Back to Journals » International Journal of Nanomedicine » Volume 12
Suppression of human arthritis synovial fibroblasts inflammation using dexamethasone-carbon nanotubes via increasing caveolin-dependent endocytosis and recovering mitochondrial membrane potential
Authors Lee YK, Kim SW, Park JY, Kang WC, Kang YJ, Khang D ![]()
Received 18 May 2017
Accepted for publication 6 July 2017
Published 10 August 2017 Volume 2017:12 Pages 5761—5779
DOI https://doi.org/10.2147/IJN.S142122
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Thomas Webster
Yeon Kyung Lee,1,* Sang-Woo Kim,1,* Jun-Young Park,1 Woong Chol Kang,2 Youn Joo Kang,3 Dongwoo Khang1,4
1Lee Gil Ya Cancer and Diabetes Institute, Gachon University, 2Department of Cardiology, Gil Medical Center, Gachon University, Incheon, 3Department of Rehabilitation Medicine, Eulji Hospital, Eulji University School of Medicine, Seoul, 4Department of Physiology, College of Medicine, Gachon University, Incheon, South Korea
*These authors contributed equally to this work
Abstract: Dexamethasone (DEX), a non-particulate glucocorticoid (GC) to inhibit anti-inflammatory response, has been widely used for the treatment of various diseases such as arthritis, cancer, asthma, chronic obstructive pulmonary disease, cerebral edema, and multiple sclerosis. However, prolonged and/or high-dose GC therapy can cause various serious adverse effects (adrenal insufficiency, hyperglycemia, Cushing’s syndrome, osteoporosis, Charcot arthropathy, etc). In this study, developed DEX-carbon nanotube (CNT) conjugates improved intracellular drug delivery via increased caveolin-dependent endocytosis and ultimately suppressed the expression of major pro-inflammatory cytokines in tumor necrosis factor-α (TNF-α)-stimulated human fibroblast-like synoviocytes (FLS) at low drug concentrations. Specifically, DEX on polyethylene-glycol (PEG)-coated CNTs induced caveolin uptake, recovered mitochondrial disruption, and inhibited reactive oxygen species production by targeting mitochondria that was released from the early endosome in TNF-α-stimulated FLS. The obtained results clearly demonstrated that DEX-PEG-coated CNTs significantly inhibited the inflammation by FLS in rheumatoid arthritis (RA) by achieving greater drug uptake and efficient intracellular drug release from the endosome, thus suggesting a mechanism of effective low-dose GC therapy to treat inflammatory diseases, including RA and osteoarthritis.
Keywords: carbon nanotubes, polyethylene-glycol, dexamethasone, arthritis, fibroblast-like synoviocytes, caveolin-dependent endocytosis
Introduction
Chronic inflammation and joint destruction are commonly found in rheumatoid arthritis (RA) as well as in other forms of arthritis.1–3 In RA, there is an obvious correlation between the regulation of multiple potent mediators, including pro-inflammatory cytokines (tumor necrosis factor-α [TNF-α], interleukin-1β [IL-1β], and interleukin-6 [IL-6]), chemokines, and matrix metalloproteinases (MMPs).4 Fibroblast-like synoviocytes (FLS) are key contributors in the pathophysiological process of arthritis and are known as a major source of inflammatory cytokines.5–7 As a critical regulator of FLS, TNF-α, in particular, activates a broad array of pro-inflammatory intracellular signal mechanisms.7,8
Increasing evidence demonstrates that oxidative stress occurs at very early stages in RA and plays a crucial role in disease progression.9,10 Reactive oxygen species (ROS), produced by oxidative stress, directly oxidizes proteins, nucleic acids, transcriptional factors, membrane phospholipids, and intracellular and extracellular components. This leads to impaired biological activity, breakdown of matrix components, and initiation of apoptosis of synoviocytes, and ROS serves as the intracellular signaling molecule that amplifies the inflammatory response in the RA joint.8 Suggested factors stimulate synoviocytes and initiate inflammatory reactions that alter the extracellular matrix of cartilaginous tissues, eventually leading to cartilage destruction.5,7 Previous studies have demonstrated that accumulation of ROS could cause physiological malfunction of FLS.8 Specifically, TNF-α can cause damage to mitochondrial membrane integrity and induce ROS generation.8,11
Glucocorticoids (GCs), including dexamethasone (DEX), show effective anti-inflammatory efficacy and have been widely used in many diseases such as RA, cancer, severe allergies, asthma, chronic obstructive lung disease, croup, brain swelling, and multiple sclerosis.12 However, their repetitive and/or high dose use is often undesirable because of serious local and systemic side effects (adrenal insufficiency, hyperglycemia, Cushing’s syndrome, osteoporosis, Charcot arthropathy, etc.). These complications often arise due to the widely distributed GC receptor throughout the body.12 Additionally, these side effects worsen when GC therapy is given over a long period of time at repeated high doses.
In recent years, nanomaterial-based GC delivery has been increasingly studied for the treatment of inflammatory disorders. For example, polymeric drug delivery systems have been shown to selectively target inflammation sites and significantly improve the therapeutic efficacy of GC in animal models.13 Specifically, nanodrug formulations of GC (ie, DEX), including liposomes, core-cross-linked micelles, and N-(2-hydroxypropyl)methacrylamide copolymer, were evaluated in an RA rat model.13 After intravenous injection, a slower drug release from polymeric carriers induced a longer duration of therapeutic activity than that of drugs with relatively faster release and resulted in better joint protection.14 However, the drug was delivered at an overdose concentration (10 mg/kg). Micelles loaded with DEX self-assembled in the amphipathic polyethylene-glycol-block-poly (ε-caprolactone) (PCL-PEG) polymer caused reduced joint swelling, bone erosion, and inflammatory cytokine expression in both the joint tissue and serum of RA rat, but exhibited poor, low drug loading (~2.2%) when conjugated with nanoparticles.15 However, previous studies did not emphasize on the outstanding intracellular mechanism used by GC liposomes and PCL-PEG.15–19
Carbon nanotubes (CNTs), primarily employed for applications in cancer treatment, are easily functionalized by surface alterations through non-covalent and covalent functionalization and, thus, are widely used as molecular carriers in both in vitro and in vivo drug delivery.20,21 Soluble CNTs functionalized by surface oxidization and coated by surfactants or amphiphilic polymers are able to be engulfed by cells via the energy-dependent endocytosis pathway.22
It is worthy to note that a few studies have demonstrated the use of CNTs in arthritis. Specifically, PEG polymers covalently attached to CNTs were used as an intra-articular delivery source for chondrocytes in osteoarthritis (OA)-induced mouse models; the coupling of pharmacological agents with an intra-articular delivery nanosystem enhanced drug residence time and increased cartilage penetration in OA joints.23 As a next step, it is imperative to reduce the side effects following repetitive short- and long-term high-dose drug use for the treatment of synovial inflammation and oxidative stress. Thus, reducing the GC dose, enhancing anti-inflammatory effects, and blocking ROS production using intra-articular delivery of nanodrugs are critical issues in GC treatment. This study demonstrated the effective mechanism of GC-CNTs for the treatment of activated FLS. Specifically, the upregulated caveolin-dependent pathways and mitochondria targeting of GC reduced activation of ROS and pro-inflammatory cytokines by FLS. The obtained results clearly support the effective immunosuppressive and anti-oxidative stress properties of PEG-coated CNT conjugated with DEX even at extremely low drug doses.
Materials and methods
Materials
Purified multi-walled CNTs (900-1260; SES, Charlevoix, MI, USA) and PEG-amine (M7N201, molecular weight of 5 kDa; NOF, Tokyo, Japan) were purchased to generate PEG-coated CNT. To conjugate with PEG-coated CNT, DEX was obtained from Sigma (D1756). Cell culture substrates, including Dulbecco’s Modified Eagle’s Medium (DMEM, 11995-065; Gibco), fetal bovine serum (FBS, 16000-044; Gibco), and antibiotics (penicillin/streptomycin, 10378016; Gibco) were purchased. To fluorescently label DEX-PEG-coated CNT, streptavidin-conjugated Alexa 488 antibody was obtained from Thermo Fisher Scientific (S11223; Waltham, MA, USA). To examine the pro-inflammatory response of FLS, the primers for TNF-α, IL-1β, IL-6, and the housekeeping gene, GAPDH, were synthesized from Bioneer (Daejeon, South Korea). The reverse transcription kit and SYBR Premix Ex Taqt kit were purchased (04913850001; Roche) to perform real-time polymerase chain reaction (PCR). The commercial kits to examine superoxide dismutase (SOD) activity (7500-100k; R&D Systems) and recombinant TNF-α (210-TA-005; R&D Systems) were purchased to determine the RA FLS response to oxidative stress. To analyze the pro-inflammatory response of FLS at the protein level, enzyme-linked immunosorbent assay (ELISA) kits for TNF-α (ab181421; Abcam), IL-1β (ab46052; Abcam), and IL-6 (ab46042; Abcam) were obtained. Anti-MMP-1 (ab38929; Abcam) and anti-MMP-3 (ab53015; Abcam) antibodies were used to perform western blot.
Preparation of nanodrug
CNTs were carboxylated and PEG was synthesized according to previously described methods.24,25 To coat CNTs with PEG, carboxylated CNTs were mixed with PEG at a weight ratio of 1:5 in 10 mL deionized water. The mixed solution was sonicated for 30 min at 4°C and dispensed into filter tubes (AmiconYM-50; Millipore, Billerica, MA, USA). The solution was then centrifuged at 4,000 rpm for 15 min and washed with deionized water at least three times. The PEG-coated CNT solution was mixed with DEX at a weight ratio of 1:1 in 2-(morpholino)ethanesulfonic acid (MES) buffer (pH 8.0). The mixtures were then incubated at 4°C for at least 24 h with shaking. The DEX-PEG-coated CNTs in MES buffer were filtered (Amicon YM-50, 100 kDa; Millipore) at 4,000 rpm at least 3 times to remove unconjugated DEX. DEX-PEG-coated CNTs were finally prepared in phosphate-buffered saline (PBS; at pH 7). The percentage of non-covalently coated DEX on PEG CNTs was determined by measuring the weight of DEX-PEG-coated CNTs after drying overnight in vacuum oven at 60°C. Drug loading was determined using the following formula: Drug loading (%) = (Weight of DEX-PEG coated CNT/Weight of PEG coated CNT) × 100. The particle size and electronic potential were analyzed (Zetasizer Nano, Malvern, UK) to determine the size distribution and surface charge of DEX-PEG-coated CNTs in PBS.
To generate streptavidin-conjugated Alexa 488 (Thermo Fisher Scientific) conjugation on DEX-PEG-coated CNTs, PEG-coated CNT solution was mixed with DEX and streptavidin-conjugated Alexa 488 in MES buffer and incubated for 24 h at 4°C in the absence of light. Unbound DEX and Alexa 488 were removed by centrifugation (AmiconYM-50 filter) and washing thoroughly with MES buffer three times. Alexa 488-labeled DEX on PEG-coated CNT (DEX-Alexa 488 PEG-coated CNT) was confirmed using an ultraviolet-visible (UV-vis) spectrometer (Libra S50; Biochrom, Terre Haute, IN, USA) at 488 nm.
