Back to Journals » Drug Design, Development and Therapy » Volume 9
Synthesis and characterization of tumor-targeted copolymer nanocarrier modified by transferrin
Authors Liu R, Wang Y, Li X, Bao W, Xia G, Chen W, Cheng J, Xu Y, Guo L, Chen B
Received 15 January 2015
Accepted for publication 18 March 2015
Published 22 May 2015 Volume 2015:9 Pages 2705—2719
DOI https://doi.org/10.2147/DDDT.S80948
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Editor who approved publication: Professor Shu-Feng Zhou
Ran Liu,1,2 Yonglu Wang,1,3 Xueming Li,3 Wen Bao,1,2 Guohua Xia,1,2 Wei Chen,3 Jian Cheng,1,2 Yuanlong Xu,3 Liting Guo,1,2 Baoan Chen1,2
1Department of Hematology (Key Department of Jiangsu Medicine), Zhongda Hospital, Medical School, 2Faculty of Oncology, Medical School, Southeast University, 3College of Pharmacy, Nanjing University of Technology, Nanjing, People’s Republic of China
Abstract: To increase the encapsulation of hydrophilic antitumor agent daunorubicin (DNR) and multidrug resistance reversal agent tetrandrine (Tet) in the drug delivery system of nanoparticles (NPs), a functional copolymer NP composed of poly(lactic-co-glycolic acid) (PLGA), poly-l-lysine (PLL), and polyethylene glycol (PEG) was synthesized and then loaded with DNR and Tet simultaneously to construct DNR/Tet–PLGA–PLL–PEG-NPs using a modified double-emulsion solvent evaporation/diffusion method. And to increase the targeted antitumor effect, DNR/Tet–PLGA–PLL–PEG-NPs were further modified with transferrin (Tf) due to its specific binding to Tf receptors (TfR), which is highly expressed on the surface of tumor cells. In this study, the influence of the diversity of formulation parameters was investigated systematically, such as drug loading, mean particle size, molecular weight, the concentration of PLGA–PLL–PEG–Tf, volume ratio of acetone to dichloromethane, the concentration of polyvinyl alcohol (PVA) in the external aqueous phase, the volume ratio of the internal aqueous phase to the external aqueous phase, and the type of surfactants in the internal aqueous phase. Meanwhile, its possible effect on cell viability was evaluated. Our results showed that the regular spherical DNR/Tet–PLGA–PLL–PEG–Tf-NPs with a smooth surface, a relatively low polydispersity index, and a diameter of 213.0±12.0 nm could be produced. The encapsulation efficiency was 70.23%±1.91% for DNR and 86.5%±0.70% for Tet, the moderate drug loading was 3.63%±0.15% for DNR and 4.27%±0.13% for Tet. Notably, the accumulated release of DNR and Tet could be sustained over 1 week, and the Tf content was 2.18%±0.04%. In cell viability tests, DNR/Tet–PLGA–PLL–PEG–Tf-NPs could inhibit the proliferation of K562/ADR cells in a dose-dependent manner, and the half maximal inhibitory concentration value (total drug) of DNR/Tet–PLGA–PLL–PEG–Tf-NPs was lower than that of DNR, a mixture of DNR and Tet, and DNR/Tet–PLGA–PLL–PEG-NPs. These results clearly indicate that the PLGA–PLL–PEG formulation is a potential drug delivery system for hydrophilic and hydrophobic drugs, and that Tf modification may increase its targeting properties.
Keywords: PLGA, PLL, PEG, daunorubicin, tetrandrine
Introduction
The use of chemotherapy in human cancer treatment is frequently limited by intrinsic or acquired multidrug resistance (MDR), which is caused by the increased expression of plasma membrane P-glycoprotein.1,2 Daunorubicin (DNR), an antibiotic, has various antitumor activities and has been widely used in the clinical treatment of acute monocytic leukemia due to its intercalation of the planar aromatic ring system between the DNA base pairs.3,4 However, its efficacy is restricted by MDR, and many tumor cells are not sensitive to DNR because of the drug efflux mediated by P-glycoprotein.4,5 Tetrandrine (Tet) is a bisbenzylisoquinoline alkaloid isolated from the root of Stephania tetrandra S. Moore,6 which is a calcium channel blocker with strong MDR reversal effects. Compared to verapamil and cyclosporin A, Tet exhibited a stronger activity to reverse drug resistance to DNR in leukemia cells.2,6–8 Hence, we propose the design of a drug delivery system (DDS) using polymer nanoparticles (NPs), in which DNR and Tet can be simultaneously encapsulated to reduce the toxic effects against normal cells and increase their therapeutic activities in a controlled release manner.
Among the DDS of polymeric NPs, poly(lactic-co-glycolic acid) (PLGA) is considered to be an ideal carrier for anticancer drugs, owing to its biodegradable and biocompatible characteristics.9,10 Drug-releasing particles and therapeutic devices made by PLGA have been approved by the United States Food and Drug Administration for clinical applications since 1989.11 A major challenge for PLGA NP in vivo tumor delivery is the lack of functional groups to increase cellular adhesion and clearance by the reticuloendothelial system. In order to overcome this challenge, a diversity of functional groups including poly(l-lysine) (PLL) and polyethylene glycol (PEG) have been introduced to PLGA polyesters by either direct conjugation12–14 or by being conjugated with additives during the construction of the functional polymer.15,16 PLL presents good targeted effects on tumor cells owing to its binding to the negatively-charged cell membrane by electrostatic absorption.17,18 On the other hand, PEG has the potential to minimize the amount of NPs that bind to plasma proteins. So, the circulation time of NPs would be enhanced and the clearance of NPs would be reduced by the reticuloendothelial system.18 Recent exciting data suggest that DDS comprising conjugates of PLGA, PLL, and PEG can significantly improve the drug encapsulation and antitumor effects in vivo and in vitro.11–,19 Given the antitumor activities of DNR and the strong MDR reversal effects of Tet, as well as the fact that PLGA–PLL–PEG-NPs represent a promising delivery system with a high drug loading and encapsulation efficiency, we designed a functional PLGA–PLL–PEG-NP simultaneously loaded with DNR and Tet (DNR/Tet–PLGA–PLL–PEG-NPs) to enhance the antitumor activity of DNR and reverse the MDR.
As we know, the performance of the polymeric NPs depends on variable factors, such as the molecular weight (Mw), as well as the composition of the polymer, the organic solvents, and the type and concentration of surfactant used.20,21 Thus, the selection of a suitable preparation technique is dependent on the physical and chemical properties of the polymers and drugs.20 Until now, there have been various fabrication methods that have been commonly used for NP preparation,20 such as the nanoprecipitation technique,22–24 single or double-emulsion solvent evaporation,25,26 and the solvent diffusion technique.27 Among these methods, the double-emulsion solvent evaporation method is popular and widely used for simultaneous hydrophilic and hydrophobic compound encapsulation, resulting in high encapsulation efficiency and featuring a relatively small particle size.20,23,28,29
More attention has been focused on the targeted therapy for cancer in recent years. Transferrin (Tf) receptors (TfR) were found to be highly expressed on the tumor cell surface in many studies.30–45 Our initial studies have shown that the liposome modified with Tf acting as doxorubicin carrier had a better targeting antitumor effect compared with the liposome carrier without Tf due to the possible binding of Tf to TfR on the tumor cell membrane. And, the incorporation of antitumor agent 5-fluorouracil into PLGA NPs significantly improved the oral bioavailability.46,47 Based on the establishment of both synthesis of Tf with PLL–PEG and TfR-targeted nanocarriers PLL–PEG–Tf-NPs, our present study was to fabricate the PLGA–PLL–PEG–Tf-NPs simultaneously loaded with DNR and Tet using a modified double-emulsion method and to perform an investigation to further prove its promising strategy for overcoming MDR in leukemia.
Materials and methods
Materials
PEG with a Mw of 4,000 Da, 4-(dimethylamino)pyridine (DMAP), anhydrous N,N-dimethylformamide hydrous (DMF), N,N-dicyclohexylcarbodiimide (DCC), and N,N-carbonyldiimidazole (CDI) were purchased from Aladdin Reagents Co., Ltd. (Shanghai, People’s Republic of China); N-carboxy-(Nε-benzyloxycarbonyl)-l-lysine anhydride (Lys[z]-NCA) was purchased from Shanghai Hanhong Chemical Co., Ltd. (Shanghai, People’s Republic of China); PLGA (50:50 lactic acid:glycolic acid) with carboxylic acid ends of Mw 30 kDa was purchased from Evonik Industries (Essen, Germany); T 60 kDa was purchased from Birmingham Polymers Inc. (Birmingham, AL, USA); PLGA of Mw 15 kDa was purchased from Shandong Medical Equipment Research Institute (Shandong, People’s Republic of China); deuterated dimethyl sulfoxide (D6-DMSO) was purchased from Cambridge Isotope Laboratories, Inc. (Tewksbury, MA, USA); hydrogen bromide in acetic acid (33 wt%, HBr/HOAc) was purchased from J&K Scientific Ltd. (Beijing, People’s Republic of China); the antitumor drugs DNR and Tet were purchased from Jinan Huifengda Chemical Co., Ltd. (Jinan, Shandong, People’s Republic of China); high-performance liquid chromatography (HPLC) grade acetonitrile was purchased from Wanqing Chemical Reagent Co., Ltd. (Nanjing, People’s Republic of China); Tf was purchased from Sigma-Aldrich Co. (St Louis, MO, USA); an ultrasonic processor (SCIENTZ-IID) was purchased from Ningbo SCIENTZ Biotechnology Co, Ltd. (Ningbo, People’s Republic of China); a proton nuclear magnetic resonance (1H NMR) spectrometer (AV-500) was purchased from Bruker Optik GmbH (Ettlingen, Germany), with D6-DMSO as the solvent and tetramethylsilane as the internal standard; an ultracentrifuge (GL-20G-H) was purchased from Shanghai Precision & Scientific Instrument Co., Ltd. (Shanghai, People’s Republic of China); and a freeze-dryer (LGJ-10C) was purchased from Beijing Sihuan Scientific Instrument Co., Ltd, Beijing, People’s Republic of China.
