Back to Archived Journals » Reports in Theoretical Chemistry » Volume 3
Switching of carbene spin states: effect of hydrogen bond donors
Authors Guha AK, Boruah A, Hazarika M, Kaman S
Received 23 February 2015
Accepted for publication 28 April 2015
Published 20 July 2015 Volume 2015:3 Pages 1—6
DOI https://doi.org/10.2147/RTC.S83255
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Jorge Llano
Ankur Kanti Guha,1 Abhijit Boruah,2 Munmi Hazarika,2 Sumi Kaman2
1Department of Chemistry, Cotton College State University, Guwahati, Assam, India; 2Department of Chemistry, Dibrugarh University, Dibrugarh, Assam, India
Abstract: Quantum chemical calculations predict that the spin states of simple carbenes can be switched over from triplet to singlet via hydrogen bonding, and thus the spin specificity of their reactions can be tuned. The stability of the singlet state of simple carbenes like: CH2 increases due to hydrogen bond formation with a single molecule of water or methanol, although the triplet state is found to be the ground state. However, the most dramatic effect of spin switch is found for diphenylcarbene (Ph2C), which becomes a ground state singlet due to formation of a hydrogen bond with the hydrogen atom of water or methanol. The present calculations reveal that the effect of hydrogen bonds on switching the spin state of carbenes is only applicable to Ph2C, as it has a very small singlet–triplet gap in its free form, ie, when it is not hydrogen bonded with water or methanol. Further, the presence of such hydrogen-bonding interaction has been verified within the realm of atoms-in-molecules analysis of the electron density.
Keywords: carbenes, spin states, quantum chemical calculations, hydrogen bonding, atoms in molecules
Introduction
Carbenes (R2C:) are divalent carbon species having two nonbonding electrons. These nonbonding electrons may have antiparallel spins (singlet state) or parallel spins (triplet state).1–6 The parent carbene CH2 has a triplet ground state with an experimental singlet–triplet (S–T) energy gap of 9.05±0.06 kcal/mol.7–9 The small difference between the S0 and T1 may be easily overcome by tuning the substituents attached to the carbene center.10 Various factors dictate the ground state multiplicity or the S–T separation of carbenes.11 Apart from the effect of substituents, solvents are found to stabilize the singlet state more than the triplet state.12–16
Products of carbene reactions are solely determined by the carbene’s spin state. For example, the singlet carbene inserts in to the O-H bonds of alcohol,17 whereas the triplet carbene undergoes insertion into C-H bonds18 (Figure 1).19 This reactivity difference of carbenes with alcohol has been extensively studied, and spin selectivity of carbene chemistry has been developed.19–22
 | Figure 1 Proposed reactions of singlet and triplet carbenes with an alcohol. |
Recently, Costa and Sander19 investigated the reaction of diphenylcarbene (DPC), Ph2C, an archetypical ground state triplet carbene, with methanol and isolated a singlet metastable complex (the complex was found to be electron spin resonance silent) in low-temperature matrices. The formation of the metastable methanol complex has been monitored and characterized by infrared spectroscopy. They also characterized the O−H insertion product, a typical product expected for singlet state carbene. Based on a different spectroscopic technique and theoretical calculations, they predicted that the singlet state of DPC can become more stable by hydrogen bonding interactions with solvents. Their results showed an interesting chemistry of switching the ground state multiplicity of carbenes by solvent interactions, and there by providing a hint toward a unique way to control the carbene’s chemical reactivity. In another report, Costa et al20 observed that the reaction of DPC with a single molecule of water switches its spin state. This prompted us to investigate the effect of hydrogen bond donors such as H2O and CH3OH on the spin states of simple alkyl and aryl carbenes, R2C: (R=H, Me, Et, Ph). Here, we present quantum chemical calculations,23–37 which reveal that the spin states of alkyl or aryl carbenes can be tuned by interaction with a hydrogen bond donor solvent or reactant. The goal of controlling the spin state of a carbene is to change the reaction path so as to achieve the good desired reaction selectivity. In order to react with an alcohol, the hydrogen bond between carbene and the solvent molecules should be broken first. Then, the carbene carbon atom can get close to and react with the alcohol. In this case, the hydrogen-bond-induced spin state change may have no effect on the real reactions. However, this hydrogen bonded interaction may prevent the dimerization of triplet carbenes by providing stability to the singlet state.
