Back to Archived Journals » Journal of Receptor, Ligand and Channel Research » Volume 7
Structure-based three-dimensional pharmacophores as an alternative to traditional methodologies
Received 7 January 2014
Accepted for publication 2 April 2014
Published 12 June 2014 Volume 2014:7 Pages 27—38
DOI https://doi.org/10.2147/JRLCR.S46845
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Anand Gaurav,1 Vertika Gautam2
1Faculty of Pharmaceutical Sciences, UCSI University, 2Department of Chemistry, Faculty of Science, University of Malaya, Kuala Lumpur, Malaysia
Abstract: Structure-based pharmacophore approaches have become widely used in drug discovery and design. This can be attributed to the development of new tools and methods over the past decade. Various tools based on different premises have been developed, including active site information in traditional pharmacophores. These tools have been widely used in virtual screening, de novo design, and lead optimization and been proven to be highly successful. Studies based on simultaneous use of structure-based pharmacophores, ligand-based pharmacophores, and docking have also come into the picture recently. Here, the development of structure-based pharmacophores as an alternative to traditional drug discovery approaches is discussed, with emphasis on the advances and latest developments in tools and success stories involving their application.
Keywords: structure-based pharmacophore, LigandScout, pocket, DS Catalyst SBP, Shape4, docking, virtual screening
Introduction
The International Union of Pure and Applied Chemistry defines a pharmacophore as “an ensemble of steric and electronic features that is necessary to ensure optimal supramolecular interactions with a specific biological target structure and to trigger (or block) its biological response”.1,2 Put simply, “a pharmacophore is a two-dimensional or three dimensional arrangement of chemical features essential for biological activity”. Over the years, pharmacophores have contributed a great deal in drug discovery efforts.3,4 They have been predominantly considered as a tool for ligand-based drug design where the structure of the receptor is not available.5 However, pharmacophores have also been used as low-resolution models for off-target effects in a structure-based design project, albeit to lesser extent.6–9 Their use as structure-based tools has been steadily increasing over the years (Figure 1).
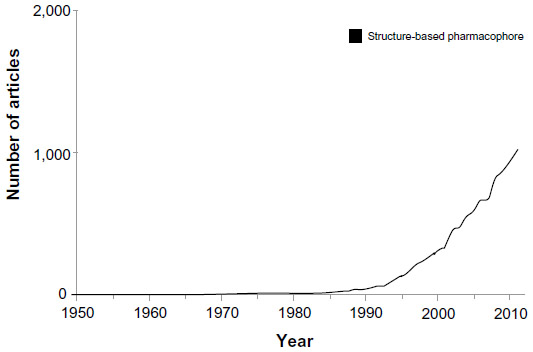 | Figure 1 Graphic representation of results obtained by searching PubMed using a keyword structure-based pharmacophore. |
Generally speaking, pharmacophores can be classified into two categories based on the method used to obtain them (Figure 2). The first category consists of models obtained by probing possible interaction points between the target and ligands. Such pharmacophores are broadly referred to as structure-based pharmacophores. Structure-based pharmacophores can themselves be obtained using two different approaches, ie, by using a complex structure of ligand and target where the three-dimensional structure of the target is available along with a certain number of ligands or by using only the three-dimensional structure of the target if the ligands for the said target are not available. The former approach involves generation of feature-based pharmacophores using H-bond formation, charge, and hydrophobic contact between ligand and receptor atoms. Further refinement of the model is performed by combining several models or by using binding data.7,9 The latter approach is sometimes referred to as a macromolecular approach and involves determination of possible interaction points in the active site. The main challenge faced in this approach is the numerous interaction points, which make the corresponding pharmacophore very complex. However, solutions to this problem are available, the most frequent being clustering of interaction point information to reduce the number of points and then derivation of a pharmacophore.10–12 The second category of pharmacophores is derived purely using the structures and binding data of ligands without considering the three-dimensional structure of receptors. Using this approach, pharmacophores are obtained by superposing a set of active molecules and extracting common chemical features that are essential for their bioactivity. Such pharmacophores are known as ligand-based pharmacophores.3,5
 | Figure 2 Classification of pharmacophore development methods. |
A survey of the literature reveals that pharmacophores has mostly been used as a ligand-based drug design tool whereby the three-dimensional structure of the receptor is not normally considered.13–16 The models thus fail to provide the detailed structural information required during lead optimization. Moreover, ligand-based pharmacophore tools, when used for virtual screening, ignore the intricate details of the binding site shape as well as interaction sites, and focus only on the key pharmacophore elements of the ligands as the query.17,18 Thus, screening based on conventional ligand-based pharmacophores give many false positives. Ligand-based pharmacophore modeling tools conventionally start with the diverse conformation generation step. Although this step has performed well in various applications, many studies have revealed that the energy of the biological conformer of a particular compound is usually well above its local energy minimum.19,20 Induced-fit theory also explains that a molecule should rearrange itself to fit into the active site of the protein, and energy spent on rearrangement is compensated by protein-ligand binding.21,22 Therefore, no protocol including energy minimization could assure the prediction of the biological conformation of small molecules. This further limits the applicability of ligand-based pharmacophores.
On the other hand, docking (a frequently used structure-based drug design approach) when used as a tool for screening also poses serious limitations, the major one being the fact that its use is highly target-dependent.23,24 Despite the framing of some general rules, it is still very difficult to know in advance which combination of docking program and scoring function will give optimal results for a particular target, and as such, it is normal practice to try in consensus a few docking/scoring combinations to identify the most suitable in each case.25,26 This makes the process slow, subjective, and less suitable for screening experiments.
Three-dimensional structure-based pharmacophores address the limitations of both ligand-based pharmacophores as well as docking. Since three-dimensional pharmacophore parameterization is not target-dependent, it handles target flexibility quite well, so is not computationally expensive. It is based on interaction residues and the shape of active sites, so overcomes the limitations of conventional pharmacophores. Many studies clearly demonstrate that inclusion of three-dimensional structural information on the receptor in the pharmacophore greatly improves its quality, especially in cases where it is used for in silico screening experiments.9,27–29
This review is an attempt to present a structure-based pharmacophore approach as an alternative to traditional computational drug design strategies like ligand-based pharmacophores and docking-based methods. Emphasis is placed on applications of structure-based pharmacophores in virtual screening based on recent publications. The paper is divided into two parts, the first elaborating the tools available for obtaining structure-based pharmacophores, and the second including an exhaustive discussion of successful application of structure-based pharmacophores from the published literature.
Structure-based pharmacophore approach: tools
As discussed in the previous section, structure-based pharmacophore modeling works directly with the three-dimensional structure of a macromolecular target or a macromolecule-ligand complex. The general protocol of structure-based pharmacophore modeling involves analysis of the complementary chemical features of the active site and their spatial relationships, and a subsequent assembling of selected features to obtain a pharmacophore. Structure-based pharmacophore modeling methods can be further classified into two subcategories, ie, macromolecule-ligand-complex-based and macromolecule (without ligand)-based. The macromolecule-ligand-complex-based approach is specialized to detect the ligand-binding site of the macromolecular target and determining the key interaction points between ligands.
