Back to Journals » International Journal of Nanomedicine » Volume 14
Synergistic Anti-Staphylococcal Activity Of Niosomal Recombinant Lysostaphin-LL-37
Authors Sadeghi S, Bakhshandeh H ![]() , Ahangari Cohan R
, Ahangari Cohan R ![]() , Peirovi A, Ehsani P, Norouzian D
, Peirovi A, Ehsani P, Norouzian D
Received 8 September 2019
Accepted for publication 7 November 2019
Published 10 December 2019 Volume 2019:14 Pages 9777—9792
DOI https://doi.org/10.2147/IJN.S230269
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Thomas Webster
Somayeh Sadeghi,1,2 Haleh Bakhshandeh,1 Reza Ahangari Cohan,1 Afshin Peirovi,1 Parastoo Ehsani,2 Dariush Norouzian1
1Department of Nano Biotechnology, New Technology Research Group, Pasteur Institute of Iran, Tehran, Iran; 2Department of Molecular Biology, Pasteur Institute of Iran, Tehran, Iran
Correspondence: Parastoo Ehsani
Department of Molecular Biology, Pasteur Institute of Iran, #69 Pasteur Avenue, Tehran 13164, Iran
Tel + 98 21 64112219
Fax + 98 21 64112803
Email [email protected]
Dariush Norouzian
Department of Nano Biotechnology, New Technology Research Group, Pasteur Institute of Iran, #69 Pasteur Avenue, Tehran 13164, Iran
Tel + 98 21 64112137
Fax + 98 21 66465132
Email [email protected]
Purpose: Staphylococcus aureus is the most common persistent pathogen in humans, so development of new formulations to combat pathogen invasion is quite necessary.
Methods: In the current study, for the first time, the synergistic activity of recombinant lysostaphin and LL-37 peptide was studied against S. aureus. Moreover, different niosomal formulations of the peptide and protein were prepared and analyzed in terms of size, shape, zeta potential, and entrapment efficiency. Also, a long-term antibacterial activity of the best niosomal formulation and free forms was measured against S. aureus in vitro.
Results: The optimal niosomal formulation was obtained by mixing the surfactants (span60 and tween60; 2:1 w/w), cholesterol, and dicetylphosphate at a ratio of 47:47:6, respectively. They showed uniform spherical shapes with the size of 565 and 325 nm for lysostaphin and LL-37, respectively. This formulation showed high entrapment efficiency for the peptide, protein, and a slow-release profile over time. Release kinetic was best fitted by Higuchi model indicating a diffusion-based release of the drugs. The lysostaphin/LL-37 niosomal formulation synergistically inhibited growth of S. aureus for up to 72 hours. However, the same amounts of free forms of both anti-microbial agents could not hold the anti-microbial effect and growth was seen in the following 72 hours. Cytotoxicity assay specified that lysostaphin/LL-37 niosomal combination had no deleterious effect on normal fibroblast cells at effective antimicrobial concentrations.
Conclusion: This study indicated that the use of lysostaphin in combination with LL-37, either in niosomal or free forms, synergistically inhibited growth of S. aureus in vitro. In addition, niosomal preparation of antimicrobial agents could provide a long-term protection against bacterial infections.
Keywords: LL-37, lysostaphin, sustained release, synergy, Staphylococcus aureus
Introduction
Antibiotic resistance is one of the major concerns in healthcare around the world because of widespread and uncontrolled usage of antibiotics. Considering high risk of antibiotic resistance on a global scale, it is estimated that antibiotic resistant bacteria could be a leading cause of death by 2050,1 unless new antimicrobial classes with novel targets and mode of actions are introduced.2 So many researchers have focused their search studies on developing new antibacterial agents.
Staphylococcus aureus is a widespread human commensal, which is currently the most common cause of healthcare-associated infections. It can cause development of different infections ranging from localized abscess to invasive infections, like skin and soft tissue infections, bacteremia, endocarditis, and osteomyelitis.3,4 Lysotaphin is one of the most recent antimicrobial agents against S. aureus due to its unique specificity, high stability, and low toxicity. Lysostaphin is a metallo-endopeptidase produced by Staphylococcus simulans. This enzyme possesses a specific antibacterial activity against staphylococcal species.4 However, single-agent treatments are not often clinically successful. So, investigations regarding administration of bactericidal agents in combination with lysostaphin are practically worthwhile. Combination of lysostaphin and antimicrobial peptides (AMP) could reduce bacterial resistance, because of the unique disruption of bacterial cell membrane and rarely reported resistance of AMPs. Moreover, some lysostaphin/AMP combinations have a synergistic antibacterial effect that is of clinical importance because the effective doses of each compound can be reduced.5,6
Among various AMPs, LL-37 as a 37-residue, cationic amphipathic α-helical peptide is an endogenous host defense peptide and plays an important role in protecting against a wide range of Gram-positive and Gram-negative bacteria. This peptide also has antifungal, antiviral, and endotoxin-binding properties and is a promoter of wound healing by affecting cell proliferation and cell differentiation.7 Therefore, there is great interest in using LL-37 as a potential agent for the treatment of chronic wound infections.8 Moreover, this peptide is significantly resistant to proteolytic degradation in solution and when bound to negatively charged membranes (mimicking bacterial membranes) which influences the life span of the peptide and its efficacy. A study conducted by Oren et al9 about the structure and organization of human antimicrobial peptide LL-37 showed that LL-37 at below MIC (minimum inhibitory concentration) causes morphological changes to cell membranes and, when MIC is reached, full lysis of the bacterial cell membranes occurs.
Some studies have shown synergism between LL-37 and other antimicrobials like human β-defensin-3, lysozyme, colistin, and imipenem against S. aureus.10,11 So, a combination of recombinant lysostaphin and LL-37 could be a potential therapy for treatment of S. aureus infections. However, the use of this combination in its free form could potentially result in loss of activity due to degradation or inactivation over time as well as probable emergence of resistant strains. Nanotechnology may be used to overcome these limitations.12
Nanotechnology is one of the best approaches used for protecting and enhancing the stability of protein or peptide drugs for a long period of time. Encapsulation of these compounds into nanovesicles may have the following benefits: (1) a tool for targeting bacteria, (2) decreasing bacterial resistance, (3) protecting antibacterial agents from inhibitors or other unfavorable conditions, and (4) acting as a long-term preservative in pharmaceutical industries.13 Liposomes and niosomes are widely used as nanovesicular structures in drug delivery systems. These structures are considered as a promising strategy for delivery of drugs in a controlled manner. Liposomes have been targeted to a wide range of bacteria for treatment of infectious diseases.14,15 Nevertheless, several significant drawbacks have been recognized for the use of liposomes as a shell carrier including high cost and high susceptibility to oxidative degradation.16 In contrast, niosomes are self-assembled non-ionic surfactants, which could form unilaminar or multilaminar vesicular structures in aquatic solutions.17 Taking into account higher stability and cost-benefit advantages of niosomes over liposomes, these nanovesicular structures are usually considered as preferred controlled delivery systems for cosmetic, food, and pharmaceutical purposes. Many peptides and proteins have been successfully encapsulated into niosomes for different applications including insulin, lysozyme,18 BSA,19 bacitracin,20 and Tat-GFP fusion protein.21
In the present study, synergistic activity of lysostaphin and synthetic LL-37 was studied against S. aureus using a checkerboard assay. Moreover, new niosomal formulations were designed and prepared for co-administration of lysostaphin and LL-37. Entrapment efficacy (EE), size distribution, and zeta potential were measured for niosomal formulations. Finally, kinetic release and antibacterial activity of the best formulation were investigated against S. aureus.