Computational details
Geometry optimization of all the molecules without any symmetry constraint were performed at B3LYP,23,24 BNL,25–27 B3LYP-D,28 LC-BLYP,29 and M06-2X30 functional using the 6-311++G(d, p) basis set frequency calculations have been performed at these levels of theory to characterize the nature of stationary points. Single point energy calculations at CCSD(T)/6-311++G(d, p)31 level of theory has been performed using the ORCA32 suite of program. However, the performance of B3LYP functional is found to better (Figure S1, supporting information) and hence, the discussion in the text is based on B3LYP level of theory. We have also calculated the S–T gap of the parent carbenes at B3LYP/Aug-cc-pVDZ. All geometry optimizations and frequency calculations have been performed using NWChem 6.1 program in gas phase.33 Quantum theory of atoms in molecules (QTAIM)34 and electron localization function (ELF)35,36 have been performed using Multiwfn 3.1 suite of program.37
Results and discussions
Table 1 lists the S–T energy separations, ΔEST, calculated at B3LYP level using the 6-311++G(d, p) and Aug-cc-pVDZ basis sets in the gas phase for four carbenes. The B3LYP/6-311++G(d, p) method gave the least absolute error in the S–T energy gaps when compared with the values computed at CCSD(T)/6-311++G(d, p) level of theory (Figure S1). The calculated values of ΔEST at B3LYP/6-311++G(d, p) level are very close to those obtained in the previous studies38,39 and are very close to the values calculated at CCSD(T)/6-311++G(d, p) level of theory. Similar results are obtained using the larger basis set (Aug-cc-pVDZ). However, the S–T gaps are slightly smaller with the Aug-cc-pVDZ basis set than with the 6-311+G(d, p) basis set. Thus, the discussion in this text is based on the B3LYP/6-311(d, p) results. The simplest carbene, methylene, has a triplet ground state with ΔEST of 11.73 kcal/mol calculated at B3LYP/6-311++G(d, p) level of theory. The experimental value of S–T separation of CH2 is 9.05 kcal/mol,7 which is 2.68 kcal/mol less than our B3LYP/6-311++G(d, p) calculated value. Increase in the basis set size to Aug-cc-pVDZ leads to a ΔEST value of 11.13 kcal/mol, which is 2.08 kcal/mol less than the reference value.
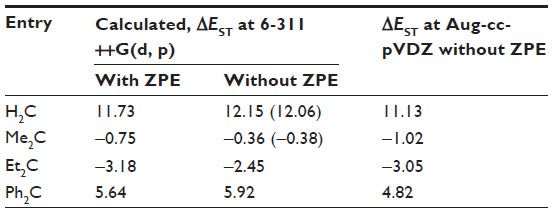 | Table 1 B3LYP calculated gas phase singlet–triplet energy separations (kcal/mol) for carbenes |
The S–T gap of dimethylcarbene, Me2C, is found to be −0.75 kcal/mol at B3LYP level of theory. This implies that the ground state of Me2C is singlet. The stability of the singlet state of Me2C can be explained by hyperconjugation. Since, singlet carbenes are isoelectronic with carbocations, hyperconjugation stabilizes the singlet state more than the triplet radical species.11 The effect of hyperconjugation is more pronounced in case of Et2C. Similarly, the conjugation of aryl groups, as in Ph2C, reduces the S–T gap of aryl carbenes.11 However, the effect of conjugation is somewhat less compared to the stabilization provided by the hyperconjugation of methyl and ethyl groups in Me2C and Et2C, respectively.
