Back to Journals » International Medical Case Reports Journal » Volume 16
Supernumerary Kidneys Associated with Disorders of Sexual Development and Cloacal Anomaly: A Case Report
Authors Mesfin T ![]() , Haji N
, Haji N ![]() , Seyoume F, Seyoum K
, Seyoume F, Seyoum K ![]() , Mesfin E
, Mesfin E ![]() , Erdachew T, Ayane D
, Erdachew T, Ayane D ![]() , Badasa G, Soboka M
, Badasa G, Soboka M
Received 5 January 2023
Accepted for publication 19 March 2023
Published 23 March 2023 Volume 2023:16 Pages 193—199
DOI https://doi.org/10.2147/IMCRJ.S403690
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Ronald Prineas
Telila Mesfin,1 Nuri Haji,1 Fikadu Seyoume,2 Kenbon Seyoum,3 Eshetu Mesfin,4 Tsegaye Erdachew,5 Daniel Ayane,5 Gadisa Badasa,5 Moisan Soboka5
1Department of Medicine, Madda Walabu University Goba General Hospital, Goba, Ethiopia; 2Department of Pediatrics, Madda Walabu University Goba General Hospital, Goba, Ethiopia; 3Department of Midwifery, Madda Walabu University Goba General Hospital, Goba, Ethiopia; 4Department of Public Health, ICAP, Finfine, Oromia, Ethiopia; 5Department of Nursing, Madda Walabu University Goba General Hospital, Goba, Ethiopia
Correspondence: Telila Mesfin, Email [email protected]
Introduction: The term “disorders of sexual differentiation” refers to a variety of issues that result in the baby’s genitalia being underdeveloped or showing characteristics shared by both sexes. Normal sexual development in utero requires a precise and coordinated spatiotemporal sequence of numerous activating and suppressing factors. Inadequate development of the bipotential gonad into an ovary or a testis is one of the most frequent causes of genital ambiguity (partial gonadal dysgenesis). One in every 50,000 babies suffers from cloacal anomalies, which makes it one of the rarest congenital malformations. The supernumerary kidney is an extremely uncommon congenital abnormality with less than 100 cases reported in the literature.
Case: We present five days old neonate admitted to the neonatal intensive care unit with a complaint of absence of anal orifice. The baby had not passed meconium within 48 hours after delivery, but the families later realized that meconium had been passing through the urethral orifice along with urine. The child was born to a 32-year-old para-four woman who claims to have been amenorrheic for the past nine months but could not recall her last regular period. On physical examination, the abdomen was grossly distended, and there was no anal opening other than just a dimple on the sacrococcygeal area, and the external genitalia appears female on inspection with labia majora well developed and no fusion.
Conclusion: Disorder of sexual differentiation is a clinically diverse set of diseases that interferes with the proper differentiation and determination of sex in the embryo and fetus. One in 50,000 live births results in cloacal abnormalities, which are extremely uncommon. Less than 100 examples of the supernumerary kidney have been documented in the literature, making it an exceptionally rare congenital anomaly.
Keywords: supernumerary kidneys, neonate, cloacal anomaly, ambiguous genitalia
Introduction
The term “disorders of sexual differentiation” refers to a wide range of problems that cause the baby’s genitalia to be poorly developed or to exhibit traits common to both sexes.1 Hermaphrodite, pseudo-hermaphrodite, and intersex are archaic and derogatory words that were once used to characterize this entity.2,3 The consensus declaration on the management of intersex diseases has substituted the term disorders of sexual development (DSD) for them.4 A precise and coordinated spatiotemporal sequence of multiple activating and repressing variables is required for normal sexual development in utero.5 DSDs might appear as any variations from the typical differentiation pattern. In the course of typical sexual development, two separate processes take place. Before the determination of the indifferent genital ridge, which switches the developing gonad into male development, during 42 days of gestation, male and female embryos cannot be distinguished.6 One of the most common reasons for genital ambiguity is an incomplete development of the bipotential gonad into either an ovary or a testis (partial gonadal dysgenesis). Particularly in premature infants, the clitoris may seem disproportionately big in comparison to the other genital components. The clitoral width should be assessed if the clitoris seems larger; newborns with values greater than 6 mm should have further testing done. Congenital adrenal hyperplasia (which accounts for >90% of cases) is the most frequent cause of ambiguous genitals, and the salt-wasting types might hasten dehydration with a subsequent fluid and electrolyte imbalance.1 In XX and under-virilized XY individuals, the external genitalia might also appear female.