Material characterization of nanodrug
Cryogenic Transmission Electron Microscopy (Cryo-TEM, F20, Tecnai) was used to visualize DEX-PEG-coated CNT on a copper grid. Next, the prepared samples were instantly frozen in ethane and preserved in liquid nitrogen, using a plunge freezing technique in Vitrobot (FEI).
Fourier transform infrared (FTIR) spectroscopy (Nicolet iS5; Thermo Fisher Scientific) with attenuated total reflectance was used to analyze attached DEX chemicals on PEG-coated CNT. Before FTIR analysis, PEG-coated CNT and DEX-PEG-coated CNT (1 mg/mL) were completely dried by a freeze drying process (at below −80°C) for 24 h using a freeze dryer (20894; CHRIST, Osterode, Germany). Each spectrum was obtained by 32 scans with a resolution of 4 cm−1.
For the drug release test, DEX-PEG-coated CNTs were prepared in PBS (pH 7.0), acetate-buffered saline (ABS, pH 5.0), and ABS with lysozyme (1 mg/mL, L1667; Sigma). DEX-PEG-coated CNTs in PBS, ABS, and ABS with lysozyme were each subjected to gentle shaking and incubation at 37°C. At each time point, the samples were centrifuged with a 100-kDa ultra-filter (Millipore) at 13,000 rpm for 15 min and 500 μL of supernatant was collected at 0.1, 0.5, 1, 48, and 288 h. The DEX supernatant was analyzed by weighing after drying at 60°C in a vacuum oven.
Cell culture
FLS were isolated by enzymatic dispersion of synovial tissues from arthritis patients, as previously described.26 Synovial tissue samples were obtained from patients with RA at the time of joint surgery. The age of arthritis patients ranged from 32 to 59 years. Tissues were subjected to monolayer culture. Informed consent was obtained from all patients, and the Ethics Committee of Kyungpook National University approved this human subject study. FLS were cultured in DMEM supplemented with non-heat-inactivated 10% FBS and 1% antibiotics at 37°C in 5% CO2. FLS at passages 3–7 were used for the experiments.
Uptake analysis
Cells were seeded in a six-well plate at a density of 4×105 cells/well and incubated overnight. The cells were pre-incubated with inhibitors 5-(N-ethyl-N-isopropyl)amiloride (EIPA, A3085), chlorpromazine (CPZ, C8138), and genistein (GEN, G6649; Sigma) for 30 min at 37°C to examine macropinocytosis and the uptake of caveolin and clathrin. The nanodrugs investigated were then incubated with cells for another 2 h. The concentration of DEX-Alexa 488-PEG-coated CNT was 0.5 μg/mL. The concentrations of EIPA, CPZ, and GEN were 25, 20, and 200 μM, respectively. Cells were then washed twice with PBS and suspended in 500 mL of PBS supplemented with 1% FBS. The Alexa 488 fluorescence obtained from single-cell suspensions was evaluated using a BD LSR II flow cytometer (Becton Dickinson Immunocytometry Systems) and analyzed using FlowJo software (Version 10.1, FlowJo, LLC). Single-cell suspensions treated with nanodrug without inhibitors served as controls.
For confocal analysis of caveolin endocytosis influence on nanodrug uptake, FLS were cultured overnight on poly-D-lysine-coated coverslips in 24-well plates. The cells were pre-incubated with GEN for 30 min at 37°C to examine changes in uptake amount of nanodrugs by caveolin-dependent endocytosis. Then, 0.5 μg/mL DEX-Alexa 488-PEG-coated CNTs were incubated with cells for another 2 h. Cells were fixed with 2% paraformaldehyde in medium overnight at 2°C. Finally, cells were mounted, visualized by confocal microscopy (LSM700; Carl Zeiss, Göttingen, Germany), and analyzed using the ZEN software (Carl Zeiss).
Intracellular trafficking
Cells were cultured overnight on poly-D-lysine-coated coverslips in 24-well plates and treated with 0.5 μg/mL of DEX-Alexa 488-PEG-coated CNT for the indicated time points. Cells were fixed with 2% paraformaldehyde in medium overnight at 4°C. Permeabilization was performed in PBS with 0.1% Triton X-100 for 15 min at room temperature. After a blocking period of 2 h with 1% bovine serum albumin (Generay Biotech Co., Shanghai, People’s Republic of China) in PBS, cells were incubated with early endosome antigen-1 (an early endosome marker, 1:200 dilution factor, polyclonal, ab2900; Abcam) and mannose 6-phosphate receptor (a late endosome marker, 1:200 dilution factor, ab2733; Abcam) antibodies overnight at 4°C in the absence of light. After washing two times with PBS, cells were incubated with Alexa Fluor 405 Goat Anti-Rabbit IgG (H+L) and Alexa Fluor 405 Goat Anti-Mouse IgG (H+L) (1:200 dilution factor, A11008, A21057; Molecular Probes) for 2 h at room temperature (RT). Mounted cells were visualized by confocal microscopy (LSM700; Carl Zeiss) and analyzed using the ZEN software (2009; Carl Zeiss).
Reactive oxygen species and superoxide dismutase analysis
The intracellular accumulation of ROS was monitored using the 2′,7′-dichlorodihydrofluorescein diacetate (DCF-DA) probe after 12 h of cell culture. Various concentrations of nanodrug-treated TNF-α-stimulated FLS were loaded with 10 μM DCF-DA and incubated at 37°C for 30 min in the dark. Cells were then cleaned and resuspended in PBS. Samples were observed under a fluorescence microplate reader at 488 nm of excitation and 530 nm of emission. SOD activity was measured using the tetrazolium-based SOD assay kit (R&D Systems), as directed by the manufacturer. DEX (1 and 0.25 μg/mL) nanodrug treated with TNF-α-stimulated FLS pellets was resuspended in cold buffer (50 mM Tris-HCL, pH 7.5, 5 mM ethylenediaminetetraacetic acid, and 1 mM 1,4-dithiothreitol) and centrifuged at 12,000 rpm for 15 min at 4°C. Sample solution was mixed with each sample and the plate was incubated at 37°C for 20 min. Samples were observed under a microplate reader at 450 nm.
Mitochondrial membrane abruption
To measure the Δψm, JC-1, a lipophilic cation-sensitive fluorescent probe for detecting Δψm, was used according to the manufacturer’s instructions (T3168; Molecular Probes). Cells were cultured on a 24-well plate at a density of 2×104 cells/well in 500 μL culture medium overnight. Cells were stimulated with TNF-α (20 ng/mL) for 12 h followed by treatment with the experimental drugs (1 μg/mL DEX, PEG-coated CNT, and 0.25 μg/mL DEX-PEG-coated CNT) for 12 h. Cells were washed and incubated with 1 μM of JC-1 at 37°C for 30 min in the absence of light. After removing the JC-1, images were captured by confocal microscopy (LSM700; Carl Zeiss) with both red and green channels. A total of 6 random, non-adjacent fields in each group were used for statistical analysis. ImageJ software was used to measure the average fluorescence intensity of the red and green fluorescence in each group. The Δψm level is represented by the JC-1 fluorescence ratio, calculated as the averaged fluorescence intensity ratio of red and green.
Real-time polymerase chain reaction
To measure cytokine expression, quantitative real-time PCR (Thermal Cycler Dice TP850; Takara Bio, Otsu, Japan) was used according to the manufacturer’s protocol. FLS were pretreated with DEX, PEG-coated CNT, and DEX-PEG-coated CNT for 2 h, followed by stimulation with TNF-α (20 ng/mL) for 12 h. Total cellular RNA was isolated from cells (1×105 cells/well in 24-well plates), using TRIZOL (15596018; Thermo Fisher Scientific). First-strand complementary DNA (cDNA) was synthesized using RT Premix (Promega). The reverse transcription conditions were carried out at 45°C for 60 min and 95°C for 5 min. Briefly, 2 μL cDNA (1 μg), 1 μL sense and antisense primer solutions (0.4 μM), 12.5 μL SYBR Premix Ex Taq (Takara Bio), and 9.5 μL dH2O were mixed to obtain a final 25 μL reaction mixture in each reaction tube. PCR was carried out with the following primers: TNF-α (forward 5′−AGA GGG CCT GTA CCT CAT CT-3′; reverse 5′-AGA GGG CCT GTA CCT CAT CT-3′), IL-1β (forward 5′-GGA TAT GGA GCA ACA AGT GG-3′; reverse 5′-CCA GCT GTA GAG TGG GCT TA-3′), IL-6 (forward 5′-CTT GCC TGG TGA AAA TCA TC-3′; reverse 5′-CTT TTT CTG CAG GAA CTG GA-3′), MMP-1 (forward 5′-AGA TTT GCC AAG AGC AGA TG-3′; reverse 5′-GTC TGC TTG ACC CTC AGA GA-3′), and MMP-3 (forward 5′-TCC CAA GCA AAT AGC TGA AG-3′; reverse 5′-CAT TTG GGT CAA ACT CCA AC-3′). GAPDH (forward 5′-TAG ACT TCG AGC AGG AGA TG-3′; reverse 5′-TTG ATC TTC ATG GTG CTA GG-3′) was used to verify that equal amounts of RNA were used for amplification.
Protein analysis (ELISA and western blot)
To analyze the anti-inflammatory efficacy of low-dose nanodrug with regard to protein level, FLS were seeded in 24-well plates. The cells were pretreated with the experimental drugs (1 μg/mL DEX, PEG-coated CNT, and 0.25 μg/mL DEX-PEG-coated CNT) for 2 h and then incubated for another 24 h with TNF-α. The culture medium was collected, and the concentrations of the cytokines were determined by ELISA using a commercial kit (Abcam), according to the manufacturer’s instructions.
To explore the inhibition of MMP protein expression, cells (5×105 cells/well in a 6-well plate) were pre-incubated for 20 min for MMP-1 and MMP-3 following 24 h stimulation with TNF-α (20 ng/mL). The cells were then rinsed twice with ice-cold PBS, and total cell lysates were gathered in 200 μL lysis buffer. The lysates were spun in a micro-centrifuge for 20 min at 4°C, and the supernatant was collected. The proteins were electrophoresed using 10% sodium dodecyl sulfate polyacrylamide gel electrophoresis and then transferred to nitrocellulose membranes. MMP-1 and MMP-3 were assayed using anti-MMP-1 and anti-MMP-3 antibodies (Abcam). Immunodetection was performed using the LAS4000 ChemiDoc imager (Fujifilm, Tokyo, Japan).
Statistical analysis
The statistical differences between mean values obtained from two sample groups were analyzed by Student’s t-test. The differences were considered significant if the P-value was less than or equal to 0.05. The statistical differences between several sample types were analyzed by analysis of variance (ANOVA) followed by the Newman-Keuls’ multiple comparison test. Asterisks (*, **, and ***) indicate the significance at P-values less than 0.05, 0.01, and 0.001, respectively.
