Odense University Hospital in the Region of Southern Denmark during January 1, 2006, to December 31, 2013, suspected of increased catecholamine secretion (n=108) (Figure 1) were screened for excessive hormonal secretion following current guidelines: patients with adrenal incidentaloma were screened with computed tomography of the adrenal glands and measurement of plasma-free metanephrines, 24-hour urinary cortisol or 1 mg overnight dexamethasone test, serum chromogranin A, and in patients with hypertension or hypokalemia, additionally plasma renin and aldosterone. As diagnostic tests for pheochromocytoma, we measured levels of either nightly urinary catecholamines or plasma metanephrines.
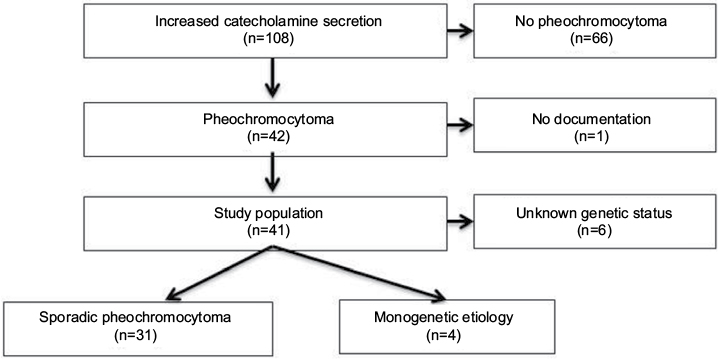 | Figure 1 Flowchart of study population. |
Criteria for inclusion in the present study were 1) biochemical and anatomical presence of pheochromocytoma, and 2) patients who had undergone surgery with removal of tumor and/or adrenalectomy of an adrenal gland. Criteria for exclusion were 1) absence of intra-adrenal tumor, 2) paragangliomas explicitly, 3) extra-adrenal secreting chromaffin tumors, 4) nonsecreting head–neck paragangliomas, 5) no operation, and 6) patients with normal levels of both urinary catecholamines and plasma metanephrines. A total of 41 patients fulfilled the inclusion criteria and were included in the study; 66 patients were excluded for one or more the aforementioned exclusion criteria. Documentation was not accessible in one patient.
The present study was an audit regarding treatment and follow-up of patients with pheochromocytoma. Patient information in the present study was used in anonymous form. This was reviewed and verified by the legal department of Odense University Hospital.
Genetic screening program
All patients with pheochromocytoma and without previously recognized monogenetic etiology were offered genetic screening in the five genes included for the systematic genetic screening program in 2006–2013. VHL, RET, SDHB, SDHC, and SDHD were analyzed with sequencing of all coding regions and subsequently analyzed for deletions and duplications. A diagnosis of multiple endocrine neoplasia type 1 relied on clinical criteria.
Grouping of patients
Patients who underwent genetic screening were divided into sporadic or nonsporadic pheochromocytoma. Sporadic pheochromocytoma was defined as 1) negative genetic screening result, 2) absence of family history of pheochromocytoma, 3) absence of metastatic disease, and 4) unilateral tumor. Three patients presented with bilateral tumors, but were still categorized as having sporadic tumors, as the other three criteria for sporadic pheochromocytoma were fulfilled. Pheochromocytomas diagnosed during the evaluation of an incidental adrenal mass were categorized as adrenal incidentalomas independent of whether patients were categorized as having monogenetic etiology or sporadic pheochromocytoma.
Data sources
We searched the PubMed database in April 2017 using the words “pheochromocytoma” and “genetic screening”. Only English-language literature was used.
Results
The study population (n=41) was initially divided into two groups: patients with monogenetic etiology known prior to the diagnosis of pheochromocytoma (n=4) and patients eligible for genetic screening (n=37).
Genetic screening
Genetic screening results were available in 31 of 37 (83.8%) patients (Figure 1). All patients screened for genetic mutations (n=31) had negative screening results. Six patients who had not been genetically screened were excluded: one patient was initially categorized as having monogenetic etiology because of suspected neurofibromatosis type 1 (NF1) diagnosis. This patient did however have bilateral vestibular schwannomas and a genetic screening identified an NF2 mutation and thus NF2. Five patients declined genetic screening, including four female patients aged 65, 76, 79, and 83 years and one male patient aged 52 years at the time of diagnosis.