Materials And Methods
Materials
Cholesterol was purchased from ACRONIS Company, USA. Sorbian monostearate (span 60), Polysorbate (Tween 60), dicetylphosphate (DCP), and Trifluoroacetic acid (TFA) were purchased from Sigma Aldrich Company, UK. All other reagents were of analytical grade.
LL-37 was purchased from Bio Basic Company, Canada. LL-37 stock solution was freshly prepared by dissolving it in deionized water to a final concentration of 10 mg/mL.
Expression Of Lysostaphin
Lysostaphin was expressed in Escherichia coli BL21 (DE3) according to a previous study. Briefly, E. coli BL21 (DE3) was transformed with pET32a plasmid encoding lysostaphin sequence by calcium chloride method.22 Transformed cells were cultured in LB broth supplemented with 100 µg/mL of ampicillin. Protein expression was induced by adding 0.5 mM isopropyl thio-β-D-galactosidase (IPTG) (Sigma Company). Expressed protein was purified by Ni-NTA affinity chromatography (Qiagen, USA). Protein concentration was determined by Bradford assay in all steps.23 The whole cell extract and soluble fraction were analyzed on a 12% SDS-PAGE gel and were stained with Coomassie brilliant blue. Purified lysostaphin was transferred to nitrocellulose membrane using a Bio-Rad transfer apparatus. Then, membrane was blocked with 5% (w/v) milk in 100 mM PBS (phosphate-buffered saline) containing 0.1% Tween-20 and was washed twice with PBS-Tween 20. The membrane was incubated with anti His-HRP conjugated antibody (1:2,000 dilution in 100 mM PBS) overnight at 4°C. After washing, the specific protein band was visualized with diaminobenzidine (DAB) and H2O2.24
RP-HPLC Method For Identification Of Lysostaphin And LL-37
To evaluate the purity of lysostaphin and LL-37, both solutions (1 mg/mL) were filter-sterilized through 0.22 μm filters. For this procedure, mobile phases A (0.1% v/v trifluoroacetic acid in water) and B (0.1% v/v trifluoroacetic acid in acetonitrile) were required. Then, filtered samples (20 µL) were injected onto C18 reverse phase HPLC column (TOSOH bioscience, 4.6×150 mm). The samples were eluted through column using a gradient program according to the manufacturer’s instruction, as follows: A/B from 65:35 to 40:60 within 25 minutes and from 40:60 to 10:90 within 1 minute, followed by maintaining at 10:90 for 4 minutes and re-equilibration of the column at 65:35 with a flow rate of 1 ml/min at 280 nm. The column temperature was set at 30°C and the total run was 50 minutes for each injected sample.
Synergistic Activity Of Lysostaphin And LL-37 By Checkerboard
Minimum inhibitory concentration (MIC) of lysostaphin and LL-37 alone and in combination against S. aureus (ATCC 6538) was evaluated by checkerboard method.25 Tested concentrations of LL-37 and lysostaphin used were equal to 250, 125, 62.5, 31.25, 15.62, 7.81, 3.9 μg/mL; and 125, 62.5, 31.25, 15.62, 7.81, 3.9, 1.96 μg/mL, respectively. The first drug (LL37) was diluted vertically while the second drug (lysostaphin) was diluted horizontally. Each well was inoculated with 105 CFU/mL of bacterial inoculum. The plate was incubated at 37°C for 24 hours.
Interaction between lysostaphin and LL-37 was determined by fractional inhibitory concentrations (FIC). FIC was obtained using the following equations:
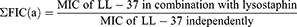

Then, FIC index (ƩFIC) was determined where; Σ FIC=FIC(a)+FIC(b).
The obtained value is interpreted as ƩFIC≤0.5 as synergistic, ƩFIC>0.5–1.0 as additive, ƩFIC>1.0 but <4.0 as indifferent, and ƩFIC≥4.0 as antagonistic.25
Preparation Of Lysostaphin/LL-37 Encapsulated Niosomes
Niosomal formulations were prepared by thin film hydration method26 using different ratios of surfactants (span 60 and tween 60; 2:1 w/w), cholesterol, and DCP, as indicated in Table 1. Briefly, the surfactants, cholesterol, and DCP were dissolved in chloroform. The solvent was removed by a rotary evaporator (Heidolph Instruments, Germany) at 60°C, 160 rpm until formation of a thin lipid film. Lysostaphin or LL-37 solutions (0.5 mg/mL) were used to dissolve this film in a water bath at 40°C, 60 rpm. The solutions were sonicated for size reduction using an ultrasonic probe homogenizer (Bandelin Sonopuls HD 4200, Germany) at 25% amplitude, for 30 seconds.
 |
Table 1 Compositions Of Different Niosomal Formulations |
Physical Characteristics Of Encapsulated Niosomes
Surface Morphology
Surface morphology of bilayer vesicles was investigated by scanning electron microscopy (NOVA NANOSEM 450 FEI model). A drop of niosomes was air dried on glass slides and was coated with 10 nm of gold for 3 minutes under argon at a pressure of 0.2 atm. After preparation, shape, and size of particles were studied using SEM at an accelerating voltage of 15 kV.
Zeta Potential And Size Distribution
Size distribution and zeta potential of niosomes, with and without lysostaphin or LL-37, were determined using Zetasizer Nano ZS (Malvern Instruments, UK) in aqueous medium at room temperature. Each sample was measured in three individual runs.
Entrapment Efficiency Measurements
Entrapped lysostaphin or LL-37 in niosomes was separated from free ones by ultracentrifugation (Eppendorf® 580R centrifuge, Germany) at 14,000 rpm for 30 minutes. The supernatant was analyzed for quantification of free lysostaphin or LL-37 using Bradford assay.23 The following equation was used to determine entrapment efficiency of lysostaphin or LL-37 in niosomes.
Fourier Transform Infrared Spectroscopy (FTIR)
The interaction between lysostaphin and LL-37 with niosomal membrane was evaluated by FTIR spectroscopy. The spectra were recorded using the PerkinElmer FTIR spectrophotometer spectrum Two (USA), equipped with ATR accessory. The samples were poured on FTIR plate and the spectra were recorded between 4,000 to 400 cm−1 with 16 scans and spectral resolution of 4 cm−1. Then, the spectra were compared to determine changes and interactions.
In Vitro Drug Release Profile
Release profiles of lysostaphin and LL-37 were determined in simulated wound fluid (SWF) containing 0.142 M sodium chloride and 0.0025 M calcium chloride in deionized water adjusted to pH 7.227 using dialysis membrane28 (MWCO 100 kDa, biotech CE tubing US/Canada); 1 mL of lysostaphin (418.4 µg/mL) and 1 mL of LL-37 (461.7 µg/mL) in encapsulated and free forms were dialysed against 50 mL of SWF solution for 72 hours at 37°C. At specific time intervals (0, 1, 2, 4, 8, 24, 48, and 72 hours), 2 mL of SWF was collected and was replaced with same volume of SWF. Concentrations of released lysostaphin and LL-37 were determined by Bradford assay.