Results and discussion
Physiochemical characterization and drug release
DEX was non-covalently conjugated with PEG-coated CNT (Figures 1A and S1), and Cryo-TEM images confirmed morphology of DEX-PEG-coated CNT (Figure 1B). The PEG-coated drug (DEX) exhibited embedded drug molecules in the PEG-coated CNT (Figure 1B).
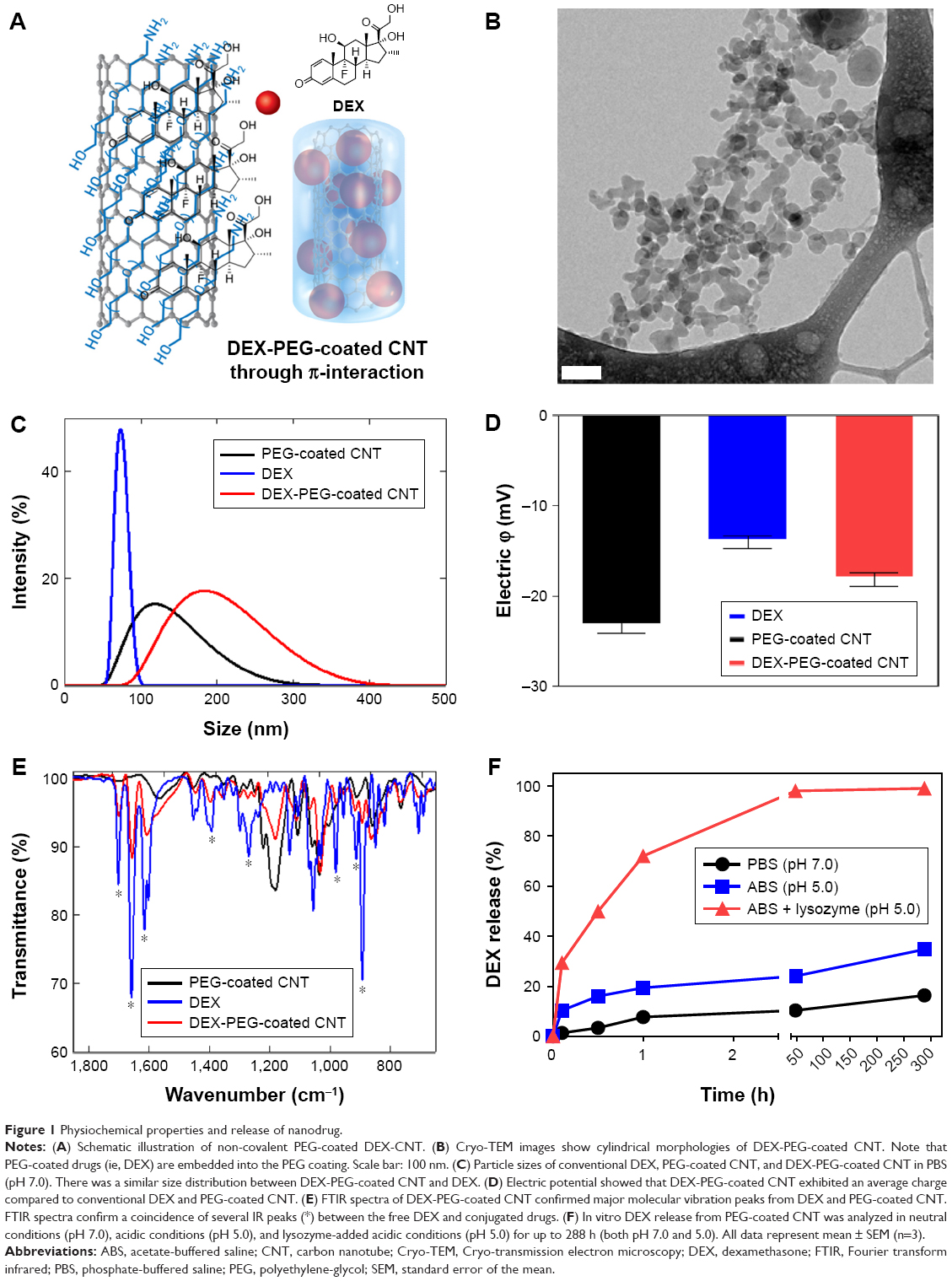 | Figure 1 Physiochemical properties and release of nanodrug. |
Average sizes of free DEX, PEG-coated CNT, and DEX-PEG-coated CNT were 80, 110, and 180 nm, respectively (Figure 1C). The polydispersity index was less than 0.5 for all tested samples (Figure S2). Electrical potential of all tested drugs indicated negative charges in neutral buffers (pH of PBS =7.2) (Figure 1D). Electrical potential analysis also identified the surface charge of DEX-PEG-coated CNT (Figure 1D). DEX-PEG-coated CNT (red) exhibited an average charge for DEX (blue) and PEG-coated CNT (blue, Figure 1D). Particle size and electric potential assessment provided the evidence that PEG-coated CNT successfully possessed DEX by non-covalent PEGylation.
The vibrational infrared (IR) peaks from DEX-PEG-coated CNT were confirmed by FTIR spectra (Figure 1E). Significant overlapped IR peaks were observed between DEX and DEX-PEG-coated CNT, and this represents the successful attachment of DEX onto PEG-coated CNT (Figure 1E). Drug loading % was 265.7%±9.3% (weight ratio of DEX/PEG-coated CNT) (n=5). DEX release from PEG-coated CNT was performed in various physiological environments. DEX release was observed for up to 288 h at different pH levels (pH 5.0 and 7.0), and an acidic pH of the lysozymes (1 mg/mL lysozyme density) by measuring the released weight of DEX (Figure 1F). In neutral conditions (PBS), the release of DEX showed no distinct increase for up to 288 h (less than 20% release) (Figure 1F). In hypoxic conditions, there are abundant cytokines in the chronically inflamed synovium,27,28 and the pH value is approximately 6.0 in the synovial fluid of arthritis joints.28 In this respect, the designed DEX-PEG-coated CNT should be stable under hypoxic conditions (acidic conditions) to sustain nano-conjugation before cell uptake. In this study, DEX showed less than 40% release in acidic conditions (ABS) after 288 h.
Although minimal drug release in the extracellular environment is generally favorable, drug transport to the specific intracellular organelles within the intracellular environment is essential.29,30 By adding lysozymes in acidic conditions, a burst release of DEX was observed (Figure 1F). Thus, it was concluded that the designed DEX-PEG-coated CNTs are extremely responsive in acidic enzymatic proteins (lysozymes, etc.) and very stable under extracellular conditions (hypoxic synovial fluid). Thus, the designed nanodrug can reach the intracellular compartments without drug loss in the synovial fluids and, thus, can maximize the effective intracellular delivery of DEX in FLS.
Uptake analysis
Endocytosis is a form of active transport in which a cell takes in outer objects by enclosing them in vesicles or vacuoles pinched off from its cytoplasmic membrane.30,31 Nanomaterials were easily internalized by cells through endocytosis.30,31 Several different types of endocytosis are involved in the uptake of nanomaterials, including macropinocytosis, clathrin-mediated endocytosis, and caveolae-mediated endocytosis.30–32 In this study, inhibitors of macropinocytosis (EIPA), clathrin-mediated endocytosis (CPZ), and caveolae-mediated endocytosis (GEN) were used to identify the endocytic mechanism that is responsible for the uptake of DEX-PEG-coated CNT. Effects of endocytic inhibitors on the internalization of DEX-PEG-coated CNT into FLS and TNF-α-activated FLS were analyzed by fluorescence-activated cell sorting (Figure 2C). It has been well documented that overproduction of inflammatory cytokines, such as TNF-α, is common in arthritis and that cytokine-activated FLS are a significant contributor to arthritis progression.5,33
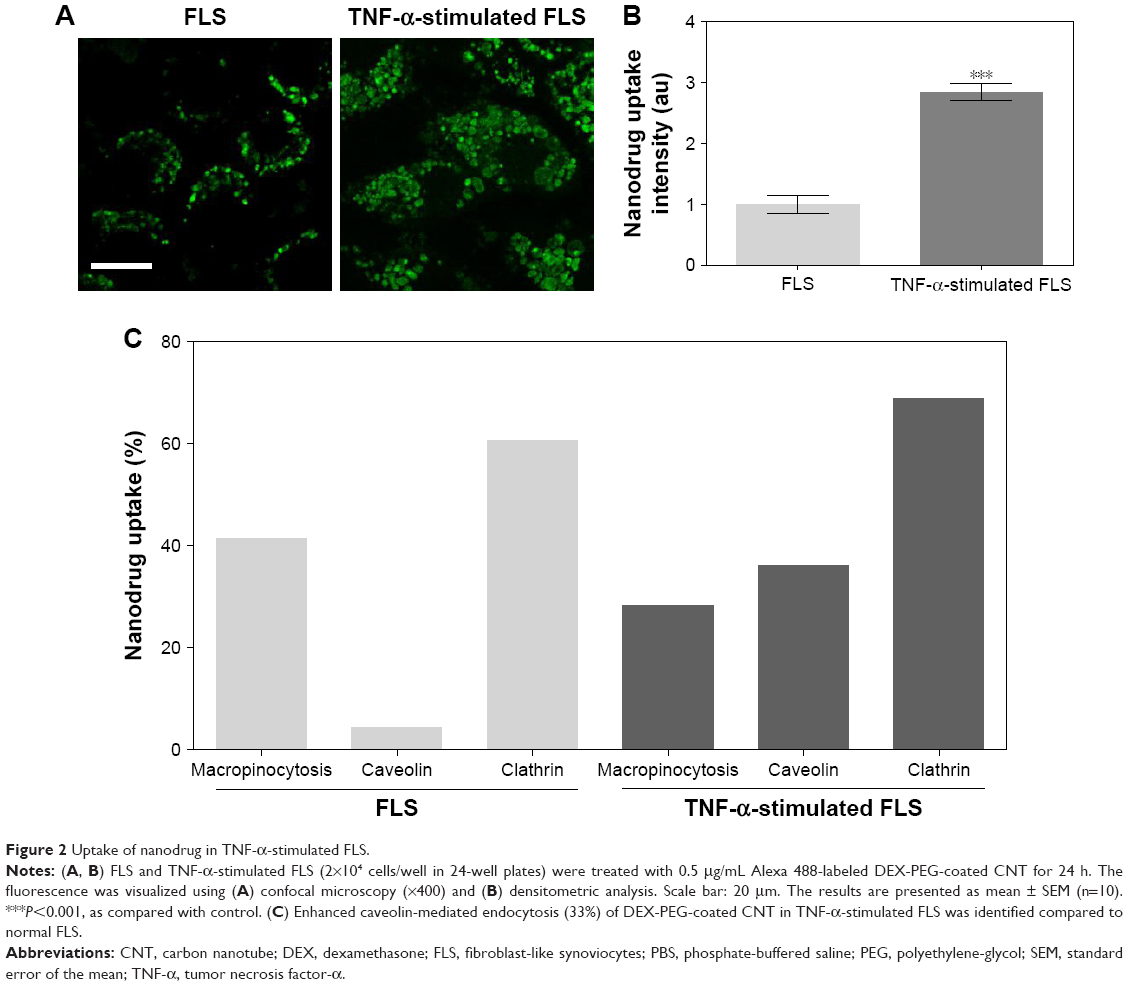 | Figure 2 Uptake of nanodrug in TNF-α-stimulated FLS. |
To investigate intracellular uptake of DEX-PEG-coated CNT in FLS, streptavidin-Alexa 488-embedded DEX-PEG-coated CNTs (DEX-Alexa 488-PEG coated CNT) were used. Non-covalent Alexa 488 conjugation on DEX-PEG-coated CNTs was confirmed by UV-vis spectrophotometer and strong absorbance was detected at 490 nm (Figure S3). It was clearly demonstrated that the TNF-α-stimulated FLS cultured with DEX-Alexa 488-PEG-coated CNTs exhibited increased uptake intensity compared to normal FLS (localized in vesicles around the nucleus after 24 h of incubation) (Figure 2A and B). Before treatment with each endocytosis inhibitor, cell viability was pre-checked by using the 3-(4, 5-dimethylthiazol-2-yl)-2, 5 diphenyltetrazolium bromide assay to ensure that there were no cytotoxic effects from treatment with endocytosis inhibitors (Figure S4).