When the three-dimensional structure of the binding site (target) is available along with a certain number of ligands, various programs are available for structure-based pharmacophore modeling; of these, Catalyst (Accelrys, Inc., San Diego, CA, USA)30 and Ligand Scout (Inte:Ligand GmbH, Vienna, Austria)31 are the most widely used, with others including energy-optimized Pharmacophore (e-Pharmacophore; Schrödinger, LLC, New York, NY, USA),32,33 Pocket version 2 (Pocket v.2; Institute of Physical Chemistry, Peking University, People’s Republic of China),7 and Snooker (Computational Drug Discovery Group, CMBI, Radboud University Nijmegen, the Netherlands).34 Herein, the technical details of these tools are discussed, along with their applicability and limitations in structure-based pharmacophore modeling. The methods of pharmacophore validation are almost the same for all tools, and include decoy set screening and receiver operating characteristic curves. An overview of these tools is shown in Table 1.
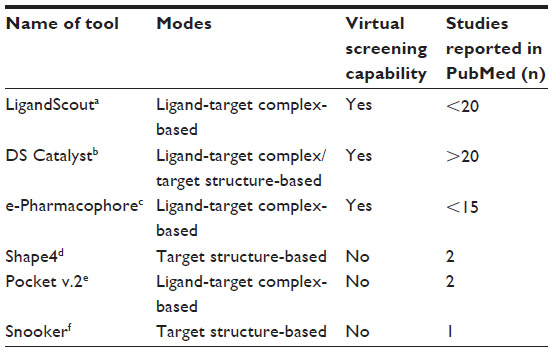 | Table 1 Overview of tools available for structure-based pharmacophore modeling |
LigandScout
The LigandScout31 (Inte:Ligand GmbH) enables users to automatically derive a feature-based pharmacophore model from a ligand-target complex structure.35–37 The ligand may be cocrystallized or docked with the target. In this program, the first step is ligand perception, which involves the assignment of ligand information on hybridization status and bond characteristics that is not present in the input data files from the Protein Data Bank. This is achieved by using an extended heuristic approach together with template-based numeric analysis. Ligand perception and interpretation are performed in two steps: the perception and correction of probable molecular topology along with ring perception; and the interpretation and subsequent assignment of hybridization states and bond types from (often ambiguous) geometric information. Feature-based pharmacophores are then generated by determining interactions between ligand and target atoms on the basis of H-bond formation, charge, and hydrophobic contact.38 These models can then be refined according to binding data or several models can be combined into one common feature pharmacophore.
LigandScout enables the user to undertake virtual screening within the program, eliminating the need to export results to other programs. However, the program also allows export of the pharmacophores into common pharmacophore format files, which can be directly imported into standard software packages. One of the positive features of LigandScout which hitherto has not been highlighted is the ease with which it can be used to derive a structure-based pharmacophore. This is especially important for new users because the whole process involves a few mouse clicks, this despite it being a very powerful tool. Despite having many useful features, LigandScout suffers from a major limitation, ie, it is not applicable in situations where only the target structure is known, ie, no ligand structures are available.38 Thus, it can be said that LigandScout is not a true structure-based pharmacophore development program.
DS Catalyst SBP
The catalyst structure-based pharmacophore implemented in Discovery Studio30 (DS Catalyst SBP; Accelrys, San Diego, CA, USA)39 is the most widely used tool for structure-based pharmacophore development, along with LigandScout.40–43 The first step in DS Catalyst SBP involves generation of interaction maps using LUDI11 (a program for de novo ligand design). In the next step, DS Catalyst SBP converts interaction maps obtained by using LUDI into Catalyst30 pharmacophoric features, ie, an H-bond acceptor, an H-bond donor, and a hydrophobe within the protein binding site.
The interaction maps can be obtained either using the target structure or the ligand-target complex structure. This enables DS Catalyst SBP to obtain structure-based pharmacophores from either a ligand-target complex structure or from a target structure (with no bound ligand).44 Another important feature of DS Catalyst SBP is that the protein structure pharmacophore features can be integrated directly with ligand features to create a complete model of the features critical for binding.45 The ability to integrate protein and ligand data can be especially powerful when complete protein structures are not available, as is the case for many kinases. Furthermore, it is also possible to add excluded volumes observed from protein structures or derived from ligand data to better correlate the model with the steric constraints imposed by the target.
As far as virtual screening is concerned, Discovery Studio has its own screening module that can be used without the need to export data to other programs. The ability to edit and cluster LUDI interactive maps of the target active site using receptor knowledge to retain only the information essential for virtual screening also helps in virtual screening.
The main limitation of the DS Catalyst SBP is that interaction maps generally consist of hundreds of Catalyst features, which means thousands of possible pharmacophoric hypotheses, and this makes the pharmacophore-based screening of a compound library computationally expensive.46 DS Catalyst SBP does not allow the export of pharmacophores into common pharmacophore format files, so restricts the import of pharmacophores to other programs. This means that pharmacophores developed using DS Catalyst SBP can neither be enhanced using other programs nor be used for screening using other platforms.
e-Pharmacophore
Another tool that has attracted the attention of researchers recently is e-Pharmacophore implemented in phase version 3.0.32,33 It uses a novel methodology and is quite different from other tools in this respect. e-Pharmacophores are structure-based pharmacophores generated by energy optimization of structure-based pharmacophores. They combine the advantages of two principal computational approaches, ie, the computational efficiency of ligand-based pharmacophore screening and the accuracy of scoring from structure-based docking. The methodology begins with refinement of the ligand pose of the ligand-receptor complex and computation of the Glide XP scoring terms. This is followed by mapping of the energies onto atoms. The next step involves generation of pharmacophore sites, followed by summation of the Glide XP energies from the atoms that comprise each pharmacophore site. Finally, the top scoring sites based on energetic ranking are used to generate a pharmacophore hypothesis that is then used for screening a database. The advantages of this method for virtual screening experiments include better database enrichments, greater diversity of retrieved actives, and faster database screening as compared with other virtual screening approaches. The e-Pharmacophore method leverages the strengths of both approaches to produce high enrichments with good diversity of active molecules. Indeed, the e-Pharmacophore method has been shown to retrieve a more diverse set of actives than other structure-based pharmacophore methods, making it a potent tool for lead hopping.47 Since e-Pharmacophore includes an integrated screening module, there is no need to export data to other screening programs. The e-Pharmacophore program has been successfully applied for structure-based pharmacophore design and virtual screening of Mycobacterium tuberculosis l-alanine dehydrogenase (MTB-l-AlaDH) inhibitors,48 vitamin D receptor inhibitors,49 histone deacetylase inhibitors,50 beta-secretase 1 inhibitors,47 and catechol-O-methyl transferase inhibitors.51 Like LigandScout, it is not applicable in situations where only the target structure is available, so is not a true structure-based pharmacophore development program. The program also lacks the option to export pharmacophores to other programs.