An adriamycin-resistant cell line (K562/ADR) was donated from the Institute of Hematology at the Chinese Academy of Medical Sciences (Tianjin, People’s Republic of China). The cells were maintained in Roswell Park Memorial Institute 1,640 medium supplemented with 10% newborn bovine serum, 10 mM HEPES buffer, 100 U/mL of penicillin, and 100 μg/mL of streptomycin at 37°C in a humidified atmosphere containing 5% CO2. In an attempt to maintain the MDR phenotype, 1 μg/mL of adriamycin was added in the medium until 1 week before the experiments.
Synthesis of PLGA–PLL–PEG copolymer
The PLGA–PLL–PEG copolymer was synthesized as previously reported.11–14,48,49
Synthesis of PLL–Cbz
A total of 4 g of Lys(z)-NCA and 60 mL of anhydrous DMF were loaded into a three-neck flask with a stirrer under a nitrogen atmosphere. The sample was stirred and then 30 μL of ethylenediamine was added to induce polymerization. After being stirred for 72 hours, 200 mL of anhydrous diethyl ether was added and then the precipitated product was dried under vacuum for 12 hours to produce PLL–carbobenzoxy (Cbz).48
Synthesis of PLGA–PLL
A total of 3 g of PLGA and 0.3 g of PLL–Cbz were added to a round-bottom flask and dissolved in anhydrous DMF under a nitrogen atmosphere. Then, 60 mg of DCC and 5 mg of DMAP were added to the flask. After being stirred for 48 hours, chloroform was added and then the solution was filtered to remove the N,N-dicyclohexylurea. The product was precipitated in methanol and isolated by vacuum filtration, washed three times with ether, and dried under vacuum for 48 hours.49 Thereafter, the dried products were dissolved in a sufficient amount of 33 wt% HBr/HOAc and stirred for 2 hours; a sufficient amount of ether was added to wash the precipitated polymer until an off-white product was obtained. The off-white polymer was dried under vacuum for 12 hours to produce the deprotected PLGA–PLL.14
Activation of PEG
Then, 4 g PEG was dissolved in dioxane and 3 g of CDI was added under nitrogen at 37°C. After being stirred for 2 hours, the unreacted CDI was removed by adding sufficient deionized water. The solution was added into a dialysis tube (Mw 3,500) and the dialysate of deionized water was changed every 2 hours. Thereafter, the solution remaining in the dialysis tube was freeze-dried for 3 days to produce activated PEG–CDI.13
Synthesis of PLGA–PLL–PEG
A total of 2 g of PEG–CDI and 4 g of PLGA–PLL were dissolved in anhydrous DMF and stirred for 48 hours under nitrogen. Then, the solution was diluted with chloroform and precipitated in methanol. The process of polymer dissolution and precipitation was repeated three times until the unconjugated PEG was removed. Thereafter, the polymer products were freeze-dried for 3 days to produce PLGA–PLL–PEG.11,12
Characterization of PLGA–PLL–PEG
The conjugations of PLGA–PLL–PEG were determined by 1H NMR spectra and recorded in D6-DMSO as a solvent and tetramethylsilane as an internal standard using a 400 MHz AV-500 spectrometer (Bruker Optik GmbH), according to previous reports.13,14
Crosslink Tf
The reaction diagram was shown in Figure 1. First, PLGA–PLL–PEG was resuspended in phosphate buffer saline (PBS) and ultrasound for 30 seconds, and it was then mixed with an appropriate amount of Tf and stirred at room temperature for 3 hours. Thereafter, the solution was refrigerated at the speed of 15,000 rpm to remove the unreacted Tf at 4°C and freeze-dried with 5% cryoprotectant to obtain blank NPs (PLGA–PLL–PEG–Tf).
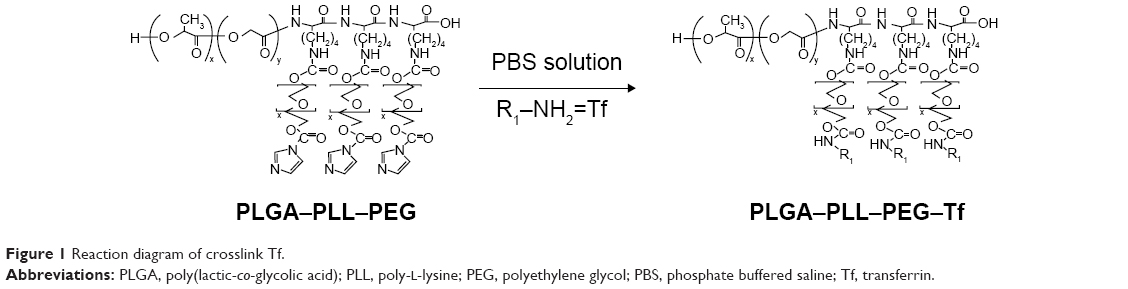 | Figure 1 Reaction diagram of crosslink Tf. |
Preparation of drug-loaded NPs (DNR/Tet–PLGA–PLL–PEG–Tf)
Based on others’ research2 and Wang (unpublished data, 2015), the molar ratio of DNR to Tet was adjusted from 1:0.22 to 1:0.85 to prepare for PLGA–PLL–PEG–Tf coloaded with DNR and Tet using the modified double-emulsion solvent evaporation/diffusion method.20
Briefly, the organic phase (O) consisting of 100 mg of blank NPs and 5 mg of Tet were dissolved in a 2 mL acetone–dichloromethane (1:1, V/V) mixture. The internal aqueous phase (W1) contained 7 mg of DNR and 0.5 mL of Tween 80 solution (6%), and the external aqueous phase (W2) consisted of 4 mL of polyvinyl alcohol (PVA) solution (1%). The internal aqueous phase was emulsified with the organic phase by sonication using an ultrasonic processor in an ice bath, and the primary emulsion (W1/O) was emulsified with the external aqueous phase by sonication to produce a double emulsion (W1/O/W2). Thereafter, 60 mL of PVA (0.3%) was added to the prepared double emulsion and stirred on a magnetic stirrer at 500 rpm overnight to evaporate and diffuse the organic solvents. To separate the drug-loaded NPs from the residual solvent, the disperse system was centrifuged for 30 minutes at 15,000 rpm and 4°C, and then washed with deionized water three times. The samples were dried by lyophilization at −50°C for 60 minutes and then at −10°C for 24 hours. The freeze-drying products were stored at 4°C and the effect of various process parameters were investigated systematically, such as the mean diameter, drug loading, Mw, concentration of blank NPs in the organic phase, concentration of PVA in the external aqueous phase, volume ratio of acetone to dichloromethane (A/D ratio), type of internal aqueous phase, and volume ratio of the internal aqueous phase to the external aqueous phase (I/E ratio). All experiments were performed by changing one of the process parameters while keeping the other parameters stable.29
Tf crosslinked product verification
First, gel permeation chromatography was used to investigate the separation situation of unreacted Tf. DNR/Tet–PLGA–PLL–PEG–Tf, and Tf dissolved in PBS was respectively run through the Rapid Resolution LC system (AKTA FPLC) (column packing: Sepharose CL-4B; mobile phase: PBS; detection wavelength: 280 nm), and then the leaching time was recorded. Thereafter, the concentration of Tf was measured by the Dual Binding (bicinchoninic acid [BCA]) kit. The BCA protein assay combines the reduction of Cu2+ to Cu1+ by protein in an alkaline medium; the first step is the chelation of copper with protein in an alkaline environment to form a light-blue complex. In the second step, BCA reacts with the reduced (cuprous) cation that was formed in step one; the intense purple-colored reaction product results from the chelation of two molecules of BCA with one cuprous ion. The BCA/copper complex is water soluble and exhibits a strong linear absorbance at 562 nm with increasing protein concentrations.
Particle size, zeta potential (ZP), and polydispersity index (PI)
Mean particle size, ZP values, and the PI of DNR/Tet–PLGA–PLL–PEG–Tf-NPs were determined by a Zetasizer 3000HS system (Malvern Instruments, Malvern, UK). The diameters and width of particle distribution were calculated using a volume distribution from the Gaussian distribution.50
Morphological evaluation under transmission electron microscopy (TEM)
The samples were placed over a copper grid coated with carbon film, stained with 0.5% phosphotungstic acid, air-dried, and examined using an H-765 TEM (Hitachi Ltd., Tokyo, Japan).
Differential scanning calorimeter (DSC) analysis
Samples of DNR, Tet, a mixture of DNR and Tet (DNR&Tet), PLGA–PLL–PEG–Tf-NPs and DNR/Tet–PLGA–PLL–PEG–Tf-NPs were heated from room temperature to 300°C at a speed of 10°C/minute; the crystalline status of the DNR and Tet inside the DNR/Tet–PLGA–PLL–PEG–Tf-NPs was investigated using DSC (DSC-204; Netzsch-Gerätebau GmbH, Selb, Germany).29
Determination of drug loading and encapsulation efficiency of DNR and Tet
Lyophilized samples (5 mg) were dissolved in 10 mL of DMSO and vortexed for 5 minutes. After centrifugation for 20 minutes at 15,300 rpm and 4°C, 20 μL of supernatant was injected into HPLC to determine the amount of DNR and Tet encapsulated in DNR/Tet–PLGA–PLL–PEG–Tf-NPs at a wavelength of 280 nm for Tet and 254 nm for DNR, and the column temperature was maintained at 37°C.48 The drug loadings were calculated by the ratio of the actual amount of DNR and Tet encapsulated to the total amounts of DNR/Tet–PLGA–PLL–PEG–Tf-NPs. The entrapment efficiencies were calculated as the actual amount of DNR and Tet encapsulated versus the theoretical amount in DNR/Tet–PLGA–PLL–PEG–Tf-NPs, respectively.20,29
Rate of drug release
The release rate of DNR and Tet from DNR/Tet–PLGA–PLL–PEG-Tf-NPs was evaluated in vitro at 37°C using the dialysis bag technique. Briefly, 100 mg of DNR/Tet–PLGA–PLL–PEG–Tf-NPs were dissolved in 100 mL of PBS and placed into a dialysis membrane bag with a Mw cutoff of 3,500 g/mol. The dialysis bag was tied and immersed in 200 mL of PBS, and then placed in a thermostatic water bath at 37°C with continuous stirring of 100 rpm. Then, a 200 μL sample was withdrawn at each predetermined sampling time, and it was replaced with the same volume of release medium in the PBS. The samples were injected into the HPLC for analysis, as indicated earlier. Meanwhile, the same molar proportion of DNR and Tet was performed as a control group in the same way.