In Vitro Antibacterial Activity
Minimum Inhibitory Concentration (MIC)
MIC values of drug loaded niosomes and free drugs were determined by Clinical Laboratory Standard Institute (CLSI) broth microdilution method with slight modification.29 Briefly, 50 µL of Mueller Hinton Broth (MHB) was added into each well of a sterile 96 well flat-bottom plate. Treatment groups included lysostaphin, LL-37, and lysostaphin/LL-37 in encapsulated and free forms and empty niosomes; 5 µL of S. aureus bacteria (107 colony forming units (CFU)/mL) was added to each well of the plate. Positive (containing MHB and bacterial suspension) and negative (containing MHB and free or encapsulated drugs without bacterial suspension) controls were also prepared. The plates were incubated at 37°C for 18 hours without shaking.
Time-Kill Assay
Antibacterial activity of lysostaphin/LL-37 encapsulated niosomes was determined against S. aureus within 72 hours using 96 well plate technique. The stock solution of either free lysostaphin or LL-37 was diluted up to a concentration of 100 µg/mL. Similarly encapsulated lysostaphin and LL-37 was diluted up to concentrations of 105 and 115 µg/mL, respectively. Microtiter plates were loaded with approximately 100 µL of test samples including: (i) free LL-37, (ii) encapsulated LL-37, (iii) free lysostaphin, (iv) encapsulated lysostaphin, (v) free lysostaphin/LL-37, (vi) encapsulated lysostaphin/LL-37, and (vii) blank niosomes. Then, 100 µL of bacterial suspension diluted to final concentration of 105 CFU/mL was added to each well. Then 96 well plate was incubated at 37°C, and optical density (OD) at 600 nm was measured at specific time intervals (0, 2, 4, 6, 24, 48, and 72 hours) using a microplate reader (EPOCH, Japan). Growth curve of S. aureus was taken as positive control.
Cytotoxicity Determination Of Lysostaphin/LL-37 Encapsulated Niosomes
(3-(4, 5-dimethylthiazol-2-yl) 2,5-diphenyl tetrazolium bromide (MTT) assay was used to determine in vitro cell viability. Mouse fibroblast L929 cells were purchased from Pasteur Institute of Iran and were cultured in RPMI 1640 containing 10% FBS at 37°C in a humidified atmosphere of 5% CO2. The cells were seeded onto 96 well plates at a concentration of 104 and were incubated for 24 hours. Free and encapsulated drugs were diluted in the culture medium. Then, the cells were exposed to increasing concentrations of lysostaphin, LL-37, and lysostaphin/LL-37 in encapsulated and free forms in triplicate for 24 hours. Blank noisome treated cells were used to assess overall viability. After incubation, 100 µL of 0.5 mg/mL of MTT solution was added into each well, and the plates were incubated for 4 hours. Then, the culture medium was removed and was replaced with 100 µL of isopropanol. Finally, absorbance of each well was measured using a microplate reader (BioTek ELx808, USA) at 570 nm. Cell viability is expressed as a percentage relative to untreated control, which was 100% viable.
Statistical Analysis
GraphPad prism software 5 was used to analyze the data. Significant differences (P≤0.05) between studied groups were determined using one-way and two-way Analysis of Variance (ANOVA). Date were presented as mean±Standard Deviation (SD).
Results And Discussion
Expression And Purification Of Lysostaphin
E. coli BL21 (DE3) lysates containing lysostaphin were prepared under native conditions. Lysostaphin was designed to have N-terminal histidine hexa peptide (6X-His) in order to be purified using a Ni-NTA agarose column. The presence of lysostaphin was analyzed by SDS-PAGE and Western Blotting (Figure 1).
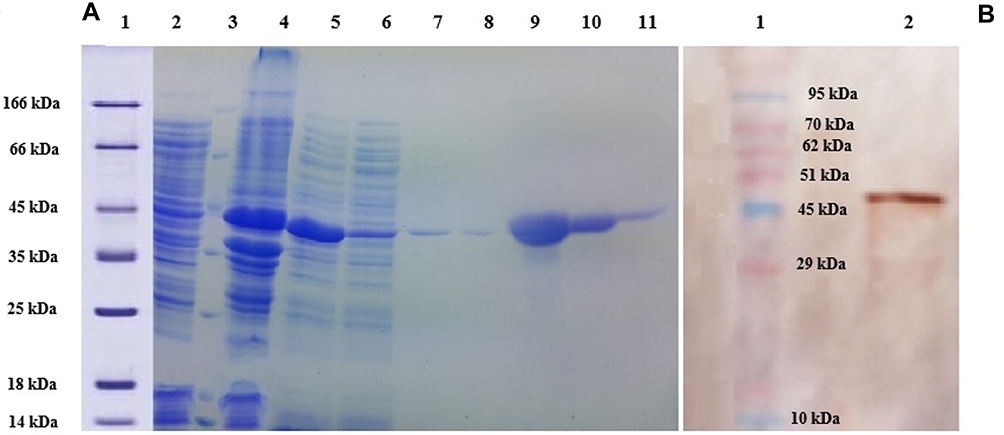 |
Figure 1 (A) SDS PAGE (12%) gel showing expression and purification of lysostaphin. Lane 1: molecular weight marker, Lane 2: uninduced cell extract, Lane 3: molecular weight marker, Lane 4: proteins from pellet after separation of readily soluble protein fraction, Lane 5: readily soluble protein extract (cytoplasmic cell proteins), Lane 6: flow through Ni-NTA agarose resin affinity chromatography, Lanes 7 and 8: Non-tagged proteins washed from affinity chromatography by 20 mM imidazole, Lanes 9–11: lysostaphin eluted by 250 mM imidazole. (B) Western Blotting analysis using anti-His-HRP conjugated antibody. Lane 1: molecular weight marker, Lane 2: lysostaphin. |
Identification Of Lysostaphin And LL-37 By RP-HPLC
Lysostaphin and LL-37 at a final concentration of 1 mg/mL were separately injected onto C18 RP-HPLC (Figure 2). In our study, lysostaphin and LL-37 were directly and easily detected at retention times of 14.83 and 11.26 minutes, respectively. HPLC analysis of lysostaphin and LL-37 indicated that they are pure.
