Back to Journals » International Medical Case Reports Journal » Volume 10
Syringomyelia presenting with unilateral optic neuropathy: a case report
Authors Ngoo QZ, Tai ELM ![]() , Wan Hitam WH
, Wan Hitam WH
Received 2 December 2016
Accepted for publication 26 January 2017
Published 14 March 2017 Volume 2017:10 Pages 87—91
DOI https://doi.org/10.2147/IMCRJ.S129239
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Scott Fraser
Qi Zhe Ngoo, Evelyn Li Min Tai, Wan Hazabbah Wan Hitam
Department of Ophthalmology, School of Medical Sciences, Universiti Sains Malaysia, Kubang Kerian, Malaysia
Purpose: In this case report, we present two cases of syringomyelia with optic neuropathy.
Findings: In Case 1, a 36-year-old Malay lady presented to our clinic with acute onset of blurring of vision in her left eye that she experienced since past 1 month. She was diagnosed with syringomyelia 12 years ago and was on conservative management. Her visual acuity was 6/6 in the right eye and counting fingers at 1 m in the left. There was a positive relative afferent pupillary defect in her left eye. Optic nerve functions of her left eye were reduced. Visual field showed a left inferior field defect. Her extraocular muscle movements were full. Magnetic resonance imaging of the brain and spine showed syringomyelia at the level of C2–C6 and T2–T9. Both of her optic nerves were normal. Her condition improved with intravenous and oral corticosteroids. In Case 2, a 44-year-old Malay lady presented to our clinic with a progressive central scotoma in her right eye that she experienced since past 1 month. She had previous history of recurrent episodes of weakness in both of her lower limbs from past 8 months. Visual acuity in her right and left eye was 6/9 and 6/6, respectively. The relative afferent pupillary defect in her right eye was positive. Optic nerve functions of her right eye were affected. Visual field showed a central scotoma in her right eye. Her extraocular muscle movements were full. Fundoscopy of her right eye showed a pale optic disc. Her left eye fundus was normal. Magnetic resonance imaging of the brain and spine showed syringomyelia at T3–T6. Both of her optic nerves were normal. A diagnosis of syringomyelia with right optic atrophy was performed. Her condition improved with intravenous and oral corticosteroids.
Conclusion: Optic neuropathy is a rare neuro-ophthalmic manifestation in patients with syringomyelia. Prompt diagnosis and timely management are essential to avoid a poor visual outcome. Intravenous corticosteroids are beneficial in the treatment of early optic neuropathy in syringomyelia.
Keywords: syringomyelia, unilateral, optic neuropathy, optic neuritis
Introduction
Syringomyelia is a chronic and progressive disease affecting the spine or craniovertebral junction in which long cavities form within the spinal cord.1 Neurological manifestations of syringomyelia include pain, motor symptoms, sensory disturbances and brainstem signs. Common ocular features are downbeat nystagmus and oscillopsia.2 Papilloedema and optic atrophy have been described as rare signs. Herein, we present two rare cases of optic neuropathy in patients with syringomyelia, which is an uncommon manifestation of the disease.
Patients’ consent
Both patients signed the written consent for the publication of this case report.
Findings
Case 1
A 36-year-old Malay lady presented to our clinic with acute onset of blurring of vision in her left eye that she experienced since past 1 month. She had been diagnosed with syringomyelia 12 years ago when she had first experienced acute bilateral lower limb weakness 1 month postdelivery. Her magnetic resonance imaging of the spine had revealed syringomyelia. Since then, she was monitored by a neurosurgical team and was being managed conservatively.
On examination in our clinic, visual acuity of her right eye was found to be 6/6 and counting fingers at 1 m in the left. There was a positive relative afferent pupillary defect in her left eye. The optic nerve functions such as light brightness, red saturation and color vision of her left eye were affected. Visual field showed a left inferior field defect. Her extraocular muscle movements were full. Both anterior and posterior segments appeared normal. Neurological examination showed bilateral lower limb weakness with intact sensation. Other examinations related to cranial nerves were normal. Magnetic resonance imaging of the brain and spine showed syringomyelia at the level of C2–C6 and T2–T9 (Figures 1 and 2). However, both of her optic nerves were normal (Figure 3).