The genital, urinary, and gastrointestinal tracts all share a single perineal outlet in cloacal malformation. This confluence typically lasts until the baby is about 5 weeks pregnant embryologically. The developing tail fold gives rise to the cloaca, a temporary structure that generally develops at the intersection of the allantois and the hindgut at three weeks.7–9 Cloacal anomalies are very rare with an incidence of one in 50,000 live births.10–12 This anomaly was regarded as incompatible with life till surgical corrections were attempted in 1950.13 The diagnosis might be made antenatally via ultrasound imaging, but it might also come as a surprise upon delivery. Identification of the cloacal defect together with the proper resuscitation and stabilization are the main goals of immediate therapy. Transfer to a multidisciplinary specialized center should come right after that.12
Less than 100 examples of the supernumerary kidney have been documented in the literature, making it an incredibly rare congenital defect.14,15 Supernumerary kidney may result from the nephrogenic cord’s uneven and aberrant division into two metanephric blastemas, which then grow into two kidneys with an incomplete or double ureteral bud.16–18 These congenital defects rarely occur all at once, and there has not been a record in the literature as of yet.
Case Presentation
We present five days old neonate admitted to the neonatal intensive care unit with a complaint of absence of anal orifice. The baby did not pass meconium for 48hrs since birth and later on, the families noticed that meconium passed through the urethral orifice together with urine. Following further evaluation, they found that there was no anal opening apart from a slight fossa on the area that was supposed to be an anal opening (Figure 1). In addition, the neonate had abdominal swelling which progressively increased since birth. Otherwise, the neonate had no fever, yellowish discoloration of skin and eyes, vomiting, and abnormal body movement.
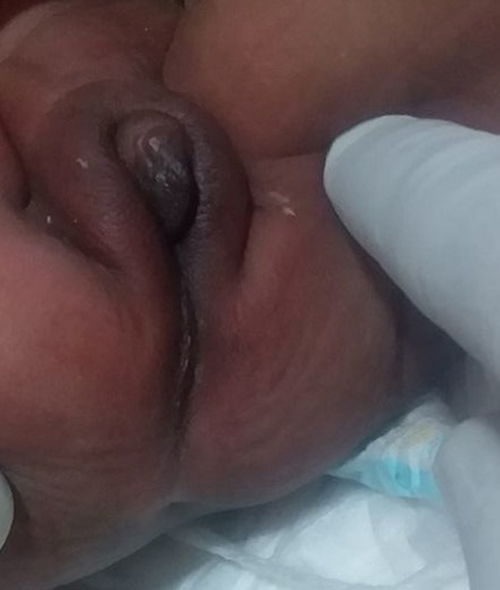 |
Figure 1 Female looking external genitalia, enlarged clitoris and absence of anal orifice. |
The baby was born to a 32-year-old para-four mother who does not remember her last normal menstrual period but claimed to be amenorrheic for the past nine months. The mother had antenatal care at the nearby health center and it was uneventful. She gave birth at the nearby health center by Spontaneous vaginal delivery after laboring for 14 hours and the birth weight was 3.2kg. The neonate cried immediately after birth with APGAR scores of 7 and 10 at 1st and 10th minutes, respectively. The mother had no chronic medical illnesses like Hypertension and diabetes mellitus. Her prior children are all healthy and alive.
On physical examination, general appearance was alert, and vital signs were pulse rate = 152 beats per minute, respiratory rate = 46 breaths per minute, temperature 36°C, and venous oxygen saturation 97% at room air. On HEENT examination, head circumference was 42 cm and anterior fontanel size was 2cm. The pertinent positive physical findings were on abdomen and Genito-urinary system. The abdomen was grossly distended, and there was no anal opening other than just a dimple on sacrococcygeal area. On the genito-urinary system, external genitalia appears female on inspection with labia majora well developed and no fusion. However, upon palpation, the labia minora fused entirely to the base of the clitoris and there is no vaginal opening. The phallus measures about 0.8cm with a central opening through which loose stool leaks while crying. The urethral opening is a common pathway for the urinary tract, genital, and rectum. There are no gonads palpable at the labioscrotal folds and inguinal area (Figure 2).