Previous studies reveal that polar solvents reduce the S–T gap of carbenes by increasing the dipole moment of the singlet state.11–16 The reduction of the S–T gap for carbenes in polar solvents has been computationally verified.12 The plot of experimentally determined S–T gaps as a function of solvent polarity was found to be linear,11 which led to the conclusion that specific carbene–solvent interaction does not play a significant role.11 However, the recent report by Costa and Sander19 and Costa et al20 clearly establishes that the spin states of carbenes can be tuned by interaction with hydrogen bond donor solvents and that the carbene–solvent interaction has a vital role to play in the ground state multiplicity of carbenes. Although the effect of solvent polarity on the S–T gap of carbenes has been theoretically explored, the explicit interaction of solvent molecules with carbenes needs to be understood.
Table 2 collects the S–T gap of carbene–solvent complexes. Interaction of H2O with CH2 reduces the S–T gap by 3.81 kcal/mol (triplet state is more stable). The most dramatic effect of solvent interaction is found in the case of Me2C…solvent complexes (the … signifies a weak interaction between atoms). The S–T gap is dramatically reduced, thus making the singlet state more stable. The effect of solvent interaction in reducing the S–T gap is also observed in the case of Ph2C. The singlet state of Ph2C…solvent complexes are found to be slightly more stable. This is in agreement with recent observation of Costa and Sander19 and Costa et al.20
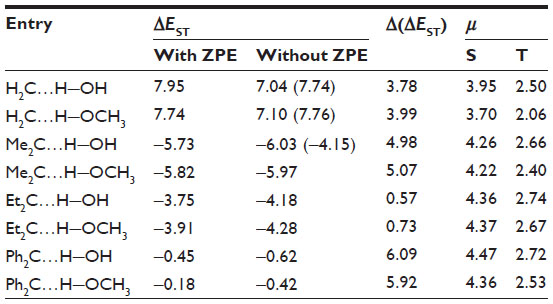 | Table 2 Gas-phase B3LYP/6-311++G(d, p) calculated singlet–triplet energy separations (in kcal/mol) and dipole moments (in debye) for carbene–solvent complexesa,b |
The interaction of hydrogen bond donors (H2O and CH3OH) increases the dipole moment of the singlet more than that of the triplet. This increase in dipole moment indicates a higher stability of the more polar singlet state over the triplet state. This is in agreement with previous theoretical studies on the preferential stabilization of the singlet state in a polar solvent due to an increase in dipole moment.11–16
Figure 2 shows the optimized geometries of the carbene…solvent complexes at the B3LYP/6-311++G(d, p) level of theory. The C…HO distances are found to be close to 2 Å, which are ~30% shorter than the sum of the van der Waals radii of carbon and hydrogen (2.85 Å).40
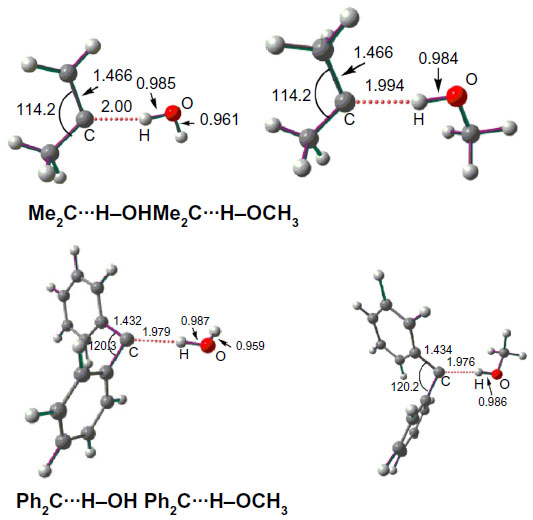 | Figure 2 B3LYP/6-311++G(d, p) optimized singlet state geometries of carbene–solvent complexes. |
The spin cross-over of Ph2C complexed with CH3OH were explored by a relaxed potential energy scan, keeping the C…HO distance fixed and optimizing all other coordinates at B3LYP/6-311++G(d, p) level. Figure 3 is a plot of the energies (kcal/mol) of the respective singlet and triplet states of the Ph2C…H-OCH3 complex. It is evident from Figure 3 that the spin cross-over from triplet to singlet takes place at C…HO distance of ~2.30 Å, which is longer than the C…HO equilibrium distance of 1.971 Å.