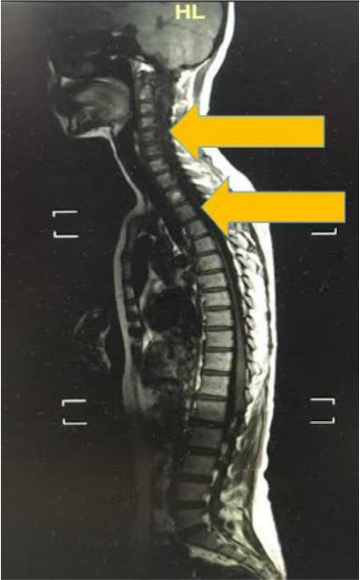 | Figure 1 Linear signal hypointensity from C2 to C6 and T2 to T9 vertebra level on T1 weighted imaging (arrow). |
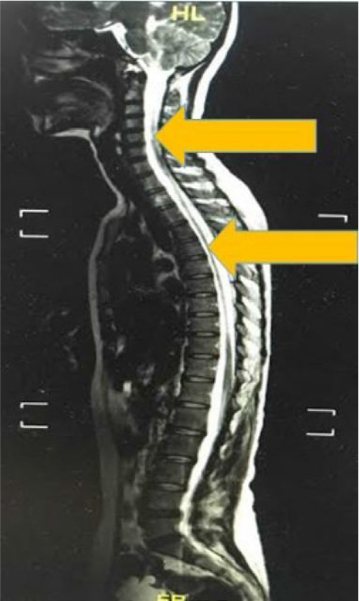 | Figure 2 Linear signal hyperintensity from C2 to C6 and T2 to T9 vertebra level on T2 weighted imaging (arrow). |
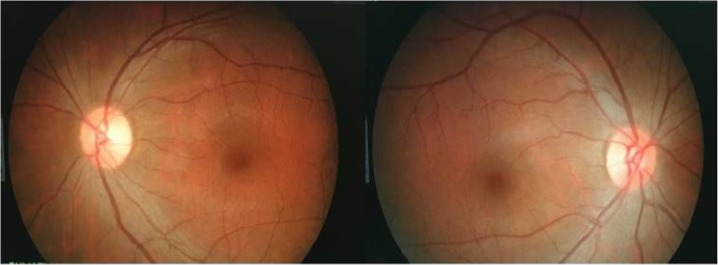 | Figure 3 Fundus photo showing both optic discs to be normal. |
A diagnosis of left retrobulbar optic neuritis was performed. She was started on intravenous methylprednisolone at a dosage of 250 mg four times a day for a period of 5 days, followed by oral prednisolone at a dosage of 45 mg (1 mg/kg/day) once daily for 9 days. Her condition dramatically improved after treatment. Visual acuity of her left eye was 6/24, and her optic nerve functions improved. She was continued with oral prednisolone with tapering dose weekly for another 1 month. Her visual acuity improved to 6/6 with good optic nerve functions. Oral prednisolone was off after 2 months. There was no complication due to the use of corticosteroids. Her vision remained good during 6 months follow-up.
Case 2
A 44-year-old Malay lady presented to our clinic with a progressive central scotoma in her right eye that she experienced since past 1 month. She had previous history of recurrent episodes of weakness in both of her lower limbs from past 8 months. She was admitted to a center for investigations; however, a proper diagnosis could not be established. She was treated conservatively. This was the first ocular presentation.
On examination, visual acuity in her right and left eye was 6/9 and 6/6, respectively. The relative afferent pupillary defect in the right eye was positive. The optic nerve functions such as light brightness, red saturation and color vision of the right eye were affected. Visual field showed a central scotoma in the right eye. Her extraocular muscle movements were full. Both anterior segments were unremarkable. Fundoscopy of her right eye showed a pale optic disc. Her left eye fundus was normal. Neurological examination showed bilateral lower limb weakness with intact sensation. Other examinations related to cranial nerves were normal. Magnetic resonance imaging of the brain and spine showed syringomyelia at the level of T3–T6 (Figures 4 and 5). However, both optic nerves were normal.
