Back to Journals » International Journal of Nanomedicine » Volume 10 » Issue 1
Styrene maleic acid micelles as a nanocarrier system for oral anticancer drug delivery – dual uptake through enterocytes and M-cells
Authors Parayath NN ![]() , Nehoff H, Müller P, Taurin S, Greish K
, Nehoff H, Müller P, Taurin S, Greish K ![]()
Received 30 April 2015
Accepted for publication 13 June 2015
Published 22 July 2015 Volume 2015:10(1) Pages 4653—4667
DOI https://doi.org/10.2147/IJN.S87681
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Thomas Webster
Neha N Parayath,1 Hayley Nehoff,1 Philipp Müller,1 Sebastien Taurin,1 Khaled Greish1,2
1Department of Pharmacology and Toxicology, University of Otago, Dunedin, New Zealand; 2Department of Oncology, Faculty of Medicine, Suez Canal University, Ismaileya, Egypt
Abstract: Drug delivery systems could potentially overcome low bioavailability and gastrointestinal toxicity, which are the major challenges for the development of oral anticancer drugs. Herein, we demonstrate the ability of styrene maleic acid (SMA) nanomicelles encapsulating epirubicin to traverse in vitro and ex vivo models of the intestinal epithelium without affecting the tissue integrity. Further, SMA micelles encapsulating a fluorescent dye dioctadecyl-3,3,3',3'-tetramethylindocarbocyanine perchlorate (DiI) showed twofold higher accumulation in the liver and spleen, 15-fold higher accumulation in the tumor, and sixfold higher accumulation in the lung as compared with the free DiI, following oral administration in a mice xenograft breast cancer model. Additionally, SMA micelles showed colocalization with microfold (M)-cells and accumulation in Peyer’s patches, which together confirms the M-cell mediated uptake and transport of SMA micelles. Our results indicate that SMA micelles, showing dual uptake by enterocytes and M-cells, are a potential tool for safe oral anticancer drug delivery.
Keywords: oral delivery, anticancer nanomedicine, enhanced permeability and retention effect, EPR
Introduction
Oral administration of drugs is widely preferred among patients due to its noninvasive nature. However, most anticancer agents are administered intravenously due to the narrow therapeutic index and the observed intra- and intersubject variability in bioavailability upon oral administration.1 Nevertheless, development of oral chemotherapeutics has gained attention, mainly, to overcome complications associated with intravenous injections, improve the patients’ quality of life, and reduce costs associated with hospitalization.2 Oral drug delivery can be tailored to maintain drug plasma concentrations above the therapeutic level, to ensure prolonged and effective exposure of cancer cells to the chemotherapeutic agent. However, the major limiting factors associated with oral administration of chemotherapeutic agents are their low bioavailability3 and severe local toxicity while traversing the gastrointestinal (GI) tract.4,5
Low bioavailability is the result of multiple factors, such as low drug stability in the wide pH range and enzyme-rich environment of the GI tract, poor penetration through the mucosal barrier, a high level of P-glycoprotein efflux, as well as presystemic metabolism in the liver after traversing the intestinal epithelium.6–8 Thus new strategies are needed to improve the oral bioavailability of anticancer drugs.
Nanosized formulations take advantage of the wide fenestrations of tumor vasculature and the impaired lymphatic drainage to selectively accumulate in the tumor tissue.9 Exploiting this enhanced permeability and retention (EPR) effect improves the therapeutic efficacy and safety of these nanosized drugs. Apart from the above characteristics, nanoparticles are widely used for drug delivery due to their ability to prolong circulatory half-life and their flexibility in accommodating tumor-targeting moieties.10 Although different nanocarriers, such as liposomes, polymeric micelles, polymer drug conjugates, self-emulsifying drug delivery systems, dendrimers, and solid lipid nanoparticles are being examined for oral delivery of anticancer drugs, so far there has been no oral anticancer nanomedicine available for clinical use. This is primarily due to poor penetration through the GI mucus.2,6
The amphiphilic nature of the styrene maleic acid (SMA) polymer has enabled the encapsulation of wide range of drugs.9,11 The SMA system is especially well-suited for translational applications, due to the low costs of the polymer, simplicity of the synthetic process, and the ability to control the loading (up to 40% w/w) and release rate of active drugs in the micelle.12
We anticipated that the amphiphilic nature of SMA micelle could enable it to pass through the mucosal barrier of the GI epithelium. In this study, we synthesized SMA micelles encapsulating epirubicin (as a model chemotherapeutic agent) to determine their ability to traverse through the in vitro and ex vivo models of the intestinal epithelium. Furthermore, a nontoxic fluorescent dye, dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate (DiI), was encapsulated in the SMA micelles to evaluate the biodistribution and uptake mechanism of the SMA micelles by the intestinal epithelium following oral administration, in a mouse xenograft breast cancer model.
Materials and methods
Materials
Cumene terminated poly (styrene-co-maleic anhydride) with a number average molecular mass (Mn) ~1,600, N-(3-dimethylaminopropyl)-N-ethylcarbodiimide hydrochloride (EDAC), fetal bovine serum, DiI, and biotin conjugated lectin from Ulex europaeus agglutinin (UEA-1) were obtained from Sigma-Aldrich Corp (St Louis, MO, USA). Antibodies were purchased from Cell Signaling Technology, Inc (Danvers, MA, USA). Antibodies used were: caveolin-1 (D46G3) XP® rabbit mAb #3267, clathrin heavy chain (D3C6) XP® rabbit mAb #4796, E-cadherin, and β-tubulin antibody #2146.
Synthesis of SMA micelles
SMA micelles were prepared as described previously.9 Briefly, the synthesis of SMA micelles was divided into three steps. The first step involved preparation of the SMA for encapsulating epirubicin or DiI. SMA was hydrolyzed under alkaline conditions by dissolving SMA in 1 M NaOH, with constant stirring at 70°C. Once soluble under alkaline conditions, the pH of the hydrolyzed SMA was adjusted to pH 7.0 with 0.1 N HCl and then, diluted to a final concentration of 10 mg/mL.
The next step involved encapsulating the epirubicin or DiI. The hydrolyzed SMA solution was adjusted to pH 5; epirubicin or DiI (15 mg of DiI dissolved in 300 μL of dimethyl sulfoxide [DMSO]) was added to the SMA solution under constant stirring. EDAC solubilized in distilled water was added to the mixture and allowed to stir for 20 minutes at pH 5. The solution was then adjusted to pH 11 with 0.1N NaOH, and stirred for 30 minutes. The pH was readjusted to 7.4 with 0.1 N HCl.
Lastly, the micelle suspension was ultrafiltered four times using a Labscale™ ultrafiltration system with a Pellicon® XL filter 10kDa (EMD Millipore, Billerica, MA, USA). The concentrated micelle solution was lyophilized to obtain the SMA-epirubicin (SMA-Epi) or SMA-DiI powder.
Characterization of SMA micelles
Loading of the SMA micelles
The loading of SMA micelles was determined by solubilizing SMA-Epi or SMA-DiI micelles at 1mg/mL in DMSO and measuring the absorbance at 480 nm and 544 nm for epirubicin and DiI, respectively, in comparison with the standard curve of the free drugs. The loading was expressed as weight % of epirubicin/DiI in the final micelle compared with the total weight of recovered SMA micelle.
Size, polydispersity index (PDI), and zeta potential determination of SMA micelles
Lyophilized SMA-Epi or SMA-DiI powder, 4 mg/mL, was solubilized in NaHCO3 (0.1M, pH 7.4) to determine the size and PDI. The same amount, 4 mg/mL, of lyophilized SMA-Epi or SMA-DiI powder was solubilized in distilled water to estimate the charge. All measurements for size distribution and zeta potential were carried out using the Malvern ZEN3600 Zetasizer Nano series (Malvern Instruments Inc., Malvern, UK) based on dynamic light scattering (DLS). Measurements were obtained from three independent experiments, each conducted in triplicate. For transmission electron microscopy (TEM), 10 μL of 7.5% SMA-Epi micelles (1 mg/mL of SMA-Epi lyophilized powder, solubilized in distilled water) was placed on the grid. Then, 1% phosphotungstic acid was added on the grid for negative staining, and the grid was visualized using a Philips CM100 BioTWIN transmission electron microscope (Philips/FEI Corporation, Eindhoven, the Netherlands).
Release rate from SMA micelles at physiological pH and in simulated gastric fluid (SGF)
Using a dialysis bag with a 12 kDa molecular weight cut off, 1.5 mL of SMA-Epi solution (1mg/mL of SMA-Epi lyophilized powder, solubilized in distilled water) was dialyzed against 15mL of distilled water (pH 7.4) or SGF (with pepsin, pH 1.6). At specified time points, 2mL of sample outside the dialysis bag was removed to measure the absorbance at 480 nm and replaced with distilled water or gastric fluid, to measure further release at physiological pH or gastric pH, respectively. Percentage of release was determined by the ratio of absorbance between solution outside the bag at defined time points and that within the bag at time zero (t=0).