For the first time, this study identified that caveolin-dependent endocytosis was upregulated in TNF-α-activated FLS compared to normal FLS (Figure 2C). Enhanced caveolin-dependent endocytosis was very significant in TNF-α-activated FLS, while macropinocytosis and clathrin-dependent pathways were major sources of endocytosis in normal FLS (Figure 2C).
To confirm the impact of upregulated caveolin-dependent endocytosis in TNF-α-stimulated FLS, caveolin-mediated endocytosis inhibitor (GEN) was used. Interestingly, DEX-PEG-coated CNT uptake was greatly inhibited by GEN in TNF-α-activated FLS, whereas there were no significant uptake changes in caveolin-blocking uptake in normal FLS (Figure 3A and B). Thus, the obtained results clearly demonstrated that upregulated caveolin-dependent endocytosis greatly enhanced uptake of DEX-PEG-coated CNT in TNF-α-activated FLS. Unlike these results, several nanodrugs (polymer13 and micelles15) harboring DEX have been reported to uptake by clathrin-dependent endocytosis. Thus, DEX-PEG-coated CNT is the first type of nanodrug that exhibited an efficient intra-cellular drug delivery via upregulated caveolin-dependent endocytosis. Considering that a greater nanodrug uptake will transport more active anti-inflammatory agents to TNF-α-activated FLS cells, it is anticipated that an increase in caveolin endocytosis of the nanodrug and TNF-α will significantly influence subsequent inflammation levels in FLS.
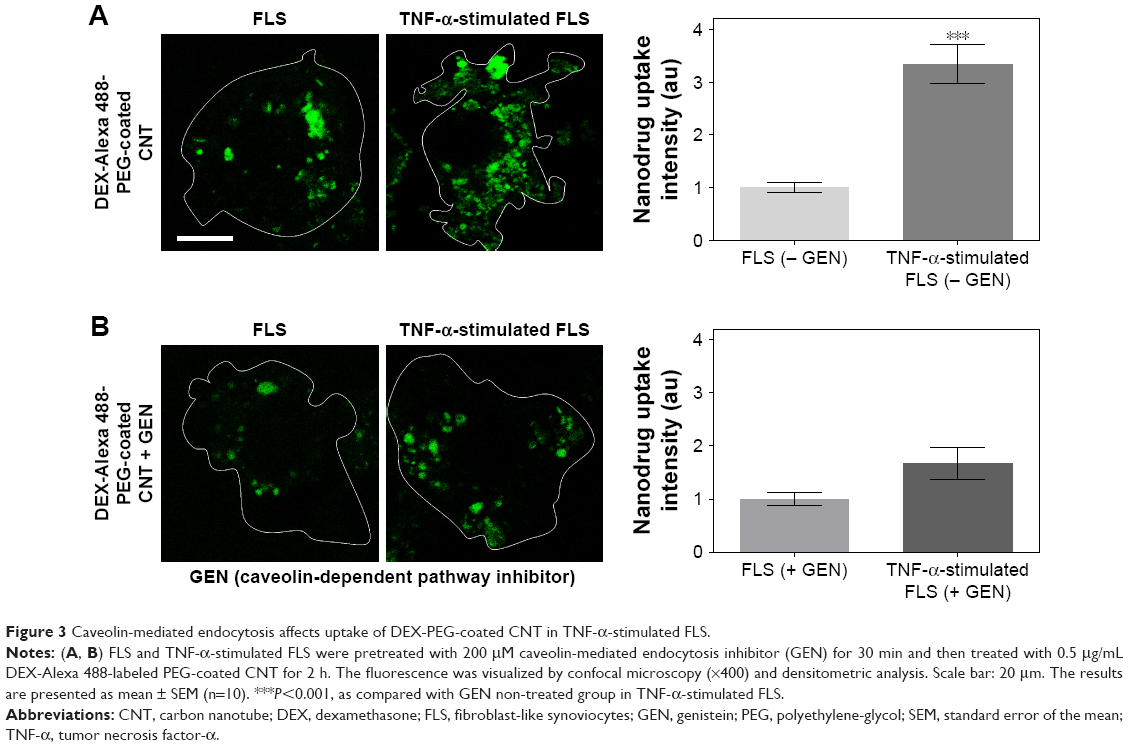 | Figure 3 Caveolin-mediated endocytosis affects uptake of DEX-PEG-coated CNT in TNF-α-stimulated FLS. |
Intracellular nanodrug delivery and changes in mitochondrial membrane function
Differing from small drug molecules, which are usually able to diffuse across cell membranes, small drug molecules attached on nanomaterials do not penetrate the cell membrane but experience vesicle-based intracellular drug delivery.30,32 For transportation of drug molecules into cells via CNTs, it was reported that conjugated drugs (both covalent and non-covalent) on CNTs follow intracellular delivery.34,35
In this study, accelerated levels of early endosome (EE; confocal image-based counted EE vesicles per cell) in TNF-α-activated FLS were observed compared to the EE time profile of normal FLS. TNF-α-stimulated FLS showed fast formation of EE vesicles and reached a maximum at approximately 2 h in EE counts compared to normal FLS (5 h) (Figure 4A–C). By contrast, co-localization signals between the late endosome (LE) stage and the nanodrug were not observed for both normal FLS and TNF-α-stimulated FLS. (Figures 4C and S5). This indicates that the EE stage did not develop into the LE stage for the tested nanodrug. In a previous study, mitochondria-targeted PEG nanoparticles exhibited enhanced significant co-localization with the EE (in 1 h), complete endosomal escape, and localization in the mitochondria of HeLa cells.36 Obtained results in this study demonstrated that most of the nanodrug was released into the cytosol after early endosomal escape (within 2 h) without a further LE stage in TNF-α-stimulated FLS.
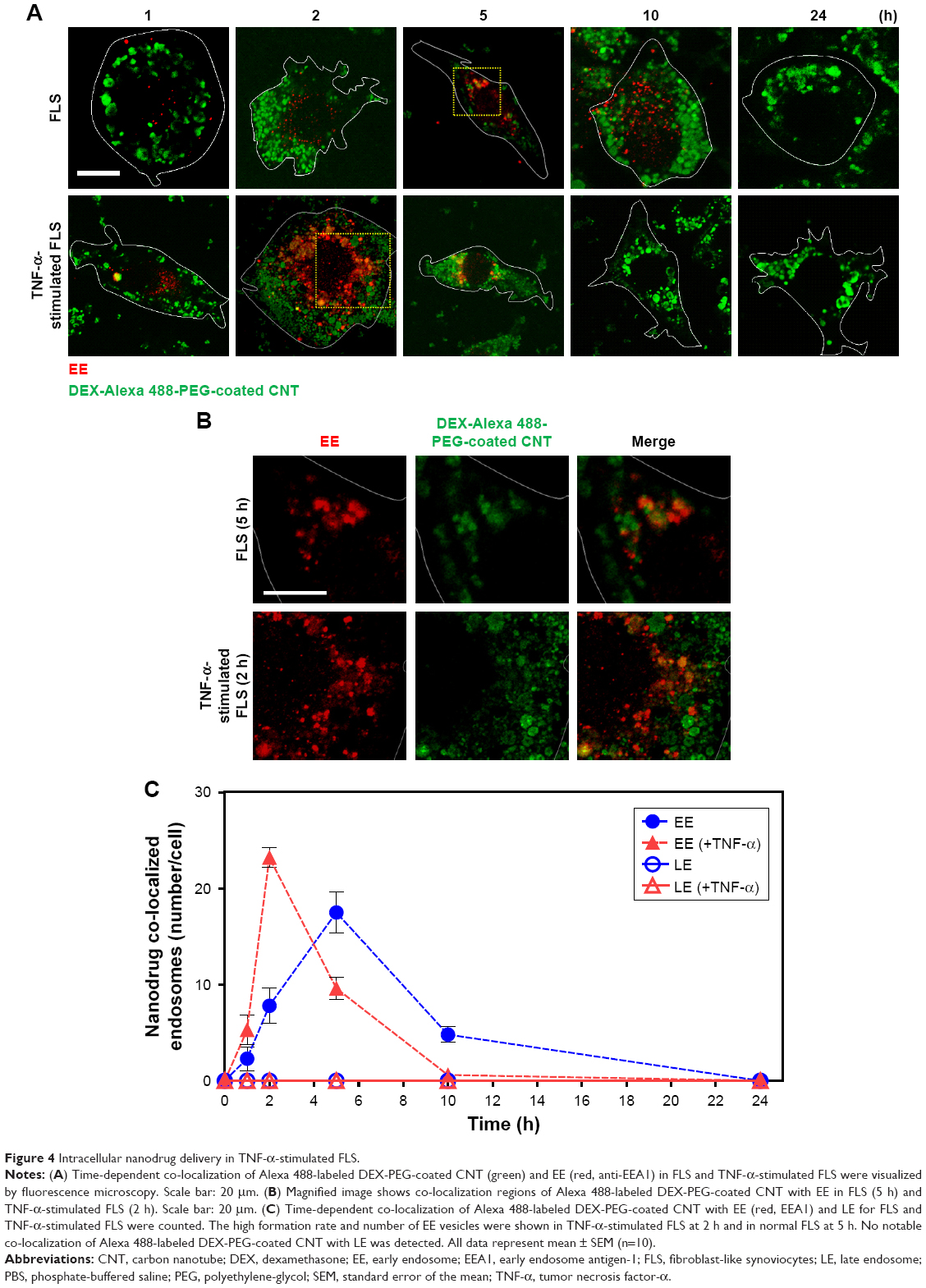 | Figure 4 Intracellular nanodrug delivery in TNF-α-stimulated FLS. |
To identify the maximum drug dose, the cell toxicity levels of DEX, PEG-coated CNT, and DEX-PEG-coated CNT in TNF-α-stimulated FLS were analyzed after exposure to various concentrations for 24 h (Figure S6). DEX-PEG-coated CNT showed no significant toxicity (greater than 80% TNF-α activated FLS viability) at a concentration below 0.5 μg/mL (PEG-coated CNT: 1 μg/mL, DEX: 1 μg/mL). Therefore, 0.5 μg/mL of DEX-PEG-coated CNT was used as the maximum drug concentration for the following in vitro experiments (Figure S6).
