Back to Journals » Drug Design, Development and Therapy » Volume 9
Structure–activity exploration of a small-molecule Lipid II inhibitor
Authors Fletcher S, Yu W ![]() , Huang J, Kwasny S, Chauhan J, Opperman T, MacKerrel Jr A, de Leeuw E
, Huang J, Kwasny S, Chauhan J, Opperman T, MacKerrel Jr A, de Leeuw E
Received 18 December 2014
Accepted for publication 10 February 2015
Published 24 April 2015 Volume 2015:9 Pages 2383—2394
DOI https://doi.org/10.2147/DDDT.S79504
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Editor who approved publication: Professor Shu-Feng Zhou
Steven Fletcher,1 Wenbo Yu,1,2 Jing Huang,1,2 Steven M Kwasny,3 Jay Chauhan,1 Timothy J Opperman,3 Alexander D MacKerell Jr,1,2 Erik PH de Leeuw4
1Department of Pharmaceutical Sciences, 2Computer-Aided Drug Design Center, University of Maryland, School of Pharmacy, Baltimore, MD, 3Microbiotix, Inc., Worcester, MA, 4Institute of Human Virology and Department of Biochemistry and Molecular Biology, University of Maryland, School of Medicine, Baltimore, MD, USA
Abstract: We have recently identified low-molecular weight compounds that act as inhibitors of Lipid II, an essential precursor of bacterial cell wall biosynthesis. Lipid II comprises specialized lipid (bactoprenol) linked to a hydrophilic head group consisting of a peptidoglycan subunit (N-acetyl glucosamine [GlcNAc]–N-acetyl muramic acid [MurNAc] disaccharide coupled to a short pentapeptide moiety) via a pyrophosphate. One of our lead compounds, a diphenyl-trimethyl indolene pyrylium, termed BAS00127538, interacts with the MurNAc moiety and the isoprenyl tail of Lipid II. Here, we report on the structure–activity relationship of BAS00127538 derivatives obtained by in silico analyses and de novo chemical synthesis. Our results indicate that Lipid II binding and bacterial killing are related to three features: the diphenyl moiety, the indolene moiety, and the positive charge of the pyrylium. Replacement of the pyrylium moiety with an N-methyl pyridinium, which may have importance in stability of the molecule, did not alter Lipid II binding or antibacterial potency.
Keywords: computer-aided drug design, Lipid II, antibiotics, phospholipid
Introduction
With the rapid increase of antibiotic resistance, there is an urgent need to develop novel classes of potent antibiotics against established molecular targets. Lipid II is essential for cell wall biosynthesis, synthesized in limited amounts,1 and has a high turnover rate, which makes it an established molecular target for antibiotics.2,3 Four different classes of peptide antibiotics that target Lipid II have been described: (1) the glycopeptides, including vancomycin and teicoplanin; (2) the depsipeptide antibiotics, including ramoplanin and enduracidins; (3) the lantibiotics, including nisin and mersacidin; and (4) cyclic peptides, including mannopeptimycins, plusbacin, and katanosin B.1,4–9 Strikingly, these molecules do not share any obvious sequence homology or structural similarity, yet all are able to specifically interact with Lipid II in the bacterial membrane environment.
The glycopeptide antibiotic vancomycin was the first compound discovered to kill bacteria by targeting Lipid II. Currently, vancomycin serves as a principal treatment for infections caused by all major Gram-positive pathogens including methicillin-resistant Staphylococcus aureus (MRSA). However, resistance to vancomycin is increasing, and a number of vancomycin-insensitive strains have been described.10
We recently reported on the functional interaction between Lipid II and defensins, a major family of natural antimicrobial peptides that protect the host’s epithelial surfaces against microbial invasion.11,12 Several studies on defensins from other species, including fungi,13 invertebrates,14 and human,15 have firmly established Lipid II as a target for this class of natural antimicrobial peptides. Based on the interaction between Lipid II and human neutrophil peptide-1, we identified low-molecular weight synthetic compounds that target Lipid II with high specificity and affinity.16 One of our lead compounds, BAS00127538, was characterized further and revealed a unique interaction with Lipid II that differs from antibiotics currently in clinical use or development. In this study, we report on the structural and functional relationships of derivatives of BAS00127538.
Materials and methods
Materials and bacterial strains
S. aureus ATCC 29213, Escherichia coli ATCC 25922, Enterococcus faecalis ATCC 29212, Streptococcus pneumonia ATCC 49619, and Acinetobacter baumannii ATCC 19606 were obtained from Microbiologics (St Cloud, MN, USA). S. aureus USA300 (MRSA), S. aureus ATCC (vancomycin intermediate-resistant S. aureus), and S. aureus NTS (vancomycin intermediate-resistant S. aureus) were generously provided by the Laboratory of Pathology, University of Maryland, School of Medicine. Vancomycin was purchased from Sigma. Compounds were obtained from various suppliers as listed in Table S1.
Computer-aided drug design – database searching
Identification of compounds similar to BAS00127538 was carried out using chemical/physiochemical similarity searches with MACCS17 and MPMFP18 fingerprints using the program MOE (Chemical Computing Group Inc.).19 The query compound, Figure 1, was chosen to account for interactions of the positively charged oxygen of BAS00127538 with the phosphate of Lipid II as indicated by molecular dynamics (MD) simulations and nuclear magnetic resonance data as discussed in our previous study.
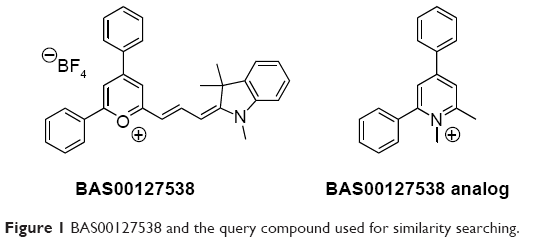 | Figure 1 BAS00127538 and the query compound used for similarity searching. |
An in-house database in the University of Maryland Computer-Aided Drug Design (CADD) Center with 5.04 million compounds was used for the similarity searching. Similar compounds with a Tanimoto index over a selected cutoff value and with drug-like characteristics that maximize bioavailability20 were selected for biological experiments.