Monogenetic etiology
Four patients carried a germ-line mutation in a gene associated with pheochromocytoma. All four patients were diagnosed with the genetic mutation prior to the diagnosis of pheochromocytoma (Tables 1 and 2). Two patients (NF1, n=2) were diagnosed with pheochromocytoma during the evaluation of adrenal incidentalomas, and two patients were diagnosed during the surveillance program for VHL disease. One patient with monogenetic etiology presented a positive family history of pheochromocytoma and VHL disease, whereas the family history of pheochromocytoma was negative in the remaining three patients. Two patients with monogenetic etiology had bilateral pheochromocytomas.
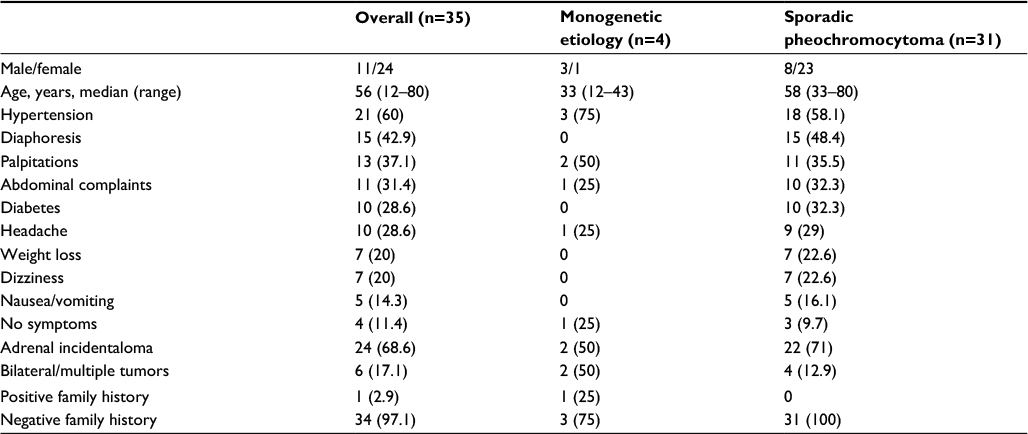 | Table 1 Clinical characterization of patients Notes: Hypertension was defined as systolic pressure ≥140 mmHg and/or diastolic pressure ≥90 mmHg and/or antihypertensive treatment. Results given as n (%). |
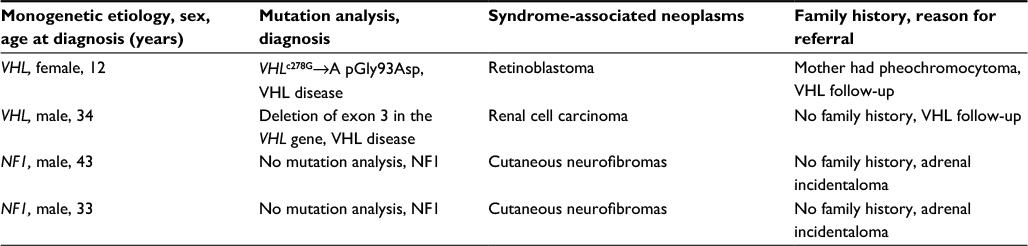 | Table 2 Genetic characteristics for patients with monogenetic etiology Abbreviations: VHL, von Hippel–Lindau; NF, neurofibromatosis. |
Sporadic pheochromocytomas
All patients screened for genetic mutation (n=31) had a negative family history of pheochromocytomas, and metastatic disease was not found in any of the patients. Three of 31 (9.7%) patients screened had bilateral tumors. These patients were however categorized as sporadic pheochromocytoma, as criteria 1–3 were fulfilled and age was high at time of diagnosis (60, 69, and 76 years). One of 31 (3.2%) had local recurrences of unilateral pheochromocytomas. This patient was 69 years old at diagnosis and 74 years at recurrence. A total of 27 of 31 (87.1%) had unilateral pheochromocytoma. Three patients, all with unilateral tumors, had genetic polymorphisms not previously described as associated with pheochromocytoma or tumor syndromes (VHL, n=1; RET, n=1; SDHD, n=1).
Adrenal incidentalomas
A total of 24 pheochromocytomas were diagnosed during evaluation of adrenal incidentalomas (monogenetic etiology, n=2; sporadic pheochromocytoma, n=22). The diameter of all adrenal incidentalomas was more than 1 cm.
Age
Patients with monogenetic etiology were all younger than 45 years at diagnosis of pheochromocytoma (12, 33, 34, and 43 years) (Table 1 and Figure 2). Patients with sporadic pheochromocytoma had a median age of 58 (range 33–80) years, and seven of 31 (22.6%) patients were younger than 45 years at diagnosis.