Cell culture
Caco-2 cells and MDA-MB-231 breast cancer cells were maintained in complete growth media (Dulbecco’s Modified Eagle’s Medium [DMEM]/ Ham’s F12, supplemented with 10% fetal bovine serum, 2mM L-glutamine, 100μg/mL penicillin, 100units/mL of streptomycin, and 2.2g/L of NaHCO3) at 37°C in a humidified atmosphere of 5% CO2. For all procedures, cells were harvested using TrypLE™ Express (Life Technologies Corp, Carlsbad, CA, USA).
In vitro cytotoxicity of SMA micelles
To ascertain that micellar encapsulation did not abolish the cytotoxicity of epirubicin, MDA-MB-231 cells were seeded into 96-well plates. After a 24-hour incubation, cells were treated for 72 hours with free epirubicin, 7.5% or 18% SMA-Epi micelles, or equivalent amounts of SMA polymer at the concentrations indicated. Following the incubation, cells were fixed using 10% trichloro-acetic acid (TCA). Cytotoxicity was determined using the sulforhodamine B assay (SRB) as previously described.13 The concentration required to decrease the cell number by 50% (IC50) was determined by nonlinear regression using GraphPad Prism software. Treatments were performed in triplicate, and data represent the mean of three independent experiments.
In vitro system for evaluating the transport across intestinal epithelium
In order to evaluate the ability of SMA-Epi micelles to traverse the intestinal epithelium, Caco-2 cells, which are known to differentiate into an enterocyte-like phenotype, were used as an in vitro model of the intestinal epithelium. Caco-2 cells were seeded at a density of 80,000 cells/cm2 onto polycarbonate Transwell filters (Costar®; Corning Inc, Corning, NY, USA) with a 0.4 μm mean pore size and diameter of 12 mm. Caco-2 cells were maintained under standard incubation conditions with the media being changed every alternate day, and the cells were used for transport experiments 21–22 days postseeding. Prior to the experiments, the transepithelial electrical resistance (TEER) of each monolayer was measured with an epithelial voltohmmeter to ascertain the monolayer integrity. Monolayers with TEER >350 Ω/cm2 were used for assays. Cell monolayers were washed with Hank’s balanced salt solution (HBSS) buffer supplemented with 1 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) (pH 7.4). SMA-Epi solution (100 μM) was added to the apical compartment. After incubating for 3 hours, TEER was measured, and media from the basolateral compartment was collected. Transport of the SMA-Epi was quantified by measuring absorbance of epirubicin in the media of the basolateral compartment at 480 nm. For transepithelial transport in the presence of endocytosis inhibitors, the cell monolayers were preincubated for 1 hour with 100 μM of dansylcadaverine or 100 μM of genistein or 50 μM of amiloride.
MTT (3-(4,5-dimethylthiazolyl-2)-2,5-diphenyltetrazolium bromide) assay
The concentrations of the endocytosis inhibitors to be used for the transport studies were determined by MTT assay. The Caco-2 cells were seeded into 96-well plates, incubated for 24 hours, and then treated for 4 hours with 50 μM, 100 μM, and 200 μM of endocytosis inhibitors. Following the incubation, the MTT assay was performed as previously described,14 and absorbance was measured at 560 nm to determine the cytotoxicity of the endocytosis inhibitors in Caco-2 cells. Treatments were performed in triplicate, and the data represent the mean of three independent experiments. The concentration that maintained the cell viability above 80% was used for the transport studies.
Western blot analysis for caveolin and clathrin expression in differentiated and undifferentiated Caco-2 cells
Caveolin and clathrin were detected through western blot analysis to compare their expression in differentiated and undifferentiated Caco-2 cells. Western blot analysis was performed on cell lysates of confluent Caco-2 (5–6 days) and Caco-2 grown on Transwell® (21 days). Cells were lysed in buffer containing 50mM Tris-HCl (pH 8), 150mM NaCl, 1% Triton™ X-100, 1% sodium dodecyl sulfate (SDS), 1mM NaF, 200 μM sodium orthovanadate, and protease inhibitors (1μg/mL leupeptin, 1μg/mL aprotinin, and 1mM PMSF). The lysates were centrifuged at 12,500rpm for 8 minutes, boiled in Laemmli buffer, and subjected to 10% polyacrylamide gel electrophoresis. Proteins were transferred to a polyvinylidene difluoride (PVDF) membrane, and the membrane was incubated overnight with primary antibodies against caveolin (1/2,500) and clathrin (1/2,000), β-tubulin (1/2,000), and E-cadherin (1/2,000) and further incubated with horseradish peroxide–conjugated secondary antibody.
Ex vivo system for evaluating the transport across intestinal epithelium
In order to evaluate the ability of SMA-Epi micelles to traverse the intestinal epithelium, we used the everted sac of rat intestinal tissue as an ex vivo model.15 The intestines were obtained from male Sprague-Dawley® rats, 4–6 weeks of age. A midline incision was made in the abdomen, and the entire small intestine was quickly excised and irrigated several times with HBSS at room temperature (RT), and immediately placed at 37°C in HBSS with a 5% CO2 and 95% O2 supply. The intestine was gently everted over a glass rod and divided into 5 cm sections of duodenum, jejunum, and ileum. These sections were ligated at both ends and filled with 1 mL HBSS. The sacs were immersed in 10 mL of SMA-Epi (100 μM) in HBSS with a 5% CO2 and 95% O2 supply. The transport of SMA-Epi across the intestinal epithelium was determined after 3 hours of incubation by measuring absorbance at 480 nm. The effect of the treatment on the tissue integrity was assessed by histology. Sections were fixed in 4% paraformaldehyde for 3 hours and then transferred to a 15% sucrose solution for overnight at 4°C, followed by 30% sucrose solution for 10 hours at 4°C. These tissue were then embedded in OCT gel and stored at −20°C. Sections of 10 μm were obtained using a Leica CM1950 cryostat (Leica Microsystems, Wetzlar, Germany) and stained with Hematoxylin. Representative images were obtained using a Nikon Ni-E upright fluorescence microscope (Nikon Corporation, Tokyo, Japan).
All ex vivo animal protocols were approved by the University of Otago Animal Ethics Committee.
In vivo oral biodistribution of SMA micelles in a subcutaneous tumor model
Female SCID® mice (5–6 weeks old) were purchased from the Animal Resources Center (Canning Vale, Australia). Mice were injected subcutaneously into the lower rear flank with MDA-MB-231 breast cancer cells (2×106 cells/0.1 mL Matrigel®). When tumors reached an average volume of ~100 mm3, mice were randomly divided into three groups. The free DiI (15 mg/kg solubilized in 1% DMSO) and SMA-DiI (15 mg/kg equivalent, solubilized in D/W) were administered through oral gavage, while the third group received SMA-DiI (15 mg/kg solubilized in distilled water [D/W]) intravenously. Animals were euthanized in a CO2 chamber 8 hours posttreatment. For histological examination, all organs were processed as described above. The sections were mounted on a glass slide, stained with Hematoxylin, and representative images were obtained using the Nikon upright fluorescence microscope. The amount of DiI distributed in each tissue was calculated based on the fluorescence intensity across ten sections of the tissue, subtracting the background autofluorescence and normalized based on the area of tissue under observation, using ImageJ software.
All in vivo procedures were approved by the Animal Ethics Committee of University of Otago.
Histochemistry of intestinal sections
To evaluate the in vivo uptake mechanism of SMA micelles, Balb/c female mice (5–6 weeks old) were administered SMA-DiI (15 mg/mL, solubilized in D/W) by oral gavage (n=4). Animals were euthanized in a CO2 chamber 4 hours later. For histological examination, organs were processed as described above. In order to investigate the role of lymphatic transport of the SMA micelles, histochemistry was performed on intestinal sections with UEA-1, a microfold (M)-cell marker. The fluorescence images of the sections were obtained to visualize the distribution of SMA-DiI. These sections were then washed twice with phosphate-buffered saline (PBS), fixed in acetone, and incubated with hydrogen peroxide (0.3%) for 20 minutes at RT. Slides were then rinsed and further incubated with biotin conjugated UEA-1 for 40 minutes at RT. Sections were rinsed with 0.05% Tween 20 in 1× PBS and incubated with streptavidin–horseradish peroxidase complex (Bio-Rad Laboratories, Hercules, CA, USA) at RT in a humidified chamber. After 30 minutes, the slides were developed with 3,30-diaminobenzidine tetrahydrochloride (DAB) as the chromogen and counterstained with Hematoxylin. Once slides were dehydrated, di-n-butyl phthalate in xylene (DPX) was applied as mounting medium, with coverslips. Five sections were analyzed from tissue obtained from each animal, and representative images were obtained.
For histochemistry of the lymphoid follicles of the intestinal tissue, the Peyer’s patches were isolated through visual examination and then frozen and sectioned as described above. These sections were imaged using the Nikon fluorescence microscope and then stained with hematoxylin and eosin (H&E).