CADD modeling and MD simulations
Molecular modeling, energy minimization, and MD simulations were performed with the program CHARMM21 using the CHARMM36 lipid22 and protein force field23,24 for Lipid II and the TIP3P water model25 along with the CHARMM General force field26–28 for the ligands. Using the final snapshot from previously published 10 ns MD simulations of the BAS00127538–Lipid II complex in aqueous solution, the aromatic rings of the Steven Fletcher (SF) analogs were aligned with those of BAS00127538. Each system was then subjected to a short energy minimization following which a 100 ps MD simulation with a short time step of 0.5 fs was carried out. Each system was then subjected to a 20 ns MD simulation run with a time step of 1 fs. Simulations were carried out in the NPT ensemble at 300 K and 1 atm with SHAKE of covalent bonds involving hydrogens, and there were no restraints in the simulations. Free energies of binding, ΔG, were estimated using the linear interaction energy method as follows:29
| (1) |
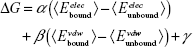
where α =0.5, β =0.16, γ cancels out as we only considered the relative free energies ΔΔG, and the unbound interaction energies were computed from 5 ns MD simulations of the compounds alone in water.
3-Lipid II purification
Short-chain water-soluble Lipid II containing a lipid tail of three isoprene units (3-Lipid II or farnesyl-Lipid II) was generated and purified essentially as described.30
Surface plasmon resonance
Surface plasmon resonance binding experiments were carried out on a BIAcore T100 system (BIAcore Inc., Piscataway, NJ, USA) at 25°C. The assay buffer was 10 mM HEPES (4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid), 150 mM NaCl, and 0.05% surfactant P20, pH 7.4 (±3 mM ethylenediaminetetraacetic acid) supplemented with 10% dimethyl sulfoxide (DMSO). 3-Lipid II (50 RUs) was immobilized on CM5 sensor chips using the amine-coupling chemistry recommended by the manufacturer. For initial determination of binding, compounds were introduced into the flow cells (30 L/min) in the running buffer at 10 μM. Resonance signals were corrected for nonspecific binding by subtracting the background of the control flow cell. After each analysis, the sensor chip surfaces were regenerated with 50 mM NaOH for 30 s at a flow rate 100 μL/min, and equilibrated with the buffer prior to next injection. For binding kinetics studies, binding isotherms were analyzed with manufacturer-supplied software for BIAcore T100.
Antibacterial activity assay
Determination of the minimal inhibitory concentrations (MICs) by dilution was carried out by broth dilution according to Clinical and Laboratory Standards Institute standards.31
Cytotoxicity
The cytotoxic concentration of antibacterial compounds that produces half maximal decrease in viability (CC50) against mammalian cells (HeLa, ATCC CCL-2.2) was determined as described.32
Macromolecular synthesis assays
The effect of the bis-indoles on the following macromolecular synthetic pathways in S. aureus NCTC 8325 was measured as previously described.33 To inhibit efflux, S. aureus NCTC 8325 was grown in the presence of 20 μg/mL of reserpine. Each data point is the average of three replicates, and the error bars represent standard deviation.
Chemical synthesis
1-Methyl-2,4-diphenyl-6-((1E,3E)-3-(1,3,3-trimethylindolin-2-ylidene)prop-1-en-1-yl)pyridin-1-ium tetrafluoroborate (“SF-5-219”)
The tetrafluoroborate salt of BAS00127538 (14 mg, 27.1 μmol) was dissolved in absolute EtOH (1 mL). A solution of methylamine in EtOH (33%; 5 μL, 40.7 μmol, 1.5 equiv) was added, followed by one drop of AcOH. The reaction mixture was heated at reflux for 16 hours, by which time the dark blue-green reaction mixture had become deep purple. All volatiles were removed in vacuo. The residue was suspended in ether (10 mL) and was briefly sonicated, and then left to stand in the fridge overnight. The next day, the purple solid was collected by vacuum filtration, washing with ether to afford the title compound (8 mg, 57%): δH (400 MHz, DMSO-d6) 8.45 (s, 1H, pyridinium), 8.20 (t, 1H, J=13.2 Hz, CH=CH=CH), 8.06–8.12 (m, 2H, Ph), 7.60–7.77 (m, 9H, Ph, pyridinium), 7.44 (d, 1H, J=7.2 Hz, indolene), 7.29 (t, 1H, J=7.8 Hz, indolene), 7.09 (d, 1H, J=8.0 Hz, indolene), 7.03 (t, 1H, J=7.4 Hz, indolene), 6.62 (d, 1H, J=13.6 Hz, CH=CH=CH), 6.03 (d, 1H, J=12.8 Hz, CH=CH=CH), 3.82 (s, 3H, NCH3), 3.36 (s, 3H, NCH3 (indolene)), 1.69 (s, 6H, C(CH3)2); m/z (atmospheric-pressure chemical ionization) 443.2 M+.
1-Isopropyl-2,4-diphenyl-6-((1E,3E)-3-(1,3,3-trimethylindolin-2-ylidene)prop-1-en-1-yl)pyridin-1-ium tetrafluoroborate (“SF-5-330”)
δH (400 MHz, DMSO-d6) 8.41 (s, 1H, pyridinium), 8.13 (t, 1H, J=13.2 Hz, CH=CH=CH), 8.06–8.09 (m, 2H, Ph), 7.60–7.67 (m, 9H, Ph, pyridinium), 7.43 (d, 1H, J=7.2 Hz, indolene), 7.27 (t, 1H, J=7.8 Hz, indolene), 7.06 (d, 1H, J=8.0 Hz, indolene), 7.01 (t, 1H, J=7.4 Hz, indolene), 6.76 (d, 1H, J=13.6 Hz, CH=CH=CH), 6.21 (d, 1H, J=12.4 Hz, CH=CH=CH), 4.91 (quintet, 1H, J=6.8 Hz, CH(CH3)2), 3.37 (s, 3H, NCH3), 1.67 (s, 6H, C(CH3)2), 1.64 (d, 6H, J=6.8 Hz, CH(CH3)2).