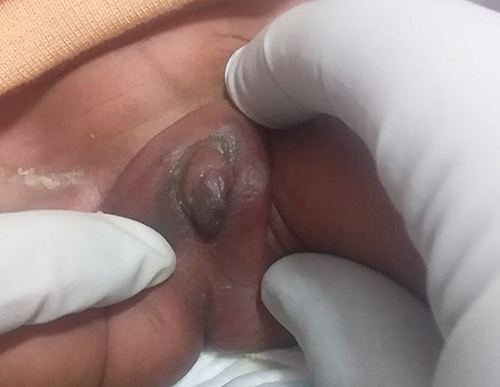 |
Figure 2 Enlarged phallus and fusion of labia minora with the base of clitoris and no vaginal orifice. |
Laboratory tests and imaging were done to explore further findings. On complete blood count, white blood cell = 20,000, neutrophil = 53%, lymphocyte = 23%, red blood cell = 5*106, hemoglobin=15gm/dl, platelet = 272*103, the blood group was A+, and random blood sugar was 120gm/dl. Plain abdominal x-ray was taken and it showed absence of air shadow in the distal large bowel (Figure 3). The abdominopelvic sonography was done to reveal any Mullerian structures and distal bowel condition. However, it only showed that unilateral (left side) supernumerary kidney, one lying in renal fossa and the other lying caudally on the left psoas muscle both having distinct capsules and blood supply with a contralateral single normal kidney. The ultrasonography did not comment on the Mullerian structures due to collapsed bladder being unable to visualize the structures (Figure 4).
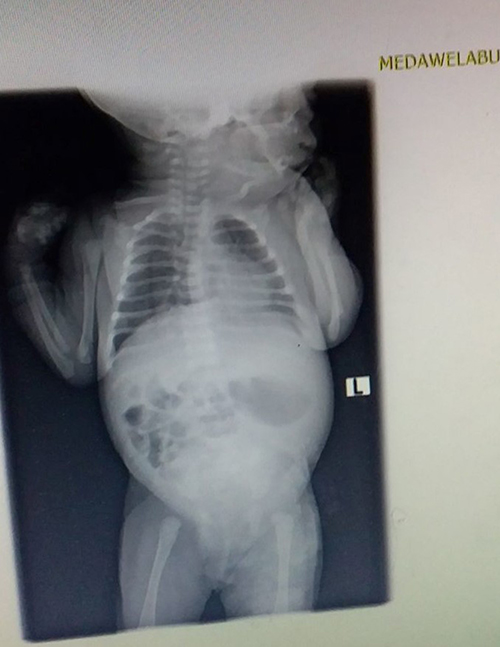 |
Figure 3 Plain abdominal x-ray showing absence of air shadow in the rectum. |
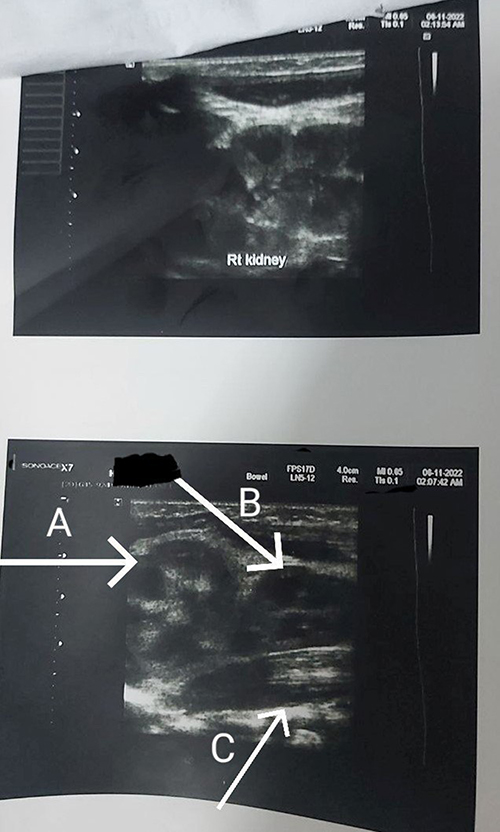 |
Figure 4 Abdominopelvic ultrasound showing left side supernumerary kidneys and right side single normal kidney ((A): left kidney (B): left supernumerary kidney lying on psoas muscle (C): left psoas muscle). |
The patient was admitted with a diagnosis of term +Normal birth weight +Appropriate for gestational age +Cloacal anomaly +Ambiguous genitalia+ Supernumerary kidney+ Early onset neonatal sepsis. The neonate was managed with calculated maintenance fluid and intravenous antibiotics, ampicillin 150mg/kg/dose Iv twice daily (BID) and gentamicin 3mg/kg/dose iv daily. On the second day of admission, the baby was referred to black lion specialized hospital for better evaluation and management.