Statistics
Groups were compared using either an unpaired t-test, one-way analysis of variance (ANOVA) or two-way ANOVA using GraphPad Prism 6 software. In all cases, the ANOVA was coupled with a Bonferroni post hoc test. P<0.05 was considered the minimal requirement for statistical significance.
Results
Characterization of SMA micelles
The SMA-Epi micelles synthesized had a recovery of 90%–95% and loading of 7.5% and 18% of epirubicin and 12.11% of DiI as determined by the weight ratio of the drug over SMA (Table 1). The mean diameter of SMA micelles, measured by DLS were 21.53±4.22 nm and 80.82±10.10 nm for 7.5% and 18% loading, respectively, and was 134.12±2.00 nm for SMA-DiI (Table 1). The mean diameter of the 7.5% SMA-Epi micelle determined by TEM was 27.5±1.31 nm, which is in agreement with the one obtained by DLS (Figure S1). The PDI of SMA micelles were 0.26±0.03 and 0.32±0.06 for 7.5% and 18% SMA-Epi, respectively, and 0.33±0.07 for SMA-DiI. The zeta potential of the SMA-Epi micelles were −0.04 and −0.11 mV for 7.5% and 18% loading of epirubicin, respectively, and −0.04 mV for SMA-DiI micelles (Table 1).
 | Table 1 Recovery, loading, size, PDI, and zeta potential of SMA micelles |
Release rate of drug from SMA micelles
The 18% SMA-Epi micelles showed less than 1% release during the first 4 hours at physiological pH (Figure 1A), while for the same incubation period the 7.5% SMA-Epi micelle showed 12% release (Figure 1A). To ascertain the stability of SMA-Epi micelles in the high-acidic environment of the stomach, the release rate of epirubicin from the 18% SMA-Epi micelle was evaluated in SGF. Although there was a burst release of 2% at 0 minutes, the release after 2 hours, which is the gastric transit time, was 12% (Figure 1B).
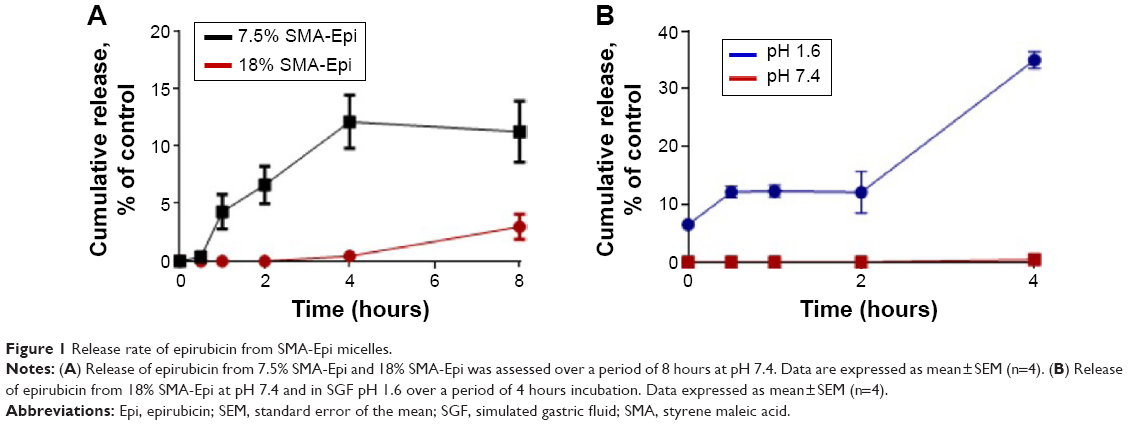 | Figure 1 Release rate of epirubicin from SMA-Epi micelles. |
In vitro cytotoxicity of SMA micelles
Doxorubicin, an anthracycline drug, is the most active and the mainstay chemotherapeutic agent for treatment of metastatic breast cancer.16,17 Epirubicin, used in this study, is an epimerized analog of doxorubicin, which is known to show less cardiotoxicity compared with doxorubicin at equal doses.18 The cytotoxicity of 7.5% and 18% SMA-Epi micelles were assessed using the breast cancer cell line MDA-MB-231 and compared with free epirubicin. The IC50 values for 7.5% and 18% SMA-Epi were 2.35 μM and 0.83 μM, respectively, while the IC50 value for free epirubicin was 0.18 μM (Figure 2). A higher loading improved the toxicity by threefold; however, the encapsulation of epirubicin decreased its cytotoxicity in vitro in comparison with the free drug.
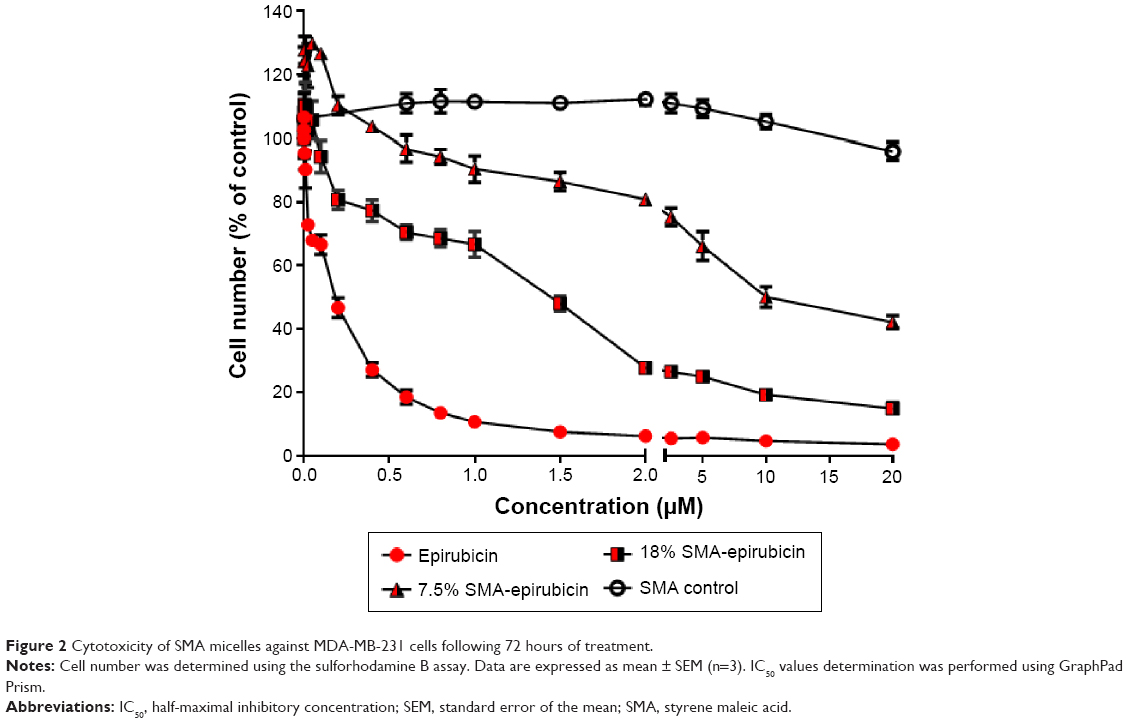 | Figure 2 Cytotoxicity of SMA micelles against MDA-MB-231 cells following 72 hours of treatment. |
Transport of SMA micelles across the in vitro model of intestinal epithelium
Transepithelial transport of SMA-Epi micelles across the differentiated Caco-2 monolayer was determined after 3 hours of incubation at 37°C. The 7.5% SMA-Epi micelles showed a higher transport (33.85 μM/cm2) as compared with the 18% SMA-Epi micelles (12.47 μM/cm2) (Figure 3A). There was no significant alteration of the TEER (P>0.05) measured after the transport experiment, ensuring the integrity of the monolayer. Particle size distribution in the HBSS solution from the lower compartment of the transwell was measured by DLS. The peak intensity observed was 80.25 nm which is similar to the size of the intact 18% SMA-Epi micelles (Figure S2). This proves that the 18% SMA-Epi micelles were transported intact as micelles.