Shape4
Shape4 (Department of Pharmaceutical Sciences, Biomanufacturing Research Institute Technology Enterprise [BRITE], North Carolina Central University, Durham, NC, USA) is another structure-based pharmacophore program designed to increase the efficiency of database searching by taking into account the topographical constraints of the target binding site and incorporating them in the pharmacophore model. This approach has been shown to help reduce the false positive rate.52,53
Shape4 employs computational geometry algorithms (Delaunay tessellation/R-shape analysis) to detect the binding site atoms and generate a negative image of the target binding site. This negative image is first represented by a set of spheres of different sizes. A variety of techniques can then be applied to represent the overall surface shape of this set of spheres. This involves various steps: the α-shape-based program is first used to detect potential binding site (pocket) atoms and the Delaunay tetrahedra formed by these atoms for a given protein structure; nCast is then used to calculate orthogonal centers defined by the vertices of the above Delaunay tetrahedra followed by generation of inner spheres around each orthogonal center, with filters potentially used to exclude the spheres that are unlikely to be useful for ligand discovery. The overall shape of this collection of spheres is then represented by Gaussian functions based on the OpenEye SHAPE library, and the shape representation is then used by Shape4 to query a database of molecules, the conformers of which are pregenerated, eg, using the program Omega. This protocol implements an efficient, basic structure-based shape-matching method for virtual screening.54–56
Shape4 is a fast, effective, and intuitive tool for structure-based pharmacophore development and virtual screening. The main feature of the Shape4 method is its applicability in situations where known inhibitors are not there. In addition, the computational hits obtained in virtual screening are found to be much more diverse than ligand-based pharmacophore or docking-based screening methods.
The main limitation of Shape4 is the fact that it does not take into consideration the ligand binding information at all. Thus, it ignores crucial information of ligand receptor interaction which may result in pharmacophores with some important pharmacophore features missing. However, this limitation helps in improving the diversity of hits obtained by virtual screening because it reduces ligand bias.
Pocket v.2
Pocket v.2 is a standalone program that has been developed based on the Pocket module in the popular de novo drug design program LigBuilder (the Institute of Physical Chemistry).57,58 It is an automatic pharmacophore development program, ie, it can derive a pharmacophore model directly from a given protein-ligand complex structure without human intervention.7 This makes it similar to LigandScout and DS Catalyst SBP. Like these programs, Pocket v.2 also reduces the key features in the pharmacophore model to a reasonable number automatically. The steps involved in pharmacophore generation by Pocket v.2 are: binding site analysis by grid generation on the region surrounding the ligand;57–61 generation of an interaction model from the grid to identify interaction features; deduction of the pharmacophore model by correlating interaction features and ligand atoms followed by clustering of features to reduce them to a reasonable number; and identification of additional binding features which are not essential for binding but can improve affinity.
The usefulness of Pocket v.2 has been established by its application to several targets, including cyclin-dependent kinase 2 (CDK2), HIV-1 protease, the estrogen receptor, and 17α-hydroxysteroid dehydrogenase.7 The pharmacophores produced by Pocket v.2 are very similar to the previously published models in all the above-mentioned cases. One notable feature of Pocket v.2 is that it can tolerate minor conformational changes on the protein side upon binding of different ligands to give a consistent pharmacophore model. Another important feature of Pocket v.2 is its ability to give similar pharmacophore models for different proteins that bind with the same ligand, thereby making it possible to categorize protein molecules according to their vital binding features. This will also be helpful for predicting potential side effects of known compounds of pharmaceutical importance. The program is available free for academic users, although it lacks many features of commercial programs, ie, it lacks a screening module and is unable to save pharmacophores in common file formats for export to other programs. Pocket v.2 also lacks the ability to generate a pharmacophore when only the target structure is available.
Snooker
Snooker is another approach which is a specifically macromolecule (without ligand)-based pharmacophore approach and applicable only for the G-protein-coupled receptor class A subfamily. The tool was designed mainly for pharmacophore modeling of G-protein-coupled receptor apoproteins.34 The modeling protocol involves the following steps: template selection, where templates are selected from previously available templates, custom-made homology models, future crystal structures, or a combination thereof; a homology model is constructed based on the alignment of the model receptor sequence with this template; rotamer sampling, where an α helix-specific rotamer library is used to add a rotamer ensemble to account for possible inaccuracy of the initial homology model; the pocket is detected by Delaunay tessellation of the Cα atom and average side chain atom positions; residues are scored on ligand binding probability by multiple sequence alignment analysis;62,63 “interaction” points are placed inside the pocket volume using the interaction geometries described in literature with densities corresponding to the residue score and rotamer probability;64 pharmacophore features are then generated with a fuzzy pharmacophore algorithm applied on the interaction points;65 and finally, ligands fulfilling all pharmacophore constraints are aligned to the pharmacophore.66
At present, this method is suitable only for pharmacophore development and design of compounds targeting the small class A subfamily of G-protein-coupled receptors. Another limitation is that the pharmacophores obtained are of low resolution and thus may require further refinement. Despite the fact that Snooker is still an approach or concept rather than a tool, it is worth mentioning that many features of other programs must be included to make this approach a widely applicable one.
Structure-based pharmacophore approach: applications
This section reviews some of the selected applications of the structure-based pharmacophore approach that have been successfully applied for ligand design, virtual screening, and lead discovery. The recent literature demonstrates that the structure-based pharmacophore approach has been a significant improvement over other structure-based methods like docking. In the future, this would be a true alternative to other methods, provided that some of the shortcomings are overcome. The applications discussed in this section clearly demonstrate that this approach is similar or better in terms of accuracy, is, faster, is applicable to diverse targets, and is relatively flexible compared with other structure-based methods. The above advantages of the structure-based pharmacophore are more perceptible when it is used for virtual screening.
In one of the most recent studies, which was designed to identify new chemical entities as carbonic anhydrase inhibitors targeting human carbonic anhydrase VII, a structure-based pharmacophore approach was used.67 Using LigandScout software, pharmacophore models were built from crystal structures of two well-known carbonic anhydrase inhibitors in complex with human carbonic anhydrase VII. The three-dimensional structures of human carbonic anhydrase VII bound with the well-known sulfonamide inhibitors, acetazolamide (3MDZ, Protein Data Bank) and ethoxzolamide (3ML5, Protein Data Bank), were chosen as the input for structure-based pharmacophore generation. Two three-dimensional pharmacophore hypotheses representing the main interactions between the enzyme and inhibitor 1 or 2 were obtained in this manner. Later, a new structure-based pharmacophore model was generated by superimposing the two structure-based hypotheses and removing the overlapped features. The model was validated and could successfully filter the most active compounds from a series of carbonic anhydrase inhibitors, thus having good discriminatory power. Subsequently, a focused library of compounds with only those compounds containing a sulfonamide moiety (6313) was prepared from the ZINC database. This library was screened against the pharmacophore model. A total of 299 compounds having high fit values were selected, and among them, 22 of those found were already known carbonic anhydrase inhibitors. This result indicated a good predictive capability of the pharmacophore model. The most interesting hits (34) were docked into the crystal structure of human carbonic anhydrase VII. As a result, two compounds, ie, 4-bromo-2-chloro-N-[4-(sulfamoylmethyl)phenyl] benzenesulfonamide and N-[4-(sulfamoylmethyl)phenyl]cyclohex-3-ene-1-carboxamide, were identified. These compounds were then tested for carbonic anhydrase inhibition activity. Both the compounds displayed significant carbonic anhydrase inhibitory effects in the nanomolar range. This study proves the worth of structure-based pharmacophores as a virtual screening and lead identification tool and also as an alternative to traditional tools used for the same purpose.