Cellular toxicity assay
Firstly, cellular toxicity to K562/ADR leukemia cells was evaluated to determine the safety and ideal concentration of both PLGA–PLL–PEG–Tf-NPs and Tet using an MTT assay. Briefly, 2×104 K562/ADR cells were seeded on a 96-well plate for 24 hours and then treated with different concentrations of PLGA–PLL–PEG–Tf-NPs, of which the maximum concentration (400 μg/mL) was far more than that used as a carrier; meanwhile, K562/ADR cells were treated with 0.5~6.0 μg/mL of Tet as well. After incubating for 24 hours, 48 hours, and 72 hours, respectively, 20 μL of MTT was added to each well and cultured for another 4 hours at 37°C. The culture medium was then removed from the wells and replaced with 150 μL of DMSO. The absorbance was measured at 570 nm using a microplate reader (Multiskan MK3; Thermo Fisher Scientific, Waltham, MA, USA) and the cell viability was calculated as follows:
ODtreated cells/ODcontrol cells ×100%. | (1) |
Thereafter, the cytotoxicity of DNR, the mixture of DNR&Tet (DNR:Tet =3.63:4.27), DNR/Tet–PLGA–PLL–PEG-NPs, and DNR/Tet–PLGA–PLL–PEG–Tf-NPs at 48 hours was determined as described earlier, and the half maximal inhibitory concentration (IC50) value was calculated with Calcusyn software (BIOSOFT, Cambridge, UK).
Statistical analysis
All data were presented as the mean ± standard deviation in triplicate, and multivariate data analysis was performed using multiple linear regressions using the SPSS software (version 17.0; IBM Corporation, Armonk, NY, USA).
Results and discussion
Structural model of Tf-modified copolymer drug-loaded NPs
Our synthetic targeted copolymer nanocarriers consist of PLGA–PLL block copolymer cores, to which we conjugated PEG arms terminated with Tf functionalities (Figure 2).
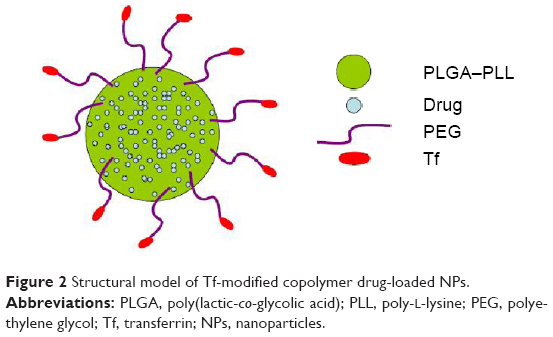 | Figure 2 Structural model of Tf-modified copolymer drug-loaded NPs. |
Characteristics of PLGA–PLL–PEG
The signal associated with the benzene ring of Cbz groups at δ=7.3 ppm was detected (Figure 3A and B), indicating the successful conjugation of PLGA–PLL–Cbz. After adding 33 wt% HBr/HOAc, a signal disappeared and this demonstrated the successful removal of Cbz in the PLGA–PLL (deprotected) (Figure 3C).49,51 The peaks associated with PEG at δ=3.51 ppm, CDI at δ=7.17 ppm, and δ=7.52 ppm were detected (Figure 3D), demonstrating the successful activation of PEG. Moreover, the peak associated with the ether bond and that presented in the conjugated PEG was also detected at δ=3.51 ppm (Figure 3E), indicating the successful coupling of PEG to PLGA–PLL.13
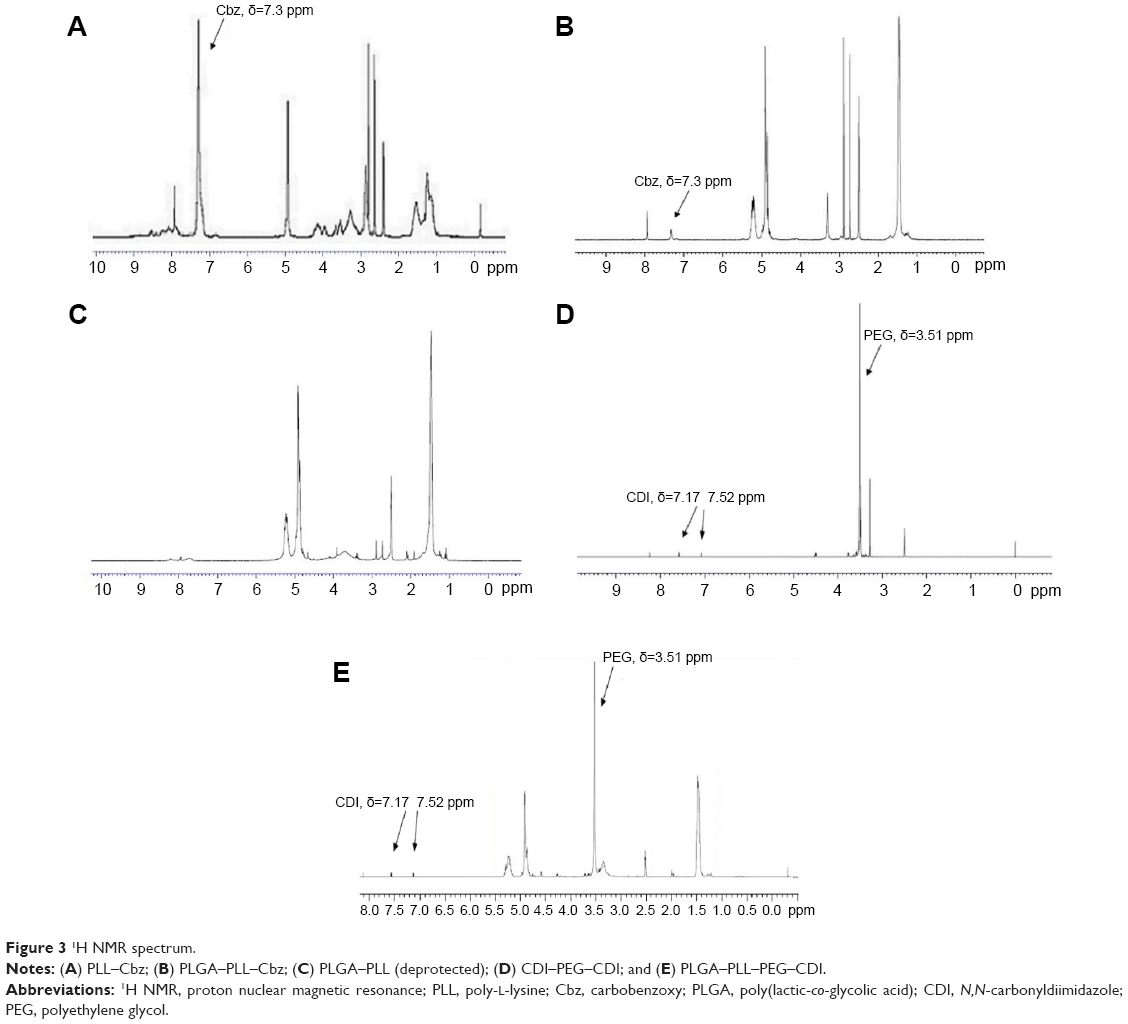 | Figure 3 1H NMR spectrum. |
Effect of formulation parameters on characteristics of PLGA–PLL–PEG–Tf-NPs
To improve the encapsulation efficiency, a modified double-emulsion solvent evaporation/diffusion method was adopted to formulate drug-loaded NPs, in which the emulsification processes are crucial factors in controlling the particle size of NPs and drug loadings of DNR and Tet in DNR/Tet–PLGA–PLL–PEG–Tf-NPs. As we know, PLGA was the basic structural and functional component of the polymer PLGA–PLL–PEG, so its Mw may influence the characteristics of PLGA–PLL–PEG. Firstly, according to our previous study, the amount of DNR was set at 7 mg and Tet at 5 mg, the aqueous phase was Tween 80 (6%), the A/D ratio was 1:1, the concentration of PLGA–PLL–PEG–Tf was 50 mg/mL, the concentration of PVA was 1% (w/v), the volume ratio of aqueous phase to oil phase (W/O ratio) was 2:1, and the I/E ratio was 1:4. We synthesized three types of PLGA–PLL–PEG with different Mw of PLGA. Our present study showed that the mean diameter of DNR/Tet–PLGA–PLL–PEG–Tf-NPs increased with the increase in Mw of PLGA (P>0.05), which can be attributed to the increasing Mw of PLGA–PLL–PEG, leading to the increased viscosity of the primary emulsion (Figure 4A). Thus, the more net shear stress decreased, the greater the mean particle size.29 We also demonstrated that the drug loading of Tet changed slightly with the increasing Mw of PLGA (P>0.05), while that of DNR increased until its Mw reached 60 kDa, and it then decreased once the Mw was >60 kDa (Figure 4A). Recently, it has been reported that the increasing Mw of PLGA–PLL–PEG could increase the length of the drug diffusion pathways from the organic phase to the external aqueous phase.28,29 Therefore, the loss of Tet through diffusion was reduced, resulting in the increase of the drug loading of Tet. On the contrary, when the Mw of PLGA reached 60 kDa, the drug loading of DNR decreased. This result could be attributed to the phenomenon that the viscosity of the emulsion solution increased with the increasing Mw of the PLGA–PLL–PEG. Therefore, the emulsion solution was difficult to break up into small droplets, as the input power of sonication remained stable.20,52
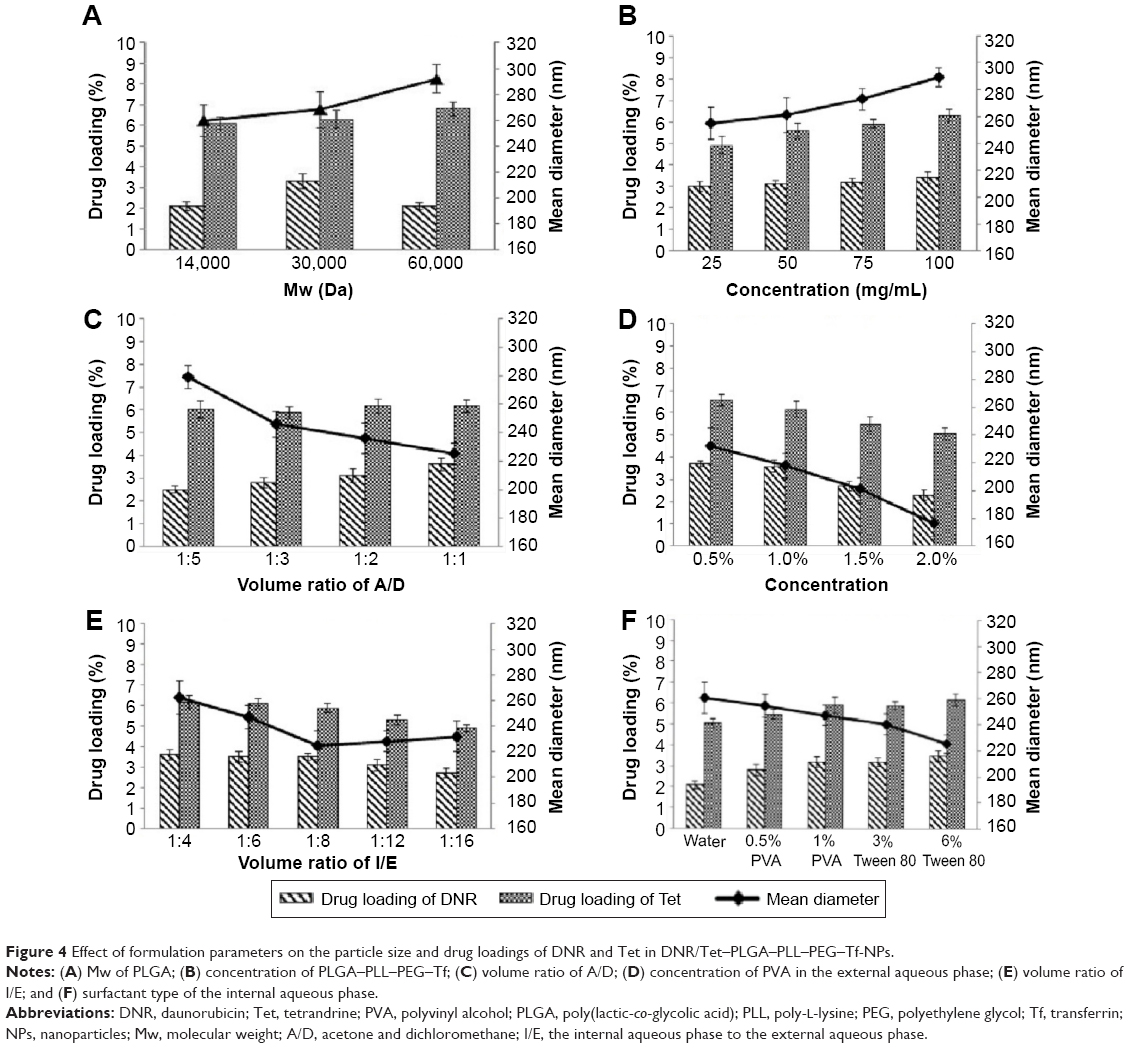 | Figure 4 Effect of formulation parameters on the particle size and drug loadings of DNR and Tet in DNR/Tet–PLGA–PLL–PEG–Tf-NPs. |
Moreover, we have investigated the effect of various concentrations of PLGA–PLL–PEG–Tf. We selected PLGA with a Mw of 30 kDa for synthetic PLGA–PLL–PEG, with DNR at 7 mg and Tet at 5 mg, Tween 80 (6%) as the aqueous phase, an A/D ratio of 1:1, a concentration of PVA of 1% (w/v), a W/O ratio of 2:1, and an I/E ratio of 1:4 were used to investigate the effect of the concentration of blank NPs ranging from 10 to 50 mg/mL. The mean diameter of DNR/Tet–PLGA–PLL–PEG–Tf-NPs increased significantly with the increasing concentration of PLGA–PLL–PEG–Tf (P<0.05) (Figure 4B). This result can be attributed to the fact that the increasing concentration of PLGA–PLL–PEG–Tf led to a high viscosity of the organic phase. Hence, the net shear stress decreased and large droplets developed.29 In addition, the PVA was not enough to cover the surface of the droplets when the concentration of PLGA–PLL–PEG–Tf increased, resulting in the coalescence of droplets and the aggregation of NPs, thereby increasing the particle size significantly.29 Moreover, the drug loading of Tet increased significantly with the increasing concentration of PLGA–PLL–PEG–Tf (P<0.05), whereas that of DNR increased only slightly (P>0.05) (Figure 4B). The reason for this phenomenon was related to the increasing viscosity as well. The drug diffusion decreased from the organic phase to the external aqueous phase with the increase in viscosity, thereby enhancing the encapsulation of Tet into the NPs.29 DNR mainly remained in the internal aqueous phase and interacted indirectly with the organic phase, thus the drug loading of DNR increased slightly.