To this date, most studies for nanodrug harboring GC have shown only in vivo anti-inflammatory efficacy without understanding the mechanism of intracellular drug delivery.15–19 Thus, there was a need to unveil a mechanism related to the inhibition of inflammation on synovial fibroblasts and FLS. TNF-α usually causes mitochondrial transmembrane disruption and induces subsequent oxidative stress signaling through the activation of ROS.8 To monitor mitochondrial transmembrane changes, membrane-permeant JC-1 dye was used. JC-1 dye exhibits potential-dependent accumulation in mitochondria, indicated by a fluorescence emission shift from green (~529 nm) to red (~590 nm). In healthy cells, JC-1 exists as a monomer in the cytosol (green), but also accumulates as aggregates in the mitochondria (red). In mitochondrial-damaged cells, JC-1 exists in a monomeric form and stains the cytosol green.37 As shown in Figure 5A and B, TNF-α (20 ng/mL) and PEG-coated CNT (1 μg/mL) induced greater levels of mitochondrial membrane disruption (green). Although free DEX did not fully recover (at a dose of 1 μg/mL) the mitochondria membrane potential, the DEX nanodrug successfully recovered the mitochondria membrane potential for TNF-α-stimulated FLS (at dose of 0.25 μg/mL) (Figure 5A and B). The recovered mitochondrial membrane potential in TNF-α-activated FLS can be contributed to the accumulation of nanodrugs into damaged mitochondria. Additionally, the greater uptake of nanodrugs into damaged mitochondria can be contributed to both caveolin-assisted endocytosis and the release of nanodrugs from the EE into the cytosol. Thus, the recovered mitochondrial membrane potential in TNF-α-activated FLS can be contributed to both caveolin-assisted endocytosis and the release of nanodrugs from the EE into the cytosol.
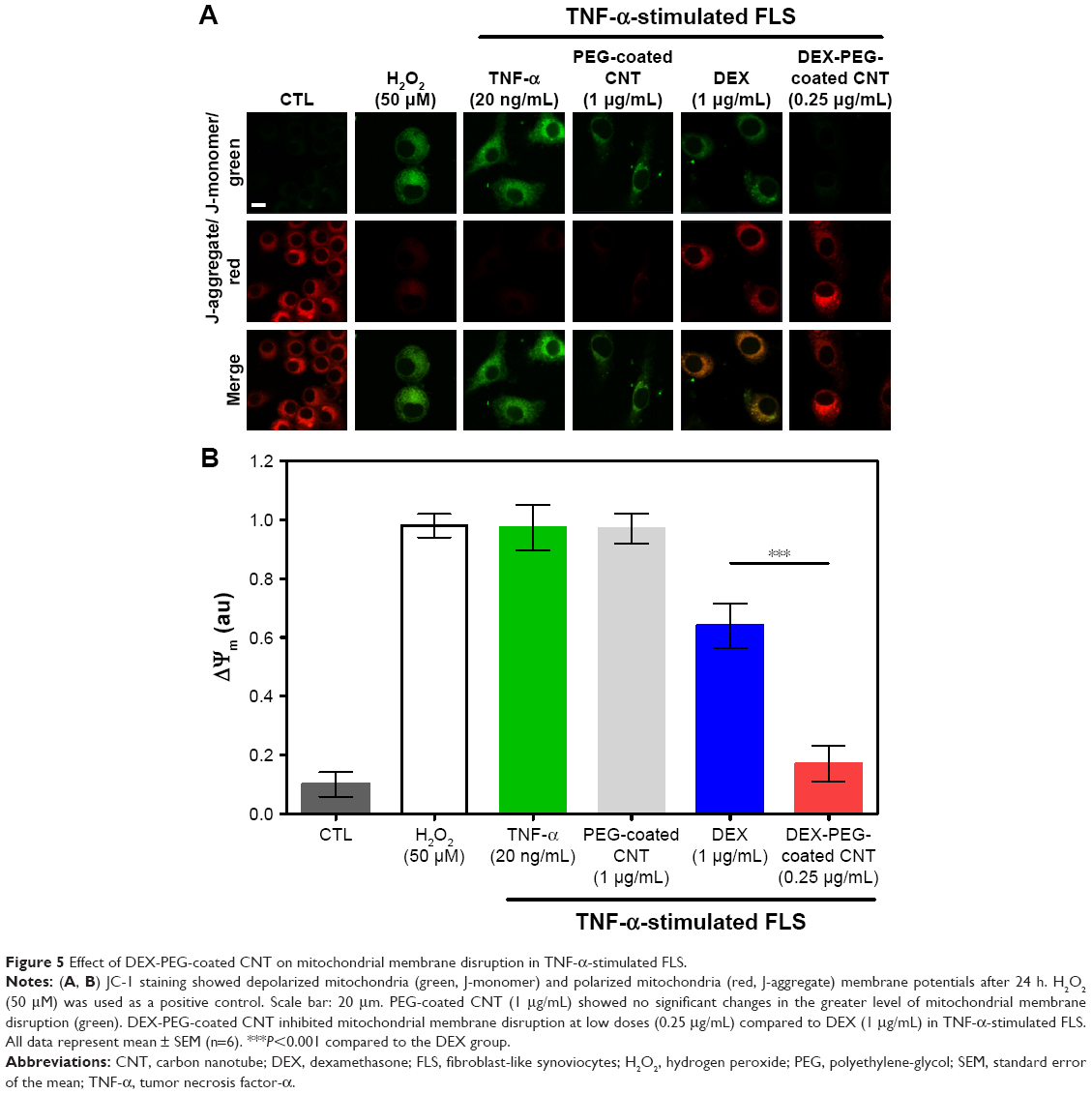 | Figure 5 Effect of DEX-PEG-coated CNT on mitochondrial membrane disruption in TNF-α-stimulated FLS. |
Inhibited ROS production and increased SOD activity
TNF-α induced ROS production by causing several pathophysiological changes in FLS cells; one of the major events is the stimulation of a pro-inflammatory response.4–6,8 Oxidative stress is a common mechanism for cell damage that has been shown to produce ROS.8 ROS are stable and highly reactive compounds that can strip electrons from cellular macromolecules and render them dysfunctional.8 Chain reactions of self-propagating free radicals (molecules that contain an unpaired electron in their outermost shell) mediate lipid peroxidation and can cause damage to the cell membrane, thereby inducing cell death.38 The accumulation of ROS depletes cellular glutathione and minimizes the defensive ability of cellular antioxidant enzymes such as SOD (physiologically and pathophysiologically).39 The connections between TNF-α and ROS are important because ROS acts on many proteins needed to regulate cellular homeostasis, including cell proliferation, survival, death, differentiation, DNA repair, and metabolism.8 Increased ROS production also influences several pathophysiological changes in cells, one being the induction of a pro-inflammatory response.38
To confirm the efficacy of DEX-PEG-coated CNT on the inhibition of ROS production, ROS was observed in TNF-α activated FLS cells, using DCF-DA. DEX-PEG-coated CNT inhibited ROS production (at a concentration of 0.0625 μg/mL) in TNF-α-activated FLS (Figure 6A). However, DEX did not have a positive effect until a concentration of 0.5 μg/mL. Furthermore, SOD activity levels showed a significant increase after treatment with DEX-PEG-coated CNT, whereas DEX did not fully recover SOD activity (at the concentration of 0.25 μg/mL) (Figure 6B). Thus, the obtained results clearly demonstrated that the low-dose nanodrug successfully inhibited TNF-α-induced ROS production and recovered SOD activity.
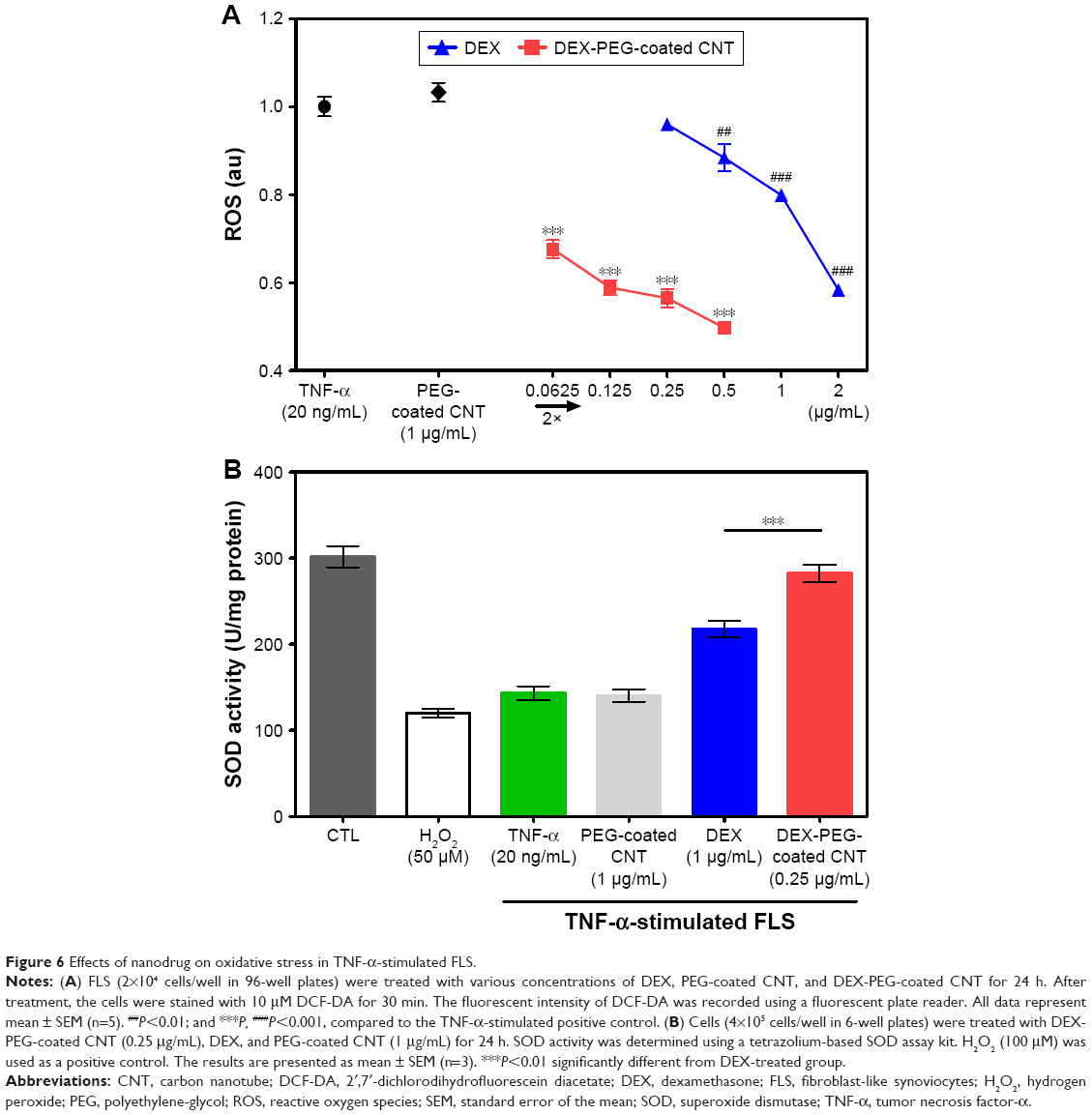 | Figure 6 Effects of nanodrug on oxidative stress in TNF-α-stimulated FLS. |
Gene expression of pro-inflammatory cytokines and MMPs
TNF-α is a major activator of inflammation and can initiate inflammatory signaling pathways, including MMPs, in FLS.4,6,40 In this study, the expression effects of DEX nanodrug on pro-inflammatory cytokines induced by TNF-α FLS were analyzed. FLS were pretreated with DEX or DEX-PEG-coated CNT for 2 h and stimulated with TNF-α for 12 h. Based on real-time PCR results, stimulated FLS expressed high levels of expression for the TNF-α, IL-1β, and IL-6 genes. Treatment with DEX and DEX-PEG-coated CNTs decreased TNF-α-induced expression of the TNF-α, IL-1β, and IL-6 genes (Figure 7A–C). Interestingly, the effective dose of conventional DEX (a drug concentration of 0.5–1 μg/mL) and a low dose of DEX-PEG-coated CNT (a drug concentration of 0.0625 μg/mL) showed similar suppressive effects on the cytokines (Figure 7A–C).