1-Isobutyl-2,4-diphenyl-6-((1E,3E)-3-(1,3,3-trimethylindolin-2-ylidene)prop-1-en-1-yl)pyridin-1-ium tetrafluoroborate (“SF-5-331”)
δH (400 MHz, DMSO-d6) 8.44 (s, 1H, pyridinium), 8.23 (t, 1H, J=13.2 Hz, CH=CH=CH), 8.08–8.10 (m, 2H, Ph), 7.62–7.71 (m, 9H, Ph, pyridinium), 7.44 (d, 1H, J=7.6 Hz, indolene), 7.28 (t, 1H, J=7.8 Hz, indolene), 7.09 (d, 1H, J=7.6 Hz, indolene), 7.03 (t, 1H, J=7.4 Hz, indolene), 6.69 (d, 1H, J=14.4 Hz, CH=CH=CH), 6.08 (d, 1H, J=12.4 Hz, CH=CH=CH), 4.30 (br, 2H, CH2CH(CH3)2), 3.37 (s, 3H, NCH3), 2.07 (m, 1H, CH2CH(CH3)2), 1.68 (s, 6H, C(CH3)2), 0.74 (br, 6H, CH2CH(CH3)2).
1-(2-Hydroxyethyl)-2,4-diphenyl-6-((1E,3E)-3-(1,3,3-trimethylindolin-2-ylidene)prop-1-en-1-yl)pyridin-1-ium tetrafluoroborate (“SF-5-332”)
δH (400 MHz, DMSO-d6) 8.42 (s, 1H, pyridinium), 8.15 (t, 1H, J=13.0 Hz, CH=CH=CH), 8.03–8.08 (m, 2H, Ph), 7.58–7.65 (m, 9H, Ph, pyridinium), 7.40 (d, 1H, J=6.8 Hz, indolene), 7.24 (t, 1H, J=7.8 Hz, indolene), 7.05 (d, 1H, J=7.6 Hz, indolene), 6.98 (t, 1H, J=7.2 Hz, indolene), 6.70 (d, 1H, J=13.6 Hz, CH=CH=CH), 5.97 (d, 1H, J=12.0 Hz, CH=CH=CH), 5.05 (m, 1H, OH), 4.41 (t, 2H, J=4.8 Hz, CH2CH2OH), 3.63 (m, 2H, CH2CH2OH), 3.38 (s, 3H, NCH3), 1.64 (s, 6H, C(CH3)2).
1-(2-(Dimethylamino)ethyl)-2,4-diphenyl-6-((1E,3E)-3-(1,3,3-trimethylindolin-2-ylidene)prop-1-en-1-yl)pyridin-1-ium tetrafluoroborate (“SF-5-334”)
δH (400 MHz, DMSO-d6) 8.44 (s, 1H, pyridinium), 8.22 (t, 1H, J=13.0 Hz, CH=CH=CH), 8.06–8.10 (m, 2H, Ph), 7.62–7.71 (m, 9H, Ph, pyridinium), 7.44 (d, 1H, J=7.6 Hz, indolene), 7.28 (t, 1H, J=7.8 Hz, indolene), 7.09 (d, 1H, J=7.6 Hz, indolene), 7.02 (t, 1H, J=7.2 Hz, indolene), 6.67 (d, 1H, J=13.6 Hz, CH=CH=CH), 6.05 (d, 1H, J=12.8 Hz, CH=CH=CH), 4.40 (t, 2H, J=6.4 Hz, CH2CH2N(CH3)2), 3.37 (s, 3H, NCH3 (indolene)), 2.57 (t, 2H, J=6.4 Hz, CH2CH2N(CH3)2), 1.90 (s, 6H, CH2CH2N(CH3)2), 1.68 (s, 6H, C(CH3)2).
Pharmacokinetic study of BAS00127538
A solution of BAS00127538 was prepared at 0.5 mg/mL in 10% DMSO and 50% polyethylene glycol in phosphate-buffered saline and administered at 1 mg/kg intravenous (tail vein) to male CD1 mice (N=3 per group). Approximately 0.02 mL of blood was collected at 5 minutes, 15 minutes, 30 minutes, 1 hour, 2 hours, 4 hours, 6 hours, 8 hours, and 14 hours posttreatment in centrifuge tubes containing 2 μL of heparin (1,000 units). Compound was quantified by liquid chromatography–tandem mass spectrometry using working solutions of 10 ng/mL, 20 ng/mL, 50 ng/mL, 100 ng/mL, 500 ng/mL, 1,000 ng/mL, 5,000 ng/mL, and 10,000 ng/mL BAS00127538 prepared in blank CD1 mouse plasma as internal standards. No adverse clinical observations were made for the duration of the experiment. Experiments were carried out by Pharmaron, Inc. (Beijing, People’s Republic of China) and have been approved by the Office of Laboratory Animal Welfare (Bethesda, MD, USA) under the identification number A5844-01.
Results and discussion
Previous studies demonstrated that BAS00127538 makes three distinct interactions with Lipid II:16 i) a phosphate–pyrylium ionic interaction, ii) the N-acetyl muramic acid–phenyl stacking, and iii) alkyl chain–indolene hydrophobic interactions. To identify Lipid II-binding analogs, we designed a query compound (Figure 1, detailed in “Materials and methods”) that was used to search the University of Maryland CADD Center in silico compound database of 5.04 million commercially available compounds using chemical fingerprint (BIT-MACCS) and physiochemical fingerprint (MPMFP). Chemical fingerprint-based searching may identify structurally similar compounds with improved biological activity, as well as lay the foundation to determine the relationship of the chemical structures to the function of the compound.34 Physiochemical fingerprint searching may identify compounds with dissimilar structures that may still have similar biological activity, potentially identifying novel lead compounds for future development. Using a similarity cutoff of 70% for the chemical fingerprint and 83% for the physiochemical fingerprint, 175 and 177 compounds were identified, respectively.
