Back to Journals » International Journal of Nanomedicine » Volume 14
Strategic approach to developing a self-microemulsifying drug delivery system to enhance antiplatelet activity and bioavailability of ticagrelor
Authors Na YG, Byeon JJ, Wang M, Huh HW, Son GH, Jeon SH, Bang KH, Kim SJ, Lee HJ, Lee HK, Cho CW ![]()
Received 10 October 2018
Accepted for publication 9 January 2019
Published 15 February 2019 Volume 2019:14 Pages 1193—1212
DOI https://doi.org/10.2147/IJN.S190426
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Thomas Webster
Young-Guk Na,1 Jin-Ju Byeon,1 Miao Wang,1 Hyun Wook Huh,1 Gi-Ho Son,1,2 Sung-Hoon Jeon,1,3 Ki-Hyun Bang,1,2 Sung-Jin Kim,1 Hye-Jin Lee,1 Hong-Ki Lee,1 Cheong-Weon Cho1
1College of Pharmacy, Chungnam National University, Daejeon, Republic of Korea; 2Korea United Pharmaceutical Co. Ltd., Sejong, Republic of Korea; 3SamA Pharmaceutical Co. Ltd., Suwon, Republic of Korea
Background: Ticagrelor (TCG) is used to inhibit platelet aggregation in patients with acute coronary syndrome, but its poor solubility and low bioavailability limit its in vivo efficacy. The purpose of this study was to manufacture an optimized TCG-loaded self-microemulsifying drug delivery system (SMEDDS) to enhance the oral bioavailability and antiplatelet activity of TCG.
Materials and methods: Solubility and emulsification tests were conducted to determine the most suitable oils, surfactants, and cosurfactants. Scheffé’s mixture design was applied to optimize the percentage of each component applied in the SMEDDS formulation to achieve optimal physical characteristics, ie, high solubility of TCG in SMEDDS, small droplet size, low precipitation, and high transmittance.
Results: The optimized TCG-loaded SMEDDS (TCG-SM) formulation composed of 10.0% Capmul MCM (oil), 53.8% Cremophor EL (surfactant), and 36.2% Transcutol P (cosurfactant) significantly improving the dissolution of TCG in various media compared with TCG in Brilinta® (commercial product). TCG-SM exhibited higher cellular uptake and permeability in Caco-2 cells than raw TCG suspension. In pharmacokinetic studies in rats, TCG-SM exhibited higher oral bioavailability with 5.7 and 6.4 times higher area under the concentration–time curve and maximum plasma concentration, respectively, than a raw TCG suspension. Antiplatelet activity studies exhibited that the TCG-SM formulation showed significantly improved inhibition of platelet aggregation compared with raw TCG at the same dose of TCG. And, a 10 mg/kg dose of raw TCG suspension and a 5 mg/kg dose of TCG-SM had a similar area under the inhibitory curve (907.0%±408.8% and 907.8%±200.5%·hours, respectively) for antiplatelet activity.
Conclusion: These results suggest that the developed TCG-SM could be successfully used as an efficient method to achieve the enhanced antiplatelet activity and bioavailability of TCG.
Keywords: ticagrelor, SMEDDS, optimization, bioavailability, platelet aggregation, antiplatelet activity
Introduction
Ticagrelor (TCG), a cyclopentyl-triazolo-pyrimidine, belongs to a class of chemically noncompetitive and reversible antagonists of the platelet P2Y12 ADP receptor.1–4 TCG was approved for use in patients with acute coronary syndrome based on a phase III study (Platelet Inhibition and Patient Outcomes), which showed a significant benefit compared with clopidogrel.5,6 However, TCG exhibits very low solubility (<10 μg/mL) at all pH values. It also has low intestinal membrane permeability corresponding to Biopharmaceutical Classification System (BCS) class IV. Because of these properties of TCG, the absolute bioavailability of TCG after oral administration is ~36%.7 Although formulations to enhance the bioavailability and antiplatelet activity of TCG, such as solid dispersion8 and cocrystallization,9 have recently been reported, few studies have been performed.
We developed a self-microemulsifying drug delivery system (SMEDDS) to overcome the above barriers and enhance the antiplatelet activity of TCG. SMEDDS have recently attracted interest for the oral delivery of insoluble drugs because of their ease of manufacture and their practical advantages in improving drug solubility and oral bioavailability.10–12 They have been applied to BCS class II or IV drugs such as atorvastatin, valsartan, lopinavir, nelfinavir, and tamoxifen citrate.12–16 A SMEDDS formulation is a stable, single-phase, and isotropic mixture of oil, surfactant, and cosurfactant without an aqueous phase. It forms an oil-in-water (o/w) microemulsion with a globule size in the range of 20–200 nm when dispersed in the gastrointestinal tract via gentle gastric motility or in an aqueous phase after oral administration.17 This self-formed microemulsion transports the drug in solubilized form. The small droplet size of the microemulsion provides large surface area for rapid dissolution and absorption of the drug, which increases the permeability of the drug through the intestinal membrane. Other potential advantages of using SMEDDS to improve the oral bioavailability of poor water-soluble drugs are their promotion of transcellular and paracellular absorption, reduction of metabolism of CYP/CYP450 by gastrointestinal enzymes, and enhancement of lymphatic transport, all of which can protect the drug from first-pass metabolism.18–21
In this study, we developed and optimized a TCG-loaded SMEDDS (TCG-SM) formulation by design of experiments using Scheffé’s mixture design based on a small number of trials and response surface methodology. We also characterized the optimized TCG-SM, evaluated its in vitro dissolution, performed cellular studies, and compared its antiplatelet activity and pharmacokinetics with those of a raw TCG suspension.