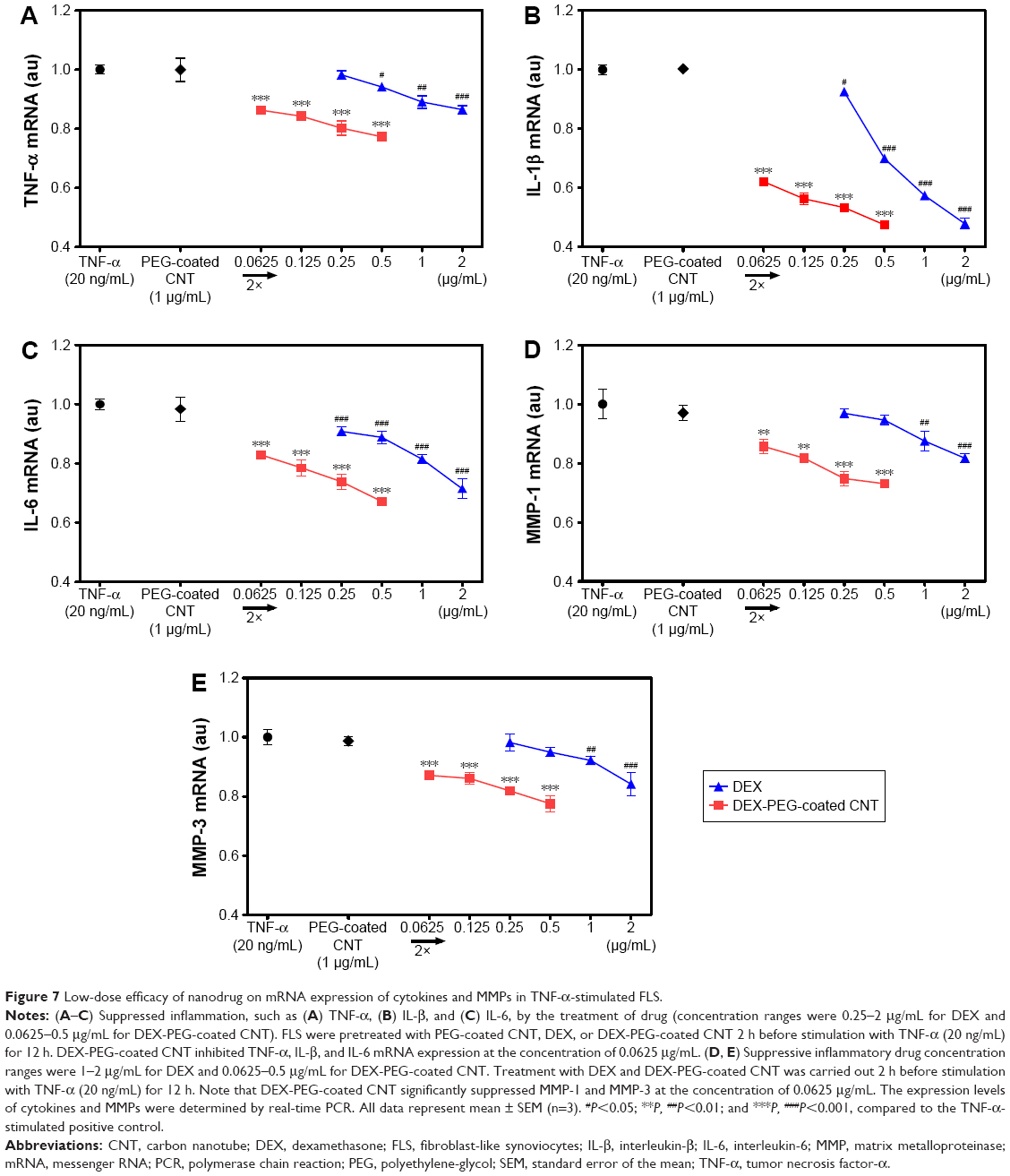 | Figure 7 Low-dose efficacy of nanodrug on mRNA expression of cytokines and MMPs in TNF-α-stimulated FLS. |
Activated pro-inflammatory cytokines stimulate the production of matrix-degrading enzymes, including MMP-1 and MMP-3, in FLS.4,26,41 Therefore, identification of a comparable effect for DEX and a low dose of DEX-PEG-coated CNT on the expression of MMP-1 and MMP-3 is essential. Real-time PCR showed that both DEX and DEX-PEG-coated CNT diminished TNF-α-induced expression of MMP-1 and MMP-3. Specifically, a low dose of DEX-PEG-coated CNT exhibited greater inhibition of MMP-1 and MMP-3 (at the concentration of 0.0625 μg/mL) than a maximum dose of DEX (at the concentration of 1–2 μg/mL for MMP-1 and MMP-3) (Figure 7D and E). Consistent with previous studies, the obtained results clearly demonstrated that a low dose of DEX conjugated with PEG-coated CNT inhibits TNF-α-induced expression of the pro-inflammatory cytokines MMP-1 and MMP-3 in FLS. Specifically, about a 20-fold lower dose of DEX-PEG-coated CNT exhibited a suppressive effect similar to that by free DEX. Thus, this study demonstrated that DEX-PEG-coated CNT possessed greater anti-inflammatory efficacy in suppressing pro-inflammatory cytokines and MMPs than conventional DEX in FLS even at low drug concentrations.
Protein expression of pro-inflammatory cytokines and MMPs
Inflammatory mediators, including TNF-α, IL-1β, and IL-6, exhibited an abundant production in the RA synovium, a high concentration in the synovium and serum in RA, and have been demonstrated to play critical roles in the pathogenesis of RA.41,42 In response to pro-inflammatory cytokines, FLS produce chemokines, which further enhance inflammation, hyperplasia, and cartilage destruction.7 To evaluate the inhibition role of DEX nanodrug on pro-inflammatory mediator expression at the protein level, we examined the effect of DEX nanodrug on the secretion of TNF-α, IL-1β, and IL-6 in TNF-α-stimulated FLS. The concentrations of the three pro-inflammatory cytokines, TNF-α, IL-1β, and IL-6, were remarkably increased in TNF-α-stimulated FLS. As shown in Figure 8A–C, treatment with a low dose of DEX-PEG-coated CNT (0.25 μg/mL) suppressed the TNF-α, IL-1β, and IL-6 production in TNF-α-stimulated FLS. Furthermore, a more significant reduction in pro-inflammatory cytokine concentration in the media was detected for the low-dose DEX-PEG-coated CNT (0.25 μg/mL) group compared with that for DEX alone (1 μg/mL) (ELISA).
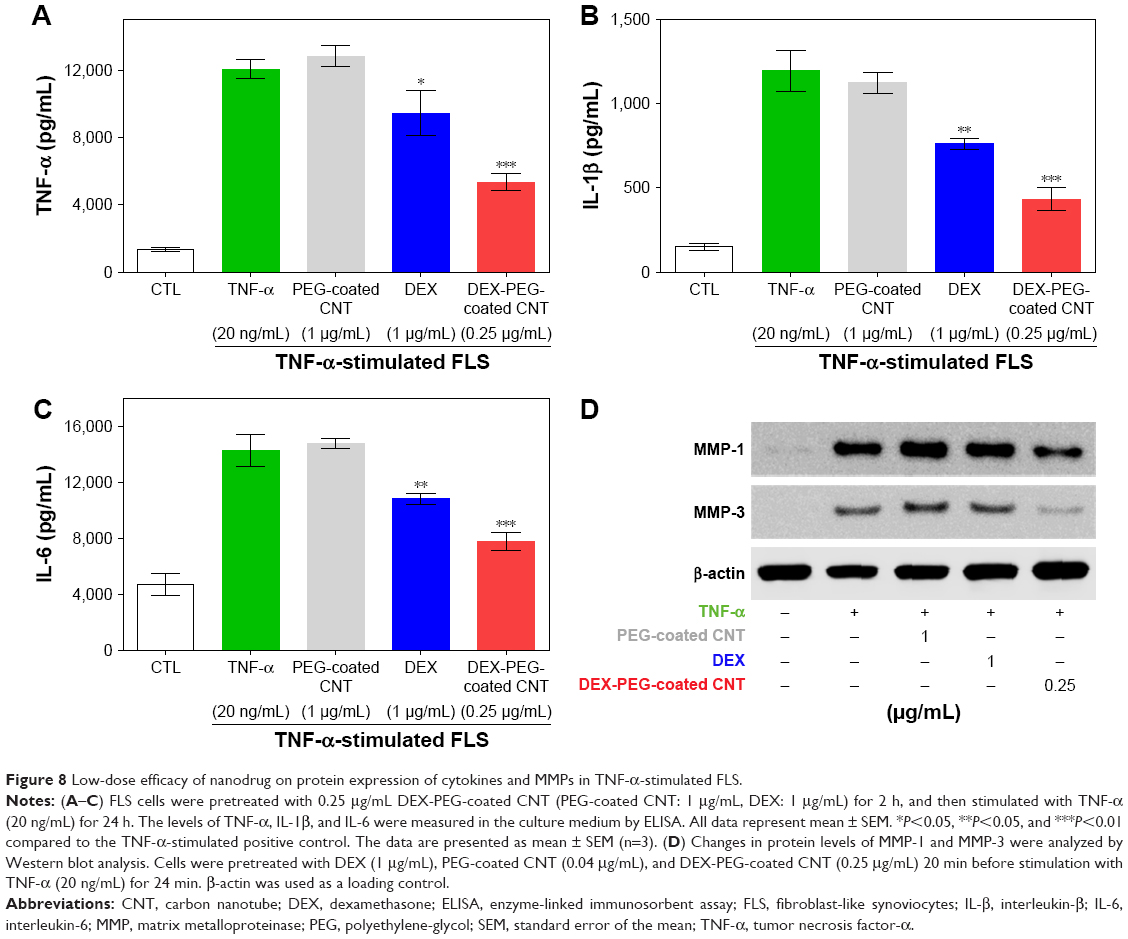 | Figure 8 Low-dose efficacy of nanodrug on protein expression of cytokines and MMPs in TNF-α-stimulated FLS. |
Additionally, the inhibitory effects of DEX nanodrug on MMP-1 and MMP-3 at the protein level induced by TNF-α FLS were analyzed by western blot analysis. FLS were pretreated with DEX (1 μg/mL) and DEX-PEG-coated CNTs (0.25 μg/mL) for 20 min and stimulated with TNF-α (20 ng/mL) for 24 h to measure the activity levels of MMP-1 and MMP-3. The obtained results clearly showed that a low dose of DEX-PEG-coated CNT (0.25 μg/mL) decreased the activation of MMP-1 and MMP-3 compared to a high dose of DEX (1 μg/mL) (Figure 8D). The obtained results demonstrated that DEX-PEG-coated CNTs exhibited TNF-α-induced pro-inflammatory cytokine production and MMP protein activation based on both mRNA and protein level analysis (Figure 8A–D).