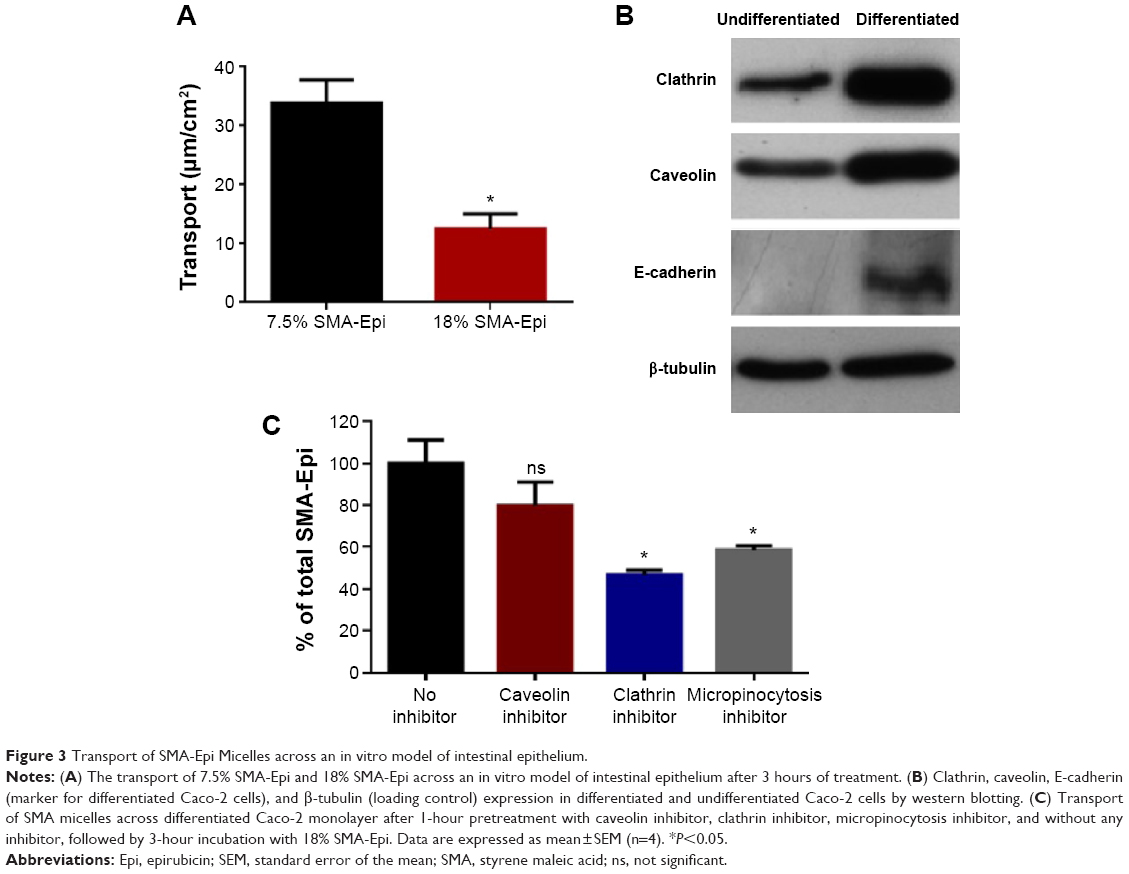 | Figure 3 Transport of SMA-Epi Micelles across an in vitro model of intestinal epithelium. |
Mechanism of transport of SMA micelles across the in vitro model of intestinal epithelium
To understand the mechanism of internalization of the SMA-Epi micelles, the differentiated Caco-2 monolayer was pretreated with endocytosis inhibitors at concentrations that maintained Caco-2 cell viability (Figure S3). Pretreatment with dansylcadaverine, an inhibitor for clathrin-dependent endocytosis, or amiloride, an inhibitor for micropinocytosis, resulted in a 54% and 32% reduction in transport of the SMA-Epi micelles, respectively (Figure 3C). However, genistein, an inhibitor of caveolin-dependent endocytosis, did not show significant reduction in transport of the SMA-Epi micelles, though both caveolin and clathrin are expressed in differentiated Caco-2 cells (Figure 3B). This indicates that the clathrin-mediated endocytosis mechanism and micropinocytosis are both involved in transepithelial transport of SMA-Epi micelles across an in vitro model of the intestinal epithelium.
Transport of SMA micelles across the ex vivo model of intestinal epithelium
The everted sac system is an efficient tool for studying the ex vivo intestinal transport of drugs.15 The jejunum sections showed 10.1% transport of 7.5% SMA-Epi and 2.71% transport of 18% SMA-Epi, while the ileum sections showed 11.96% transport of 7.5% SMA-Epi and 4.03% transport of 18% SMA-Epi (Figure 4A). No transport across the duodenum was detected within the detection limit of epirubicin absorbance at 480 nm. Thus, the 7.5% SMA-Epi micelles showed a higher transport as compared with the 18% SMA-Epi micelles (Figure 4A), in agreement with the differentiated Caco-2 monolayer. The ileum sections also showed higher transport as compared with the jejunum sections for both 7.5% and 18% SMA-Epi micelles, though the difference was not statistically significant (Figure 4A). The rat intestinal tissue morphology was not altered by the SMA-Epi transport as shown by hematoxylin staining (Figure 4B).
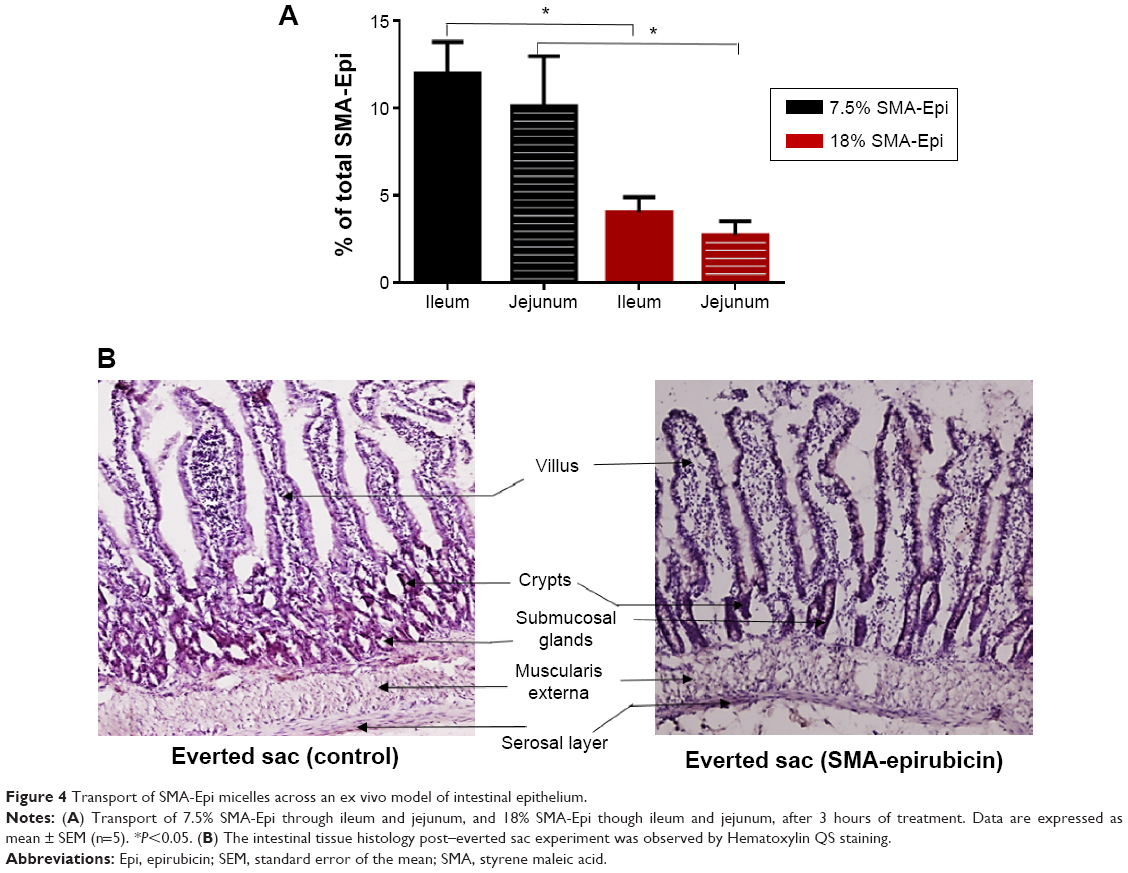 | Figure 4 Transport of SMA-Epi micelles across an ex vivo model of intestinal epithelium. |
In vivo oral biodistribution of SMA micelles
DiI, a nontoxic, fluorescent dye, was encapsulated into SMA micelles to evaluate its biodistribution across different organs in a mouse xenograft breast cancer model. Liver, spleen, and lung sections displayed fluorescence following oral and intravenous administration of SMA-DiI, while no fluorescence was observed with oral administration of free DiI. The kidney sections exhibited fluorescence only following intravenous administration of SMA-DiI and oral administration of free DiI. The intestinal tissue showed fluorescence lining the villi following oral administration of SMA-DiI, while a completely diffused fluorescence pattern was observed with orally administered free DiI. The intestinal sections for intravenously administrated SMA-DiI showed sharp fluorescence at the center of the villi. For the myocardial tissue, only the intravenously administered SMA-DiI showed fluorescence (Figure 5A).