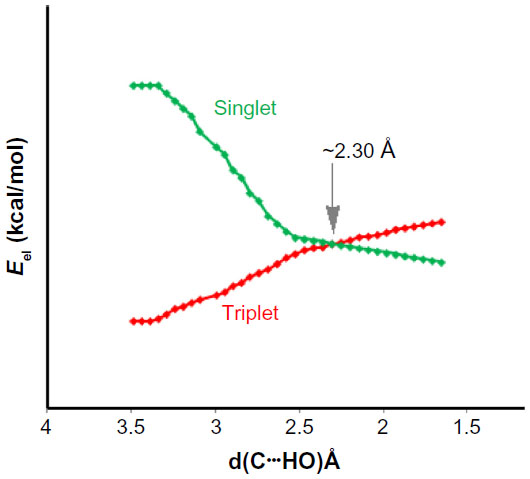 | Figure 3 Spin cross-over for Ph2C…H−OCH3 complex calculated at the B3LYP/6-311++G(d, p) level without zero-point energy correction. |
The formation of hydrogen bonds has been confirmed by applying the QTAIM34 and the ELF35,36 analysis (Figure 4 and Table 3). These topological analyses reveal a bond path and a (3, −1) bond critical point between the carbenic carbon and the hydrogen atom of the H−O bond of H2O or CH3OH (Figure 4). The Laplacian of the electron density, ∇2ρ, is positive and the local electronic energy density, H(r), is negative, which is a characteristic feature of a polar covalent bond.34 The ELF35,36 values at the bond critical points of these complexes are also significant. All these topological features confirm the presence of such interaction.
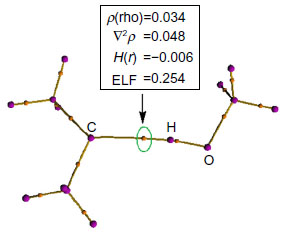 | Figure 4 Molecular graph of the Me2C…H−OCH3 complex showing the topological parameters at the C…HO bond critical point. All values are in atomic units. |
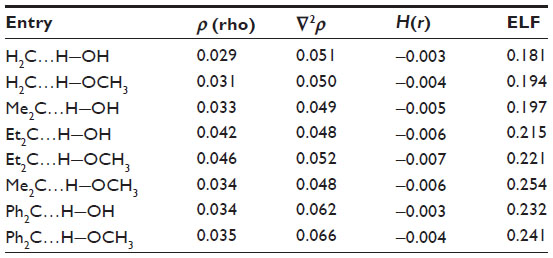 | Table 3 Topological analysis of the C…H−O bond for the complexes |
Conclusion
In conclusion, quantum chemical calculations showed that the interaction of hydrogen bond donor molecules can modify the S–T gap of alkyl or aryl carbenes. This interaction may lead to a higher stability of the singlet state over the triplet state by increasing the dipole moment of the former. This increase in dipole moment results in more charge reorganization in the singlet state, rendering them more stable. A simple carbene like CH2 shows a decrease in S–T gap of ~4.0 kcal/mol due to formation of hydrogen bonds with water or methanol, although the triplet state is still the ground state. The most dramatic effect of hydrogen bond formation on the spin state of carbenes is observed for DPC, for which the singlet state becomes the ground state. Thus, it appears that hydrogen bond-induced spins witch over is applicable to DPCs among the four carbenes considered in this study. This might be due to the fact that the S–T gap of the parent Ph2C is very small (~5 kcal/mol) and may get reduced upon hydrogen bond formation to such an extent that the hydrogen bonded singlet Ph2C becomes the ground state. This observation is in line with the recent experimental finding of Costa and Sander19 and Costa et al.20 The existence of this hydrogen-bonded interactions has been verified by our topological studies of the electron density. The outcome of this study is encouraging in the sense that a single hydrogen bond donor molecule may switch the ground state spin multiplicity of those carbenes with a smaller S–T gap, and thus may prevent their dimerization in solutions of polar solvents.