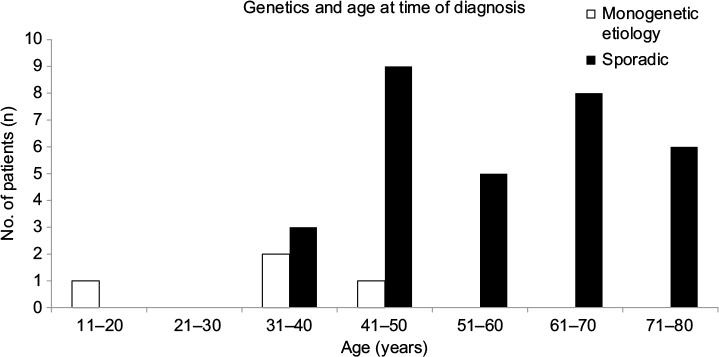 | Figure 2 Genetics and age at diagnosis of pheochromocytoma (n=35). |
Discussion
In the present study, four (11.4%) patients harbored a germ-line mutation predisposing them to the pheochromocytoma. In addition, one patient initially thought to have NF1 was diagnosed with NF2. While pheochromocytoma is a rare but well-known manifestation in NF1, this has not previously been described in NF2. The genetic mutations were all identified prior to the patients developing pheochromocytoma. Screening the remaining subjects with newly diagnosed pheochromocytoma did not detect any new mutation-positive individuals. The prevalence of genetic mutations in the present study was lower than previous studies.11–13 Paragangliomas are reported to be more frequently associated with monogenetic aetiology.11,13 The exclusion of these tumors could explain the lower prevalence of genetic mutations in the present study. A recent systematic review of 31 studies and 5,031 patients found the frequency of germ-line mutations in sporadic pheochromocytoma and paraganglioma to be approximately 11%–13%.14 Their definition of sporadic tumors was very similar to the criteria in the present study, including 1) a negative family history of pheochromocytoma or paraganglioma, 2) the absence of syndromic features, 3) a lack of bilateral disease, and 4) the absence of metastatic disease,14 and a recent consensus statement summarized gene panels of pheochromocytomas and paragangliomas based on current evidence to include ten basic genes (genes extensively validated in the literature predominantly associated with familial diseases and syndromic features), 15 extended genes (the basic genes included; genes proven to be functionally relevant, but with frequency less than 1%), and a further 12 comprehensive genes (genes with low number of events and limited data) as candidates for targeted genetic screening.15
Similar genetic screening programs to the present work were performed in several studies in patients with pheochromocytoma. Erlic et al found predisposed germ-line mutations in 188 of 989 (19%) patients with clinical presentation of pheochromocytoma and nonsyndromic pheochromocytoma.12 Mannelli et al found predisposed germ-line mutations in 161 of 501 (32.1%) patients with pheochromocytoma and/or paraganglioma and in 102 of 341 (29.9%) patients when paragangliomas were excluded.13 A screening for mutations in the RET, VHL, SDHB, and SDHD identified predisposed germ-line mutations in 66 of 271 (24.4%) patients with nonsyndromic pheochromocytoma and no family history and in 52 of 241 (21.6%) patients when paragangliomas were excluded.11
Rapid advances in genetic screening methods have identified additional susceptibility genes in pheochromocytoma, including SDHA, SDHC, SDHAF2, FH, EGLN1/PHD2, KIF1B, HIF2A, TMEM127, and MAX, the latter two with frequency of less than 2%.4 The screening of these new genes was not included and represents a limitation to the present retrospective study. However, several of these genes have not yet been fully validated, and information on prevalence, penetrance, and phenotype is very limited. Welander et al did however establish that 7% of apparently sporadic cases carried a germ-line mutation when applying a more comprehensive genetic screening panel.16 Future studies will illuminate which of the susceptibility genes should be included in systematic genetic screening for pheochromocytomas. In the present study, three patients had bilateral tumors and one patient had recurrence of unilateral tumor but negative genetic screening results. Based on the current literature, we think it would be relevant to offer these patients new genetic screening in our clinic to investigate for rarer genetic causes of pheochromocytoma with a panel of all known susceptibility genes.
In the present study, none of the patients with negative genetic screening presented with a positive family history of pheochromocytoma or other tumors. Still, these patients can harbor germ-line mutations in other susceptibility genes.16 Identifying a genetic predisposition has important implications for the patient, as it allows for individual surveillance programs, genetic counseling, and predictive testing of family members. The guidelines for genetic screening are continuously updated, and patients with negative genetic screening in the present retrospective study with, eg, adrenergic or noradrenergic presentation of pheochromocytomas will in future presumably be offered genetic screening with a panel of all known susceptibility genes.