In another study, a very simple manual approach to development of structure-based pharmacophores for highly cyclooxygenase (COX)-2 selective inhibitors using Catalyst 3.0 was described recently.68 The first step involved identification of common features shared by 16 diverse COX-2 inhibitor ligands without considering activity values.30 The simple feature-based pharmacophore obtained in this manner consisted of an H-bond acceptor, two hydrophobic groups, and an aromatic ring, which was consistent with available structure-activity relationship data. The pharmacophore model was then transformed into a structure-based pharmacophore by adding exclusion spheres representing important residues of the COX-2 binding site based on human COX-2, modeled from crystallized murine COX-2 in complex with SC-558 (Protein Data Bank code 6COX). Such a pharmacophore is known to reduce the number of hits, and also the number of false positives in a hit list, by a factor of 2–5 in virtual screening.69 The resulting pharmacophore was used to screen the Maybridge database. The first 300 hits were then selected for further analysis. From bioavailability, scoring, and flexibility criteria, 40 compounds were retained, and eight of these were assayed on the basis of their high scoring values. Five compounds preferentially inhibited COX-2, two had a pharmacological profile similar to rofecoxib and one was a potent COX-2 selective compound with a original scaffold. BTB 09656 (6,8-dibromo-3-carbethoxycoumarin) was identified as the most potent COX-2 inhibitor among the selected compounds. The impressive performance of structure-based pharmacophore in getting promising hits through virtual screening has been illustrated in this study.
The results of a study reported by Zhang et al clearly demonstrate the ability of structure-based pharmacophores to identify actives from large databases. In this case, Pocket v.2 was used to obtain a structure-based pharmacophore model for the main proteinase of severe acute respiratory syndrome coronavirus (SARS-CoV).70 The experimental structure of the SARS-CoV main proteinase complexed with its peptide inhibitor CMK (Protein Data Bank ID 1UK4), along with the predicted structures of the SARS-CoV main proteinase with six drugs were used for creating pharmacophore models. The Pocket module in the LigBuilder program (the Institute of Physical Chemistry) was used to obtain the pharmacophore models. A set of seven eight-point pharmacophore models was developed in this manner. Based on these models, a common four-point pharmacophore distance pattern was extracted. This pharmacophore distance pattern was subsequently used to search the National Cancer Institute three-dimensional database containing 250, 251 compounds. Thirty existing drugs containing the pharmacophore query were obtained as hits, among them six compounds (azauridine, pyrazofurin, ribavirin, 2′,3′-dideoxycytidine, dideoxyguanosine, and 5-bromo-2′-deoxycytidine) that already showed anti-SARS-CoV activity experimentally, thereby establishing the ability of a pharmacophore to identify antiviral structural elements as well as to screen a large database rapidly.
In another study, structure-based pharmacophores for angiotensin-converting enzyme 2 (ACE2) inhibitors were developed and used successfully for virtual screening to get hits with micromolar half maximal inhibitory concentration (IC50) values. The active site of ACE2 in complex with a bound ACE2 inhibitor (MLN-4760) was first studied using LigandScout.71 Multiple chemical features were detected and mapped onto the ligand functional groups, and alternative hydrogen bond donor and/or acceptor sites were also considered simultaneously on the protein within the limits of geometric constraints. Exclusion volume spheres were added to the structure-based models against the coordinates defined by protein side chain atoms in order to characterize inaccessible areas for any potential ligand. Further model refinement, such as addition of shape constraints, was carried out using Catalyst version 4.9 software. The ACE2 pharmacophore hypothesis was used to screen the ACE inhibitor database and the Derwent World Drug Index comprising 63,307 drugs and pharmacologically active compounds. The Catalyst program was used for screening purposes, and 25 compounds were shortlisted for biological testing based on the screening results. Some of the molecules demonstrated micromolar activity in the ACE2 inhibition assay. The most potent ACE2 inhibitor among the compounds identified was 4S-16659 [(E)-3-[5-(5-chloro-2-methylphenyl)furan-2-yl]-2-[(5-methyl-1H-1,2,4-triazol-3-yl)sulfanyl]prop-2-enoic acid].
Shape pharmacophore models are some of the structure-based pharmacophores that have been explored and validated recently for virtual screening and lead discovery. In one such study, shape pharmacophore models were validated for virtual screening of anesthetic compounds.72 The three-dimensional structure of apoferritin (poferritin complexed with isoflurane [1XZ3] and halothane complexed with apoferritin [1XZ1])73 was used as the basis for development of several shape pharmacophore models using the Shape 4 program. The models were validated for their application in virtual screening by demonstrating that they can efficiently recover known anesthetic agents from a database of diverse compounds. It was observed that the shape pharmacophore scores a significant linear correlation with the measured apoferritin site binding affinities of several known anesthetic compounds. The shape pharmacophore models derived were quite remarkable in their ability to recover known anesthetic molecules from a diverse set of decoys randomly sampled from the Asinex database. Thus, shape pharmacophore models can qualitatively distinguish anesthetic molecules from other unrelated compounds. These results are consistent with the fact that the ferritin binding pocket is largely hydrophobic, so shape and size play a dominant role in binding. The further advantage of a faster screening speed was also observed in this study.
Structure-based pharmacophore models obtained using single-target/ligand-target structures have proved their worth as an alternative to the traditional approaches. This is particularly evident by their role in the virtual screening process, where they have taken center stage in many studies. However, such models are likely to miss some important interaction information. Recently, some studies have reported the multicomplex-based comprehensive pharmacophore mapping technique, where multiple protein-ligand complexes available in the Protein Data Bank are used to devise the pharmacophore map. These pharmacophores are able to identify even more critical interaction information than the conventional structure-based pharmacophores and can prove more useful in virtual screening. Studies using such models successfully are discussed in the following paragraphs.