Twenty-one of the top ranking compounds similar to query were obtained and tested for antibacterial activity. In addition, seven compounds were selected to examine functionality of the indolene moiety (Table S1). Out of the 28 compounds that were tested, three compounds (ASN10791182, 4400-0093, and 56133428) showed antibacterial activity against S. aureus (Table 1). We next compared the antibacterial activity of BAS00127538, ASN10791182, 4400-0093, and 51633428 against an extended panel of bacterial species (Table S2). As previously reported, BAS00127538 is potently active against Gram-positive species.16 In particular, BAS00127538 showed activity against S. aureus (MIC 0.5), irrespective of vancomycin- or methicillin resistance. BAS00127538 was also active against the Gram-negative bacteria E. coli and A. baumannii,35 with MICs of 8 μg/mL and 2 μg/mL, respectively. The antibacterial activity of ASN10791182, 4400-0093, 56133428, and BAS00127537 was significantly reduced as compared to that of BAS00127538, in particular against S. aureus and A. baumannii (Table S2). Compounds ASN10791182, 4400-0093, and 56133428 were tested further for cytotoxicity and their ability to bind to Lipid II. Compounds ASN10791182 and 4400-0093 showed a 30-fold and 70-fold reduction, respectively, in Lipid II-binding affinity compared to BAS00127538, whereas 56133428 and BAS00127537 Lipid II binding was reduced ~15-fold (Table 1). Reduction in Lipid II-binding affinities coincided with a reduction in cytotoxicity (approximately fivefold for ASN10791182, greater than tenfold for 4400-0093, approximately twofold for 56133428) as well as antibacterial activity (32-fold for ASN10791182 and 4400-0093, 16-fold for 56133428) compared to BAS00127538 (Table 1). None of the other compounds showed antibacterial activity (Table S1). Of these compounds, only Z56760026 and BAS00127537 bound Lipid II (Kd: 3.7±3.5×10−5 M and 71±15.3×10−5 M; Table S1). The lack of activity of these compounds indicates that the nonpolar aromatic tail on the active compounds is functionally important. This finding is consistent with our molecular model indicating these moieties to interact with the hydrophobic isoprenyl units of Lipid II.16
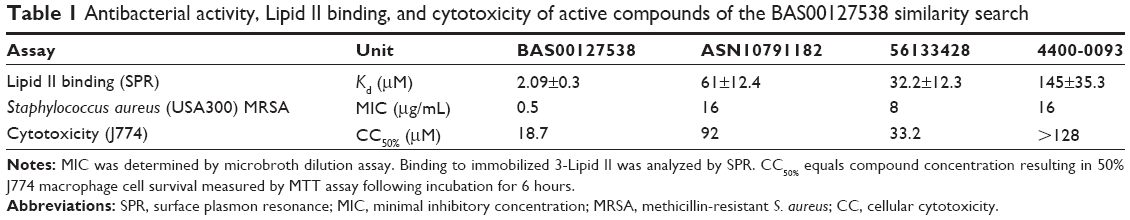 | Table 1 Antibacterial activity, Lipid II binding, and cytotoxicity of active compounds of the BAS00127538 similarity search |
Due to the presence of a pyrylium moiety in BAS00127538, it is possible that the compound is reactive toward nucleophiles, including water, amines, and thiols. We tested the chemical stability of BAS00127538 directly by determining its pharmacokinetic profile in vivo. BAS00127538 was administered as a single dose by intravenous injection, and compound plasma concentration was determined by liquid chromatography–tandem mass spectrometry over time. Compound was rapidly cleared in vivo (half-life of 22.7 minutes) and was no longer detected after 1 hour (Figure 2). Based on this observation, the pharmacokinetic parameters were calculated (Table 2). The observed maximum plasma concentration was 101 ng/mL and decreased rapidly over time. The mean residence time of the unchanged drug in circulation was 0.226 hour with an area under the curve AUCinf of 27.9 h ng/mL (Table 2). Volume of distribution at equilibrium (Vss) was 12.2 L/kg with total plasma clearance (CL) of 711 mL/min/kg.
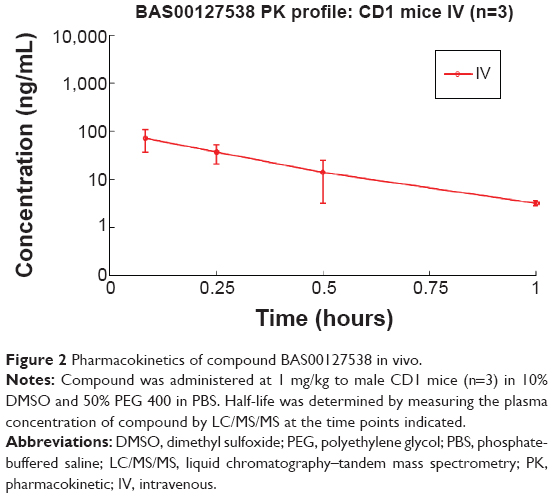 | Figure 2 Pharmacokinetics of compound BAS00127538 in vivo. |
 | Table 2 Pharmacokinetic properties of BAS00127538 |
Based on the structure of the BAS00127538–Lipid II complex, the potential reactivity of pyrylium moieties, and the in vivo data above, we designed five BAS00127538 derivatives to increase chemical stability in the scaffold (Figure 3). Our initial design strategy was twofold: i) convert pyrylium to pyridinium to diminish potential reactivity and maintain ionic interaction with phosphate, and ii) increase hydrogen bonding between inhibitor and Lipid II. The five molecules in Figure 3 were modeled onto Lipid II and subjected to MD simulations. In addition, a sixth compound in which the pyridinium moiety was substituted with an acid group was designed as a negative control as the negative charge of the acid moiety would have unfavorable interactions with the phosphate of Lipid II based. During the 20 ns MD simulations, three of the compounds were observed to dissociate from Lipid II during the first 2 ns; these included SF-5-219, SF-5-330 and as anticipated, the negative control compound containing the acid moiety. For the remaining compounds, including BAS00127538, their binding free energies with Lipid II were computed. The binding free energies of BAS00127538, SF-5-331, SF-5-332, and SF-5-334 were estimated by linear interaction energy method. As shown in Table 3, BAS00127538 is predicted to be the strongest binding ligand, and SF-331 the second strongest.