The inclusion of patients with pheochromocytoma diagnosed during the evaluation of adrenal incidentaloma could have affected the prevalence of genetic mutations in the present study. A total of 24 of 35 (68.6%) patients included were diagnosed due to evaluation of adrenal incidentalomas. Our result is consistent with the findings of Motta-Ramirez et al, who reviewed records of 335 adrenalectomies and found that 19 of 33 (57.6%) pheochromocytomas were adrenal incidentalomas.17 In comparison, an autopsy review from 1981 of 54 autopsy-proven pheochromocytomas found that 41 of 54 (76%) patients had not been diagnosed with pheochromocytoma while alive.18 Other studies reported adrenal incidentalomas in 29 of 192 (15.1%) patients, including both pheochromocytoma and paraganglioma19 or pheochromocytomas exclusively found as adrenal incidentalomas in 15 of 150 (10%) patients.20 In contrast to other studies,19,20 all patients referred to our department with adrenal incidentaloma were systematically screened for pheochromocytoma, and this could have increased the observed prevalence of pheochromocytomas found as adrenal incidentalomas in the present study. In the present study, two patients were categorized as having adrenal incidentaloma, though harboring germ-line mutations predisposing to the pheochromocytoma. The patients (both with NF1) were classified as having adrenal incidentalomas, as it was not the predisposed germ-line mutation or due to surveillance follow-up programs that led to further investigation of pheochromocytoma, but because of an incidentally discovered adrenal mass. Due to the wider use of imaging over the last few years combined with more detailed imaging, increased discovery of incidental adrenal masses is to be expected, making genetic evaluation of adrenal incidentalomas of great importance.21 To our knowledge, no studies have evaluated genetic screening of adrenal incidentalomas or incidental pheochromocytomas exclusively.
Due to the limited number of patients, statistical analysis was not possible; however, all patients with monogenetic etiology were younger than 45 years at diagnosis, which supports the view that age <45 years is a predictor of monogenetic etiology. Conversely, seven patients with apparently sporadic pheochromocytoma were younger than 45 years at diagnosis, and of these, two patients had not previously described genetic polymorphisms in VHL or RET.
In the present study, two of five patients with bilateral tumors had monogenetic etiology (NF1, n=1; VHL, n=1), even though this group accounted for only 11.4% of patients overall. This result could support bilateral tumors being a predictor of monogenetic etiology. Bilateral tumors are more often seen in patients with RET mutations.22 As no patients with RET mutations were identified, the prevalence of bilateral tumors in our study was expected to be low.
In the present study, we examined genetic variants as biomarkers. The use of molecular pathological epidemiology (MPE) is emerging and provides an opportunity to investigate the inherent heterogeneity of neoplasms and pathogenic processes, such as pheochromocytomas.23 MPE examines links between various exposures and the molecular pathology of diseases. In pheochromocytomas, MPE could be applied specifically to germ-line variants and establish more precise genetic screening panels. In the present study, three patients with sporadic pheochromocytomas had polymorphisms not previously described as associated with pheochromocytomas. In these cases, evaluation of the genetic results could benefit from MPE. To our knowledge, no studies have investigated the potential use of MPE in genetic screening for pheochromocytomas.
Strengths and limitations may apply in the present study. Our department works as the regional center for patients with pheochromocytoma, making the prevalence of monogenetic etiology among pheochromocytomas representative of our region, covering a population of 1.2 million inhabitants. A small population size could lead to higher uncertainty in the results presented, and makes it difficult to do statistical analysis, which hinders the generalizability of our results. As this was a retrospective cohort study, we as investigators had limited control of data collection, and we cannot exclude the possibility that data are incomplete. Selection bias and confounders can occur.
In the present study, the disease causing genetic mutation was identified in the patients prior to the diagnosis of pheochromocytoma. Additional studies are needed to determine if genetic screening should include all patients diagnosed with pheochromocytomas and should be extended to new genes.
Disclosure
These results have previously been presented as a poster at the American Urological Association Meeting, May 6–10, 2016, San Diego, CA, USA. The authors report no conflicts of interest in this work.