Arooj et al illustrated the application of a multiple pharmacophore approach to identify structurally diverse hits as chymase inhibitors.74 X-ray crystallographic data for chymase in complex with different inhibitors were used to generate four structure-based pharmacophore models. Pharmacophores were generated using the Pharmacophore module of Discovery Studio version 3.0 (Accelrys Software, Inc., San Diego, CA, USA) with default parameters. Four different crystal structures for chymase cocrystallized with four different inhibitors (3N7O, 1T31, 3SON, and 2HVX) were used. One ligand-based pharmacophore model was also developed from experimentally known inhibitors. After successful validation, all pharmacophore models were employed in database screening to retrieve hits with novel chemical scaffolds. Four structurally diverse compounds were selected as final hits. The top scoring hit was KM 09155 (2-[[4-methyl-5-[2-oxo-2-(phenylamino)ethyl]sulfanyl-1,2,4-triazol-3-yl]methylsulfanyl]-N-phenylacetamide). The identified ligands are expected to bind to all possible bioactive conformations available in the active site of the enzyme.
Another study describing the multicomplex/structure-based pharmacophore for CDK2 was recently reported.75 In this case, a comprehensive pharmacophore map was generated based on a collection of 124 crystal structures of the human CDK2-inhibitor complex. This ensemble of structures is expected to contain almost all the chemical features important for CDK2-inhibitor interactions.76 A comparison with previously reported ligand-based pharmacophores showed that the ligand-based models are just a subset of this comprehensive map. The developed model not only successfully discriminated between known CDK2 inhibitors and the molecules of the focused inactive dataset, but was also capable of correctly predicting the activities of a wide variety of CDK2 inhibitors in an external active dataset. The superiority of multicomplex/structure-based pharmacophores over ligand-based methods has been further demonstrated by the results of this study.
The applicability of multicomplex-based pharmacophore models for screening large databases has also been described by Moser et al, who developed a receptor-based pharmacophore model of soluble epoxide hydrolase.77 Thirteen crystal structures (Protein Data Bank numbers 1S8O, 1VJ5, 1ZD2, 1ZD3, 1ZD4, 1ZD5, 3ANS, 3ANT, 3I1Y, 3I28, 3KOO, 3OTQ, and 3PDC) of soluble epoxide hydrolase were used for this purpose. The Protein Ligand Interaction Fingerprints assistant together with the QueryGenerator tool of the Molecular Operating Environment package (Chemical Computing Group Inc., Montreal, QC, Canada) was used to model the shape of the binding pocket as excluded volumes.78 The clusters of receptor pharmacophore annotation points within the binding pocket were examined, and corresponding features were created, leading to a total of nine features. The pharmacophore was then used to perform a virtual screening on the “merged libraries” database provided by Asinex for novel compounds. They identified 3,191 unique hits which were further subjected to Lipinski, ADMET filters, and docking to get lead compounds. Evaluation of the leads led to identification of the most potent lead compound, ie, (3-chloro-phenyl)-carbamic acid 3-(3-nitro-benzenesulfonylamino)-propyl ester, with an IC50 value of 3.2 μM. As in previous examples, multicomplex/structure-based pharmacophores have been used as the principal tool to screen a large database in order to reach a smaller dataset. However, unlike previous examples, the smaller dataset was further screened by docking and other tools to obtain the lead compounds.
The examples discussed above highlight the principal role played by structure-based pharmacophores in the process of virtual screening and lead generation, thereby providing an alternative to docking and other traditional methods. Despite the success of structure-based pharmacophores in virtual screening and the lead discovery process, its combined application along with docking and other tools has led to improved performance in database screening. The following paragraphs provide an insight into some studies where the combined approach has been used.
In a study designed to discover new structures as inhibitors of di-zinc metallo-b-lactamase, virtual screening based on structure-based pharmacophores followed by docking was used.79 Pharmacophore models were generated based on succinic acid crystallized with IGF2 mRNA-binding protein 1 (IMP-1) (Protein Data Bank code 1JJT) and mercaptocarboxylate ligand crystallized with IMP-1 (Protein Data Bank code 1DD6). Catalyst 4.6 was used for pharmacophore generation as well as a database search. Seventy-four hits were obtained from the database search, and the inhibitory effect was tested for 13 of these compounds. Different criteria were used to select compounds for experimental determination of the IC50 value. First, they needed to fit into the active site of the enzyme; second they needed to fit the pharmacophore elements; and finally, had to be relatively small. In addition, the ligands were required to have different types of groups with metal binding ability. The ligand with the highest IC50 value was found to be 2,5-diphenyl-furan-3,4-dicarboxylic acid. The experimental results showed that this way of selecting ligands worked reasonably well. However, the docking score alone should be used with caution, because it might lead to too many false positives.
Another study used the combined approach of structure-based pharmacophores for virtual screening followed by docking for identification of Trypanosoma brucei leucyl-tRNA synthetase (TbLeuRS) inhibitors.80 The crucial interaction patterns between TbLeuRS and Leu-AMS (stable analog of endogenous substrate Leu-AMP) were analyzed using LigandScout.31 Four structure-based pharmacophore models were generated and subsequently evaluated by a set of 20 molecules including substrate analog Leu-AMS, ten known LeuRS inhibitors, and nine random compounds from the SPECS database. The pharmacophore models were used to screen SPECS database, and a number of 2-pyrrolinones were discovered to be TbLeuRS inhibitors. The most potent compound was 5-(3-fluoro-phenyl)-3-hydroxy-1-[2-(1H-indol-3-yl)-ethyl]-4-(4-methoxy-benzoyl)-1,5-dihydro-pyrrol-2-one, with an IC50 of 170.3 μM. Based on this lead compound, new molecules were designed, synthesized, and evaluated by docking and experimental evaluation, with promising results.
Some of the studies employing both structure-based pharmacophores and ligand-based pharmacophores in the screening process have clearly established the superiority of the former over the latter for this purpose. Karkola et al performed one such study on the 17β-hydroxysteroid dehydrogenase type 1 (17β-HSD1) enzyme, a potential anticancer target.81 Structure-based pharmacophores were generated based on: the crystal structure of 17β-HSD1; the crystal structure of 17β-HSD1 with bound estradiol (Protein Data Bank entry code 1FDT); and the interactions of a potent inhibitor and the enzyme. The first pharmacophore was generated by applying the Catalyst protocol of Discovery Studio 2.1 using the 17β-HSD1 crystal structure, while the other two pharmacophores were generated by applying the LigandScout program’s automatic pharmacophore generation protocol using the 17β-HSD1 crystal structure bound to estradiol and a potent inhibitor. LigandScout was used for the latter two cases because Catalyst cannot generate pharmacophores based on ligand receptor interactions. The Maybridge HitFinder™ Collection database with generated conformers was searched to find compounds that fitted the pharmacophoric requirements. Twenty compounds were obtained using pharmacophore-based screening. The compounds were further screened using docking and different scoring functions. The screening protocol was successful in identification of compounds having diverse structures quite different from the ligand complexed with 17β-HSD1. This study also involved generation of a ligand-based pharmacophore. However, the structure-based model proved more informative and superior for screening purposes. The hits obtained when a structure-based pharmacophore was used for screening were more diverse than those obtained using a ligand-based pharmacophore. The application of docking following pharmacophore-based database screening helped to rank the hits accurately and as per their reported activity.