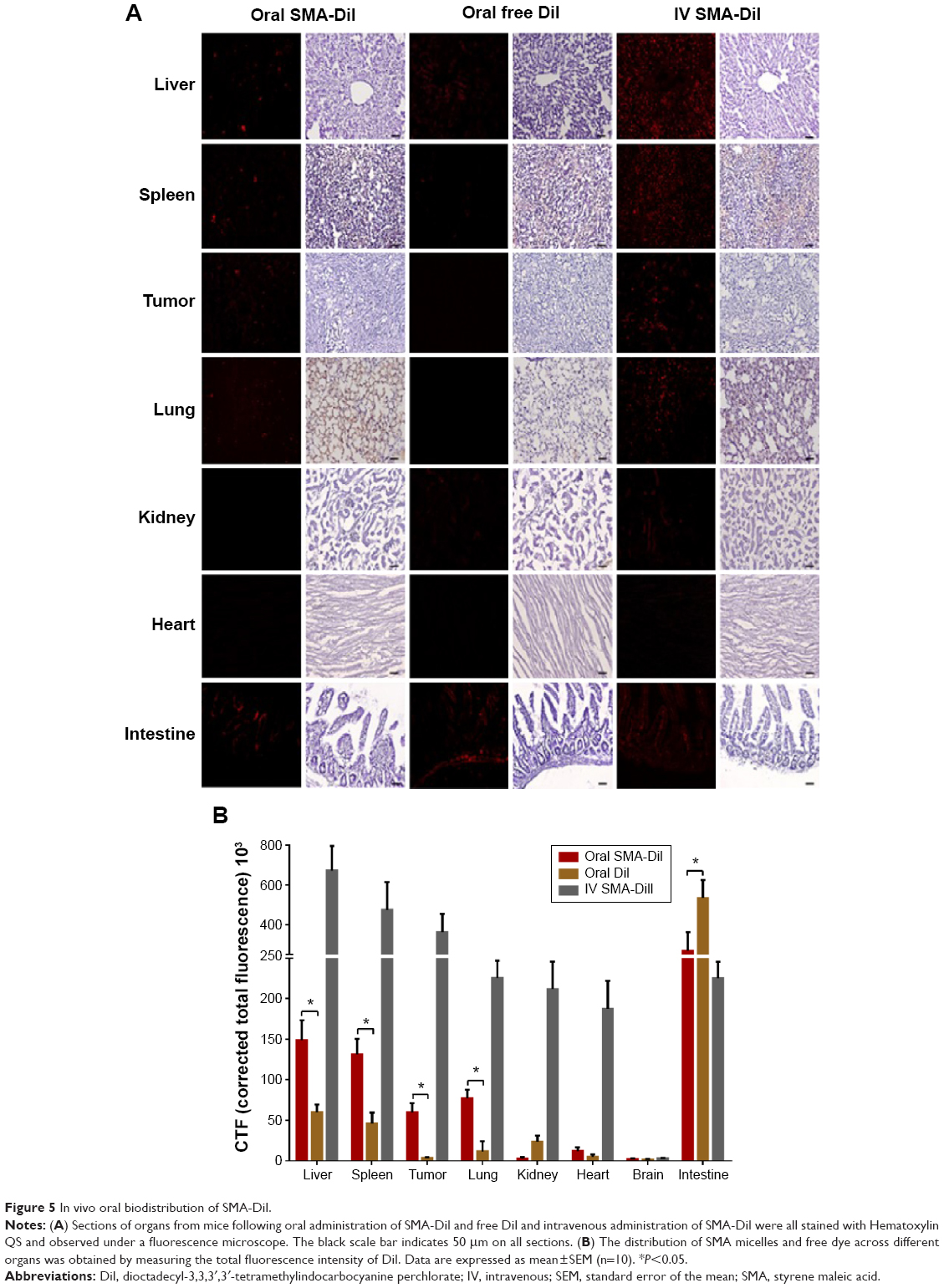 | Figure 5 In vivo oral biodistribution of SMA-DiI. |
The biodistribution of SMA micelles after oral administration followed a similar pattern to that after intravenous administration of SMA micelles, with a high accumulation in liver, spleen, and lung compared with other organs (Figure 5B). In comparison with oral administration of free DiI, the oral administration of SMA-DiI showed approximately twofold higher accumulation in liver and spleen, and six- and 15-fold higher accumulation in the lung and tumor, respectively.
In vivo uptake mechanism of SMA micelles through intestinal epithelium
The histochemistry of the intestinal sections showed that the fluorescence of SMA-DiI overlapped the M-cells stained with UEA-1, though the DiI fluorescence was not restricted to M-cells (Figure 6). This indicated the involvement of M-cells in the uptake of SMA-DiI through the lining of intestinal villi. Also, accumulation of SMA-DiI was observed underneath the epithelial lining of Peyer’s patches isolated from the intestinal tissue and stained with Hematoxylin (Figure 7).
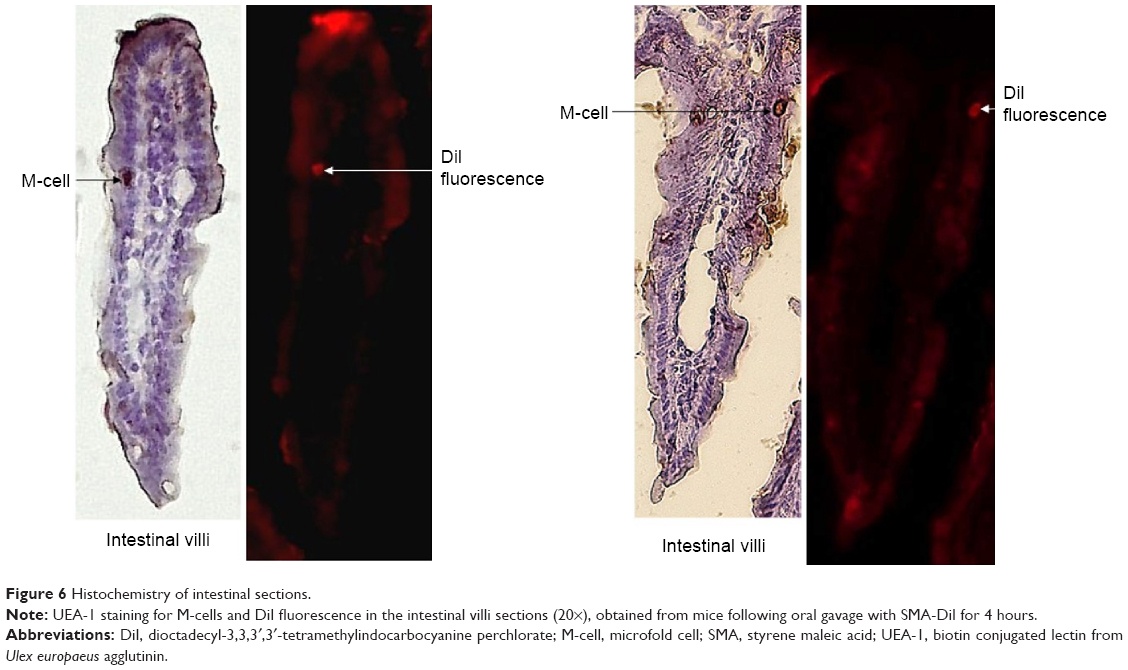 | Figure 6 Histochemistry of intestinal sections. |
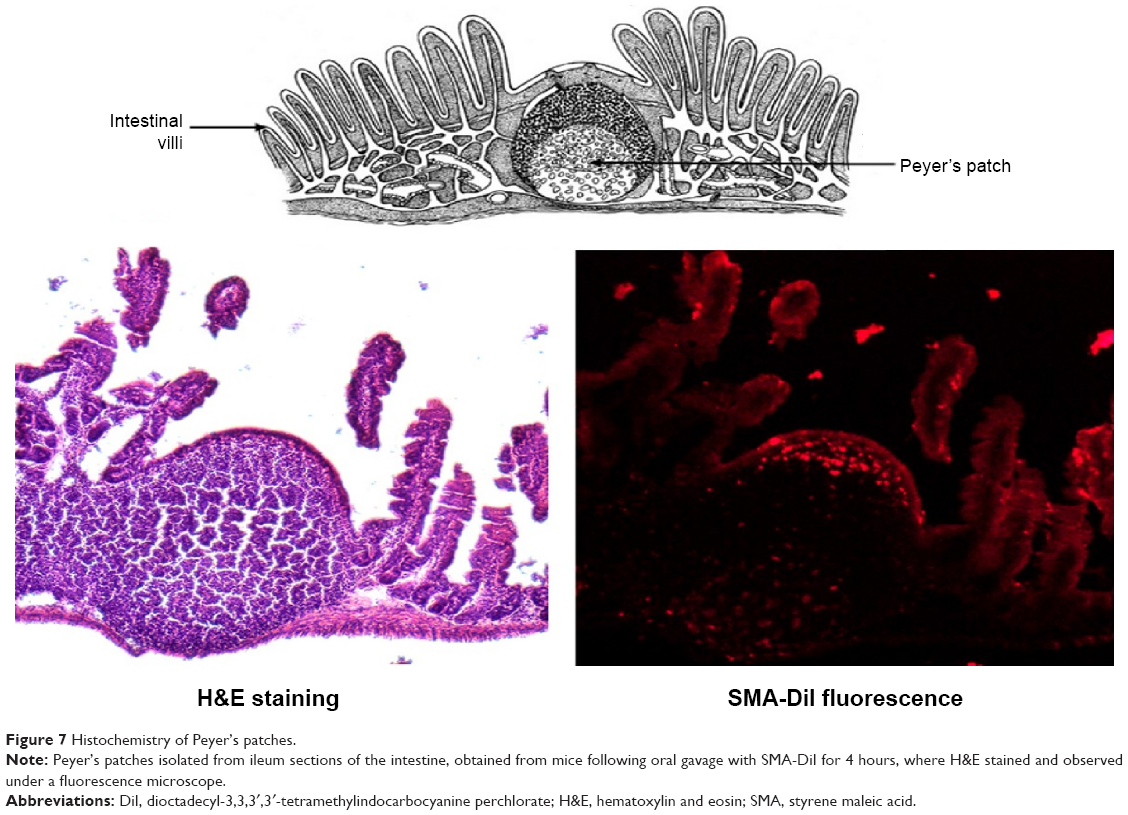 | Figure 7 Histochemistry of Peyer’s patches. |
Discussion
Oral delivery of anticancer drugs is advantageous compared with intravenous infusions as it reduces patient discomfort, improves patient quality of life, and could maintain therapeutic plasma drug concentrations for extended durations.2,6 The use of nanocarriers to orally deliver anticancer drugs can protect the drug from the harsh GI environment, reduce local toxicity, and enhance the oral bioavailability of the drug. Many nanoformulations have proved beneficial in improving the bioavailability and reducing toxicity of the anticancer agents.19–22 However, so far, no oral anticancer nanomedicine has been approved for clinical use.