References
Welander J, Soderkvist P, Gimm O. Genetics and clinical characteristics of hereditary pheochromocytomas and paragangliomas. Endocr Relat Cancer. 2011;18(6):R253–R276. | ||
Lenders JW, Pacak K, Walther MM, et al. Biochemical diagnosis of pheochromocytoma: which test is best? JAMA. 2002;287(11):1427–1434. | ||
Tsirlin A, Oo Y, Sharma R, Kansara A, Gliwa A, Banerji MA. Pheochromocytoma: a review. Maturitas. 2014;77(3):229–238. | ||
Lenders JW, Duh QY, Eisenhofer G, et al. Pheochromocytoma and paraganglioma: an endocrine society clinical practice guideline. J Clin Endocrinol Metab. 2014;99(6):1915–1942. | ||
Jett K, Friedman JM. Clinical and genetic aspects of neurofibromatosis 1. Genet Med. 2010;12(1):1–11. | ||
Binderup ML, Bisgaard ML, Harbud V, et al. Von Hippel-Lindau disease (vHL): national clinical guideline for diagnosis and surveillance in Denmark. Dan Med J. 2013;60(12):B4763. | ||
Sabbagh A, Pasment E, Imbard A, et al. NF1 molecular characterization and neurofibromatosis type 1 genotype-phenotype correlation: the French experience. Hum Mutat. 2013;34(11):1510–1518. | ||
Jafri M, Maher ER. The genetics of phaeochromocytoma: using clinical features to guide genetic testing. Eur J Endocrinol. 2012;166(2):151–158. | ||
Aron D, Terzolo M, Cawood TJ. Adrenal incidentalomas. Best Pract Res Clin Endocrinol Metab. 2012;26(1):69–82. | ||
Young WF Jr. The incidentally discovered adrenal mass. N Engl J Med. 2007;356(6):601–610. | ||
Neumann HP, Bausch B, McWhinney SR, et al. Germ-line mutations in nonsyndromic pheochromocytoma. N Engl J Med. 2002;346(19):1459–1466. | ||
Erlic Z, Rybicki L, Peczkowska M, et al. Clinical predictors and algorithm for the genetic diagnosis of pheochromocytoma patients. Clin Cancer Res. 2009;15(20):6378–6385. | ||
Mannelli M, Castellano M, Schiavi F, et al. Clinically guided genetic screening in a large cohort of Italian patients with pheochromocytomas and/or functional or nonfunctional paragangliomas. J Clin Endocrinol Metab. 2009;94(5):1541–1547. | ||
Brito JP, Asi N, Bancos I, et al. Testing for germline mutations in sporadic pheochromocytoma/paraganglioma: a systematic review. Clin Endocrinol (Oxf). 2015;82(3):338–345. | ||
Toledo RA, Burnichon N, Cascon A, et al. Consensus statement on next-generation-sequencing-based diagnostic testing of hereditary phaeochromocytomas and paragangliomas. Nat Rev Endocrinol. 2017;13(4):233–247. | ||
Welander J, Andreasson A, Juhlin CC, et al. Rare germline mutations identified by targeted next-generation sequencing of susceptibility genes in pheochromocytoma and paraganglioma. J Clin Endocrinol Metab. 2014;99(7):E1352–E1360. | ||
Motta-Ramirez GA, Remer EM, Herts BR, Gill IS, Hamrahian AH. Comparison of CT findings in symptomatic and incidentally discovered pheochromocytomas. AJR Am J Roentgenol. 2005;185(3):684–688. | ||
Sutton MG, Sheps SG, Lie JT. Prevalence of clinically unsuspected pheochromocytoma: Review of a 50-year autopsy series. Mayo Clinic Proc. 1981;56(6):354–360. | ||
Amar L, Servais A, Gimenez-Roqueplo AP, Zinzindohoue F, Chatellier G, Plouin PF. Year of diagnosis, features at presentation, and risk of recurrence in patients with pheochromocytoma or secreting paraganglioma. J Clin Endocrinol Metab. 2005;90(4):2110–2116. | ||
Kudva YC, Young WF, Thompson GB, Grant CS, van Heerden JA. Adrenal incidentaloma: an important component of the clinical presentation spectrum of benign sporadic adrenal pheochromocytoma. Endocrinologist. 1999;9(2):77–80. | ||
Smith-Bindman R, Miglioretti DL, Johnson E, et al. Use of diagnostic imaging studies and associated radiation exposure for patients enrolled in large integrated health care systems, 1996-2010. JAMA. 2012;307(22):2400–2409. | ||
Amar L, Bertherat J, Baudin E, et al. Genetic testing in pheochromocytoma or functional paraganglioma. J Clin Oncol. 2005;23(34):8812–8818. | ||
Ogino S, Nishihara R, VanderWeele TJ, et al. The role of molecular pathological epidemiology in the study of neoplastic and non-neoplastic diseases in the era of precision medicine. Epidemiology. 2016;27(4):602–611. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.