In a report by Saxena et al, application of energy-optimized pharmacophores and docking were described for designing novel MTB-l-AlaDH inhibitors. They used the e-Pharmacophore program implemented in phase version 3.0 to generate a pharmacophore hypothesis for MTB-l-AlaDH inhibitors.48 Since no inhibitors were reported for l-alanine dehydrogenase, co-factor NAD+ bound with the protein along with pyruvate as the substrate was used for generation of a pharmacophore hypothesis. In the first step, the ligand NAD+ was docked into the active site of the protein l-AlaDH from MTB (Protein Data Bank code 2VHW) using Glide XP. The best conformer of the docked compound along with the binding pose (Xpdes file of the Glide XP output) was then used to obtain pharmacophores using the e-Pharmacophore program. This pharmacophore model was then used as a query for screening of the Asinex database containing 500,000 unique structure records. This was followed by ranking the hits by docking using Glide XP. Five top hit compounds with a good docking score and interaction pattern were obtained finally. In vitro enzymatic inhibition studies of these five ligands yielded two compounds with micromolar IC50 values. The top scoring lead compound, 2,2′-(4,4′-biphenyldiyldiimino)bis[6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol], demonstrated the best IC50 value of 35.54 μM. As seen in previous examples, the application of structure-based pharmacophores for virtual screening followed by docking of hits to rank them proved to be a useful strategy for virtual screening and lead discovery.
In all the studies involving use of structure-based pharmacophores, the hits obtained showed structural diversity and the lead compounds identified contained functional groups quite different from the original ligand of the ligand-target complex. This is an important advantage offered by the structure-based pharmacophore approach with compared with the traditional approaches.
Conclusion
Recent advances in the development of tools for structure-based pharmacophores have brought this approach into focus for medicinal chemists who are now using it widely for de novo drug design, lead discovery, and virtual screening. The flexibility of the manner in which this approach can be used makes this technique ideal for situations where other traditional drug design approaches do not provide answers. The tools, which are available now, are very simple and indeed involve a few clicks only, thus making the process of pharmacophore generation very rapid. The advantages offered by structure-based pharmacophores are most evident in cases where they are used for virtual screening and lead discovery. Virtual screening carried out using structure-based pharmacophores is much faster than docking-based screening, while the accuracy is similar, with fewer cases of complete failures. Thus, structure-based pharmacophore approaches have succeeded as a viable alternative to docking and other structure-based approaches in virtual screening. However, as shown by many studies, screening of large databases using structure-based pharmacophores should be followed by docking studies of the most interesting hits to select final lead compounds. Therefore, it is worth mentioning that despite having emerged as a viable alternative to docking for virtual screening, structure-based pharmacophores when combined with docking lead to more accurate results than pharmacophore-based screening, while being much faster than docking-based screening. Despite all the advantages offered by structure-based pharmacophores, there are some challenges which need to be addressed in the future. Some of these challenges include consideration of receptor flexibility and selection of appropriate pharmacophore features from the large pool available. In conclusion, the tools for structure-based pharmacophore discovery continue to evolve, and are having a steadily wider impact as an alternative to traditional approaches. They are now an essential part of lead discovery, and every computer-aided drug discovery scientist should be familiar with them.
Disclosure
The authors report no conflicts of interest in this work.
References
Moss GP. Glossary of Terms Used in Medicinal Chemistry (IUPAC Recommendations 1998). International Union of Pure and Applied Chemistry: Recommendations on Organic and Biochemical Nomenclature, Symbols and Terminology. 2013. Available from: http://www.chem.qmul.ac.uk/iupac/medchem/ix.html. Accessed May 27, 2014. | |
Wermuth CG, Langer T. Pharmacophore identification. In: Kubinyi H, editor. 3D-QSAR in Drug Design. Theory, Methods, and Applications. Leiden, The Netherlands: ESCOM Science Publishers; 1993. | |
Kurogi Y, Guner OF. Pharmacophore modeling and three-dimensional database searching for drug design using catalyst. Curr Med Chem. 2001;8(9):1035–1055. | |
Mustata G, Follis AV, Hammoudeh DI, et al. Discovery of novel Myc-Max heterodimer disruptors with a three-dimensional pharmacophore model. J Med Chem. 2009;52(5):1247–1250. | |
Langer T, Krovat EM. Chemical feature-based pharmacophores and virtual library screening for discovery of new leads. Curr Opin Drug Discov Devel. 2003;6(3):370–376. | |
Carlson HA, Masukawa KM, Rubins K, et al. Developing a dynamic pharmacophore model for HIV-1 integrase. J Med Chem. 2000;43(11):2100–2114. | |
Chen J, Lai L. Pocket v.2: further developments on receptor-based pharmacophore modeling. J Chem Inf Model. 2006;46(6):2684–2691. | |
Fox T, Haaksma EE. Computer based screening of compound databases: 1. Preselection of benzamidine-based thrombin inhibitors. J Comput Aided Mol Des. 2000;14(5):411–425. | |
Steindl TM, Schuster D, Laggner C, Langer T. Parallel screening: a novel concept in pharmacophore modeling and virtual screening. J Chem Inf Model. 2006;46(5):2146–2157. | |
Baroni M, Cruciani G, Sciabola S, Perruccio F, Mason JS. A common reference framework for analyzing/comparing proteins and ligands. Fingerprints for Ligands and Proteins (FLAP): theory and application. J Chem Inf Model. 2007;47(2):279–294. | |
Bohm HJ. The computer program LUDI: a new method for the de novo design of enzyme inhibitors. J Comput Aided Mol Des. 1992;6(1):61–78. | |
Wolber G, Seidel T, Bendix F, Langer T. Molecule-pharmacophore superpositioning and pattern matching in computational drug design. Drug Discov Today. 2008;13(1–2):23–29. | |