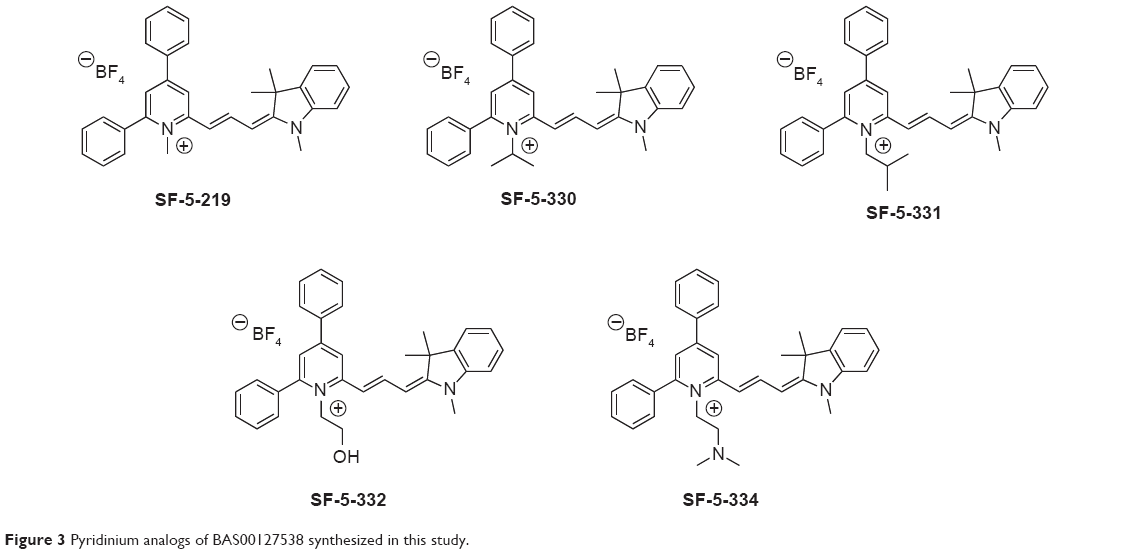 | Figure 3 Pyridinium analogs of BAS00127538 synthesized in this study. |
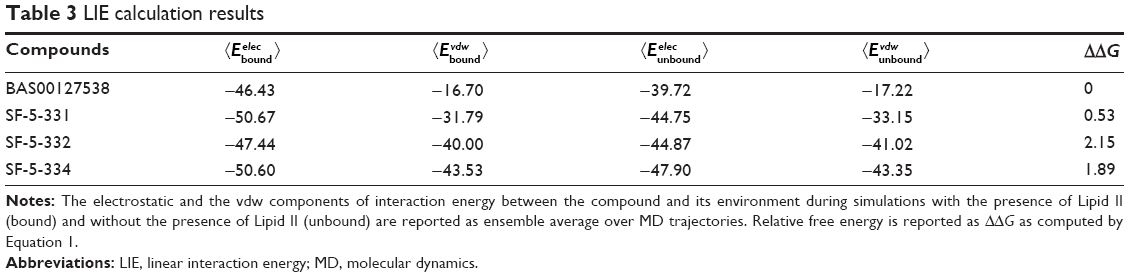 | Table 3 LIE calculation results |
Subsequently, all five compounds, SF-5-219, SF-5-330, SF-5-331, SF-5-332, and SF-5-334, were synthesized according to Figure 4 to challenge the modeling and to evaluate the compounds experimentally. First, we examined the antibacterial activities, Lipid II binding, and cytotoxicity of the BAS00127538 derivatives (Table 4). The modeling results were generally predictive of the relative antibacterial activities of the pyridinium analogs (Table 4), with the exception of the isobutyl analog, a result that may indicate an alternative binding mode for that compound. All analogs were shown to bind to Lipid II in the surface plasmon resonance measurements,16 with more variability in the MICs. Based on their broad-range antibacterial activity, SF-5-330 and SF-5-331 were selected to determine their mechanism of action (Figure 5). BAS00127538 most potently inhibited cell wall synthesis (IC50 of 0.19 μg/mL vs 0.42 μg/mL, 0.39 μg/mL, and 0.62 μg/mL for DNA, protein, or lipid synthesis, respectively) in accordance with our previous findings.16 SF-5-330 and SF-5-331 inhibited all pathways at low concentrations.
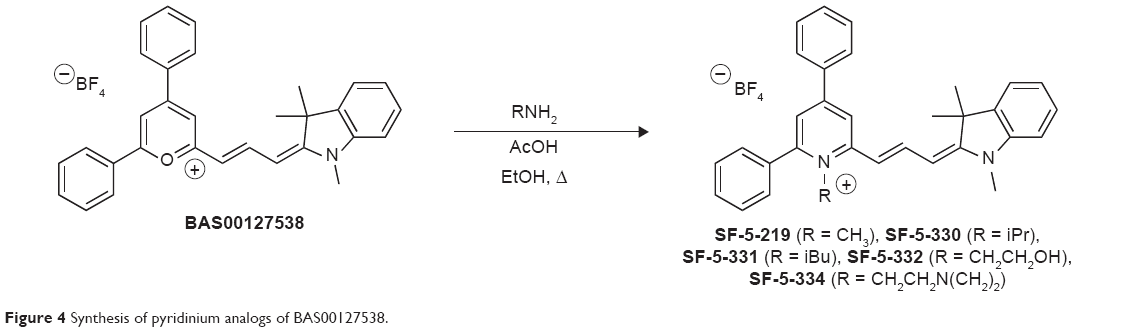 | Figure 4 Synthesis of pyridinium analogs of BAS00127538. |
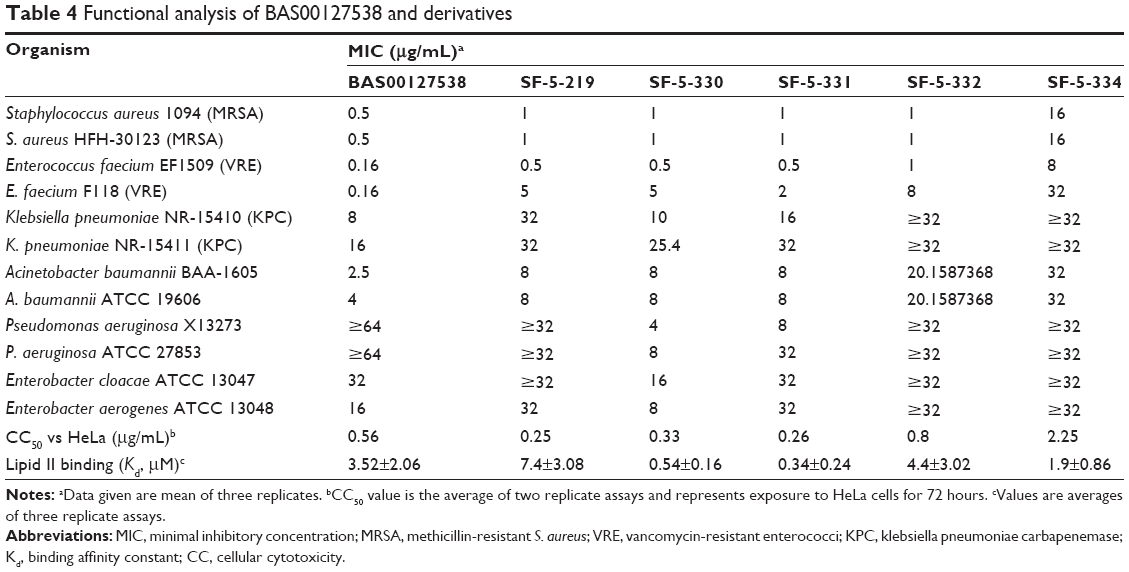 | Table 4 Functional analysis of BAS00127538 and derivatives |
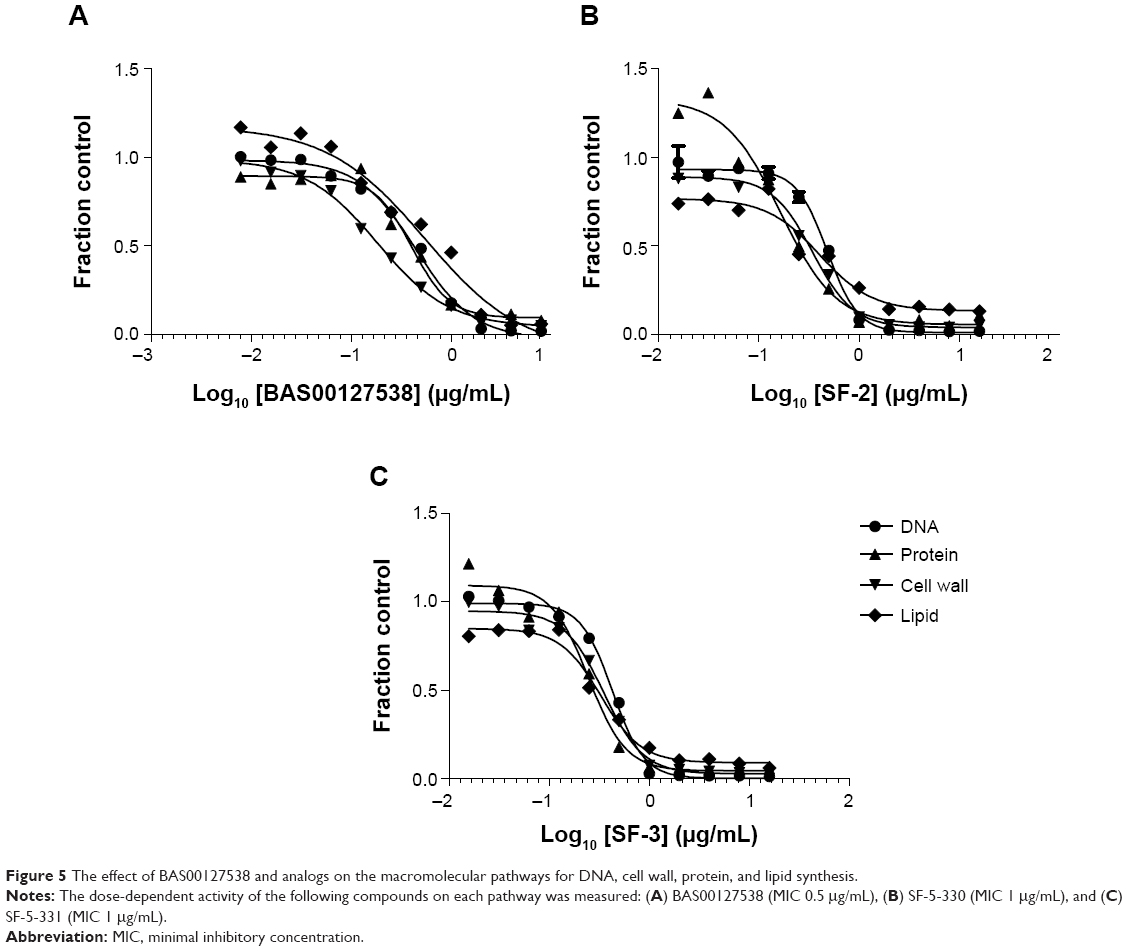 | Figure 5 The effect of BAS00127538 and analogs on the macromolecular pathways for DNA, cell wall, protein, and lipid synthesis. |
Conclusion