Disclosure
The authors report no conflicts of interest in this work.
References
Kirmse W. Carbene Chemistry. New York, NY: Academic Press; 1971. | |
Jones M, Moss RA, editors. Carbenes. Vols 1 and 2. New York, NY: Wiley Interscience; 1973 and 1975. | |
Brinker UH, editor. Advanced in Carbene Chemistry. Vol 1. Greenwich, CT: JAI Press; 1994. | |
Brinker UH, editor. Advanced in Carbene Chemistry. Vol 2. Stamford, CT: JAI Press; 1998. | |
Brinker UH, editor. Advanced in Carbene Chemistry. Vol 3. Amsterdam: Elsevier; 2001. | |
Moss RA, Platz MS, Jones M Jr, editors. Reactive Intermediate Chemistry. Hoboken, NJ: Wiley-Interscience; 2004. | |
McKellar ARW, Bunker PR, Sears TJ, Evenson KM, Saykally RJ, Langhoff SR. Far infrared laser magnetic resonance of singlet methylene: Singlet–triplet perturbations, singlet–triplet transitions, and the singlet–triplet splitting. J Chem Phys. 1983;79(11):5251–5264. | |
Balasubramanian K, McLean AD. The singlet–triplet energy separation in silylene. J Chem Phys. 1986;85(9):5117–5119. | |
Allen WD, Schaefer HF. geometrical structures, force constants, and vibrational spectra of SiH, SiH2, SiH3, and SiH4. Chem Phys. 1986; 108(2):243–274. | |
Baird NC, Taylor KF. Multiplicity of the ground state and magnitude of the T1-S0 gap in substituted carbenes. J Am Chem Soc. 1978;100(5):1333–1338. | |
Hirai K, Itoh T, Tomioka H. Persistent triplet carbenes. Chem Rev. 2009;109(8):3275–3332 and references therein. | |
Wang Y, Hadad CM, Toscano JP. Solvent dependence of the 2-naphthyl(carbomethoxy)carbene singlet−triplet energy gap. J Am Chem Soc. 2002;124(8):1761–1767. | |
Sitzman EV, Langan JG, Eisenthal KB. Intermolecular effects on intersystem crossing studied on the picosecond timescale: the solvent polarity effect on the rate of singlet-to-triplet intersystem crossing of diphenylcarbene. J Am Chem Soc. 1984;106(6):1868–1869. | |
Langan JG, Sitzmann EV, Eisenthal KB. Picosecond laser studies on the effect of structure and environment on intersystem crossing in aromatic carbenes. Chem Phys Lett. 1984;110(5):521–527. | |
Sitzmann V, Langan JG, Griller D, Eisenthal KB. Effects of solvent polarity and structure on intersystem crossing in diphenylcarbenes. A picosecond laser study on dimesitylcarbene. Chem Phys Lett. 1989; 161(4–5):353–360. | |
Wang J, Kubicki J, Gustafson TL, Platz MS. The dynamics of carbene solvation: an ultrafast study of p-biphenylyltrifluoromethylcarbene. J Am Chem Soc. 2008;130(7):2304–2313. | |
Hadel LM, Platz MS, Scaiano JC. Study of hydrogen atom abstraction reactions of triplet diphenylcarbene in solution. J Am Chem Soc. 1984; 106(2):283–287. | |
Kirmse W, Kilian J, Steenken S. Carbenes and the oxygen-hydrogen bond: spectroscopic evidence for protonation of diarylcarbenes to give diarylcarbenium ions. J Am Chem Soc. 1990;112(17):6399–6400. | |
Costa P, Sander W. Hydrogen bonding switches the spin state of diphenylcarbene from triplet to singlet. Angew Chem Int Ed. 2014;53(20):5122–5125. | |
Costa P, Olivia MF, Garcia ES, Sander W. The highly reactive benzhydryl cation isolated and stabilized in water ice. J Am Chem Soc. 2014;136(44):15625–15630. | |