Adhikari N, Halder AK, Mondal C, Jha T. Ligand based validated comparative chemometric modeling and pharmacophore mapping of aurone derivatives as antimalarial agents. Curr Comput Aided Drug Des. 2013;9(3):417–432. | |
Barbaro R, Betti L, Botta M, et al. Synthesis, biological evaluation, and pharmacophore generation of new pyridazinone derivatives with affinity toward alpha(1)- and alpha(2)-adrenoceptors. J Med Chem. 2001;44(13):2118–2132. | |
Gozalbes R, Barbosa F, Nicolai E, Horvath D, Froloff N. Development and validation of a pharmacophore-based QSAR model for the prediction of CNS activity. ChemMedChem. 2009;4(2):204–209. | |
Sakkiah S, Arullaperumal V, Hwang S, Lee KW. Ligand-based pharmacophore modeling and Bayesian approaches to identify c-Src inhibitors. J Enzyme Inhib Med Chem. 2014;29(1):69–80. | |
Khedkar SA, Malde AK, Coutinho EC, Srivastava S. Pharmacophore modeling in drug discovery and development: an overview. Med Chem. 2007;3(2):187–197. | |
Kirubakaran P, Muthusamy K, Singh KH, Nagamani S. Ligand-based pharmacophore modeling; atom-based 3D-QSAR analysis and molecular docking studies of phosphoinositide-dependent kinase-1 inhibitors. Indian J Pharm Sci. 2012;74(2):141–151. | |
Bostrom J. Reproducing the conformations of protein-bound ligands: a critical evaluation of several popular conformational searching tools. J Comput Aided Mol Des. 2001;15(12):1137–1152. | |
Vieth M, Hirst JD, Brooks CL 3rd. Do active site conformations of small ligands correspond to low free-energy solution structures? J Comput Aided Mol Des. 1998;12(6):563–572. | |
Kirchmair J, Laggner C, Wolber G, Langer T. Comparative analysis of protein-bound ligand conformations with respect to catalyst’s conformational space subsampling algorithms. J Chem Inf Model. 2005;45(2):422–430. | |
Yankeelov JA Jr, Koshland DE Jr. Evidence for conformation changes induced by substrates of phosphoglucomutase. J Biol Chem. 1965;240:1593–1602. | |
Kellenberger E, Rodrigo J, Muller P, Rognan D. Comparative evaluation of eight docking tools for docking and virtual screening accuracy. Proteins. 2004;57(2):225–242. | |
Warren GL, Andrews CW, Capelli AM, et al. A critical assessment of docking programs and scoring functions. J Med Chem. 2006;49(20):5912–5931. | |
Bissantz C, Folkers G, Rognan D. Protein-based virtual screening of chemical databases. 1. Evaluation of different docking/scoring combinations. J Med Chem. 2000;43(25):4759–4767. | |
Charifson PS, Corkery JJ, Murcko MA, Walters WP. Consensus scoring: a method for obtaining improved hit rates from docking databases of three-dimensional structures into proteins. J Med Chem. 1999;42(25):5100–5109. | |
Guner O, Clement O, Kurogi Y. Pharmacophore modeling and three dimensional database searching for drug design using catalyst: recent advances. Curr Med Chem. 2004;11(22):2991–3005. | |
Joseph-McCarthy D, Thomas BE, Belmarsh M, Moustakas D, Alvarez JC. Pharmacophore-based molecular docking to account for ligand flexibility. Proteins. 2003;51(2):172–188. | |
Krovat EM, Fruhwirth KH, Langer T. Pharmacophore identification, in silico screening, and virtual library design for inhibitors of the human factor Xa. J Chem Inf Model. 2005;45(1):146–159. | |
Discovery Studio [computer program]. Version 4.0. San Diego, CA, USA: Accelrys Inc. | |
Wolber G, Langer T. LigandScout: 3-D pharmacophores derived from protein-bound ligands and their use as virtual screening filters. J Chem Inf Model. 2005;45(1):160–169. | |
Dixon SL, Smondyrev AM, Knoll EH, Rao SN, Shaw DE, Friesner RA. PHASE: a new engine for pharmacophore perception, 3D QSAR model development, and 3D database screening: 1. Methodology and preliminary results. J Comput Aided Mol Des. 2006;20(10–11):647–671. | |
Dixon SL, Smondyrev AM, Rao SN. PHASE: a novel approach to pharmacophore modeling and 3D database searching. Chem Biol Drug Des. 2006;67(5):370–372. | |
Sanders MP, Verhoeven S, de Graaf C, et al. Snooker: a structure-based pharmacophore generation tool applied to class A GPCRs. J Chem Inf Model. 2011;51(9):2277–2292. | |
Chowdhury A, Sen S, Dey P, et al. Computational validation of 3-ammonio-3-(4-oxido-1H-imidazol-1-ium-5-yl) propane-1, 1-bis (olate) as a potent anti-tubercular drug against mt-MetAP. Bioinformation. 2012;8(18):875–880. | |
Khan AH, Prakash A, Kumar D, Rawat AK, Srivastava R, Srivastava S. Virtual screening and pharmacophore studies for FTase inhibitors using Indian plant anticancer compounds database. Bioinformation. 2010;5(2):62–66. | |
Khan MT, Wuxiuer Y, Sylte I. Binding modes and pharmacophore modelling of thermolysin inhibitors. Mini Rev Med Chem. 2012;12(6):515–533. | |
Sanders MP, Barbosa AJ, Zarzycka B, et al. Comparative analysis of pharmacophore screening tools. J Chem Inf Model. 2012;52(6):1607–1620. | |
Kirchhoff PD, Brown R, Kahn S, Waldman M, Venkatachalam CM. Application of structure-based focusing to the estrogen receptor. J Comput Chem. 2001;22(10):993–1003. | |
Sutter J, Li J, Maynard AJ, Goupil A, Luu T, Nadassy K. New features that improve the pharmacophore tools from Accelrys. Curr Comput Aided Drug Des. 2011;7(3):173–180. | |
Al-Balas QA, Amawi HA, Hassan MA, Qandil AM, Almaaytah AM, Mhaidat NM. Virtual lead identification of farnesyltransferase inhibitors based on ligand and structure-based pharmacophore techniques. Pharmaceuticals. 2013;6(6):700–715. | |
Almasri IM, Taha MO, Mohammad MK. New leads for DPP IV inhibition: structure-based pharmacophore mapping and virtual screening study. Arch Pharm Res. 2013;36(11):1326–1337. | |
Liu C, He G, Jiang Q, Han B, Peng C. Novel hybrid virtual screening protocol based on molecular docking and structure-based pharmacophore for discovery of methionyl-tRNA synthetase inhibitors as antibacterial agents. Int J Mol Sci. 2013;14(7):14225–14239. | |
Steindl T, Langer T. Influenza virus neuraminidase inhibitors: generation and comparison of structure-based and common feature pharmacophore hypotheses and their application in virtual screening. J Chem Inf Model. 2004;44(5):1849–1856. | |
Schuster D, Langer T. The identification of ligand features essential for PXR activation by pharmacophore modeling. J Chem Inf Model. 2005;45(2):431–439. | |
Barillari C, Marcou G, Rognan D. Hot-spots-guided receptor-based pharmacophores (HS-Pharm): a knowledge-based approach to identify ligand-anchoring atoms in protein cavities and prioritize structure-based pharmacophores. J Chem Inf Model. 2008;48(7):1396–1410. | |
Muthusamy K, Singh KhD, Chinnasamy S, et al. High throughput virtual screening and E-pharmacophore filtering in the discovery of new BACE-1 inhibitors. Interdiscip Sci. 2013;5(2):119–126. | |