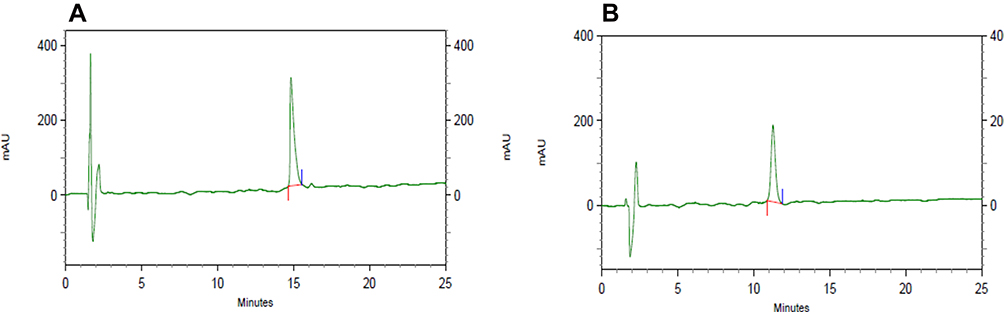 |
Figure 2 RP-HPLC chromatograms for identification of (A) lysostaphin and (B) LL-37. Lysostaphin and LL-37 showed a retention time of 14.83 and 11.26 minutes, respectively. |
Synergistic Activity Testing Of Lysostaphin And LL-37
Combination therapies dramatically reduce the chance of resistant species being selected provided that two different mechanisms of action are used for the drugs.30 Previous studies have shown that lysostaphin with specific antimicrobial peptides has a synergistic effect against S. aureus,5,6 due to the fact that lysostaphin renders S. aureus more susceptible to a wide range of antimicrobial peptides. Moreover, the synergistic effect of LL-37 with conventional antibiotics was elucidated in previous studies. A study conducted by Geitani et al10 revealed that LL-37 significantly decreases MICs of imipenem (a β-lactam antibiotic) and colistin (cyclic polypeptides) against Pseudomonas aeruginosa isolates. Similar results were observed with human β-defensin-3 peptide and LL-37 against S. aureus.11 Therefore, a new field of research in the fight against drug-resistant staphylococci is emerging. However, any synergistic effects observed in vitro should be evaluated in vivo to confirm that the desired interaction is repeated in animal infection models, because this is not necessarily generalized.31
Checkerboard assay was performed to confirm the synergistic activity of lysostaphin and LL-37. In this study, LL-37 alone was not able to achieve desirable bacterial inhibition, as indicated by a high MIC value (250 µg/mL). But, in the presence of lysostaphin, the antibacterial effect of LL-37 increased and led to a significant inhibition of S. aureus growth. As previously described, when ƩFIC is less than 0.5, a synergistic effect is observed between two antimicrobial drugs. The calculated fractional inhibitory concentration index (ƩFIC) for S. aureus ATCC6538 using lysostaphin and LL-37 is 0.25, which indicates a synergistic effect between lysostaphin and LL-37 in inhibition of bacterial growth. Thus, it can be speculated that synergistic bactericidal activity of lysostaphin/LL-37 combination could result from the ability of cell wall degradation by lysostaphin and greater accessibility of LL-37 to the cell membrane31,32 (Figure 3). Nevertheless, the actual mechanism of synergy between lysostaphin and LL-37 is not yet clearly known.
 |
Figure 3 Mechanisms involved in antibacterial synergistic effect of lysostaphin on peptidoglycan and LL-37 on cytoplasmic membrane of S. aureus. LL-37 (PDB ID: 2K6O) and lysostaphin (PDB ID: 4LXC) 3-D structures were retrieved from Protein Data Bank (PDB). |
Preparation Of Lysostaphin/LL-37 Encapsulated Into Niosomes
Niosomal dispersions consisting of lipid phase were prepared using different weight ratios of surfactants, cholesterol, and DCP (Table 1). Results showed that all the formulations obtained from the mixture of surfactants (span 60, tween 60) and cholesterol formed vesicles with different sizes. It was shown that a combination of span and tween would lead to production of niosomes with highly stable membranes.33 Moreover, entrapment efficiency for prepared niosomes using a combination of span 60 and tween 60 is higher than those prepared using merely span 60 or tween 60. Junyaprasert et al34 concluded that niosomes prepared with a ratio of 2:1 span 60 and tween 60 (S2 T1) were a suitable formulation which may have resulted from the higher rigidity of their vesicle membrane. This formulation had the highest entrapment efficiency and stability after storage for 4 months at different temperatures compared to other formulations with different ratios of span 60 and tween 60. So, in our study niosomes obtained from the mixture of span 60 and tween 60 at a ratio of 2:1 w/w were used for further investigation.
Characteristic Of Niosomes Encapsulated With Lysostaphin/LL-37
Morphology, Vesicular Size, And Zeta Potential Of Prepared Niosomes
Niosomes showed relatively smooth and spherical structures having homogenous dispersion with an approximate length of 20 nm based on field emission SEM images, as shown in Figure 4. The size of niosomes obtained by SEM was much smaller than that obtained by Nano Zetasizer. This was because Zetasizer Nano ZS system measures size (hydrodynamic diameter), dispersion, molecular weight, and zeta potential of the nanoparticles, while SEM measures exact diameter of each particle.
 |
Figure 4 Field emission SEM image of niosomal formulation containing surfactants (span 60, tween 60):cholesterol:DCP at 47:47:6% weight ratio. Niosomes had relatively smooth and unique spherical structures. |
Particle size, zeta potential, and the percentage of lysostaphin and LL-37 encapsulated into niosomes are presented in Table 2. Obtaining a particle size of less than 500 nm has been the goal of many studies, because particles more than 500 nm in size progressively tend to be precipitated during a few days. As shown in Table 2, F1–F3 niosomal formulations are similar to F4–F6, respectively, except that F1–F3 were loaded with lysostaphin and F4–F6 were loaded with LL-37. According to the table, decreasing the amount of cholesterol from 67 to 27% significantly reduces the particle size (P<0.05), except for the F3 formulation in lysostaphin encapsulated niosomes. However, no significant difference (P<0.05) was observed between F2 and F3 formulations of this group. This result is in accordance with previous studies showing that an increment in the cholesterol amount led to an increase in the size of vesicle.35,36
 |
Table 2 The Particle Size, Zeta Potential, And Entrapment Efficiency Of Blank And Encapsulated Niosomes |
In this study, the zeta potential of blank niosomes containing DCP was negative, ranging from −8.05 to −16.70 mV, indicating an electrostatic repulsion between the particles led to good stability of the formulations. It was due to the fact that DCP as a charge inducer renders negative charge to vesicular structure and increases stability by preventing aggregation of anionic niosomes.37 When lysostaphin or LL-37 was added to the system, zeta potential changed. As shown in Table 2, zeta potentials of encapsulated lysostaphin or LL-37 significantly (P<0.05) increased in all three formulations, meaning that an amount of lysostaphin or LL-37 is adsorbed on surface of the particles. Investigations have reported that zeta potential of anionic nanoparticles loaded with positively charged peptides increases compared to blank ones. For example, Manosroi et al20 showed that the zeta potential of blank anionic niosomes containing tween 61: cholesterol: DCP was −41.3±1.69 mV while anionic niosomes entrapped with positively charged bacitracin had a zeta potential of +22.68±1.31 mV. Zeta potential of bacitracin loaded nanoparticles increased about 63 mV by encapsulation of bacitracin. This might be due to cationic nature of bacitracin.