Kirmse W. In: Brinker UH, editor. Advances in Carbene Chemistry.Vol 1. Greenwich, CT: JAI Press; 1994:1–57. | |
Kirmse W. In: Brinker UH, editor. Advances in Carbene Chemistry. Vol 3. Amsterdam: Elsevier Science BV; 2001:1–51. | |
Becke AD. Density-functional thermochemistry. III. The role of exact exchange. J Chem Phys. 1993;98(7):5648–5652. | |
Lee C, Yang W, Parr RG. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys Rev B Condens Matter. 1988;37(2):785–789. | |
Baer R, Neuhauser D. Density functional theory with correct long-range asymptotic behavior. Phys Rev Lett. 2005;94(4):043002. | |
Livshits E, Baer R. A well-tempered density functional theory of electrons in molecules. Phys Chem Chem Phys. 2007;9(23):2932–2941. | |
Livshits E, Baer R. A density functional theory for symmetric radical cations from bonding to dissociation. J Phys Chem A. 2008;112(50):12789–12791. | |
Grimme S, Antony J, Ehrlich S, Krieg H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J Chem Phys. 2010;132(15):154104–154119. | |
Iikura H, Tsuneda T, Yanai T, Hirao K. A long-range correction scheme for generalized-gradient-approximation exchange functionals. J Chem Phys. 2001;115(8):3540–3544. | |
Zhao Y, Truhlar DG. A new local density functional for main-group thermochemistry, transition metal bonding, thermochemical kinetics, and noncovalent interactions. J Chem Phys. 2006;125(19):194101–194118. | |
Pople JA, Head-Gordon M, Raghavachari K. Quadratic configuration interaction. A general technique for determining electron correlation energies. J Chem Phys. 1987;87(10):5968–5975. | |
Neese F. ORCA – An Ab Initio, Density Functional and Semiempirical Program Package (version 3.0.2). University of Bonn, 2008. | |
Valiev M, Bylaska EJ, Govind N, et al. NWChem: version 6.1: a comprehensive and scalable open-source solution for large scale molecular simulations. Comput Phys Commun. 2010;181(9):1477–1489. | |
Bader RFW. Atoms in Molecules: A Quantum Theory. Oxford, UK: Oxford University Press; 1990. | |
Silvi B, Savin A. Classification of chemical bonds based on topological analysis of electron localization functions. Nature. 1994;371(20):683–686. | |
Becke AD, Edgecombe KE. A simple measure of electron localization in atomic and molecular systems. J Chem Phys. 1990;92(9):5397–5403. | |
Tian L. Multiwfn: A Multifunctional Wavefunction Analyzer (version 3.1). Available from: http://Multiwfn.codeplex.com. Accessed May 22, 2015. | |
Sulzbach HM, Bolton E, Lenoir D, Schleyer PvR, Schaefer HF III. Tetra-tert-butylethylene: an elusive molecule with a highly twisted double bond. can it be made by carbene dimerization? J Am Chem Soc. 1996;118(41):9908–9914. | |
Richards CA Jr, Kim S-J, Yamaguchi Y, Schaefer HF III. Dimethylcarbene: a singlet ground state? J Am Chem Soc. 1995; 117(40):10104–10107. | |
Huheey JE, Keiter EA, Keiter RL. Inorganic Chemistry: Principles of Structure and Reactivity, 4th ed. Singapore: Pearson Education Pte Ltd; 2004. |
Supplementary material
Cartesian coordinates of all the molecules calculated at B3LYP/6-311++G(d, p) level.
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.