To understand more findings from the optimum formulation protocols, other formulation parameters were also investigated as follows.
A/D volume ratio
Our previous study observed that using a mixed solvent of acetone and dichloromethane can significantly reduce the size of NPs, so we examined the A/D ratio from 1:1 to 1:5, with 7 mg of DNR and 5 mg of Tet, PLGA with a Mw of 30 kDa, Tween 80 (6%) as the aqueous phase, a concentration of PVA of 1% (w/v), a W/O ratio of 2:1, an I/E ratio of 1:4, and a concentration of PLGA–PLL–PEG–Tf of 50 mg/mL. As shown in Figure 4C, the mean diameter reduced significantly as the A/D volume ratio increased (P<0.05). Acetone can diffuse freely from the organic phase into the external aqueous phase due to the character of its partial water miscibility. The interfacial tension decreased significantly with the increase of the A/D volume ratio, which resulted from the rapid partitioning of acetone into the external aqueous phase. Thus, the particle size of the emulsion droplets decreased significantly.20 On the other hand, the drug loading of DNR increased significantly (P<0.05) with the increase of the A/D volume ratio, whereas that of Tet changed only slightly (P>0.05). The results could be attributed to fact that acetone diffused freely into the external aqueous phase, which led to the local supersaturation of regions near the interface. Therefore, polymer precipitation and solidification were induced to prevent the rapid dispersion of DNR to the external aqueous phase.21,53,54 The drug loading of Tet changed inconspicuously because the increase of the A/D volume ratio could not affect the partitioning of Tet in the organic phase. Though acetone diffused to the external aqueous phase, Tet still remained in the organic phase and interacted with the PLGA–PLL–PEG–Tf.
Concentration of PVA in the external aqueous phase
Emulsifier in preparation of double emulsion is to make the drugs loaded in NPs stable. PVA was widely used. We selected PLGA with a Mw of 30 kDa, 7 mg of DNR and 5 mg of Tet, Tween 80 (6%) as the aqueous phase, an A/D ratio of 1:1, the W/O ratio was 2:1, the I/E ratio was 1:4, and the concentration of PLGA–PLL–PEG–Tf was 50 mg/mL. As shown in Figure 4D, the mean diameter decreased significantly with the increasing concentration of PVA in the external aqueous phase (P<0.05). This result was attributed to the fact that a high concentration of PVA could prevent the coalescence of droplets during the evaporation of the organic solvent. Thus, the mean diameter of NPs decreased significantly. Moreover, PVA molecules could incorporate into the surface of NPs after the evaporation of the organic solvent,55,56 which could form a hydrated layer and prevent the aggregation of NPs.29,57 However, the viscosity of the external aqueous phase increased with the increasing concentration of PVA, which resulted in difficulties with the sonication operation when the input power was same.58 As shown in Figure 4D, the drug loadings of the two drugs decreased significantly with the increasing concentration of PVA (P<0.05), which probably resulted from the decrease of the mean particle size. When the concentration of PVA increased, the dispersion of the drugs into the external aqueous phase increased significantly during the emulsification procedure, resulting in a decrease of drug partitions remaining in the emulsion droplets. Therefore, fewer drug molecules could interact with the polymer molecules of PLGA–PLL–PEG, and the drug loadings of DNR and Tet decreased significantly.20,29
I/E volume ratio
We chose PLGA with a Mw of 30 kDa, DNR at 7 mg and Tet at 5 mg, Tween 80 (6%) as the aqueous phase, an A/D ratio of 1:1, a W/O ratio of 2:1, and a concentration of blank NPs of 50 mg/mL. As shown in Figure 4E, when the I/E volume ratio decreased, the mean diameter decreased and then reached a plateau. This result could be attributed to two competitive effects induced by the I/E volume ratio. First, the amount of PVA increased with the decreasing volume ratio of I/E, which resulted in a decrease in the interfacial tension and led to a decrease in the mean diameter. Meanwhile, the net shear stress reduced with the increased volume of whole system, which led to an increase in the mean diameter.59 When the latter effect dominated the former, the mean diameter decreased. Otherwise, the mean diameter increased as the latter effect dominated the former. As shown in Figure 4E, the drug loadings of DNR and Tet decreased significantly with a decrease of the I/E volume ratio (P<0.05), which resulted from the increased amount of drug diffusion to the external aqueous phase during the emulsification procedure. Fewer drugs remained in the internal aqueous phase to interact with the polymer of PLGA–PLL–PEG during solvent evaporation, thereby decreasing the drug loadings of DNR and Tet.20
Surfactants of the internal aqueous phase
As seen from Figure 4F, the mean diameter and drug loading were significantly affected by the type of surfactant (P<0.05), which is an important factor in maintaining the stability of the primary emulsion. In our present study, DNR/Tet–PLGA–PLL–PEG–Tf-NPs with a small particle size and higher drug loadings were obtained when the concentration of Tween 80 was 6%, which was selected as the surfactant of the internal aqueous phase.
Freeze-drying cryoprotectant
Cryoprotectant was an important parameter for the redispersible nanoformulations.60,61 In order to preserve the original particle size of the NPs, different types of cryoprotectant were investigated. The mean particle size of the DNR/Tet–PLGA–PLL–PEG–Tf-NPs was 224.5±15.8 nm before freeze-drying. As shown in Figure 5, the mean diameter of the NPs increased significantly when compared with that before freeze-drying, when either sucrose, mannitol, or lactose was used as the cryoprotectant (P<0.05). It was also demonstrated that the smallest particle size of the DNR/Tet–PLGA–PLL–PEG–Tf-NPs was obtained as the cryoprotectant was trehalose, which was capable of preserving the particle size without any aggregation or adhesion.