Entrapment efficiency
Entrapment efficiency of lysostaphin and LL-37 in niosomes with different cholesterol amounts are presented in Table 2. Results of analysis showed that (P<0.05), F5 formulation with 47% cholesterol showed the highest percentage, at 92.34±0.04 for LL-37 encapsulated form. So, this formulation was chosen for further evaluation. However, no significant difference (P<0.05) was observed among three formulations in lysostaphin encapsulated niosomes. Yet, as seen in Table 2, the F2 formulation has a smaller size of about 500 nm than the other two formulations. Therefore, this formulation was used as the optimal formulation for further investigation. Obviously, by increasing the cholesterol percentage up to 47%, the amount of entrapment raised. However, entrapment efficiency declined by further increasing the cholesterol amount. Similar results have been reported by other studies.19,38 This indicates that a level of cholesterol beyond a certain level causes disruption of bilayer structure, resulting in loss of drug entrapment.39 So, by choosing an optimum ratio of surfactant to cholesterol, more loaded drugs in niosomal vesicles are obtained.
According to the theory proposed by Colas et al,13 positively charged antimicrobial peptide nisin tends to be more entrapped in anionic nanoliposomes. So, it may be concluded that the high EE of lysostaphin and LL-37 observed in our study was due to the interaction between positively charged lysostaphin and LL-37 with negatively charged anionic niosomes. However, if the nanostructure is neutral and protein drug is positively/negatively charged, less EE will be expected. As an example, insulin (negatively charged protein) has been encapsulated into neutral niosomal vesicles containing span 60 and cholesterol by about 40%.40 However, the entrapment efficiency of insulin into positively charged liposomes was higher than that of neutral liposomes.41
Fourier Transform Infrared Spectroscopy (FTIR)
FTIR spectra of both unloaded and loaded niosomes were performed in order to study the possible interaction of lysostaphin and LL37 with the lipid membrane (Figure 5). In our study, the intensity at about 1642 cm−1, resulting from c=c and c=o stretching vibrations of lysostaphin and LL-37, was increased in encapsulated niosomes in comparison with blank niosomes. However, no significant changes in peaks were observed after encapsulating lysostaphin and LL-37, as no bonds between lysostaphin or LL-37 and niosomes were formed.
 |
Figure 5 FTIR spectra of (A) blank niosomes and (B) niosomes containing 1:1 volume ratio of encapsulated lysostaphin and encapsulated LL-37. |
Measuring Release Of Lysostaphin And LL-37 From Niosomal Nanoparticles
The rate of drug release from drug delivery systems is critical and should be studied in order to achieve an optimal system with desirable release characteristics. Release of lysostaphin and LL-37 from niosomes occurred in a sustained manner during the study. Encapsulated LL-37 or encapsulated lysostaphin (niosomes containing 461.7 µg/mL of LL-37 or 418.4 µg/mL of lysostaphin) were released around 84.3±2.53 and 56.51±3.50% in the first 24 hours, respectively (Figure 6). However, release of lysostaphin/LL-37 from niosomal formulation (niosomes containing 1:1 volume ratio of encapsulated lysostaphin and encapsulated LL-37) occurred at approximately 63.5±0.77% for 24 hours and was followed by a slow and sustained release over the entire release period of 72 hours. On the contrary, almost all contents of free lysostaphin/LL-37 were quickly released from the dialysis bag after a short time, as depicted in Figure 7A. This kind of release profile has also been observed for BSA release from niosomal formulations.19
 |
Figure 6 In vitro release profile of (A) lysostaphin and (B) LL-37 from niosomal formulation containing surfactants (span 60, tween 60):cholesterol:DCP at 47:47:6% weight ratio in 50 mL of SWF (pH 7.2) at 37°C for 72 hours. |
 |
Figure 7 Overall release of lysostaphin/LL-37 from niosomal formulation in SWF releasing medium. (A) In vitro release profile of lysostaphin and LL-37 from niosomal formulation containing surfactants (span 60, tween 60):cholesterol:DCP at 47:47:6% weight ratio in 50 mL of SWF (pH 7.2) at 37°C for 72 hours. (B) gradient SDS PAGE (12–20%) gel showing release profile of lysostaphin and LL-37 into SWF releasing medium after 24 hours. Lane 1: molecular weight marker, Lane 2: BSA (Sigma Aldrich, USA), Lane 3: lysostaphin (Sigma Aldrich, USA), Lane 4: Lysozyme (Sigma Aldrich, USA), Lane 5: LL-37 (Bio Basic Inc, Canada), Lane 6: free lysostaphin/LL-37, Lane 7: lysostaphin/LL-37 released after 24 hours. SWF releasing medium was concentrated 50 times prior to loading. |
This study supports conclusions made by other researchers that release profiles indicate a typical biphasic pattern in which sustained release can last for a long time with a low initial burst.7,42 As illustrated in Figure 7B, SDS-PAGE analysis of the amount of lysostaphin and LL-37 released after 24 hours is presented with a 50-fold concentration of SWF releasing medium. After 24 hours, the release profile of lysostaphin and LL-37 from niosomal formulations appeared to reach a plateau, probably due to Maximum Drug Depletion Quantity (MDDQ) phenomenon.43 This is because initial burst release (during the first 24 hours) from niosomes may be due to the protein and peptide available on the surface and inside of the niosomal structure. Subsequently, due to the intense hydrophobic interaction between the lipid nanostructure and drug, a part of the incorporated drugs remains inside the lipid core, thereby reducing release and reaching the plateau. Similar release patterns have also been observed in other nanostructures such as solid lipid nanoparticles.43,44
Release of molecules across a bilayer membrane depends on composition and fluidity of the bilayer membrane. Electrostatic interactions between drug and surfactants is important, especially when the drug exists in an ionized form at physiologic pH.26
To understand the drug release mechanism, a linear form of various kinetic models was plotted based on release data. Some of the most important release kinetic models are as follows: zero-order (cumulative percent drug release vs time), first-order (log cumulative percent drug remaining vs time), Higuchi (cumulative percent drug release vs square root of time), and Korsmeyer Peppas (log percent cumulative drug release vs log time). The r2 calculated for linear curve was obtained by regression analysis to determine the release kinetics of the optimum formulation (F2 for encapsulated lysostaphin and F5 for encapsulated LL-37). A kinetic model with a regression coefficient near to 1 is a desirable model for the release profile of that formulation. Because there were no statistically significant differences between r2 Higuchi and Korsmeyer Peppas models for lysostaphin encapsulated niosomes, it can be concluded that the drug release mechanism for all three dosage forms in SWF medium (lysostaphin, LL-37, and lysostaphin/LL-37 encapsulated niosomes) can be best fitted by the Higuchi model. Based on this model, drug release from the vesicles might be attributed to the diffusion mechanism (Table 3).45