Discussion
When chromosomal, gonadal, and anatomical sex development occurs atypically, it is referred to as disorders of sex development (DSD). This can have an impact on 1:1000 to 1:1500 people, with the most extreme ambiguity being seen in up to 1:5000 born.19,20 Disorder of sexual differentiation is a clinically diverse set of diseases that interferes with the proper differentiation and determination of sex in the embryo and fetus. While certain disorders may show up later in life, others may appear from birth as ambiguous genitalia.21 The gonads of males and females are not distinguished within the genital bud until the sixth week. The gonad in the testis starts to differentiate under the control of the Sry/Sox9 set and other genes. Sertoli cells develop from support cells, while Leydig cells develop from steroidogenic cells over the course of about 60 days. The first oocytes begin to emerge from the gonad predestined to become an ovary after 77 to 84 days. The granulosa is made up of supporting cells, and the theca is made up of steroidogenic cells.22 The Wolffian ducts in the seminal vesicles, ductus deferens, and epididymis, as well as the Müllerian ducts, are differentiated as a result of the expression of the SRY gene on the Y chromosome. This cascade results in the formation of a testis, which secretes testosterone (Leydig cells) and anti-Müllerian hormone (AMH) (Sertoli cells). Additionally, insulin-like factor 3 (INSL3), which influences testicular descent, is produced by Leydig cells.23 In the absence of the Y chromosome, the Müllerian ducts develop the uterus, fallopian tubes, and upper third of the vagina, while the Wolff ducts regress.19 For healthy sexual development in utero, a precise spatiotemporal sequence of several activating and repressing factors is necessary.5 Any abnormalities from the standard differentiation pattern could manifest as disorders of sexual development.
Given that it is frequently an unexpected occurrence, recognition of ambiguous genitalia at birth could happen in any obstetric facility or environment.24,25 The results produce a difficult diagnostic issue and a complicated psychosocial scenario. The neonate we present has normal looking labia majora and fused labia minora with the base of clitoris. The phallus is enlarged measuring 0.8 cm having central opening through which urine and loose meconium pass. The external genitalia looks like female whilst having virilization.
The cause of persistent cloaca, also known as cloacal anomalies, which are abnormalities of the urogenital sinus and anorectum, is not yet recognized.12 These disorders are uncommon, although it can be challenging to determine their exact occurrence because of classification issues and the inclusion of other rectal anomalies in certain estimations. Yet, it is estimated that there are roughly 1:50,000 live births globally with cloacal abnormalities.26 Some passage of meconium happens during the first 12 hours of delivery, and 95% of premature babies and 99% of term babies pass meconium within 48 hours. If the anal orifice is nonexistent or misplaced, a physical examination alone can generally diagnose an imperforate anus. To rule out any potential related cardiac, renal, or spine defects (VACTREL), all neonates with anorectal or cloacal malformations should be examined.1 The most severe and complicated anorectal (ARM) and urogenital malformations, accounting for 10% of all ARM, are cloacal anomalies, which are rare congenital deformities. Individuals with persistent cloaca are always female because they have a single perineal opening for rectum, urethra, and vagina.27 The same is true for our case. She has one common opening for vagina, urethra and rectum. The external urethral opening serves as a common outlet to the perineum for these three structures.