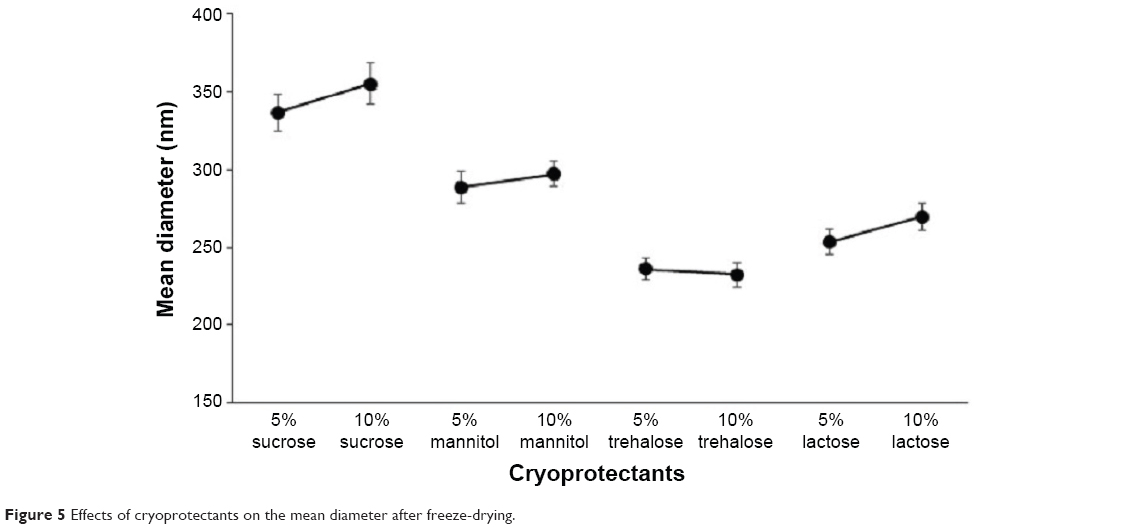 | Figure 5 Eeffects of cryoprotectants on the mean diameter after freeze-drying. |
Optimization of DNR/Tet–PLGA–PLL–PEG–Tf-NPs
The mean particle size of DNR/Tet–PLGA–PLL–PEG–Tf-NPs increased with the increasing Mw of PLGA and the concentration of PLGA–PLL–PEG–Tf, whereas it decreased with the increasing concentration of PVA and the volume ratio of A/D. The results of multiple linear regression analysis demonstrated that the concentration of PLGA–PLL–PEG–Tf, the volume ratio of A/D, and the concentration of PVA had significant influences on the mean diameter of DNR/Tet–PLGA–PLL–PEG–Tf-NPs (P<0.05).
The drug loading of DNR was positively correlated with the concentration of PLGA–PLL–PEG–Tf, the A/D volume ratio, and the I/E volume ratio, whereas it was negatively correlated with PVA concentration (P<0.5). Moreover, the drug loading of Tet was positively correlated with the Mw of PLGA, the concentration of PLGA–PLL–PEG–Tf, and the volume ratio of I/E, whereas it was negatively correlated with the concentration of PVA. The results of the multiple linear regression analysis illustrated that the volume ratio of A/D, the concentration of PVA, the volume ratio of I/E, and the surfactant of the internal aqueous phase were the dominant factors (P<0.05) in controlling the drug loading of DNR, whereas the concentration of the PLGA–PLL–PEG–Tf, the concentration of PVA, the volume ratio of I/E, and the surfactant of the internal aqueous phase were the dominant factors (P<0.05) in controlling the drug loading of Tet.
Taking the high drug loadings of the two drugs, the small particle size, and the convenience of the experimental operations into account, the Mw of PLGA (30 kDa), the concentration of PLGA–PLL–PEG–Tf (50 mg/mL), the volume ratio of A/D (1:1), the concentration of PVA (1%), the volume ratio of I/E (1:8), and surfactant of the internal aqueous phase (6% Tween 80) were chosen for the optimization of DNR/Tet–PLGA–PLL–PEG–Tf-NPs.
Tf crosslinked product verification
The Tf content of DNR/Tet–PLGA–PLL–PEG–Tf-NPs was determined using the BCA protein detection kit, and it was 2.18%±0.04% (w/w). The results of gel permeation chromatography showed that the time of Tf elution was 52.0±6.0 minutes (Figure 6A), while that of DNR/Tet–PLGA–PLL–PEG–Tf-NP elution was 22.0±3.0 minutes (Figure 6B), which was shorter than that of Tf. After centrifugation with 15,000 rpm, the solution of DNR/Tet–PLGA–PLL–PEG-NPs was light green and that of DNR/Tet–PLGA–PLL–PEG–Tf-NPs was purple (Figure 7), indicating that Tf is covalently attached to the NPs, not adsorbed by the electrostatic effect.
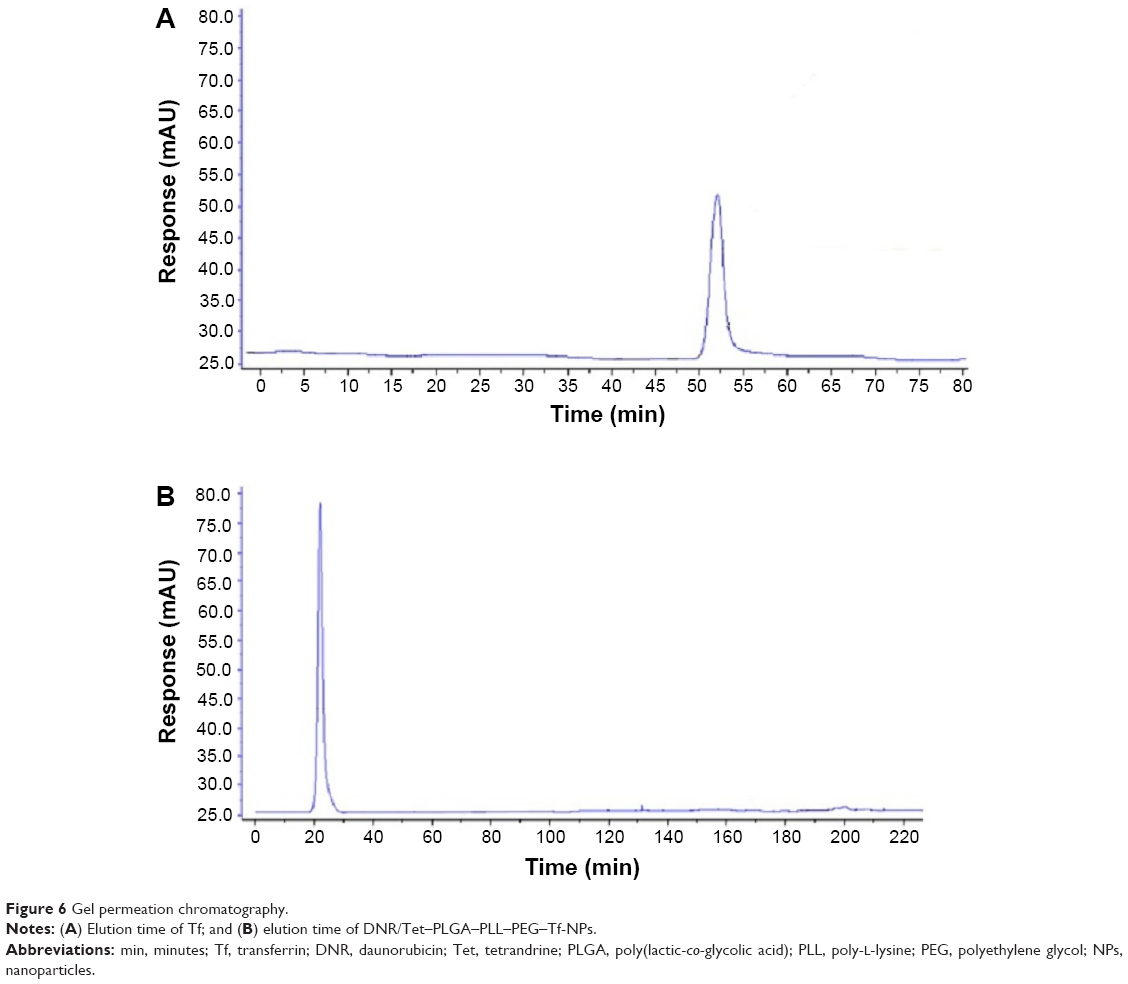 | Figure 6 Gel permeation chromatography. |
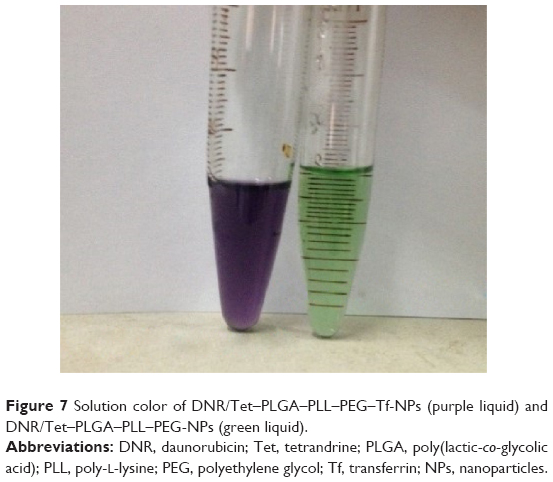 | Figure 7 Solution color of DNR/Tet–PLGA–PLL–PEG–Tf-NPs (purple liquid) and DNR/Tet–PLGA–PLL–PEG-NPs (green liquid). |
Encapsulation efficiency and drug loading
The encapsulation efficiency was 70.23%±1.91% for DNR and 86.5%±0.70% for Tet, and the drug loadings were 3.63%±0.15% for DNR and 4.27%±0.13% for Tet. The molar ratio of DNR to Tet was approximately 1:1 in the NPs, which was an appropriate proportion for reversing MDR. There was no significant difference between the encapsulation efficiencies and the drug loading of the three batches of PLGA–PLL–PEG–Tf-NPs. These results further demonstrated that the formulation process was stable and reproducible.
Particle morphology, ZP, and PI
As shown in Figures 8A, B and 9, the mean particle size of the NPs was 213.0±12.0 nm, and they featured a spherical shape and a smooth surface; histogram analysis of the TEM image showed that mean particle size was 212.2±18.0 nm (Figure 8B), which indicated that the size distribution was narrow. The ZP is a key indicator of the stability of colloidal dispersions, and it has been demonstrated that NPs with a weak negative charge could exhibit favorable in vivo behavior, including an enhanced permeability and retention effect-based tumor disposition.44 Therefore, the NPs with −19.16±0.11 mV were suitable for the enhanced permeability and retention effect-based efficient tumor disposition. Furthermore, PI is a measure of the distribution of the molecular mass in a given polymer sample, and it indicates the distribution of individual molecular masses in a batch of polymers. The PI was 0.075±0.011 in our present study, which is suitable for DDS.