Previously, SMA micellar formulations encapsulating doxorubicin, administered intravenously in the S-180 sarcoma mouse model have shown elevenfold higher tumor accumulation along with a significant reduction in cardiac and bone marrow toxicity as compared with the free drug.9 In the current study, we evaluated the potential of SMA micelles as a nanocarrier system for oral anticancer drug delivery. The SMA-Epi micelles had the appropriate size and charge to evade renal clearance.23,24 The release rate of epirubicin from SMA micelles demonstrated that the 18% SMA-Epi micelles were more stable as compared with 7.5% SMA-Epi micelles at the physiological pH, which is likely a result of formation of a stronger hydrophobic core, due to the interactions between the styrene moiety and epirubicin.9 Similar observations were reported for self-assembled nanoparticles of epirubicin.25 This can prove advantageous in tailoring the release rate of the drug from the SMA micelles, based on the quantity of the drug encapsulated into the micelle.12 The relatively higher release rate of epirubicin in the acidic conditions of gastric fluid could be attributed to the protonation of the maleate component of the SMA polymer, which plays a critical role in micellar stability. At alkaline pH, the micelle becomes slowly ionized, which increases its hydrodynamic volume, possibly due to the repulsive force between negative COO− groups. At acidic pH, the unionized maleic acid leads to a smaller hydrodynamic volume of the micelle.26 This effect could result in the release of epirubicin that is not in hydrophobic interaction with the styrene core, resulting in burst release. Another factor could be the protonation of the amine group of epirubicin, resulting in the escape of the ionized (NH3+) drug from the hydrophobic micellar core at acidic pH. At neutral pH, there could exist an electrostatic interaction between the NH3+ of epirubicin and COO− of SMA. This probable electrostatic interaction can be neutralized under highly acidic conditions, resulting in an increased release rate of epirubicin from the SMA micelle. The acidic pH of stomach can hamper the stability of polymeric drug delivery systems.27 However, our synthesized SMA micelles, showed a release of less than 12% in SGF for the initial 2 hours, which is the time required for gastric transit. This indicates an advantage of SMA micelles over other nanocarriers, such as liposomes, which display poor stability in highly acidic pH due to aggregation of the constituent phospholipids, which leads to reduced oral bioavailability.6,28,29 Surface modifications are thus required for these lipid nanocarriers to retain their stability.30
Epirubicin encapsulated in the SMA micelles retained its cytotoxic effect in MDA-MB-231 breast cancer cells. Epirubicin, like other anthracyclines, enters the cell through active transportation by SLC22A16 or via diffusion.31,32 However, drugs encapsulated in SMA micelles rely on a time- and energy-dependent endocytosis process for uptake of drugs into the cell,33 thus limiting the uptake as compared with that of the free drug, in turn often leading to a higher IC50 value as compared with the free drug. Conversely, under in vivo circumstances, the SMA micelles would exhibit higher accumulation in tumor tissue compared with free drug, due to the EPR effect.9 Thus, SMA encapsulated drug would have better in vivo efficacy than would free drug as observed in other anthracycline nanomedicines.34 For example, Doxil®, a Food and Drug Administration (FDA)-approved doxorubicin nanomedicine, was shown to have an IC50 value of 2.5 μg/mL as compared with free doxorubicin having an IC50 value of 0.1 μg/mL, in vitro.35 Nevertheless, in vivo evaluation showed that mice had 50% larger tumors when treated with free doxorubicin as compared with mice treated with the equivalent dose of Doxil.36 Among the two micellar formulations, the 7.5% SMA-Epi micelle showed lower cytotoxicity as compared with the 18% SMA-Epi micelle. The uptake of 18% SMA-Epi micelles through endocytosis would lead to higher accumulation of the drug in the cells as compared with the drug accumulation through uptake of the 7.5% SMA-Epi micelle. Thus, even though the 7.5% SMA-Epi micelles show higher release of drug compared with that of 18% SMA-Epi micelle, the cytotoxic effect of the 18% SMA-Epi may be higher due to higher accumulation of drug in the cells, through uptake of the higher loading 18% SMA-Epi micelles.
The SMA-Epi micelles, which showed appropriate physiochemical characteristics, were further evaluated for their ability to traverse the intestinal epithelium. The SMA micelles synthesized with 7.5% and 18% loading showed efficient transport across the in vitro intestinal epithelium model, indicating uptake of SMA micelles by enterocyte-resembling cells. The higher transport of 7.5% SMA-Epi micelles could be attributed to the cumulative transport of the faster released drug from the 7.5% loaded SMA micelle, which can diffuse through the cells of the monolayer.37 In contrast, the 18% SMA-Epi micelle is transported across the Caco-2 monolayer as an intact micelle. This is supported by the size distribution of the transported particles from 18% SMA-Epi micelles, which showed that the population of particles with maximum intensity were of the same size as the size of 18% SMA-Epi micelles. However, the transport levels shown by both SMA micelles were higher than the transport reported in the literature for other nanoformulations encapsulating epirubicin. For example, PSC 833 liposomes show transport of 2.5 nM/cm2 of epirubicin.38 while dipalmitoyl phosphatidylcholine liposomes show transport of 2.25 nM/cm2 of epirubicin,39 both of which are lower than the transport shown by SMA micelles. In the ex vivo model of the intestinal epithelium, the 7.5% SMA-Epi micelles showed higher transport as compared with the 18% SMA-Epi micelles, a result that is consistent with the in vitro model of the intestinal epithelium. SMA micelles showed two- and tenfold higher transport across the ex vivo model of intestinal epithelium compared with other nanoformulations, such as chitosan encapsulating docetaxel nanoparticles (molecular weight of 20,000 g/mol), which have a diameter of 153±9.0 nm,40 and mannosamine HCl liposomes encapsulating acyclovir, which have a diameter of 371±7 nm.41 Notably, the intestinal tissue morphology was intact after the transport of the SMA micelles, demonstrating that the transportation of SMA micelles was not related to alteration of the tissue integrity.
Particles sized between 50 and 200 nm are generally internalized through clathrin-mediated endocytosis,42,43 and this was true for the SMA-Epi micelles. However, the process of internalization of nanoparticles is dependent to a great extent on the cell type.44 In polarized differentiated Caco-2 cells, although both caveolin and clathrin are expressed, the expression of caveolin is more pronounced on the basolateral side.45 Thus, SMA micelles traversed the intestinal epithelium through a transcellular route mediated by clathrin-dependent endocytosis and micropinocytosis. Certain nanoformulations, such as dendrimers with molecular weight 3,256 Da and chitosan polymer drug conjugates with molecular weight 31 and 170 kDa, exploit the paracellular route for traversing the intestinal epithelium.46 Chitosan nanoparticles interact with the filamentous (F)-actin present in the intestinal lamina, leading to depolarization of ZO-1 protein, which is an integral part of tight junctions.47 Paracellular transport of nanoparticles across the intestinal epithelium through tight junction modulation can disrupt the barrier function of the intestinal epithelium between gut flora and the intestinal immune system, leading to undesirable inflammatory reactions.48 In this respect, the SMA micelles could be a safer oral drug delivery system than other systems.