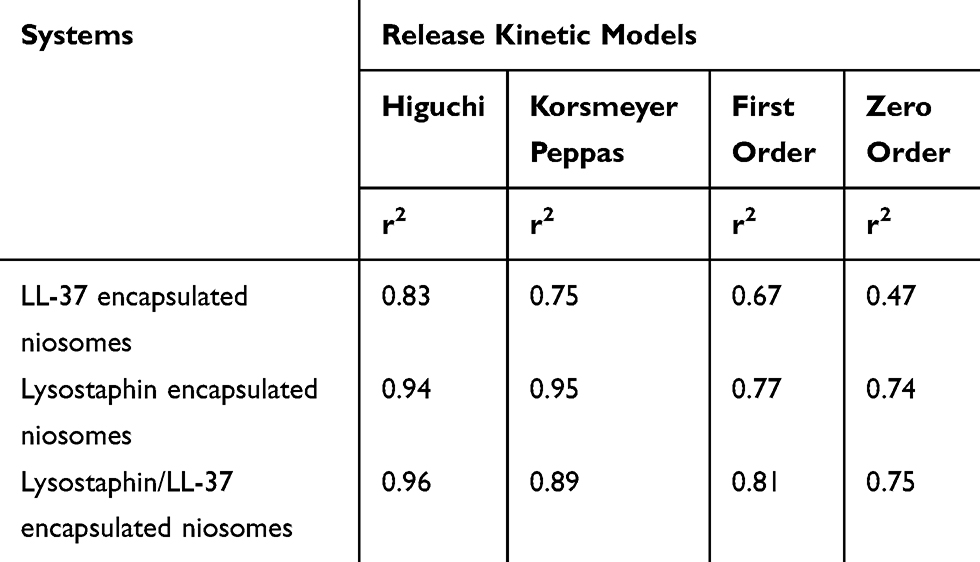 |
Table 3 Release Kinetic Models Obtained By Regression Analysis |
In Vitro Antibacterial Assay Of Lysostaphin/LL-37 Encapsulated Niosomes
Evaluation Of Minimum Inhibitory Concentrations
MICs of lysostaphin, LL-37, and lysostaphin/LL-37 in encapsulated and free forms are presented in Table 4. As depicted in Table 4, MIC of free lysostaphin obtained for S. aureus ATCC 6538 was much higher than previously reported MIC of lysostaphin against S. aureus ATCC 29,213, which was equal to 0.06 µg/mL.46 This may be due to the following reasons: (i) applying an ultrasound process for breaking the cell expressing lysostaphin, (ii) purity of lysostaphin, (iii) type of the bacterial strains used, and (iv) a Trx-His-S-enterokinase sequence in vector pET32a expressed along with lysostaphin and may have an effect on activity of lysostaphin. Likewise, MIC for LL-37 in encapsulated and free forms against S. aureus ATCC 6538 was much higher than MIC reported by Lukas Boge for lipid crystalline nanoparticles loaded with LL-37, which was >16 µg/mL.47 Mainly attributing to the use of ultrasound process during preparation of niosomal nanoparticles, whereas lipid crystalline nanoparticles are spontaneously formed by hydration of specific amphiphilic molecules. In addition, strains used in these two studies were different.
 |
Table 4 MIC Values Of Free And Encapsulated Lysostaphin And LL-37 Against S. Aureus |
As expected, blank niosomes had no detectable antibacterial activity. It is worth noting that most studies related to noisomal encapsulated antibacterial agents have emphasized on increased activity of encapsulated drugs.36 On the contrary, in our study, encapsulated forms had MIC higher than free ones, resulting from negative charge of anionic niosomes and negative charge of bacterial membranes, repulsion of niosomes may have occurred resulting in less antibacterial activity.48 Abbaszadegan et al49 synthesized silver nanoparticles with different surface charges and investigated their antibacterial activity against S. aureus. They showed that negatively charged nanoparticles had less antibacterial activity than positively or neutrally charged nanoparticles. It is noteworthy that reduction of antibacterial activity of encapsulated drugs can be attributed to partial denaturation of lysostaphin/LL-37 in ultrasound process during noisome preparation, leading to a decrease in the drug activity.50 However, as expected, lysostaphin/LL-37 encapsulated niosomes had lower MIC than lysostaphin encapsulated or LL-37 encapsulated ones due to synergistic antibacterial activity of lysostaphin and LL-37 confirmed by checkerboard assay.
Inhibitory Effect Of Free And Encapsulated Lysostaphin/LL-37
In this study, antibacterial activity of lysostaphin/LL-37 in encapsulated and free forms was assessed as a function of time against S. aureus (Figure 8). A slow and long-lasting inhibition was observed in niosomal formulations during 72 hours incubation whereas free drugs were used up in early hours, and then the S. aureus population began to increase. These different antibacterial activity patterns related to free and encapsulated drugs comply with previous reports. In these studies, growth inhibition as indicated by an increased lag phase, reduced growth rate, or reduced final OD of bacterial strains were observed and encapsulated drugs were gradually released over time, leading to a “lower and longer” antibacterial activity pattern.17,51,52 This effect may be due to the fact that the vesicles, apart from interacting with the outer membrane of the bacteria, can release a large amount of drug close to the bacterial surface, creating a gradient of drug concentration facilitating intracellular drug delivery.53 As shown in Figure 8, the inhibitory effect was statistically significant between free and encapsulated drugs in all six dosage forms at interval times of 24, 48, and 72 hours. From this point of view, encapsulation of drugs used an optimized niosomal formulation enhanced antistaphylococcal effect of drugs without absolute killing of the bacteria. Surprisingly, the inhibitory effect of encapsulated lysostaphin/LL-37 was higher than encapsulated lysostaphin or encapsulated LL-37 alone, suggesting that encapsulated lysostaphin/LL-37 as a combined formulation synergistically enhanced antistaphylococcal activity in an in vitro time-kill study. Likewise, Fumakia et al7 observed a similar inhibition using solid lipid nanoparticles co-encapsulated with LL-37 and A1 to control the development of S. aureus infection.
 |
Figure 8 Antibacterial activity of free and encapsulated drugs against S. aureus measured by optical density as a function of time. Concentration of either free lysostaphin or LL-37 was adjusted at 100 µg/mL. While concentrations of lysostaphin and LL-37 in encapsulated forms were equal to 105 and 115 µg/mL, respectively. As depicted in the figure, encapsulated lysostaphin/LL-37 showed a long-time and enhanced antistaphylococcal activity against S. aureus. |
In Vitro Cytotoxicity
Since AMPs are more suitable to be used as topical dosage forms, cytotoxicity assay was carried out on a mouse fibroblast cell line (L929 cell line, ATCC® CCL-1TM). The effect of the same concentration of the formulations on fibroblast cell viability was determined by MTT assay. As seen in Figure 9, the viability of free and encapsulated LL-37 was very low at their MIC concentrations. However, cytotoxicity of lysostaphin and lysostaphin/LL-37 in encapsulated and free forms was not observed at concentrations of MICs or higher than MICs of respective bacterium. Surprisingly, the cytotoxicity of niosomal forms was substantially higher than that of free forms (P<0.05). This might be due to differences in intracellular trafficking or different cellular uptakes of niosomal and free forms.36 It is noteworthy that the surfactants used in this study were generally regarded as safe (GRAS).54,55 Therefore, more cytotoxicity of niosomal forms cannot be attributed to the type of surfactants used in preparation of niosomes.
 |
Figure 9 In vitro cytotoxicity studies using mouse fibroblast L929 cells. (A) lysostaphin in encapsulated and free forms and blank niosomes, (B) LL-37 in encapsulated and free forms and blank niosomes, (C) lysostaphin/LL-37 in encapsulated and free forms and blank niosomes. Values represent mean±SD, n=3. *P<0.05, **P<0.01, ***P< 0.001. The values written in the x-axis of part (C) are related to lysostaphin/LL-37 concentrations, respectively. No cytotoxicity was observed for lysostaphin/LL-37 in encapsulated and free forms at concentrations of MIC or higher than MIC. |
Conclusion
We have successfully demonstrated, for the first time, the synergistic effect of lysostaphin and LL-37 against S. aureus. The results of the present study revealed that lysostaphin/LL-37 encapsulated niosomes could offer a prolonged antibacterial activity with a lower dose requirement. Our study confirmed the development of a delivery system containing different antimicrobial agents can be considered as a promising vehicle to combat severe infections in the human population.