Conclusion
As addressed in previous studies, it has been unclear for the exact mechanism of internalization and intracellular trafficking of nanodrugs for the treatment of inflammatory associated diseases.15–19 In this study, authors identified that DEX-PEG-coated CNTs were taken up by TNF-α-stimulated RA FLS via upregulated caveolin-mediated endocytosis, induced greater nanodrug uptake, and quickly released therapeutic agents in the EE compartments. Meanwhile, released therapeutic agents accumulated in the mitochondria, which in turn successfully suppressed TNF-α-induced ROS production and recovered mitochondrial membrane disruption at very low doses of DEX-PEG-coated CNTs. In addition, the significant inhibition of pro-inflammatory cytokines was exhibited by TNF-α-stimulated FLS in the DEX-PEG-coated CNT group. Consequently, the overall results suggest that the pro-inflammatory, cytokine-stimulated, and FLS-targeted DEX-PEG-coated CNTs possess a greater potential for low drug dose therapy in the treatment of arthritis than current available therapies. Thus, it is highly feasible that DEX-PEG-coated CNTs can be used as a clinical option to reduce the side effects sustained from the repeated and prolonged use of DEX over a long period of time.
Acknowledgments
This research was supported by grants from the National Research Foundation of Korea (2014R1A2A1A11052615, 2014R1A5A2009242, 2016R1C1B1009561, and 2017R1D1A1B03031983) and the Korea Health Technology R&D Project through the KHIDI, funded by the Ministry of Health & Welfare (HI14C1802).
Disclosure
The authors report no conflicts of interest in this work.
References
McInnes IB, Schett G. The pathogenesis of rheumatoid arthritis. N Engl J Med. 2011;365(23):2205–2219. | ||
Mobasheri A, Rayman MP, Gualillo O, Sellam J, van der Kraan P, Fearon U. The role of metabolism in the pathogenesis of osteoarthritis. Nat Rev Rheumatol. 2017;13(5):302–311. | ||
Perretti M, Cooper D, Dalli J, Norling LV. Immune resolution mechanisms in inflammatory arthritis. Nat Rev Rheumatol. 2017;13(2):87–99. | ||
McInnes IB, Schett G. Cytokines in the pathogenesis of rheumatoid arthritis. Nat Rev Immunol. 2007;7(6):429–442. | ||
Bartok B, Firestein GS. Fibroblast-like synoviocytes: key effector cells in rheumatoid arthritis. Immunol Rev. 2010;233(1):233–255. | ||
Bottini N, Firestein GS. Duality of fibroblast-like synoviocytes in RA: passive responders and imprinted aggressors. Nat Rev Rheumatol. 2013;9(1):24–33. | ||
Noss EH, Brenner MB. The role and therapeutic implications of fibroblast-like synoviocytes in inflammation and cartilage erosion in rheumatoid arthritis. Immunol Rev. 2008;223:252–270. | ||
Blaser H, Dostert C, Mak TW, Brenner D. TNF and ROS crosstalk in inflammation. Trends Cell Biol. 2016;26(4):249–261. | ||
Mapp PI, Grootveld MC, Blake DR. Hypoxia, oxidative stress and rheumatoid arthritis. Br Med Bull. 1995;51(2):419–436. | ||
Hitchon CA, El-Gabalawy HS. Oxidation in rheumatoid arthritis. Arthritis Res Ther. 2004;6(6):265–278. | ||
Nathan C, Cunningham-Bussel A. Beyond oxidative stress: an immunologist’s guide to reactive oxygen species. Nat Rev Immunol. 2013;13(5):349–361. | ||
Schäcke H, Döcke WD, Asadullah K. Mechanisms involved in the side effects of glucocorticoids. Pharmacol Ther. 2002;96(1):23–43. | ||
Liu XM, Quan LD, Tian J, Laquer FC, Ciborowski P, Wang D. Syntheses of click PEG-dexamethasone conjugates for the treatment of rheumatoid arthritis. Biomacromolecules. 2010;11(10):2621–2628. | ||
Quan L, Zhang Y, Crielaard BJ, et al. Nanomedicines for inflammatory arthritis: head-to-head comparison of glucocorticoid-containing polymers, micelles, and liposomes. ACS Nano. 2014;8(1):458–466. | ||
Wang Q, Jiang J, Chen W, Jiang H, Zhang Z, Sun X. Targeted delivery of low-dose dexamethasone using PCL-PEG micelles for effective treatment of rheumatoid arthritis. J Control Release. 2016;230:64–72. | ||
van den Hoven JM, Hofkens W, Wauben MH, et al. Optimizing the therapeutic index of liposomal glucocorticoids in experimental arthritis. Int J Pharm. 2011;416(2):471–477. | ||
Phillips NC, Thomas DP, Knight CG, Dingle JT. Liposome-incorporated corticosteroids. II. Therapeutic activity in experimental arthritis. Ann Rheum Dis. 1979;38(6):553–557. | ||
Koning GA, Schiffelers RM, Wauben MH, et al. Targeting of angiogenic endothelial cells at sites of inflammation by dexamethasone phosphate-containing RGD peptide liposomes inhibits experimental arthritis. Arthritis Rheum. 2006;54(4):1198–1208. | ||
Turjeman K, Barenholz Y. Liposomal nano-drugs based on amphipathic weak acid steroid prodrugs for treatment of inflammatory diseases. J Drug Target. 2016;24(9):805–820. | ||