Following oral administration, a notable difference in biodistribution of free DiI and SMA-DiI was observed. SMA-DiI showed higher accumulation in tissues with wide vasculatures, such as the liver, spleen, and tumor as compared with free DiI, which was similar that in previous studies on SMA doxorubicin micelles administered intravenously.9 This difference in biodistribution of free DiI and SMA-DiI indicates that the SMA-DiI was transported across the intestinal epithelium as an intact micelle. The micellar system, due to its large size, would accumulate in liver and spleen sinusoids, which is a common phenomenon for most nanosized constructs. This could also be true for hydrophobic DiI dye reaching the circulation, which may possibly be transported through interaction with a carrier lipid or protein molecule. In this case, the liver and spleen sinusoids would allow extravasation of this large-sized dye associated with the lipid or protein carrier. The observed accumulation in the kidney could be due to entrapment of large molecular sized particles in the acute angles between the specialized renal vessels.49
Interestingly, a significantly high accumulation of SMA-DiI was observed in the lung tissue as compared with the free DiI. Nanoparticles that enter the lymphatic system of the intestine are carried through the thoracic duct and the right lymphatic duct into the subclavian vein, which drains the deoxygenated blood to right atrium.50 The pulmonary artery then carries the deoxygenated blood along with the nanoparticles to the lung lobes. The nanoparticles, which are not retained in the lung, enter the systemic circulation for further distribution, eventually accumulating in the tumor, liver, and spleen. The lymphatic uptake of SMA micelles has the advantage of evading first-pass metabolism by the liver.51
To further confirm the lymphatic uptake of SMA micelles, the intestinal sections were stained with an M-cell marker, UEA-1 lectin, which binds to α-fucose residues on the surface of M-cells.51 M-cells are specialized cells that take up foreign particles and transfer them to the lymphoid follicles underneath the intestinal epithelium. The SMA micelles showed colocalization with M-cells stained with UEA-1 lectin. These M-cells are known to show transcytosis of microparticles to the underlying lymphoid tissue.52 Microscopic observation of the Peyer’s patches isolated from the intestinal tissue showed accumulation of SMA-DiI fluorescence beneath the epithelial lining of these Peyer’s patches, confirming the uptake and transport of the SMA micelles through the M-cells to the underlying lymphoid tissue. Ileum sections are known to have large numbers of Peyer’s patches as compared with jejunum and duodenum sections.53 The higher transport of SMA micelles through the ileum sections as compared with other sections of the intestine further emphasize SMA micellar uptake through the M-cells along with the enterocytes. The M-cell uptake of SMA micelles reduces the lysosomal degradation of the nanoparticles, as M-cells have fewer lysosomes compared with enterocytes.51 This transport pathway can favor the use of SMA delivery for oral vaccine delivery.54
Conclusion
This study demonstrates that SMA micelles can traverse in vitro and ex vivo models of intestinal epithelium without affecting tissue integrity, thus reducing the possibility of any toxicity toward the intestinal epithelium. The transport of SMA micelles was found to be mediated through M-cell uptake along with enterocytes, which could potentiate increased bioavailability of the drug encapsulated in the micelles. Altogether our data confirms the advantages of the use of SMA micelles as a nanocarrier system for oral drug delivery.
Acknowledgments
This work was supported by a 2014 University of Otago Research Grant (grant number 110273.01.R.LM) to KG. The authors would like to thank Professor Hamid Ghandehari for his kind gift of Caco-2 cells and Professor Rhonda Rosengren, Dr Sarah Baird, and Dr Gregory Giles for editing the paper.
Disclosure
The authors report no conflicts of interest with this work.
References
Smorenburg CH, Sparreboom A. Oral anticancer agents. In: Pinedo HM, Smorenburg CH, editors. Drugs Affecting Growth of Tumours. Basel: Birkhäuser Verlag; 2006:153–166. | ||
Mei L, Zhang Z, Zhao L, et al. Pharmaceutical nanotechnology for oral delivery of anticancer drugs. Adv Drug Deliv Rev. 2013;65(6):880–890. | ||
Stuurman FE, Nuijen B, Beijnen JH, Schellens JH. Oral anticancer drugs: mechanisms of low bioavailability and strategies for improvement. Clin Pharmacokinet. 2013;52(6):399–414. | ||
Rose PG, Blessing JA, Mayer AR, Homesley HD. Prolonged oral etoposide as second-line therapy for platinum-resistant and platinum-sensitive ovarian carcinoma: a Gynecologic Oncology Group study. J Clin Oncol. 1998;16(2):405–410. | ||
Graziano MJ, Pilcher GD, Walsh KM, Kasali OB, Radulovic L. Preclinical toxicity of a new oral anticancer drug, CI-994 (acetyldinaline), in rats and dogs. Invest New Drugs. 1997;15(4):295–310. | ||
Thanki K, Gangwal RP, Sangamwar AT, Jain S. Oral delivery of anticancer drugs: challenges and opportunities. J Control Release. 2013;170(1):15–40. | ||
Sparreboom A, van Asperen J, Mayer U, et al. Limited oral bioavailability and active epithelial excretion of paclitaxel (Taxol) caused by P-glycoprotein in the intestine. Proc Natl Acad Sci U S A. 1997;94(5):2031–2035. | ||
Malingré MM, Beijnen JH, Schellens JH. Oral delivery of taxanes. Invest New Drugs. 2001;19(2):155–162. | ||
Greish K, Sawa T, Fang J, Akaike T, Maeda H. SMA-doxorubicin, a new polymeric micellar drug for effective targeting to solid tumours. J Control Release. 2004;97(2):219–230. | ||
Miyata K, Christie RJ, Kataoka K. Polymeric micelles for nano-scale drug delivery. React Funct Polym. 2011;71(3):227–234. | ||
Daruwalla J, Nikfarjam M, Greish K, et al. In vitro and in vivo evaluation of tumor targeting styrene-maleic acid copolymer-pirarubicin micelles: Survival improvement and inhibition of liver metastases. Cancer Sci. 2010;101(8):1866–1874. | ||
Nehoff H, Parayath N, Taurin S, Greish K. The influence of drug loading on caveolin-1 mediated intracellular internalization of doxorubicin nanomicelles in vitro. J Nanomed Nanotechnol. 2014;5(197):2. | ||
Vichai V, Kirtikara K. Sulforhodamine B colorimetric assay for cytotoxicity screening. Nat Protoc. 2006;1(3):1112–1116. | ||
Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods. 1983;65(1–2):55–63. | ||
Barthe L, Woodley JF, Kenworthy S, Houin G. An improved everted gut sac as a simple and accurate technique to measure paracellular transport across the small intestine. Eur J Drug Metab Pharmacokinet. 1998;23(2):313–323. | ||
Norris B, Pritchard KI, James K, et al. Phase III comparative study of vinorelbine combined with doxorubicin versus doxorubicin alone in disseminated metastatic/recurrent breast cancer: National Cancer Institute of Canada Clinical Trials Group Study MA8. J Clin Oncol. 2000;18(12):2385–2394. | ||
Paridaens R, Biganzoli L, Bruning P, et al. Paclitaxel versus doxorubicin as first-line single-agent chemotherapy for metastatic breast cancer: a European Organization for Research and Treatment of Cancer randomized study with cross-over. J Clin Oncol. 2000;18(4):724–733. | ||
Brambilla C, Rossi A, Bonfante V, et al. Phase II study of doxorubicin versus epirubicin in advanced breast cancer. Cancer Treat Rep. 1986;70(2):261–266. | ||