Acknowledgments
This project was financially supported by the Pasteur Institute of Iran.
Disclosure
The authors declare that they have no conflicts of interest in this work.
References
1. López-Igual R, Bernal-Bayard J, Rodríguez-Patón A, Ghigo J-M, Mazel D. Engineered toxin–intein antimicrobials can selectively target and kill antibiotic-resistant bacteria in mixed populations. Nat Biotechnol. 2019;1:755–760.
2. Bumann D. Has nature already identified all useful antibacterial targets? Curr Opin Microbiol. 2008;11:387–392. doi:10.1016/j.mib.2008.08.002
3. Niemirowicz K, Piktel E, Wilczewska AZ, et al. Core–shell magnetic nanoparticles display synergistic antibacterial effects against Pseudomonas aeruginosa and Staphylococcus aureus when combined with cathelicidin LL-37 or selected ceragenins. Int J Nanomed. 2016;11:5443. doi:10.2147/IJN.S113706
4. Browder HP, Zygmunt WA, Young J, Tavormina P. Lysostaphin: enzymatic mode of action. Biochem Biophys Res Commun. 1965;19:383–389. doi:10.1016/0006-291X(65)90473-0
5. Desbois AP, Gemmell CG, Coote PJ. In vivo efficacy of the antimicrobial peptide ranalexin in combination with the endopeptidase lysostaphin against wound and systemic meticillin-resistant Staphylococcus aureus (MRSA) infections. Int J Antimicrob Agents. 2010;35:559–565. doi:10.1016/j.ijantimicag.2010.01.016
6. Polak J, Della Latta P, Blackburn P. In vitro activity of recombinant lysostaphin-antibiotic combinations toward methicillin-resistant Staphylococcus aureus. Diagn Micr Infec Dis. 1993;17:265–270. doi:10.1016/0732-8893(93)90034-5
7. Fumakia M, Ho EA. Nanoparticles encapsulated with LL37 and serpin A1 promotes wound healing and synergistically enhances antibacterial activity. Mol Pharm. 2016;13:2318–2331. doi:10.1021/acs.molpharmaceut.6b00099
8. Duplantier AJ, van Hoek ML. The human cathelicidin antimicrobial peptide LL-37 as a potential treatment for polymicrobial infected wounds. Front Immunol. 2013;4:143. doi:10.3389/fimmu.2013.00143
9. Oren Z, Lerman JC, Gudmundsson GH, Agerberth B, Shai Y. Structure and organization of the human antimicrobial peptide LL-37 in phospholipid membranes: relevance to the molecular basis for its non-cell-selective activity. Biochem J. 1999;341:501. doi:10.1042/bj3410501
10. Geitani R, Moubareck CA, Touqui L, Sarkis DK. Cationic antimicrobial peptides: alternatives and/or adjuvants to antibiotics active against methicillin-resistant Staphylococcus aureus and multidrug-resistant Pseudomonas aeruginosa. BMC Microbiol. 2019;19:54. doi:10.1186/s12866-019-1416-8
11. Alaiwa MHA, Reznikov LR, Gansemer ND, et al. pH modulates the activity and synergism of the airway surface liquid antimicrobials β-defensin-3 and LL-37. PNAS. 2014;111:18703–18708. doi:10.1073/pnas.1422091112
12. Laridi R, Kheadr E, Benech R-O, Vuillemard J, Lacroix C, Fliss I. Liposome encapsulated nisin Z: optimization, stability and release during milk fermentation. Int Dairy J. 2003;13:325–336. doi:10.1016/S0958-6946(02)00194-2
13. Colas J-C, Shi W, Rao VM, Omri A, Mozafari MR, Singh H. Microscopical investigations of nisin-loaded nanoliposomes prepared by Mozafari method and their bacterial targeting. Micron. 2007;38:841–847.
14. Sanderson NM, Jones MN. Targeting of cationic liposomes to skin‐associated bacteria. Pest Sci. 1996;46:255–261. doi:10.1002/(SICI)1096-9063(199603)46:3<255::AID-PS345>3.0.CO;2-Y
15. Jones MN. Use of liposomes to deliver bactericides to bacterial biofilms. Methods Enzymol. 2005;391:211–228.
16. Cortesi R, Esposito E, Corradini F, et al. Non-phospholipid vesicles as carriers for peptides and proteins: production, characterization and stability studies. Int J Pharm. 2007;339:52–60. doi:10.1016/j.ijpharm.2007.02.024
17. Kopermsub P, Mayen V, Warin C. Nanoencapsulation of nisin and ethylenediaminetetraacetic acid in niosomes and their antibacterial activity. AJSR. 2012;4:457.
18. Rochdy Haj-Ahmad R, Ali Elkordy A, Shu Chaw C. In vitro characterisation of Span™ 65 niosomal formulations containing proteins. Curr Drug Deliv. 2015;12:628–639. doi:10.2174/1567201812666150511095432
19. Moghassemi S, Hadjizadeh A, Omidfar K. Formulation and characterization of bovine serum albumin-loaded niosome. AAPS PharmSciTech. 2017;18:27–33. doi:10.1208/s12249-016-0487-1
20. Manosroi A, Khanrin P, Werner RG, Götz F, Manosroi W, Manosroi J. Entrapment enhancement of peptide drugs in niosomes. J Microencapsul. 2010;27:272–280. doi:10.3109/02652040903131293
21. Manosroi A, Lohcharoenkal W, Götz F, Werner RG, Manosroi W, Manosroi J. Cellular uptake enhancement of Tat-GFP fusion protein loaded in elastic niosomes. J Biomed Nanotechnol. 2011;7:366–376. doi:10.1166/jbn.2011.1300
22. Farhangnia L, Ghaznavi-Rad E, Mollaee N, Abtahi H. Cloning, expression, and purification of recombinant Lysostaphin from Staphylococcus simulans. Jundishapur J Microbiol. 2014;7:e10009.
23. Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi:10.1016/0003-2697(76)90527-3
24. Towbin H, Staehelin T, Gordon J. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. PNAS. 1979;76:4350–4354. doi:10.1073/pnas.76.9.4350
25. van Vuuren SF, Nkwanyana MN, de Wet H. Antimicrobial evaluation of plants used for the treatment of diarrhoea in a rural community in northern Maputaland, KwaZulu-Natal, South Africa. BMC Complem Altern M. 2015;15:53. doi:10.1186/s12906-015-0570-2
26. Tavano L, Muzzalupo R, Picci N, de Cindio B. Co-encapsulation of antioxidants into niosomal carriers: gastrointestinal release studies for nutraceutical applications. Colloids Surf B Biointerfaces. 2014;114:82–88. doi:10.1016/j.colsurfb.2013.09.058
27. Lutz JB, Zehrer CL, Solfest SE, Walters S-A. A new in vivo test method to compare wound dressing fluid handling characteristics and wear time. Ostomy Wound Manag. 2011;57:28.