One of the urogenital system’s rarest congenital defects is a supernumerary kidney.28 Other anomalies like ureteral atresia, vaginal atresia, horseshoe kidney, complete duplication of the urethra and penis with an ectopic ureteral opening into the vagina or introitus, imperforate anus, ventricular septal defects, meningomyelocele, and coarctation of the aorta may also be present in conjunction with the extra kidney.29 The extra kidney may be attached to the normal kidney through loose areolar tissue or may be completely independent from it.18 It is challenging to define a methodology for diagnosis due to the great variety of combined congenital defects and the relative rarity of such instances; as a result, overdiagnosis with numerous pointless imaging tests is carried out. Therefore, it can be difficult to diagnose patients who have extra kidneys. The majority of these instances were diagnosed utilizing a variety of radiological imaging methods in the literature, including CT and MRI scans, CT angiography, IVP, ultrasound, and scans using dimercaptosuccinic acid (DMSA) and diethylene triamine pentaacetate (DTPA). Although some writers claim that IVP, CT, and ultrasound are sufficient for the identification of extra kidneys.30 In our case, abdominopelvic ultrasound revealed two left kidneys, one situated in renal fossa and the other lying caudally on left psoas muscle both having distinct capsule and blood supply with contralateral single normal kidney. However, it did not comment on the mullerian structures due to empty bladder unable to visualize the structures.
To our knowledge, there is no literature report on congenital anomalies containing ambiguous genitalia, cloacal anomalies and supernumerary kidneys all at once. Even though supernumerary kidney is among rarest congenital urological anomalies, the majority of case reports focused on the location and numbers of the kidneys than its association with the above anomalies. Therefore, it is worth reporting to advance clinical medicine and evidence-based management of patients with the conditions.
Conclusion
Disorder of sexual differentiation is a clinically diverse set of diseases that interferes with the proper differentiation and determination of sex in the embryo and fetus. Cloacal anomaly is the most severe and rarest form of anorectal malformations. The supernumerary kidney is an extremely uncommon congenital abnormality with less than 100 cases reported in the literature. Therefore, it is worth reporting to advance clinical medicine and evidence-based management of patients with the conditions.
Data Sharing Statement
Data on the case clinical information, informed consent form, and images are available for review from the corresponding author upon reasonable request.
Consent
Written informed consent was taken from the patient’s parents for publication of her condition and accompanying images.
Disclosure
The authors declare that there are no conflicts of interest in this work.
References
1. Kliegman RM, Toth H, Bordini BJ, Basel D. Nelson Pediatric Symptom-Based Diagnosis E-Book. Elsevier Health Sciences; 2022.
2. Hughes IA, Houk C, Ahmed SF, et al. Consensus statement on management of intersex disorders. J Pediatr Urol. 2006;2(3):148–162. doi:10.1016/j.jpurol.2006.03.004
3. Hughes IA, Nihoul-Fékété C, Thomas B, et al. Consequences of the ESPE/LWPES guidelines for diagnosis and treatment of disorders of sex development. Best Pract Res Clin Endocrinol Metab. 2007;21(3):351–365. doi:10.1016/j.beem.2007.06.003
4. Mehmood KT, Rentea RM. Ambiguous genitalia and disorders of sexual differentiation. In: StatPearls. StatPearls Publishing; 2022.