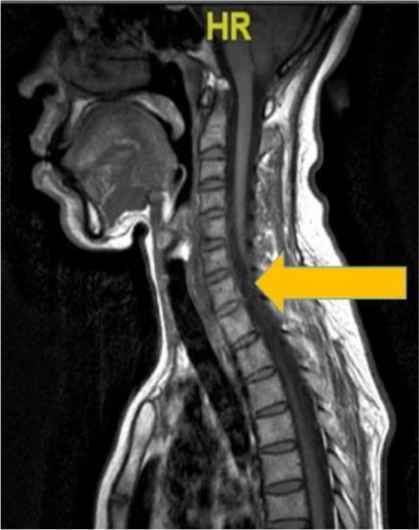 | Figure 4 Linear signal hypointensity from T3 to T6 vertebra level on T1 weighted imaging (arrow). |
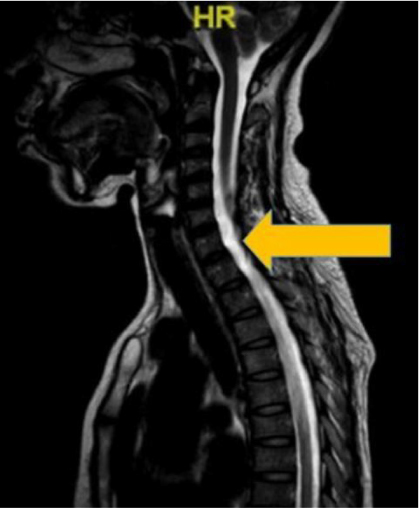 | Figure 5 Linear signal hyperintensity from T3 to T6 vertebra level on T2 weighted imaging (arrow). |
She was diagnosed with syringomyelia with right optic atrophy. She was started on intravenous methylprednisolone at a dosage of 250 mg four times a day for 5 days, followed by oral prednisolone at a dosage of 40 mg (1 mg/kg/day) once daily for 9 days. Her condition improved after 2 weeks of treatment, with reduction in the right eye central scotoma. However, due to the preexisting optic atrophy, visual acuity in her right eye was 6/9. Her optic nerve functions persistently reduced compared to her left eye. She was continued with oral prednisolone with tapering dose weekly. Subsequent follow-up at 1 month after diagnosis showed that her ocular condition was stable. Her limb weakness improved slightly, but she remained wheelchair bound. There was no complication of treatment. Her condition was stable at 6 months follow-up.
Discussion
Syringomyelia is a chronic disease, in which fluid-filled cavities in the spinal cord called syrinx expand and elongate over time. In this process, they destroy the nerve fibers of the spinal cord that lead to a variety of neurological manifestations, depending on the size and location of the syrinx.3 Optic neuritis is an extremely rare neuro-ophthalmic manifestation in syringomyelia. Previous published reports have only noted the occurrence of optic neuritis when syringomyelia occurs in patients with multiple sclerosis.4,5 Herein, we report two cases of optic neuritis in patients with syringomyelia; one patient had an acute episode of optic neuritis, whereas the other had already developed optic atrophy, which may have been preceded by a subacute attack of optic neuritis, in view of the diminished optic nerve function tests and central scotoma in that eye.
Although these two cases of syringomyelia were idiopathic, Chiari type-1 malformation has been reported as one of the etiologic factors in syringomyelia.6 It has been postulated that idiopathic syringomyelia is similar to that seen in Chiari type-1 malformation in terms of its pathogenesis.7 This malformation, also known as hindbrain hernia, is a structural abnormality in the region of craniovertebral junction, causing cerebellar tonsils to go below the foramen magnum and obstruct cerebrospinal fluid circulation.6 The rise in spinal subarachnoid pressure subsequently causes fluid to move upward inside the syrinx and into the brainstem, leading to a condition known as syringobulbia.8 There may be paralysis of the cranial nerves, which explains the diplopia experienced by some patients. Patients can present with diplopia, oscillopsia, nystagmus, anisocoria and ptosis in cases of hindbrain-related syringomyelia.2,9 Another syringomyelia-associated neuro-ophthalmic condition is Horner syndrome, which shows compression along the course of the sympathetic pathway by a lower cervical syrinx.10
The optic disc pallor occurring in many patients with syringomyelia is most likely due to the chronicity of the intracranial hypertension. Intracranial hypertension leads to papilloedema, which when long-standing, results in optic atrophy and generally irreversible reduction in optic nerve functions. Optic atrophy in patients with syringomyelia can be due to increased intracranial pressure caused by closure of the foramina at the roof of the fourth ventricle.11 Due to the underlying anatomical basis of the diagnosis of this condition, it is almost impossible to diagnose it when patients present with visual complaints in the absence of neurological symptoms. The introduction of magnetic resonance imaging has improved the pickup rate of this condition; nevertheless, a high index of suspicion is required for the clinician to perform this imaging not just for the brain, as is common practice in optic neuritis, but also for the spine.
The management of syringomyelia depends on the initial presentation and its progression. Since the natural history of syringomyelia in any individual patient is not immediately apparent, a conservative approach can be recommended, with close follow-up and periodic examination using magnetic resonance imaging. When there is progression in the neurological deficit, definitive treatment is required. Craniovertebral decompression may result in improvement in neurological symptoms and signs.12,13 This is probably due to release of pressure and traction on cranial nerves. However, till date, due to the rarity and variability of associated neuro-ophthalmic manifestations, there is no clear evidence of the management of these conditions in syringomyelia.