Although no definite structure–activity relationship could be determined, some conclusions on the structural features that may influence the antimicrobial activity of compounds like BAS00127538 can be drawn by the comparison among the structures and activities of the compounds.
- The molecular modeling of BAS00127538 and derivatives indicates a binding mode that is consistent with the nuclear magnetic resonance data.16
- Lipid II binding correlates with the presence of the following: 1) Positive charge of the pyrylium; for BAS00127538, this charge is predicted to interact with the phosphate moiety of Lipid II.16 The pyridinium derivatives (SF analogs) bind Lipid II with high affinity. Compound 56133428 binds to Lipid II with higher affinity and is more bactericidal compared to ASN10791182, which does not carry the positive charge. 2) Hydrophobicity of the indolene moiety; for BAS00127538, this moiety is predicted to interact with the first residues of the isoprenyl chain of Lipid II.16 These isoprenyl residues of Lipid II are assumed to reside in the interface between the phospholipid head groups and fatty acid chains, thus making it drug accessible.1,5 BAS00127538 and the SF analogs bind Lipid II with the highest affinity, and all contain the indolene moiety. Compound 56133428 does not contain the indolene and shows reduced Lipid II-binding affinity. 3) Phenyl rings; for BAS00127538, the two phenyl rings interact with the N-acetyl muramic acid moiety of Lipid II.16 All of the SF analogs, ASN10791182 and 56133428, are active compounds and contain two phenyl rings projected from the core of the small molecules, whereas 4400-0093 does not. Elaboration of these phenyl rings with para-ethoxy groups as in BAS00127537 reduces Lipid II-binding affinity and negates bacterial killing. Compound Z56760026 (Table S1) binds Lipid II but does not kill S. aureus.