5. Witchel SF. Disorders of sex development. Best Pract Res Clin Obstet Gynaecol. 2018;48:90–102. doi:10.1016/j.bpobgyn.2017.11.005
6. Warne GL, Zajac JD. Disorders of sexual differentiation. Endocrinol Metab Clin North Am. 1998;27(4):945–967. doi:10.1016/S0889-8529(05)70049-9
7. Qi BQ, Williams A, Beasley S, et al. Clarification of the process of separation of the cloaca into rectum and urogenital sinus in the rat embryo. J Pediatr Surg. 2000;35(12):1810–1816. doi:10.1053/jpsu.2000.19265
8. Cilento BG Jr, Benacerraf BR, Mandell J. Prenatal diagnosis of cloacal malformation. Urology. 1994;43(3):386–388. doi:10.1016/0090-4295(94)90086-8
9. Adams MC, Ludlow J, Brock JOHNWIII, et al. Prenatal urinary ascites and persistent cloaca: risk factors for poor drainage of urine or meconium. J Urol. 1998;160(6 Part 1):2179–2181. doi:10.1016/S0022-5347(01)62288-2
10. Zaccara A, Gatti C, Silveri M, et al. Persistent cloaca: are we ready for a correct prenatal diagnosis? Urology. 1999;54(2):367. doi:10.1016/S0090-4295(99)00135-1
11. Alexander F, Kay R. Cloacal anomalies: role of vesicostomy. J Pediatr Surg. 1994;29(1):74–76. doi:10.1016/0022-3468(94)90528-2
12. Fernando MA, Creighton SM, Wood D. The long-term management and outcomes of cloacal anomalies. Pediatric Nephrol. 2015;30:759–765. doi:10.1007/s00467-014-2875-7
13. Petrikovsky BM, Walzak MP Jr, D’Addario PF. Fetal cloacal anomalies: prenatal sonographic findings and differential diagnosis. Obstet Gynecol. 1988;72(3):464–468.
14. Sureka B, Mittal MK, Mittal A, et al. Supernumerary kidneys – a rare anatomic variant. Surg Radiol Anat. 2014;36(2):199–202. doi:10.1007/s00276-013-1135-z
15. McAninch JW. Supernumerary kidneys; 2022.
16. Oto A, Kerimoğlu U, Eskiçorapçi S, et al. Bilateral supernumerary kidney: imaging findings. JBR-BTR. 2002;85(6):300–303.
17. Tada Y, Kokado Y, Hashinaka Y, et al. Free supernumerary kidney: a case report and review. J Urol. 1981;126(2):231–232. doi:10.1016/S0022-5347(17)54457-2
18. N’guessan G, Stephens FD. Supernumerary kidney. J Urol. 1983;130(4):649–653. doi:10.1016/S0022-5347(17)51385-3
19. Gomez-Lobo V. Multidisciplinary care for individuals with disorders of sex development. Curr Opin Obstet Gynecol. 2014;26(5):366. doi:10.1097/GCO.0000000000000101
20. Thyen U, Lanz K, Holterhus P-M, et al. Epidemiology and initial management of ambiguous genitalia at birth in Germany. Horm Res Paediatr. 2006;66(4):195–203. doi:10.1159/000094782
21. Hyun G, Kolon TF. A practical approach to intersex in the newborn period. Urol Clin. 2004;31(3):435–443. doi:10.1016/j.ucl.2004.04.008
22. Caldas BC, Lopes AA, Farias MPCBE. Ambiguous genitalia. In: Endocrinology and Diabetes. Springer; 2022:197–206.
23. Ferlin A, Foresta C. Insulin‐like factor 3: a novel circulating hormone of testicular origin in humans. Ann NY Acad Sci. 2005;1041(1):497–505. doi:10.1196/annals.1282.074
24. Meyers-Seifer CH, Charest NJ. Diagnosis and management of patients with ambiguous genitalia. Semin Perinatol. 1992;16:332–339.
25. Izquierdo G, Glassberg KI. Gender assignment and gender identity in patients with ambiguous genitalia. Urology. 1993;42(3):232–242. doi:10.1016/0090-4295(93)90610-M
26. Hendren WH. Cloaca, the most severe degree of imperforate anus: experience with 195 cases. Ann Surg. 1998;228(3):331. doi:10.1097/00000658-199809000-00006
27. Caldwell BT, Wilcox DT. Long-term urological outcomes in cloacal anomalies. In: Seminars in Pediatric Surgery. Elsevier; 2016.
28. Mejia M, Limback J, Ramirez A, et al. A case of supernumerary kidney. Cureus. 2018;10(12). doi:10.7759/cureus.3686
29. Wu J, Garcia J. Supernumerary kidney with Wilms’ tumor. Wis Med J. 1971;70(10):211–216.
30. Abid BH, Moalla N, Zmerli S. Pelvic kidney apropos of 50 cases. Ann Urol Paris. 1989;23:11–16.
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.