Ji SR, Liu C, Zhang B, et al. Carbon nanotubes in cancer diagnosis and therapy. Biochim Biophys Acta. 2010;1806(1):29–35. | ||
Lu F, Gu L, Meziani MJ, et al. Advances in bioapplications of carbon nanotubes. Adv Mater. 2009;21(2):139–152. | ||
Prato M, Kostarelos K, Bianco A. Functionalized carbon nanotubes in drug design and discovery. Acc Chem Res. 2008;41(1):60–68. | ||
Sacchetti C, Liu-Bryan R, Magrini A, Rosato N, Bottini N, Bottini M. Polyethylene-glycol-modified single-walled carbon nanotubes for intra-articular delivery to chondrocytes. ACS Nano. 2014;8(12):12280–12291. | ||
Liu Z, Tabakman SM, Chen Z, Dai H. Preparation of carbon nanotube bioconjugates for biomedical applications. Nat Protoc. 2009;4(9):1372–1382. | ||
Lee YK, Choi J, Wang W, et al. Nullifying tumor efflux by prolonged endolysosome vesicles: development of low dose anticancer-carbon nanotube drug. ACS Nano. 2013;7(10):8484–8497. | ||
Nam EJ, Kang JH, Sung S, et al. A matrix metalloproteinase 1-cleavable composite peptide derived from transforming growth factor β-inducible gene h3 potently inhibits collagen-induced arthritis. Arthritis Rheum. 2013;65(7):1753–1763. | ||
Edmonds SE, Blake DR, Morris CJ, Winyard PG. An imaginative approach to synovitis – the role of hypoxic reperfusion damage in arthritis. J Rheumatol Suppl. 1993;37:26–31. | ||
Edmonds SE, Ellis G, Gaffney K, Archer J, Blake DR. Hypoxia and the rheumatoid joint: immunological and therapeutic implications. Scand J Rheumatol Suppl. 1995;101:163–168. | ||
Nichols JW, Bae YH. Odyssey of a cancer nanoparticle: from injection site to site of action. Nano Today. 2012;7(6):606–618. | ||
Iversen TG, Skotland T, Sandvig K. Endocytosis and intracellular transport of nanoparticles: present knowledge and need for future studies. Nano Today. 2011;6(2):176–185. | ||
Sahay G, Alakhova DY, Kabanov AV. Endocytosis of nanomedicines. J Control Release. 2010;145(3):182–195. | ||
Mayor S, Pagano RE. Pathways of clathrin-independent endocytosis. Nat Rev Mol Cell Biol. 2007;8(8):603–612. | ||
Feldmann M, Maini RN. Lasker Clinical Medical Research Award. TNF defined as a therapeutic target for rheumatoid arthritis and other autoimmune diseases. Nat Med. 2003;9(10):1245–1250. | ||
Bianco A, Kostarelos K, Prato M. Applications of carbon nanotubes in drug delivery. Curr Opin Chem Biol. 2005;9(6):674–679. | ||
Liu Z, Robinson JT, Tabakman SM, Yang K, Dai H. Carbon materials for drug delivery & cancer therapy. Mater Today. 2011;14(7–8):316–323. | ||
Marrache S, Dhar S. Engineering of blended nanoparticle platform for delivery of mitochondria-acting therapeutics. Proc Natl Acad Sci U S A. 2012;109(40):16288–16293. | ||
Cossarizza A, Baccarani-Contri M, Kalashnikova G, Franceschi C. A new method for the cytofluorimetric analysis of mitochondrial-membrane potential using the J-aggregate forming lipophilic cation 5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazolcarbocyanine iodide (Jc-1). Biochem Biophys Res Commun. 1993;197(1):40–45. | ||
Simon HU, Haj-Yehia A, Levi-Schaffer F. Role of reactive oxygen species (ROS) in apoptosis induction. Apoptosis. 2000;5(5):415–418. | ||
Knight JA. Review: free radicals, antioxidants, and the immune system. Ann Clin Lab Sci. 2000;30(2):145–158. | ||
Komatsu N, Takayanagi H. Inflammation and bone destruction in arthritis: synergistic activity of immune and mesenchymal cells in joints. Front Immunol. 2012;3:77. | ||
Firestein GS. Evolving concepts of rheumatoid arthritis. Nature. 2003;423(6937):356–361. | ||
Zangerle PF, De Groote D, Lopez M, et al. Direct stimulation of cytokines (IL-1 beta, TNF-alpha, IL-6, IL-2, IFN-gamma and GM-CSF) in whole blood: II. Application to rheumatoid arthritis and osteoarthritis. Cytokine. 1992;4(6):568–575. |
Supplementary materials
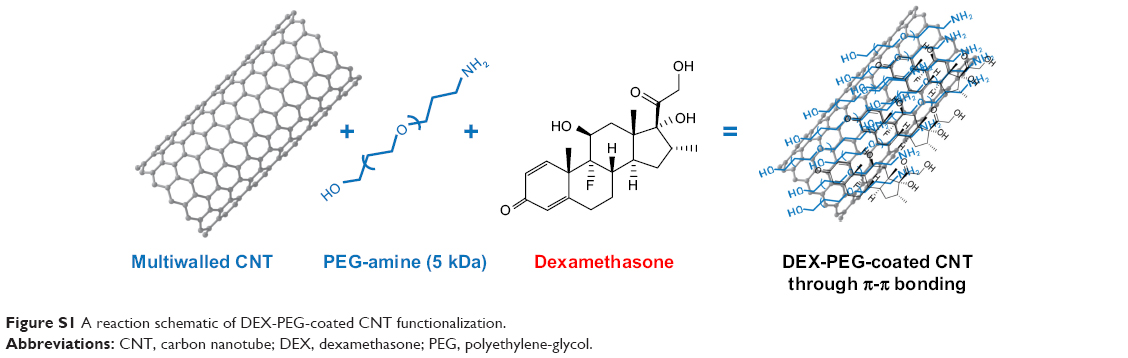 | Figure S1 A reaction schematic of DEX-PEG-coated CNT functionalization. |
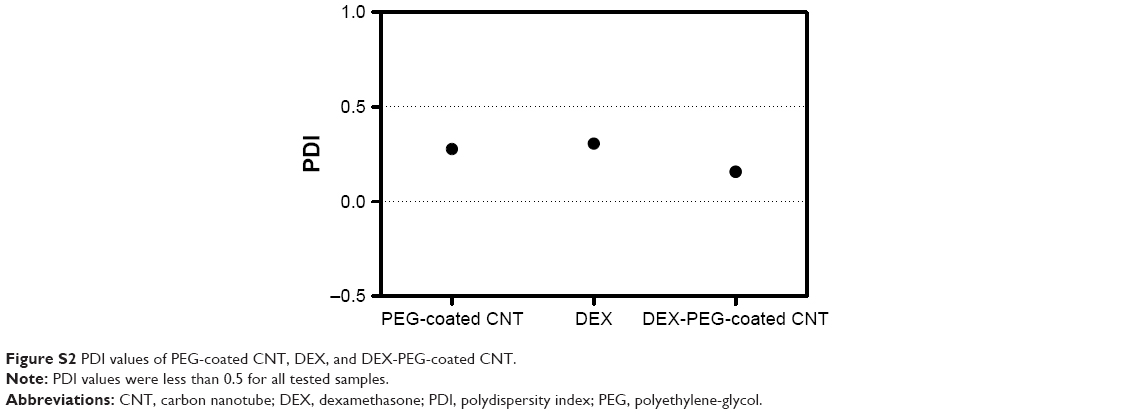 | Figure S2 PDI values of PEG-coated CNT, DEX, and DEX-PEG-coated CNT. |
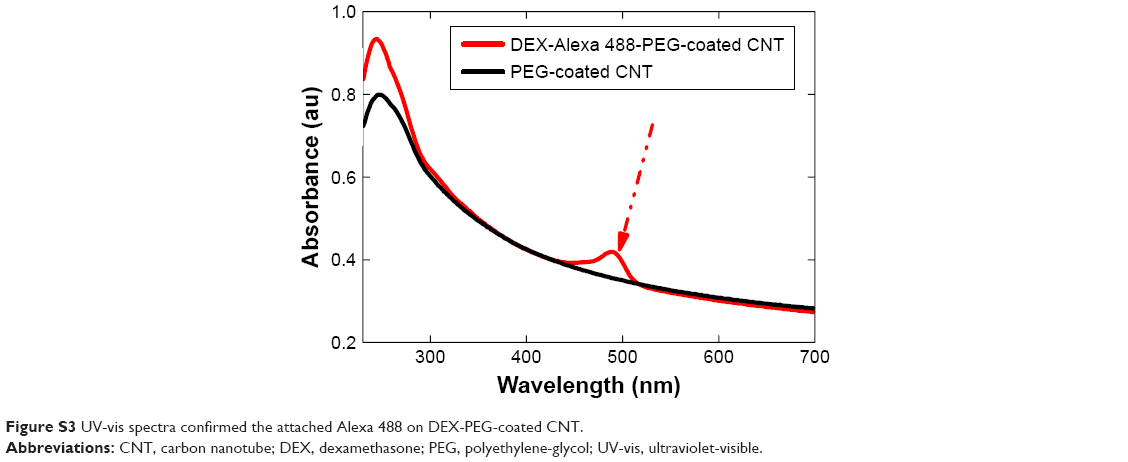 | Figure S3 UV-vis spectra confirmed the attached Alexa 488 on DEX-PEG-coated CNT. |
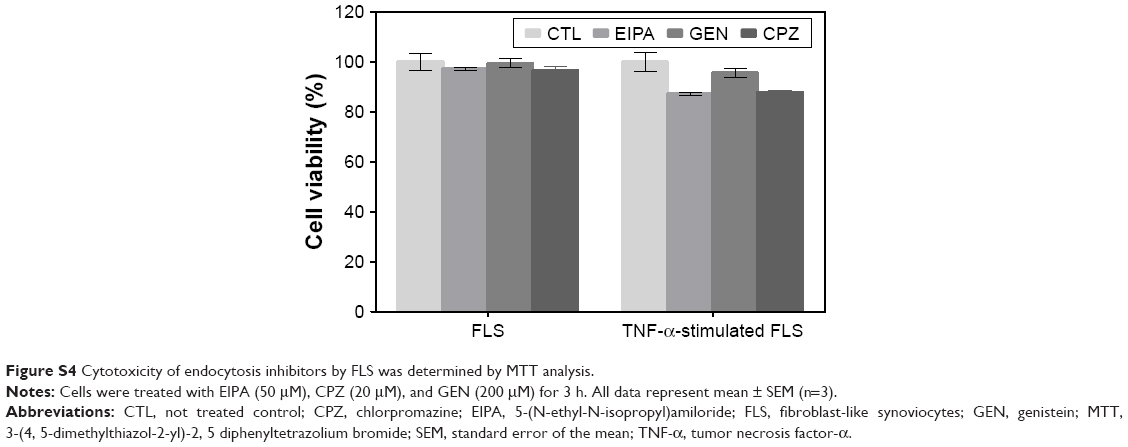 | Figure S4 Cytotoxicity of endocytosis inhibitors by FLS was determined by MTT analysis. |
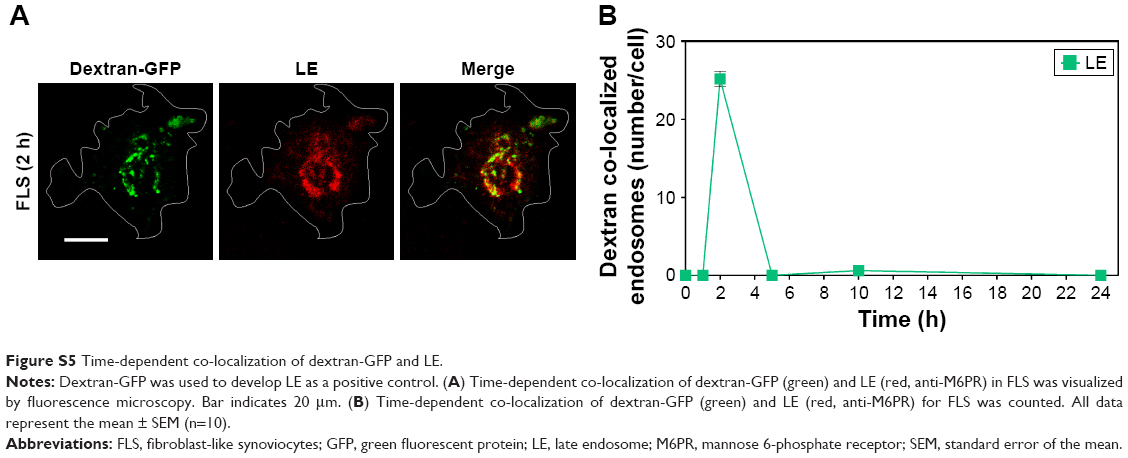 | Figure S5 Time-dependent co-localization of dextran-GFP and LE. |
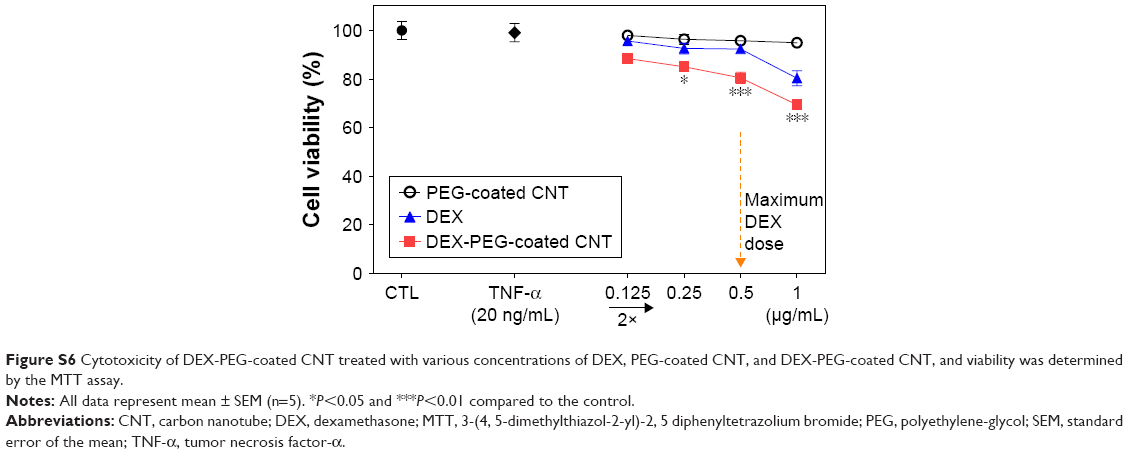 | Figure S6 Cytotoxicity of DEX-PEG-coated CNT treated with various concentrations of DEX, PEG-coated CNT, and DEX-PEG-coated CNT, and viability was determined by the MTT assay. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.