Saxena S, Devi PB, Soni V, Yogeeswari P, Sriram D. Identification of novel inhibitors against Mycobacterium tuberculosis l-alanine dehydrogenase (MTB-AlaDH) through structure-based virtual screening. J Mol Graph Model. 2014;47(0):37–43. | |
Nagamani S, Kesavan C, Muthusamy K. E-Pharmacophore mapping and docking studies on Vitamin D receptor (VDR). Bioinformation. 2012;8(15):705–710. | |
Kalyaanamoorthy S, Chen YP. Energy based pharmacophore mapping of HDAC inhibitors against class I HDAC enzymes. Biochim Biophys Acta. 2013;1834(1):317–328. | |
Jatana N, Sharma A, Latha N. Pharmacophore modeling and virtual screening studies to design potential COMT inhibitors as new leads. J Mol Graph Model. 2013;39:145–164. | |
Ebalunode JO, Ouyang Z, Liang J, Zheng W. Novel approach to structure-based pharmacophore search using computational geometry and shape matching techniques. J Chem Inf Model. 2008;48(4):889–901. | |
Dong X, Ebalunode JO, Yang SY, Zheng W. Receptor-based pharmacophore and pharmacophore key descriptors for virtual screening and QSAR modeling. Curr Comput Aided Drug Des. 2011;7(3):181–189. | |
Binkowski TA, Naghibzadeh S, Liang J. CASTp: Computed Atlas of Surface Topography of proteins. Nucleic Acids Res. 2003;31(13):3352–3355. | |
Dundas J, Ouyang Z, Tseng J, Binkowski A, Turpaz Y, Liang J. CASTp: computed atlas of surface topography of proteins with structural and topographical mapping of functionally annotated residues. Nucleic Acids Res. 2006;34(Web Server issue):W116–W118. | |
Edelsbrunner H, Facello M, Liang J. On the definition and the construction of pockets in macromolecules. Pac Symp Biocomput. 1996: 272–287. | |
Wang R, Gao Y, Lai L. LigBuilder: a multi-purpose program for structure-based drug design. J Mol Model. 2000;6(7–8):498–516. | |
Yuan Y, Pei J, Lai L. LigBuilder 2: a practical de novo drug design approach. J Chem Inf Model. 2011;51(5):1083–1091. | |
Wang R, Liu L, Lai L, Tang Y. SCORE: a new empirical method for estimating the binding affinity of a protein-ligand complex. J Mol Model. 1998;4(12):379–394. | |
Stahl M, Rarey M. Detailed analysis of scoring functions for virtual screening. J Med Chem. 2001;44(7):1035–1042. | |
Tao P, Lai L. Protein ligand docking based on empirical method for binding affinity estimation. J Comput Aided Mol Des. 2001;15(5):429–446. | |
Kumar S, Nussinov R. Relationship between ion pair geometries and electrostatic strengths in proteins. Biophys J. 2002;83(3):1595–1612. | |
Sanders MP, Fleuren WW, Verhoeven S, et al. ss-TEA: entropy based identification of receptor specific ligand binding residues from a multiple sequence alignment of class A GPCRs. BMC Bioinformatics. 2011;12:332. | |
Renner S, Schneider G. Fuzzy pharmacophore models from molecular alignments for correlation-vector-based virtual screening. J Med Chem. 2004;47(19):4653–4664. | |
Breu V, Hashido K, Broger C, et al. Separable binding sites for the natural agonist endothelin-1 and the non-peptide antagonist bosentan on human endothelin-A receptors. Eur J Biochem. 1995;231(1):266–270. | |
Shi L, Javitch JA. The binding site of aminergic G protein-coupled receptors: the transmembrane segments and second extracellular loop. Annu Rev Pharmacol Toxicol. 2002;42:437–467. | |
De Luca L, Ferro S, Damiano FM, et al. Structure-based screening for the discovery of new carbonic anhydrase VII inhibitors. Eur J Med Chem. 2014;71:105–111. | |
Michaux C, de Leval X, Julemont F, Dogne JM, Pirotte B, Durant F. Structure-based pharmacophore of COX-2 selective inhibitors and identification of original lead compounds from 3D database searching method. Eur J Med Chem. 2006;41(12):1446–1455. | |
Greenidge PA, Carlsson B, Bladh LG, Gillner M. Pharmacophores incorporating numerous excluded volumes defined by X-ray crystallographic structure in three-dimensional database searching: application to the thyroid hormone receptor. J Med Chem. 1998;41(14):2503–2512. | |
Zhang XW, Yap YL, Altmeyer RM. Generation of predictive pharmacophore model for SARS-coronavirus main proteinase. Eur J Med Chem. 2005;40(1):57–62. | |
Rella M, Rushworth CA, Guy JL, Turner AJ, Langer T, Jackson RM. Structure-based pharmacophore design and virtual screening for novel angiotensin converting enzyme 2 inhibitors. J Chem Inf Model. 2006;46(2):708–716. | |
Ebalunode JO, Dong X, Ouyang Z, Liang J, Eckenhoff RG, Zheng W. Structure-based shape pharmacophore modeling for the discovery of novel anesthetic compounds. Bioorg Med Chem. 2009;17(14):5133–5138. | |
Liu R, Loll PJ, Eckenhoff RG. Structural basis for high-affinity volatile anesthetic binding in a natural 4-helix bundle protein. FASEB J. 2005;19(6):567–576. | |
Arooj M, Sakkiah S, Kim S, Arulalapperumal V, Lee KW. A combination of receptor-based pharmacophore modeling and QM techniques for identification of human chymase inhibitors. PLoS One. 2013;8(4):e63030. | |
Zou J, Xie HZ, Yang SY, Chen JJ, Ren JX, Wei YQ. Towards more accurate pharmacophore modeling: multicomplex-based comprehensive pharmacophore map and most-frequent-feature pharmacophore model of CDK2. J Mol Graph Model. 2008;27(4):430–438. | |
Legraverend M, Tunnah P, Noble M, et al. Cyclin-dependent kinase inhibition by new C-2 alkynylated purine derivatives and molecular structure of a CDK2-inhibitor complex. J Med Chem. 2000;43(7):1282–1292. | |
Moser D, Achenbach J, Klingler FM, Estel la B, Hahn S, Proschak E. Evaluation of structure-derived pharmacophore of soluble epoxide hydrolase inhibitors by virtual screening. Bioorg Med Chem Lett. 2012;22(21):6762–6765. | |
Vilar S, Cozza G, Moro S. Medicinal chemistry and the molecular operating environment (MOE): application of QSAR and molecular docking to drug discovery. Curr Top Med Chem. 2008;8(18):1555–1572. | |
Olsen L, Jost S, Adolph HW, Pettersson I, Hemmingsen L, Jorgensen FS. New leads of metallo-beta-lactamase inhibitors from structure-based pharmacophore design. Bioorg Med Chem. 2006;14(8):2627–2635. | |
Zhao Y, Wang Q, Meng Q, et al. Identification of Trypanosoma brucei leucyl-tRNA synthetase inhibitors by pharmacophore- and docking-based virtual screening and synthesis. Bioorg Med Chem. 2012;20(3):1240–1250. | |
Karkola S, Alho-Richmond S, Wahala K. Pharmacophore modelling of 17beta-HSD1 enzyme based on active inhibitors and enzyme structure. Mol Cell Endocrinol. 2009;301(1–2):225–228. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.