- Lipid II binding correlates with antibacterial activity and cell wall specificity.
- Replacement of the pyrylium oxygen with a nitrogen to afford the chemically more stable pyridinium framework was successful and maintains broad-range antibacterial activity, in particular for the isobutyl analog SF-5-331; however, this modification also results in loss of pathway specificity and increases the cytotoxicity-to-MIC ratio.
- Pharmacokinetic analysis of BAS00127538 upon single-dose intravenous injection indicates that compound concentration over the MIC for S. aureus can be achieved at 1 mg/kg.
In summary, these studies focused on the potentially active scaffold of BAS00127538 pointed out the functional importance of the positions of the phenyl groups, the positively charged pyrylium/pyridinium, and hydrophobicity of the indole side chain in the substitution pattern. Optimization at these positions may lead to the development of small-molecule antibiotic targeting Lipid II with broad-spectrum activity.
Acknowledgments
This work was supported by National Institutes of Health grant AI092033 and a Maryland Innovation Initiative award to EdL. Further support was provided through the University of Maryland CADD Center. The funders had no role in study design, data collection and analysis, or preparation of the manuscript.
Disclaimer
The data as presented in this manuscript submitted to Drug Design, Development and Therapy are original and have not been published by any journal. Some of the data were presented at the 2014 Interscience Conference of Antimicrobial Agents and Chemotherapy meeting, Washington, DC, USA as a poster. The accompanying poster abstract can be found at http://www.icaaconline.com/php/icaac2014abstracts/data/papers/2014/F-978.htm. A full copy of the poster can be provided upon request.
Disclosure
The authors report no conflicts of interest in this work. US patent #8,796,323 has been issued (ADM and EdL, Inventors). This does not alter our adherence to all Drug Design, Development and Therapy policies on sharing data and materials.
References
Breukink E, de Kruijff B. Lipid II as a target for antibiotics. Nat Rev Drug Discov. 2006;5:321–332. | ||
Labischinski H, Goodell EW, Goodell A, Hochberg ML. Direct proof of a “more-than-single-layered” peptidoglycan architecture of Escherichia coli W7: a neutron small-angle scattering study. J Bacteriol. 1991;173:751–756. | ||
McCloskey MA, Troy FA. Paramagnetic isoprenoid carrier lipids. 2. Dispersion and dynamics in lipid membranes. Biochemistry. 1980;19:2061–2066. | ||
McCafferty DG, Cudic P, Frankel BA, Barkallah S, Kruger RG, Li W. Chemistry and biology of the ramoplanin family of peptide antibiotics. Biopolymers. 2002;66:261–284. | ||
Bauer R, Dicks LM. Mode of action of lipid II-targeting lantibiotics. Int J Food Microbiol. 2005;101:201–216. | ||
Brotz H, Bierbaum G, Reynolds PE, Sahl HG. The lantibiotic mersacidin inhibits peptidoglycan biosynthesis at the level of transglycosylation. Eur J Biochem. 1997;246:193–199. | ||
McCafferty DG, Cudic P, Yu MK, Behenna DC, Kruger R. Synergy and duality in peptide antibiotic mechanisms. Curr Opin Chem Biol. 1999;3:672–680. | ||
Ruzin A, Singh G, Severin A, et al. Mechanism of action of the mannopeptimycins, a novel class of glycopeptide antibiotics active against vancomycin-resistant gram-positive bacteria. Antimicrob Agents Chemother. 2004;48:728–738. | ||
Maki H, Miura K, Yamano Y. Katanosin B and plusbacin A(3), inhibitors of peptidoglycan synthesis in methicillin-resistant Staphylococcus aureus. Antimicrob Agents Chemother. 2001;45:1823–1827. | ||
Howden BP, Davies JK, Johnson PD, Stinear TP, Grayson ML. Reduced vancomycin susceptibility in Staphylococcus aureus, including vancomycin-intermediate and heterogeneous vancomycin-intermediate strains: resistance mechanisms, laboratory detection, and clinical implications. Clin Microbiol Rev. 2010;23:99–139. | ||
Ganz T. Defensins: antimicrobial peptides of innate immunity. Nat Rev Immunol. 2003;3:710–720. | ||
de Leeuw E, Li C, Zeng P, et al. Functional interaction of human neutrophil peptide-1 with the cell wall precursor lipid II. FEBS Lett. 2010;584:1543–1548. | ||