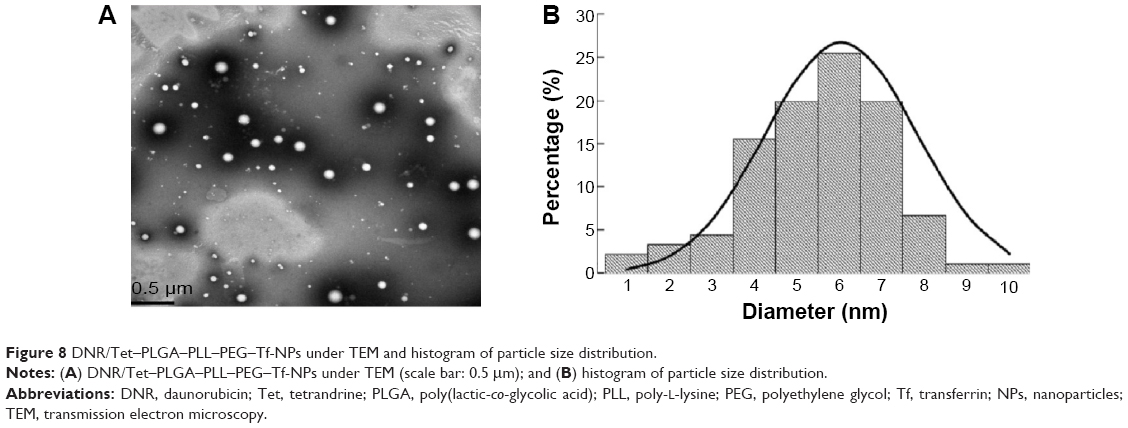 | Figure 8 DNR/Tet–PLGA–PLL–PEG–Tf-NPs under TEM and histogram of particle size distribution. |
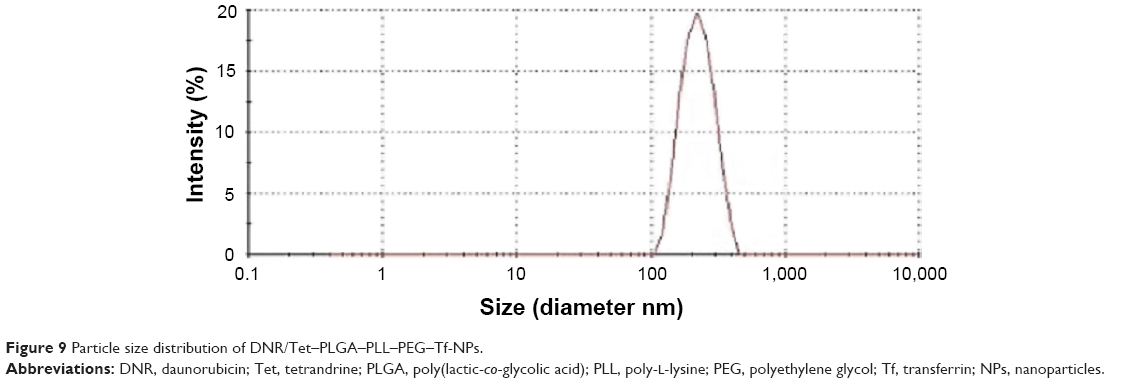 | Figure 9 Particle size distribution of DNR/Tet–PLGA–PLL–PEG–Tf-NPs. |
DSC analysis of the DNR/Tet–PLGA–PLL–PEG–Tf-NPs
The results of the DSC thermogram analysis showed the physical state of DNR and Tet in the DNR/Tet–PLGA–PLL–PEG–Tf-NPs (Figure 10); an endothermic peak of melting at 224°C was present for Tet (green line) and that at 198°C was exhibited for DNR (blue line). Conversely, an endothermic peak of melting at 203°C was exhibited for the physical mixture samples of DNR and Tet (purple line). However, the endothermic peaks were not detected in the DNR/Tet–PLGA–PLL–PEG–Tf-NPs (yellow line). These results demonstrated that the physical status of the DNR and Tet in DNR/Tet–PLGA–PLL–PEG–Tf-NPs was a disordered crystalline or amorphous phase, or it was a solid solution state.20,62
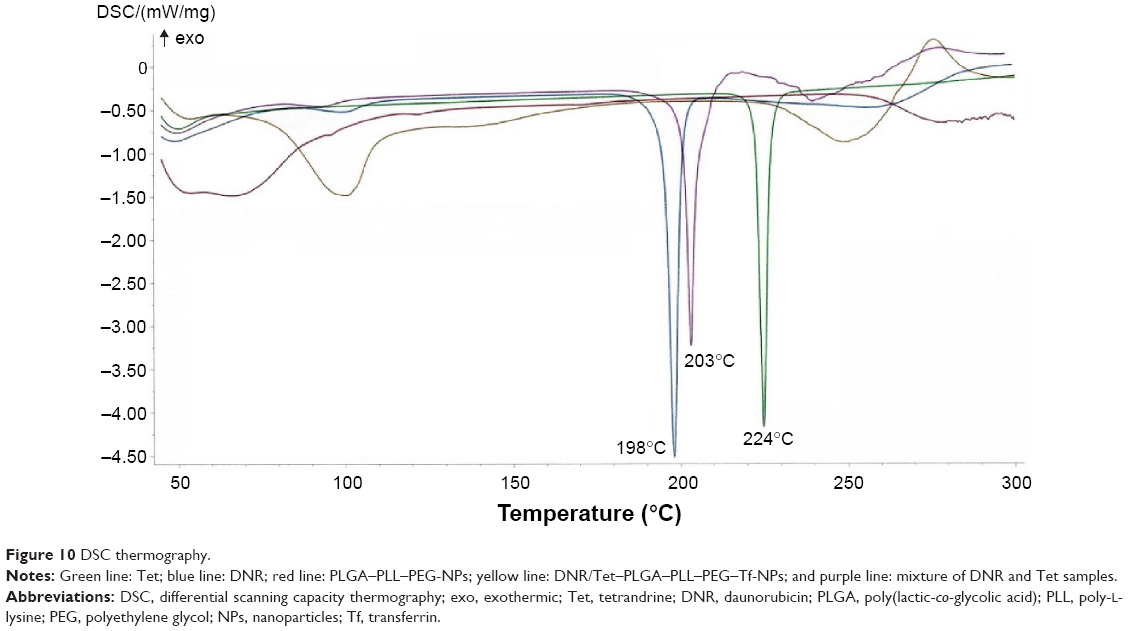 | Figure 10 DSC thermography. |
In vitro release
The release profiles of the DNR and Tet from DNR/Tet–PLGA–PLL–PEG–Tf-NPs were shown in Figure 11. Approximately 97% of the nonencapsulated DNR and Tet were released in 3 hours, while only ~50% of Tet and 60% of DNR were released from the DNR/Tet–PLGA–PLL–PEG–Tf-NPs in 24 hours. Thus, it could be inferred that the release of DNR and Tet from DNR/Tet–PLGA–PLL–PEG–Tf-NPs was significantly prolonged when compared with the nonencapsulated drugs. Notably, the release of Tet loaded in DNR/Tet–PLGA–PLL–PEG–Tf-NPs was always lower than that of DNR from the four accumulated release curves, which probably resulted from the poor water solubility of Tet. Compared with the initial release before 24 hours, the release rates of DNR and Tet were relatively low, which was probably attributed to the slow degradation of the polymer matrix. Interestingly, less than 80% of the drugs were released from the DNR/Tet–PLGA–PLL–PEG–Tf-NPs in 1 week; thus, it could be concluded that the degradation of the PLGA–PLL–PEG–Tf-NPs was comparably slower in the release medium.
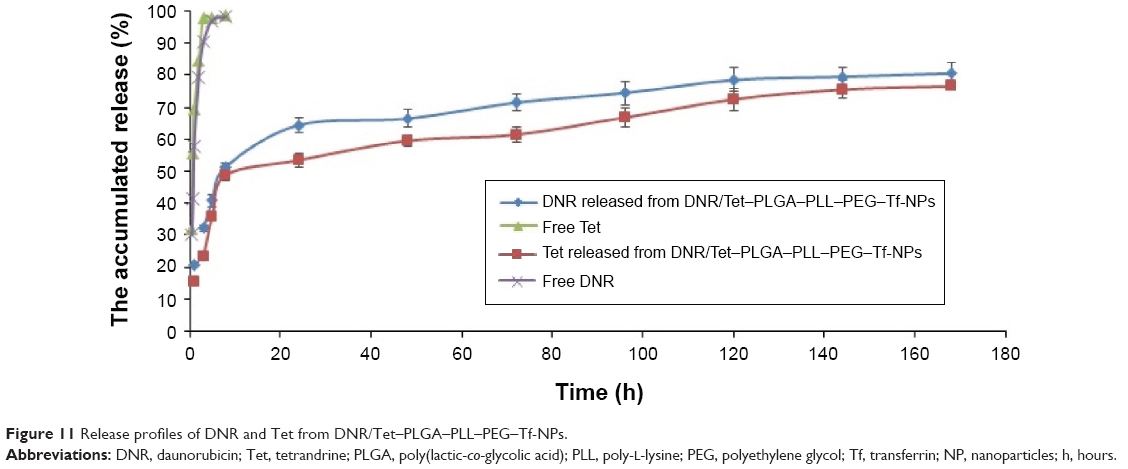 | Figure 11 Release profiles of DNR and Tet from DNR/Tet–PLGA–PLL–PEG–Tf-NPs. |
Cytotoxicity assays of DNR/Tet–PLGA–PLL–PEG–Tf-NPs
The MTT assay was conducted to elucidate the potential biological effects of DNR/Tet–PLGA–PLL–PEG–Tf-NPs on K562/ADR cells. As shown in Figure 12, when the concentration of PLGA–PLL–PEG–Tf-NPs was <400 μg/mL, there were no significant differences when compared to the control group (P>0.05). Notably, when the concentration of Tet was <2 μg/mL, the cell proliferation rate was >90% (Figure 13A). These results suggested that PLGA–PLL–PEG–Tf-NPs (<400 μg/mL) and low doses of Tet (<2 μg/mL) showed no obviously significant cytotoxicity on K562/ADR cells.