Iqbal J, Sarti F, Perera G, Bernkop-Schnürch A. Development and in vivo evaluation of an oral drug delivery system for paclitaxel. Biomaterials. 2011;32(1):170–175. | ||
Bradley MO, Swindell CS, Anthony FH, et al. Tumor targeting by conjugation of DHA to paclitaxel. J Control Release. 2001;74(1–3):233–236. | ||
Dube A, Nicolazzo JA, Larson I. Chitosan nanoparticles enhance the plasma exposure of (-)-epigallocatechin gallate in mice through an enhancement in intestinal stability. Eur J Pharm Sci. 2011;44(3):422–426. | ||
Wiwattanapatapee R, Carreño-Gómez B, Malik N, Duncan R. Anionic PAMAM dendrimers rapidly cross adult rat intestine in vitro: a potential oral delivery system? Pharm Res. 2000;17(8):991–998. | ||
Alexis F, Pridgen E, Molnar LK, Farokhzad OC. Factors affecting the clearance and biodistribution of polymeric nanoparticles. Mol Pharm. 2008;5(4):505–515. | ||
Elsabahy M, Wooley KL. Design of polymeric nanoparticles for biomedical delivery applications. Chem Soc Rev. 2012;41(7):2545–2561. | ||
Li L, Gao FP, Tang HB, et al. Self-assembled nanoparticles of cholesterol-conjugated carboxymethyl curdlan as a novel carrier of epirubicin. Nanotechnology. 2010;21(26):265601. | ||
Iyer AK, Greish K, Fang J, Murakami R, Maeda H. High-loading nanosized micelles of copoly(styrene-maleic acid)-zinc protoporphyrin for targeted delivery of a potent heme oxygenase inhibitor. Biomaterials. 2007;28(10):1871–1881. | ||
Hörter D, Dressman JB. Influence of physicochemical properties on dissolution of drugs in the gastrointestinal tract. Adv Drug Deliv Rev. 2001;46(1–3):75–87. | ||
Derycke AS, de Witte PA. Liposomes for photodynamic therapy. Adv Drug Deliv Rev. 2004;56(1):17–30. | ||
Shi G, Guo W, Stephenson SM, Lee RJ. Efficient intracellular drug and gene delivery using folate receptor-targeted pH-sensitive liposomes composed of cationic/anionic lipid combinations. J Control Release. 2002;80(1–3):309–319. | ||
Iwanaga K, Ono S, Narioka K, et al. Oral delivery of insulin by using surface coating liposomes: Improvement of stability of insulin in GI tract. Int J Pharm. 1997;157(1):73–80. | ||
Glück S. Adjuvant chemotherapy for early breast cancer: optimal use of epirubicin. Oncologist. 2005;10(10):780–791. | ||
Thorn CF, Oshiro C, Marsh S, et al. Doxorubicin pathways: pharmacodynamics and adverse effects. Pharmacogenet Genomics. 2011;21(7):440. | ||
Sahay G, Alakhova DY, Kabanov AV. Endocytosis of nanomedicines. J Control Release. 2010;145(3):182–195. | ||
Zhao Q, Han B, Wang Z, Gao C, Peng C, Shen J. Hollow chitosan-alginate multilayer microcapsules as drug delivery vehicle: doxorubicin loading and in vitro and in vivo studies. Nanomedicine. 2007;3(1):63–74. | ||
Ten Hagen TL, Van Der Veen AH, Nooijen PT, Van Tiel ST, Seynhaeve AL, Eggermont AM. Low-dose tumor necrosis factor-alpha augments antitumor activity of stealth liposomal doxorubicin (DOXIL) in soft tissue sarcoma-bearing rats. Int J Cancer. 2000;87(6):829–837. | ||
Brouckaert P, Takahashi N, van Tiel ST, et al. Tumor necrosis factor-alpha augmented tumor response in B16BL6 melanoma-bearing mice treated with stealth liposomal doxorubicin (Doxil) correlates with altered Doxil pharmacokinetics. Int J Cancer. 2004;109(3):442–448. | ||
Di L, Artursson P, Avdeef A, et al. Evidence-based approach to assess passive diffusion and carrier-mediated drug transport. Drug Discov Today. 2012;17(15–16):905–912. | ||
Lo Y, Liu F, Cherng J. Effect of PSC 833 liposomes and Intralipid on the transport of epirubicin in Caco-2 cells and rat intestines. J Control Release. 2001;76(1–2):1–10. | ||
Lo YL. Phospholipids as multidrug resistance modulators of the transport of epirubicin in human intestinal epithelial Caco-2 cell layers and everted gut sacs of rats. Biochem Pharmacol. 2000;60(9):1381–1390. | ||
Saremi S, Atyabi F, Akhlaghi SP, Ostad SN, Dinarvand R. Thiolated chitosan nanoparticles for enhancing oral absorption of docetaxel: preparation, in vitro and ex vivo evaluation. Int J Nanomedicine. 2011;6:119–128. | ||
Pukanud P, Peungvicha P, Sarisuta N. Development of mannosylated liposomes for bioadhesive oral drug delivery via M cells of Peyer’s patches. Drug Deliv. 2009;16(5):289–294. | ||
Vollrath A, Schallon A, Pietsch C, et al. A toolbox of differently sized and labeled PMMA nanoparticles for cellular uptake investigations. Soft Matter. 2013;9(1):99–108. | ||
Watson P, Jones AT, Stephens DJ. Intracellular trafficking pathways and drug delivery: fluorescence imaging of living and fixed cells. Adv Drug Deliv Rev. 2005;57(1):43–61. | ||
Iversen TG, Skotland T, Sandvig K. Endocytosis and intracellular transport of nanoparticles: present knowledge and need for future studies. Nano Today. 2011;6(2):176–185. | ||
Vogel U, Sandvig K, van Deurs B. Expression of caveolin-1 and polarized formation of invaginated caveolae in Caco-2 and MDCK II cells. J Cell Sci. 1998;111(Pt 6):825–832. | ||
El-Sayed M, Rhodes CA, Ginski M, Ghandehari H. Transport mechanism(s) of poly (amidoamine) dendrimers across Caco-2 cell monolayers. Int J Pharm. 2003;265(1–2):151–157. | ||
Schipper NG, Olsson S, Hoogstraate JA, deBoer AG, Vårum KM, Artursson P. Chitosans as absorption enhancers for poorly absorbable drugs 2: mechanism of absorption enhancement. Pharm Res. 1997;14(7):923–929. | ||
Turner JR. Intestinal mucosal barrier function in health and disease. Nat Rev Immunol. 2009;9(11):799–809. | ||
Yu T, Greish K, McGill LD, Ray A, Ghandehari H. Influence of geometry, porosity, and surface characteristics of silica nanoparticles on acute toxicity: their vasculature effect and tolerance threshold. ACS Nano. 2012;6(3):2289–2301. | ||
Yáñez JA, Wang SW, Knemeyer IW, Wirth MA, Alton KB. Intestinal lymphatic transport for drug delivery. Adv Drug Delivery Rev. 2011;63(10–11):923–942. | ||
Kunisawa J, Kurashima Y, Kiyono H. Gut-associated lymphoid tissues for the development of oral vaccines. Adv Drug Deliv Rev. 2012;64(6):523–530. | ||
Ling SS, Magosso E, Khan NA, Yuen KH, Barker SA. Enhanced oral bioavailability and intestinal lymphatic transport of a hydrophilic drug using liposomes. Drug Dev Ind Pharm. 2006;32(3):335–345. | ||
Ermund A, Schütte A, Johansson ME, Gustafsson JK, Hansson GC. Studies of mucus in mouse stomach, small intestine, and colon. I. Gastrointestinal mucus layers have different properties depending on location as well as over the Peyer’s patches. Am J Physiol Gastrointest Liver Physiol. 2013;305(5):G341–G347. | ||
Shakweh M, Ponchel G, Fattal E. Particle uptake by Peyer’s patches: a pathway for drug and vaccine delivery. Expert Opin Drug Deliv. 2004;1(1):141–163. |
Supplementary materials
 | Figure S1 Size distribution of 7.5% SMA-Epi was determined by TEM at concentration of 1 mg/mL in water with 1% phosphotungstic acid for staining. |
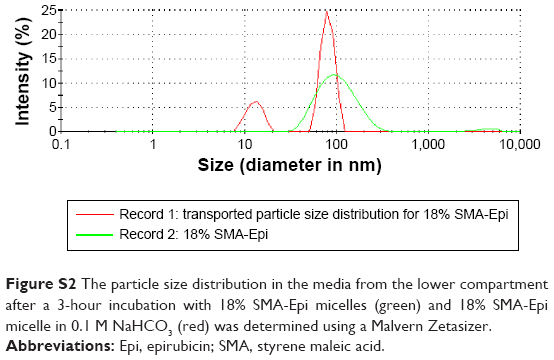 | Figure S2 The particle size distribution in the media from the lower compartment after a 3-hour incubation with 18% SMA-Epi micelles (green) and 18% SMA-Epi micelle in 0.1 M NaHCO3 (red) was determined using a Malvern Zetasizer. |
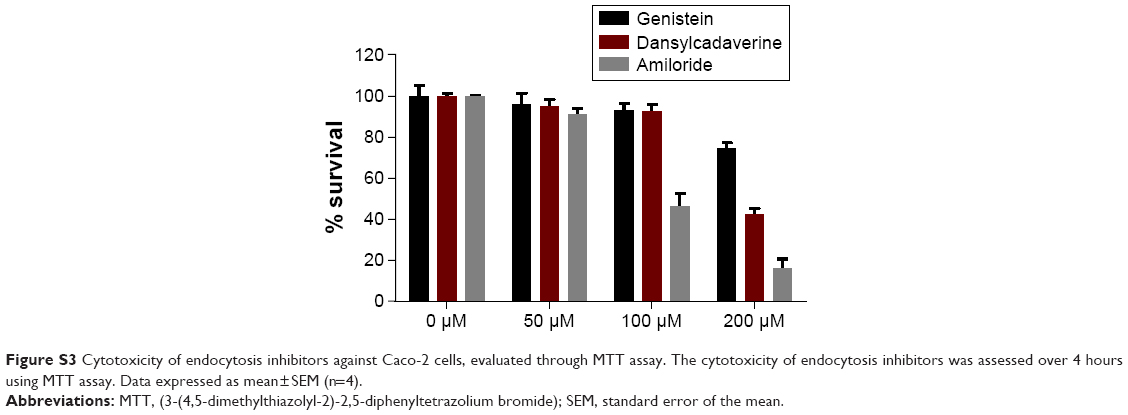 | Figure S3 Cytotoxicity of endocytosis inhibitors against Caco-2 cells, evaluated through MTT assay. The cytotoxicity of endocytosis inhibitors was assessed over 4 hours using MTT assay. Data expressed as mean±SEM (n=4). |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.