Schneider T, Kruse T, Wimmer R, et al. Plectasin, a fungal defensin, targets the bacterial cell wall precursor Lipid II. Science. 2010;328:1168–1172. | ||
Schmitt P, Wilmes M, Pugnière M, et al. Insight into invertebrate defensin mechanism of action: oyster defensins inhibit peptidoglycan biosynthesis by binding to lipid II. J Biol Chem. 2010;285:29208–29216. | ||
Sass V, Schneider T, Wilmes M, et al. Human beta-defensin 3 inhibits cell wall biosynthesis in Staphylococci. Infect Immun. 2010;78:2793–2800. | ||
Varney KM, Bonvin AM, Pazgier M, et al. Turning defense into offense: defensin mimetics as novel antibiotics targeting lipid II. PLoS Pathog. 2013;9:e1003732. | ||
Brown RD, Martin YC. The information content of 2D and 3D structural descriptors relevant to ligand-receptor binding. J Chem Inf Comput Sci. 1997;37:1–9. | ||
Xue L, Godden JW, Stahura FL, Bajorath J. Design and evaluation of a molecular fingerprint involving the transformation of property descriptor values into a binary classification scheme. J Chem Inf Comput Sci. 2003;43:1151–1157. | ||
Lichtenstein SJ, Dorfman M, Kennedy R, Stroman D. Controlling contagious bacterial conjunctivitis. J Pediatr Ophthalmol Strabismus. 2006;43:19–26. | ||
Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev. 2001;46:3–26. | ||
Brooks BR, Brooks CL 3rd, Mackerell AD Jr, et al. CHARMM: the biomolecular simulation program. J Comput Chem. 2009;30:1545–1614. | ||
Klauda JB, Venable RM, Freites JA, et al. Update of the CHARMM all-atom additive force field for lipids: validation on six lipid types. J Phys Chem B. 2010;114:7830–7843. | ||
MacKerell AD, Bashford D, Bellott M, et al. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J Phys Chem B. 1998;102:3586–3616. | ||
Best RB, Zhu X, Shim J, et al. Optimization of the additive CHARMM all-atom protein force field targeting improved sampling of the backbone φ, ψ and side-chain χ1 and χ2 dihedral angles. J Chem Theory Comput. 2012;8:3257–3273. | ||
Jorgensen WL. Transferable intermolecular potential functions for waters, alcohols, and ethers. Application to liquid water. J Am Chem Soc. 1981;103:335. | ||
Vanommeslaeghe K, Hatcher E, Acharya C, et al. CHARMM general force field: a force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J Comput Chem. 2010;31:671–690. | ||
Vanommeslaeghe K, Mackerell AD Jr. Automation of the CHARMM General Force Field (CGenFF) I: bond perception and atom typing. J Chem Inf Model. 2012;52:3144–3154. | ||
Vanommeslaeghe K, Raman EP, MacKerell AD Jr. Automation of the CHARMM General Force Field (CGenFF) II: assignment of bonded parameters and partial atomic charges. J Chem Inf Model. 2012;52:3155–3168. | ||
Åqvist J, Medina C, Samuelsson JE. A new method for predicting binding affinity in computer-aided drug design. Protein Eng. 1994;7:385–391. | ||
Breukink E, van Heusden HE, Vollmerhaus PJ, et al. Lipid II is an intrinsic component of the pore induced by nisin in bacterial membranes. J Biol Chem. 2003;278:19898–19903. | ||
CLSI. Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria That Grow Aerobically; Approved Standard – Eighth Edition. Wayne, PA: CLSI; 2009. | ||
Butler MM, Lamarr WA, Foster KA, et al. Antibacterial activity and mechanism of action of a novel anilinouracil-fluoroquinolone hybrid compound. Antimicrob Agents Chemother. 2007;51:119–127. | ||
Cotsonas King A, Wu L. Macromolecular synthesis and membrane perturbation assays for mechanisms of action studies of antimicrobial agents. Curr Protoc Pharmacol. 2009;Chapter 13:Unit13A17. | ||
Macias AT, Mia MY, Xia G, Hayashi J, MacKerell AD Jr. Lead validation and SAR development via chemical similarity searching; application to compounds targeting the pY+3 site of the SH2 domain of p56lck. J Chem Inf Model. 2005;45:1759–1766. | ||
de Leeuw EP. Efficacy of the small molecule inhibitor of Lipid II BAS00127538 against Acinetobacter baumannii. Drug Des Devel Ther. 2014;8:1061–1064. |
Supplementary materials
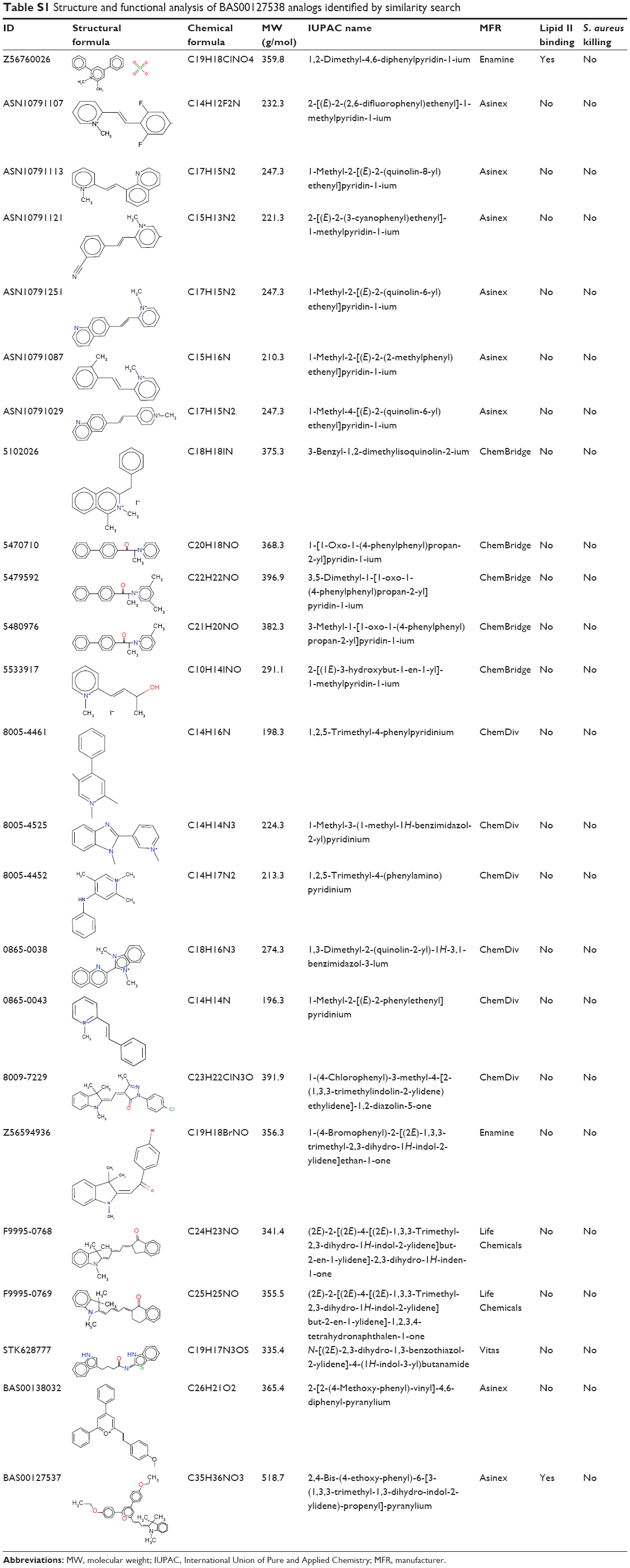 | Table S1 Structure and functional analysis of BAS00127538 analogs identified by similarity search |
 | Table S2 Antibacterial activity of active BAS00127538 similars |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.