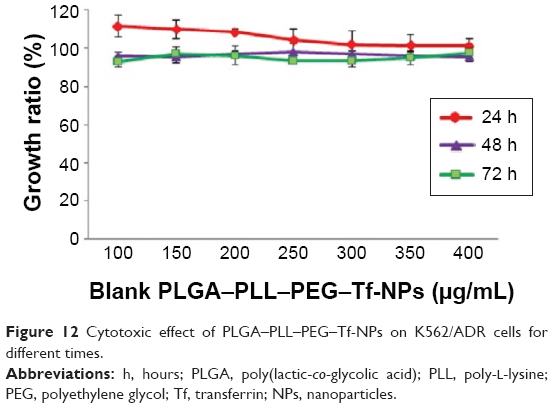 | Figure 12 Cytotoxic effect of PLGA–PLL–PEG–Tf-NPs on K562/ADR cells for different times. |
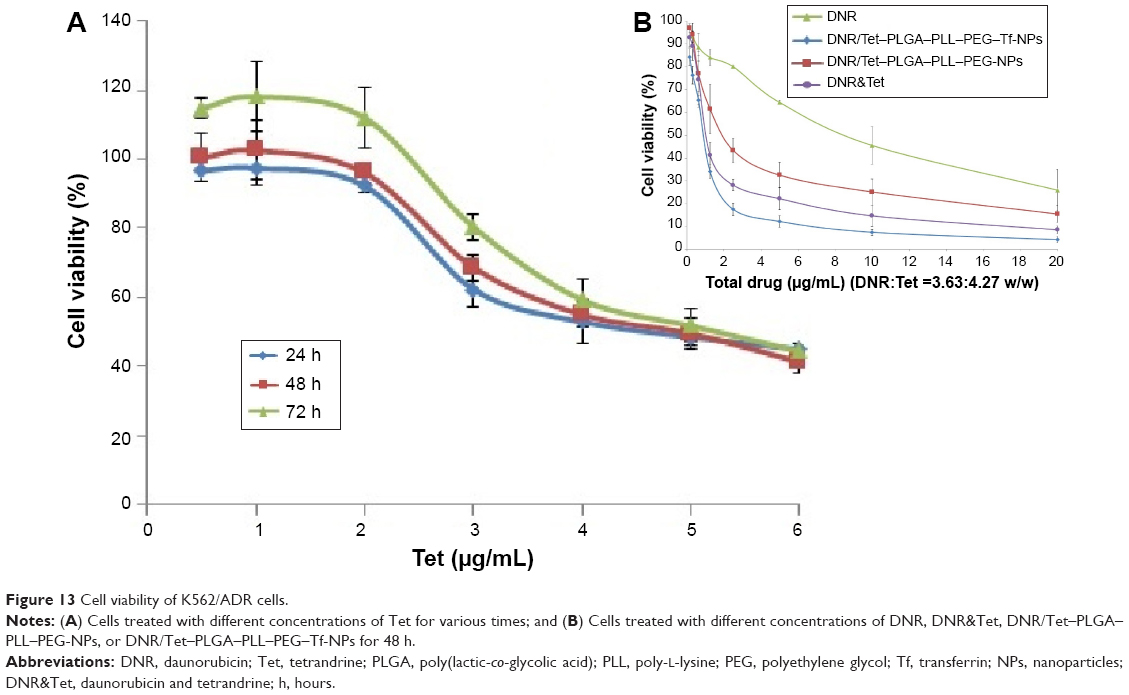 | Figure 13 Cell viability of K562/ADR cells. |
The dose-dependent curve showed that the loss of cell viability was notable in a dose-dependent manner when the K562/ADR cells were exposed to DNR, DNR&Tet, DNR/Tet–PLGA–PLL–PEG-NPs, and DNR/Tet–PLGA–PLL–PEG–Tf-NPs for 48 hours (Figure 13B). The IC50 value of DNR&Tet at 48 hours was much lower than that of DNR (1.47±0.15 μg/mL versus 10.03±0.69 μg/mL, respectively; P<0.05). However, the IC50 value of DNR/Tet–PLGA–PLL–PEG–Tf-NPs was significantly decreased (0.87±0.01 μg/mL; P<0.05). These data showed that Tet could enhance the cytotoxicity of DNR effectively and that DNR/Tet–PLGA–PLL–PEG–Tf-NPs showed stronger cytotoxicity when compared with the free drugs and DNR/Tet–PLGA–PLL–PEG-NPs (2.61±0.46 μg/mL; P<0.05). Therefore, we cannot exclude the possibility that this may be attributed to the sustained release of DNR from DNR/Tet–PLGA–PLL–PEG–Tf-NPs and the tumor-targeted effect by Tf, which should be further demonstrated in vitro and in vivo.
Conclusion
It was concluded that DNR (hydrophilic) and Tet (hydrophobic) could be simultaneously encapsulated into PLGA–PLL–PEG–Tf-NPs with a relatively small mean diameter and high drug loading using a modified double-emulsion technique. Tf can be covalently attached to PEG arms to produce PLGA–PLL–PEG–Tf-NPs, and it is not adsorbed by the electrostatic effect. The smaller mean diameter could be achieved through the optimization of the concentration of PLGA–PLL–PEG–Tf, the ratio of A/D volume, and the concentration of PVA. Meanwhile, the drug loadings could be enhanced by the optimization of the concentration of PLGA–PLL–PEG–Tf, the concentration of PVA, the ratio of I/E volume, and the surfactant of the internal aqueous phase. Furthermore, the inhibition of DNR/Tet–PLGA–PLL–PEG–Tf-NPs on leukemia K562/ADR cells occurred in a dose-dependent manner. Thus, the systematic investigation reported in our study indicated that the optimized PLGA–PLL–PEG–Tf-NP formulation can be potentially used for hydrophilic and hydrophobic chemotherapeutic DDS, which may be a promising strategy for overcoming MDR in leukemia or other drug-resistant tumors.
Acknowledgments
This work was supported by the National Nature Science Foundation of China (number: 81170492), the National Nature Science Foundation of China (number: 81370673), the National High Technology Research and Development Program 863 (number: 2012AA022703), the Anhui Provincial Natural Science Foundation (number: 1208085QH168), the Graduate Research and Innovation Program of Jiangsu Province (number: CXZZ11-0177), the National Innovative Research Program for College Students (number: 1210286098), the Jiangsu Province Health Department Medical Science and Technology Development Fund Project (number: H201229), and the Key Department of Jiangsu Medicine (number: 2011-12).
Disclosure
The authors report no conflicts of interest in this work.
References
Ling X, He Y, Zhang G, Zhou Y, Yan B. Increased P-glycoprotein expression in mitochondria is related to acquired multidrug resistance in human hepatoma cells depleted of mitochondrial DNA. Int J Oncol. 2012;40(1):109–118. | ||
Liu ZL, Hirano T, Tanaka S, Onada K, Oka K. Persistent reversal of P-glycoprotein-mediated daunorubicin resistance by tetrandrine in multidrug-resistant human T lymphoblastoid leukemia MOLT-4 cells. J Pharm Pharmacol. 2003;55(11):1531–1537. | ||
Liu Y, Li BH, Sun XM, Lin AH, Wang DB. Effect of selenium on the interaction between daunorubicin and cardiac myosin. Biol Trace Elem Res. 2012;147(1–3):240–245. | ||
Gewirtz DA. A critical evaluation of the mechanisms of action proposed for the antitumor effects of the anthracycline antibiotics adriamycin and daunorubicin. Biochem Pharmacol. 1999;57(7):727–741. | ||
Fan L, Li F, Zhang H, et al. Co-delivery of PDTC and doxorubicin by multifunctional micellar nanoparticles to achieve active targeted drug delivery and overcome multidrug resistance. Biomaterials. 2010;31(21):5634–5642. | ||
Xu WL, Shen HL, Ao ZF, et al. Combination of tetrandrine as a potential-reversing agent with daunorubicin, etoposide and cytarabine for the treatment of refractory and relapsed acute myelogenous leukemia. Leuk Res. 2006;30(4):407–413. | ||
Chen B, Cheng J, Shen M, et al. Magnetic nanoparticle of Fe3O4 and 5-bromotetrandrin interact synergistically to induce apoptosis by daunorubicin in leukemia cells. Int J Nanomedicine. 2009;4:65–71. | ||
Wang F, Zhang W, Guo L, et al. Gambogic acid suppresses hypoxia-induced hypoxia-inducible factor-1α/vascular endothelial growth factor expression via inhibiting phosphatidylinositol 3-kinase/Akt/mammalian target protein of rapamycin pathway in multiple myeloma cells. Cancer Sci. 2014;105(8):1063–1070. | ||
Chen Y, Bathula SR, Yang Q, Huang L. Targeted nanoparticles deliver siRNA to melanoma. J Invest Dermatol. 2010;130(12):2790–2798. | ||
Van de Ven H, Vermeersch M, Matheeussen A, et al. PLGA nanoparticles loaded with the antileishmanial saponin β-aescin: factor influence study and in vitro efficacy evaluation. Int J Pharm. 2011; 420(1):122–132. | ||
Zhou J, Patel TR, Fu M, Bertram JP, Saltzman WM. Octa-functional PLGA nanoparticles for targeted and efficient siRNA delivery to tumors. Biomaterials. 2012;33(2):583–591. | ||
Bertram JP, Williams CA, Robinson R, Segal SS, Flynn NT, Lavik EB. Intravenous hemostat: nanotechnology to halt bleeding. Sci Transl Med. 2009;1(11):11ra22. | ||
Bertram JP, Rauch MF, Chang K, Lavik EB. Using polymer chemistry to modulate the delivery of neurotrophic factors from degradable microspheres: delivery of BDNF. Pharm Res. 2010;27(1):82–91. | ||
Bertram JP, Jay SM, Hynes SR, Robinson R, Criscione JM, Lavik EB. Functionalized poly(lactic-co-glycolic acid) enhances drug delivery and provides chemical moieties for surface engineering while preserving biocompatibility. Acta Biomater. 2009;5(8):2860–2871. | ||
Fahmy TM, Samstein RM, Harness CC, Mark Saltzman W. Surface modification of biodegradable polyesters with fatty acid conjugates for improved drug targeting. Biomaterials. 2005;26(28):5727–5736. | ||
Keegan ME, Falcone JL, Leung TC, Saltzman WM. Biodegradable microspheres with enhanced capacity for covalently bound surface ligands. Macromolecules. 2004;37(26):9779–9784. | ||
Arnold LJ, Dagan A, Gutheil J, Kaplan NO. Antineoplastic activity of poly(L-lysine) with some ascites tumor cells. Proc Natl Acad Sci U S A. 1979;76(7):3246–3250. | ||
Mislick KA, Baldeschwieler JD. Evidence for the role of proteoglycans in cation-mediated gene transfer. Proc Natl Acad Sci U S A. 1996; 93(22):12349–12354. | ||
Liu P, Yu H, Sun Y, Zhu M, Duan Y. A mPEG-PLGA-b-PLL copolymer carrier for adriamycin and siRNA delivery. Biomaterials. 2012;33(17):4403–4412. | ||
Liu J, Qiu Z, Wang S, Zhou L, Zhang S. A modified double-emulsion method for the preparation of daunorubicin-loaded polymeric nanoparticle with enhanced in vitro anti-tumor activity. Biomed Mater. 2010;5(6):065002. | ||
Sahana DK, Mittal G, Bhardwaj V, Kumar MN. PLGA nanoparticles for oral delivery of hydrophobic drugs: influence of organic solvent on nanoparticle formation and release behavior in vitro and in vivo using estradiol as a model drug. J Pharm Sci. 2008;97(4):1530–1542. | ||
Barichello JM, Morishita M, Takayama K, Nagai T. Encapsulation of hydrophilic and lipophilic drugs in PLGA nanoparticles by the nanoprecipitation method. Drug Dev Ind Pharm. 1999;25(4):471–476. | ||