28. Hua S. Comparison of in vitro dialysis release methods of loperamide-encapsulated liposomal gel for topical drug delivery. Int J Nanomedicine. 2014;9:735. doi:10.2147/IJN
29. LiPuma JJ, Rathinavelu S, Foster BK, et al. In vitro activities of a novel nanoemulsion against Burkholderia and other multidrug-resistant cystic fibrosis-associated bacterial species. Antimicrob Agents Chemother. 2009;53:249–255. doi:10.1128/AAC.00691-08
30. Zhao X, Drlica K. Restricting the selection of antibiotic-resistant mutants: a general strategy derived from fluoroquinolone studies. Clin Infect Dis. 2001;33:S147–S156. doi:10.1086/cid.2001.33.issue-s3
31. Desbois AP, Coote PJ. Bactericidal synergy of lysostaphin in combination with antimicrobial peptides. Eur J Clin Microbiol Inf Dis. 2011;30:1015–1021. doi:10.1007/s10096-011-1188-z
32. Kang J, Dietz MJ, Li B. Antimicrobial peptide LL-37 is bactericidal against Staphylococcus aureus biofilms. PLoS One. 2019;14:e0216676. doi:10.1371/journal.pone.0216676
33. Liu T, Guo R. Investigation of PEG 6000/Tween 80/Span 80/H 2 O niosome microstructure. Colloid Polym Sci. 2007;285:711–713. doi:10.1007/s00396-006-1627-z
34. Junyaprasert VB, Singhsa P, Suksiriworapong J, Chantasart D. Physicochemical properties and skin permeation of Span 60/Tween 60 niosomes of ellagic acid. Int J Pharm. 2012;423:303–311. doi:10.1016/j.ijpharm.2011.11.032
35. Varshosaz J, Pardakhty A, Hajhashemi V-I, Najafabadi AR. Development and physical characterization of sorbitan monoester niosomes for insulin oral delivery. Drug Deliv. 2003;10:251–262. doi:10.1080/drd_10_4_251
36. Moazeni E, Gilani K, Sotoudegan F, et al. Formulation and in vitro evaluation of ciprofloxacin containing niosomes for pulmonary delivery. J Microencapsul. 2010;27:618–627. doi:10.3109/02652048.2010.506579
37. Khaksa G, D’Souza R, Lewis S, Udupa N. Pharmacokinetic study of niosome encapsulated insulin. Indian J Exp Biol. 2000;38(9):901–905.
38. Gurrapu A, Jukanti R, Bobbala SR, Kanuganti S, Jeevana JB. Improved oral delivery of valsartan from maltodextrin based proniosome powders. Adv Powder Technol. 2012;23:583–590. doi:10.1016/j.apt.2011.06.005
39. Agarwal R, Katare O, Vyas S. Preparation and in vitro evaluation of liposomal/niosomal delivery systems for antipsoriatic drug dithranol. Int J Pharm. 2001;228:43–52. doi:10.1016/S0378-5173(01)00810-9
40. Pardakhty A, Moazeni E, Varshosaz J, Hajhashemi V, Najafabadi AR. Pharmacokinetic study of niosome-loaded insulin in diabetic rats. DARU. 2011;19:404.
41. Hwang SH, Maitani Y, Takayama K, NAGAI T. High entrapment of insulin and bovine serum albumin into neutral and positively-charged liposomes by the remote loading method. Chem Pharm Bull (Tokyo). 2000;48:325–329. doi:10.1248/cpb.48.325
42. Xie J, Ng WJ, Lee LY, Wang CH. Encapsulation of protein drugs in biodegradable microparticles by co-axial electrospray. J Colloid Interface Sci. 2008;317:469–476. doi:10.1016/j.jcis.2007.09.082
43. Müller RH, MaÈder K, Gohla S. Solid lipid nanoparticles (SLN) for controlled drug delivery–a review of the state of the art. Eur J Pharm Biopharm. 2000;50:161–177. doi:10.1016/S0939-6411(00)00087-4
44. Ebrahimi HA, Javadzadeh Y, Hamidi M, Jalali MB. Repaglinide-loaded solid lipid nanoparticles: effect of using different surfactants/stabilizers on physicochemical properties of nanoparticles. DARU. 2015;23:46. doi:10.1186/s40199-015-0128-3
45. Dash S, Murthy PN, Nath L, Chowdhury P. Kinetic modeling on drug release from controlled drug delivery systems. Acta Pol Pharm. 2010;67:217–223.
46. Yang X-Y, Li C-R, Lou R-H, et al. In vitro activity of recombinant lysostaphin against Staphylococcus aureus isolates from hospitals in Beijing, China. J Med Microbiol. 2007;56:71–76. doi:10.1099/jmm.0.46788-0
47. Boge L. Lipid-Based Liquid Crystals as Drug Delivery Vehicles for Antimicrobial Peptides. Department of Chemistry and Chemical Engineering, Chalmers University of Technology; 2018.
48. Lewies A, Wentzel JF, Jordaan A, Bezuidenhout C, Du Plessis LH. Interactions of the antimicrobial peptide nisin Z with conventional antibiotics and the use of nanostructured lipid carriers to enhance antimicrobial activity. Int J Pharm. 2017;526:244–253. doi:10.1016/j.ijpharm.2017.04.071
49. Abbaszadegan A, Ghahramani Y, Gholami A, et al. The effect of charge at the surface of silver nanoparticles on antimicrobial activity against gram-positive and gram-negative bacteria: a preliminary study. J Nanomater. 2015;16:53.
50. Were LM, Bruce BD, Davidson PM, Weiss J. Size, stability, and entrapment efficiency of phospholipid nanocapsules containing polypeptide antimicrobials. J Agric Food Chem. 2003;51:8073–8079. doi:10.1021/jf0348368
51. Kopermsub P, Mayen V, Warin C. Potential use of niosomes for encapsulation of nisin and EDTA and their antibacterial activity enhancement. Food Res Int. 2011;44:605–612. doi:10.1016/j.foodres.2010.12.011
52. Were LM, Bruce B, Davidson PM, Weiss J. Encapsulation of nisin and lysozyme in liposomes enhances efficacy against Listeria monocytogenes. J Food Prot. 2004;67:922–927. doi:10.4315/0362-028X-67.5.922
53. Amusa AS, Satish J, Gopalakrishna P. In vitro activities of fluoroquinolones entrapped in non-ionic surfactant vesicles against ciprofloxacin-resistant bacteria strains. IJPSDR. 2012;1:5.
54. Newman SP. Principles of metered-dose inhaler design. Respir Care. 2005;50:1177–1190.
55. Niven R. Feasibility studies with recombinant human granulocyte colony-stimulating factor. Lung Biol Health Dis. 1997;107:413–452.
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.