In both of our patients, even the one with optic atrophy, commencement of systemic corticosteroids was associated with improvement in visual function. The patients’ improved vision function may be related to the effect of corticosteroids on reducing the size of the syrinx, as has been demonstrated by Matsuda et al.14 The Optic Neuritis Treatment Trials found that although high-dose intravenous corticosteroids followed by oral maintenance produce a more rapid recovery of vision among patients with acute optic neuritis, corticosteroids did not improve the final visual acuity or optic nerve functions.15 Extrapolating the findings of this trial to our study, this corticosteroid regime may also have utility in the management of optic neuropathy in syringomyelia. In our patient with a delayed presentation to hospital, optic atrophy had already developed, suggesting that unlike the typical optic neuritis, which tends to resolve spontaneously and thus may be managed conservatively, optic neuritis in patients with syringomyelia should be aggressively treated.
Conclusion
Optic neuropathy is a rare neuro-ophthalmic manifestation in patients with syringomyelia. Prompt diagnosis and timely management are essential to avoid a poor visual outcome. Intravenous corticosteroids are beneficial in the treatment of early optic neuropathy in syringomyelia.
Disclosure
The authors declare that they have no conflicts of interest in this work.
References
National Institute of Neurological Disorders and Stroke. Syringomyelia fact sheet. 2016. Available from: https://www.ninds.nih.gov/Disorders/Patient-Caregiver-Education/Fact-Sheets/Syringomyelia-Fact-Sheet. Accessed March10, 2017. | ||
Rowlands A, Sgouros S, Williams B. Ocular manifestations of hindbrain-related syringomyelia and outcome following craniovertebral decompression. Eye (Lond). 2000;14(Pt 6):884–888. | ||
Guinto G, Abdo M, Aréchiga N, Zepeda E. Different types of syringomyelia and their management: Part I. Contemp Neurosurg. 2009;31(20):1–7. | ||
Deguchi K, Takeuchi H, Yamada A, et al. [Multiple sclerosis with syringomyelia – case report]. No To Shinkei. 1994;46(1):65–69. Japanese [with English abstract]. | ||
Das A, Puvanendran K. Syringomyelia and complex regional pain syndrome as complications of multiple sclerosis. Arch Neurol. 1999;56(8):1021–1024. | ||
Leung V, Magnussen JS, Stoodley MA, Bilston LE. Cerebellar and hindbrain motion in Chiari malformation with and without syringomyelia. J Neurosurg Spine. 2016;24(4):546–555. | ||
Heiss JD, Snyder K, Peterson MM, et al. Pathophysiology of primary spinal syringomyelia. J Neurosurg Spine. 2012;17(5):367–380. | ||
Del Maestro M, De Paulis D, Ricci A, Di Cola F, Galzio R. Syringobulbia associated with posterior fossa meningioma: a review of the literature. Childs Nerv Syst. 2014;30(10):1749–1752. | ||
Barrows JL, Cogan DG. Ocular manifestations of the Arnold-Chiari malformation. AMA Arch Neurol Psychiatry. 1954;72:116. | ||
Moreno TA, El-Dairi MA, Cabrera MT. Isolated Horner syndrome and syringomyelia in a child. J AAPOS. 2012;16(6):569–570. | ||
Alpers BJ, Comroe BI. Syringomyelia with choked disc. J Nerv Ment Dis. 1931;73:577. | ||
Dyste GN, Menezes AH, VanGilder JC. Symptomatic Chiari malformations. An analysis of presentation, management, and long-term outcome. J Neurosurg. 1989;71(2):159–168. | ||
Talacchi A, Meneghelli P, Borghesi I, Locatelli F. Surgical management of syringomyelia unrelated to Chiari malformation or spinal cord injury. Eur Spine J. 2016;25(6):1836–1846. | ||
Matsuda M, Tabata K, Miki J, Ishigame H, Asano M, Ikeda S. Multiple sclerosis with secondary syringomyelia. An autopsy report. J Neurol Sci. 2001;184(2):189–196. | ||
Beck RW, Cleary PA, Anderson MM Jr, et al; The Optic Neuritis Study Group. A randomized, controlled trial of corticosteroids in the treatment of acute optic neuritis. N Engl J Med. 1992;326(9):581–588. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.