Lo CT, Van Tassel PR, Saltzman WM. Poly(lactide-co-glycolide) nanoparticle assembly for highly efficient delivery of potent therapeutic agents from medical devices. Biomaterials. 2010;31(13):3631–3642. | ||
Anand P, Nair HB, Sung B, et al. Design of curcumin-loaded PLGA nanoparticles formulation with enhanced cellular uptake, and increased bioactivity in vitro and superior bioavailability in vivo. Biochem Pharmacol. 2010;79(3):330–338. | ||
Zambaux MF, Bonneaux F, Gref R, et al. Influence of experimental parameters on the characteristics of poly(lactic acid) nanoparticles prepared by a double emulsion method. J Control Release. 1998;50(1–3):31–40. | ||
Rosca ID, Watari F, Uo M. Microparticle formation and its mechanism in single and double emulsion solvent evaporation. J Control Release. 2004;99(2):271–280. | ||
Murakami H, Kobayashi M, Takeuchi H, Kawashima Y. Preparation of poly(DL-lactide-co-glycolide) nanoparticles by modified spontaneous emulsification solvent diffusion method. Int J Pharm. 1999;187(2):143–152. | ||
Song X, Zhao Y, Wu W, et al. PLGA nanoparticles simultaneously loaded with vincristine sulfate and verapamil hydrochloride: systematic study of particle size and drug entrapment efficiency. Int J Pharm. 2008;350(1–2):320–329. | ||
Song X, Zhao Y, Hou S, et al. Dual agents loaded PLGA nanoparticles: systematic study of particle size and drug entrapment efficiency. Eur J Pharm Biopharm. 2008;69(2):445–453. | ||
Kondo K, Noguchi M, Mukai K, et al. Transferrin receptor expression in adenocarcinoma of the lung as a histopathologic indicator of prognosis. Chest. 1990;97(6):1367–1371. | ||
Whitney JF, Clark JM, Griffin TW, Gautam S, Leslie KO. Transferrin receptor expression in nonsmall cell lung cancer. Histopathologic and clinical correlates. Cancer. 1995;76(1):20–25. | ||
Prutki M, Poljak-Blazi M, Jakopovic M, Tomas D, Stipancic I, Zarkovic N. Altered iron metabolism, transferrin receptor 1 and ferritin in patients with colon cancer. Cancer Lett. 2006;238(2):188–196. | ||
Sciot R, Paterson AC, van Eyken P, Callea F, Kew MC, Desmet VJ. Transferrin receptor expression in human hepatocellular carcinoma: an immunohistochemical study of 34 cases. Histopathology. 1988;12(1): 53–63. | ||
Ryschich E, Huszty G, Knaebel HP, Hartel M, Büchler MW, Schmidt J. Transferrin receptor is a marker of malignant phenotype in human pancreatic cancer and in neuroendocrine carcinoma of the pancreas. Eur J Cancer. 2004;40(9):1418–1422. | ||
Walker RA, Day SJ. Transferrin receptor expression in non-malignant and malignant human breast tissue. J Pathol. 1986;148(3):217–224. | ||
Lloyd JM, O’Dowd T, Driver M, Tee DE. Demonstration of an epitope of the transferrin receptor in human cervical epithelium – a potentially useful cell marker. J Clin Pathol. 1984;37(2):131–135. | ||
Seymour GJ, Walsh MD, Lavin MF, Strutton G, Gardiner RA. Transferrin receptor expression by human bladder transitional cell carcinomas. Urol Res. 1987;15(6):341–344. | ||
Tomita Y, Nishiyama T, Sato S, Fujiwara M. [Expression of transferrin receptor on transitional cell cancer]. Hinyokika Kiyo. 1991;37(1):11–16. Japanese. | ||
Gatter KC, Brown G, Trowbridge IS, Woolston RE, Mason DY. Transferrin receptors in human tissues: their distribution and possible clinical relevance. J Clin Pathol. 1983;36(5):539–545. | ||
Pileri S, Gobbi M, Rivano MT, Martinelli G. Immunohistological study of transferrin receptor expression in non-Hodgkin’s lymphoma. Br J Haematol. 1984;58(3):501–508. | ||
Medeiros LJ, Picker LJ, Horning SJ, Warnke RA. Transferrin receptor expression by non-Hodgkin’s lymphomas. Correlation with morphologic grade and survival. Cancer. 1988;61(9):1844–1851. | ||
Barnett D, Wilson GA, Lawrence AC, Buckley GA. Transferrin receptor expression in the leukaemias and lymphoproliferative disorders. Clin Lab Haematol. 1987;9(4):361–370. | ||
Habeshaw JA, Lister TA, Stansfeld AG, Greaves MF. Correlation of transferrin receptor expression with histological class and outcome in non-Hodgkin lymphoma. Lancet. 1983;1(8323):498–501. | ||
Recht LD, Griffin TW, Raso V, Salimi AR. Potent cytotoxicity of an antihuman transferrin receptor-ricin A-chain immunotoxin on human glioma cells in vitro. Cancer Res. 1990;50(20):6696–6700. | ||
Soyer HP, Smolle J, Torne R, Kerl H. Transferrin receptor expression in normal skin and in various cutaneous tumors. J Cutan Pathol. 1987;14(1):1–5. | ||
Li X, Xu Y, Chen G, Wei P, Ping Q. PLGA nanoparticles for the oral delivery of 5-Fluorouracil using high pressure homogenization-emulsification as the preparation method and in vitro/in vivo studies. Drug Dev Ind Pharm. 2008;34(1):107–115. | ||
Li X, Ding L, Xu Y, Wang Y, Ping Q. Targeted delivery of doxorubicin using stealth liposomes modified with transferrin. Int J Pharm. 2009;373(1–2):116–123. | ||
Ahn CH, Chae SY, Bae YH, Kim SW. Synthesis of biodegradable multi-block copolymers of poly(L-lysine) and poly(ethylene glycol) as a non-viral gene carrier. J Control Release. 2004;97(3):567–574. | ||
Lavik EB, Hrkach JS, Lotan N, Nazarov R, Langer R. A simple synthetic route to the formation of a block copolymer of poly(lactic-co-glycolic acid) and polylysine for the fabrication of functionalized, degradable structures for biomedical applications. J Biomed Mater Res. 2001;58(3):291–294. | ||
Wang Y, Li X, Wang L, Xu Y, Cheng X, Wei P. Formulation and pharmacokinetic evaluation of a paclitaxel nanosuspension for intravenous delivery. Int J Nanomedicine. 2011;6:1497–1507. | ||
Deng C, Chen X, Yu H, Sun J, Lu T, Jing X. A biodegradable triblock copolymer poly(ethylene glycol)-b-poly(L-lactide)-b-poly(L-lysine): synthesis, self-assembly, and RGD peptide modification. Polymer. 2007;48(1):139–149. | ||
Takenaga M, Yamaguchi Y, Kitagawa A, et al. Optimum formulation for sustained-release insulin. Int J Pharm. 2004;271(1–2):85–94. | ||
Cohen-Sela E, Chorny M, Koroukhov N, Danenberg HD, Golomb G. A new double emulsion solvent diffusion technique for encapsulating hydrophilic molecules in PLGA nanoparticles. J Control Release. 2009;133(2):90–95. | ||
Mora-Huertas CE, Fessi H, Elaissari A. Polymer-based nanocapsules for drug delivery. Int J Pharm. 2010;385(1–2):113–142. | ||
Galindo-Rodriguez S, Allémann E, Fessi H, Doelker E. Physicochemical parameters associated with nanoparticle formation in the salting-out, emulsification-diffusion, and nanoprecipitation methods. Pharm Res. 2004;21(8):1428–1439. | ||
Nandi A, Khakhar DV, Mehra A. Coalescence in surfactant-stabilized emulsions subjected to shear flow. Langmuir. 2001;17(9): 2647–2655. | ||
Tesch S, Schubert H. Influence of increasing viscosity of the aqueous phase on the short-term stability of protein stabilized emulsions. J Food Eng. 2002;52(3):305–312. | ||
Budhian A, Siegel SJ, Winey KI. Haloperidol-loaded PLGA nanoparticles: systematic study of particle size and drug content. Int J Pharm. 2007;336(2):367–375. | ||
Mainardes RM, Evangelista RC. PLGA nanoparticles containing praziquantel: effect of formulation variables on size distribution. Int J Pharm. 2005;290(1–2):137–144. | ||
Chung NO, Lee MK, Lee J. Mechanism of freeze-drying drug nanosuspensions. Int J Pharm. 2012;437(1–2):42–50. | ||
Beirowski J, Inghelbrecht S, Arien A, Gieseler H. Freeze-drying of nanosuspensions, 1: freezing rate versus formulation design as critical factors to preserve the original particle size distribution. J Pharm Sci. 2011;100(5): 1958–1968. | ||
Dubernet C. Thermoanalysis of microspheres. Thermochim Acta. 1995;248:259–269. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.