Materials and methods
Materials
TCG was supplied by Korea United Pharm Inc. (Seoul, Republic of Korea). Tween 20 (polyoxyethylene sorbitan monolaurate), Tween 80 (polyoxyethylene sorbitan monooleate), olive oil, castor oil, oleic acid, mineral oil, sunflower oil, cotton seed oil, linseed oil, Span 80 (sorbitan monooleate), and polyethylene glycol 400 (PEG 400) were purchased from Samchun Chemical Co., Ltd (Pyeongtaek, Republic of Korea). Labrafac CC (medium-chain triglycerides), Lauroglycol FCC (propylene glycol monolaurate, type I), Lauroglycol 90 (propylene glycol monolaurate, type II), Peceol (glycerol monooleates, type 40), Labrafac Lipophile WL 1349 (medium-chain triglycerides), Labrafil M 2125 CS (linoleoyl macrogol-6 glycerides), Labrafil M 1944 CS (oleoyl macrogol-6 glycerides), Capryol PGMC (propylene glycol monocaprylate, type I), Capryol 90 (propylene glycol monocaprylate, type II), Labrasol (caprylocaproyl macrogol-8 glycerides), and Transcutol P (diethylene glycol monoethyl) were kindly provided by Gattefossé Co. (Lyon, France). Capmul MCM (glyceryl caprylate/caprate) was provided by ABITEC Corporation (Cleveland, OH, USA). Lecithin, isopropyl palmitate, and isopropyl myristate were purchased from Tokyo Chemical Industries Co., Ltd. (Toshima, Japan). Span 83 (sorbitan sesquioleate), Span 85 (sorbitan trioleate), tetraglycol, propylene glycol, DMSO, MTT, and ethylenediaminetetraacetic acid were purchased from Sigma-Aldrich (St Louis, MO, USA). Pluronic L64 (triblock copolymer-EO13PO30EO13) and Cremophor EL (polyoxyl 35 hydrogenated castor oil) were obtained as a gift from BASF Co. (Ludwigshafen, Germany). The HPLC-grade acetonitrile and methanol were purchased from JT Baker (Phillipsburg, NJ, USA). All other chemicals and solvents used in this study were of analytical grade. Distilled water was used throughout the experiments.
HPLC analysis
The HPLC analysis of TCG was carried out by modifying a method described previously.22 An Agilent 1100 series LC model (Agilent Technologies Inc., Santa Clara, CA, USA), equipped with a pump, autosampler, thermostat, and UV-Vis detector was used for analysis. An Xterra RP18 column (250×4.6 mm, 5 μm; Waters, Milford, MA, USA) was used for the analysis of TCG. The isocratic mobile phase consisted of acetonitrile and 50 nM ammonium acetate buffer (58:42, v/v). The pH of the mobile phase was adjusted to 8.2 with 6 M ammonium hydroxide. The flow rate was 1.0 mL/min, sample injection volume was 20 μL, and the column temperature kept constant at 40°C. The wavelength for ultraviolet detection of TCG was 254 nm.
Preliminary screening of excipients for SMEDDS
To select a suitable oil, surfactant, and cosurfactant for the SMEDDS of TCG, the solubility of TCG in various excipients was determined by equilibrium solubility. Equilibrium solubility studies on TCG were carried out in various oils (Capryol 90, Capryol PGMC, Capmul MCM, Labrafil M 1944 CS, Labrafil M 2125 CS, Labrafac Lipophile WL 1349, Peceol, Lauroglycol 90, Lauroglycol FCC, Labrafac CC, isopropyl myristate, isopropyl palmitate, linseed oil, cotton seed oil, sunflower oil, mineral oil, oleic acid, castor oil, and olive oil), surfactants (Cremophor EL, Pluronic L64, Labrasol, Tween 80, and Tween 20), and cosurfactants (propylene glycol, tetraglycol, Transcutol P, and PEG 400). Briefly, an excess amount of the drug was added to different vials containing 1 g of each excipient and mixed for 72 hours to reach saturation at 25°C using an angle rotator (model AG; FinePCR, Gunpo, Republic of Korea) with 15 rpm. The saturated mixtures were centrifuged at 15,000 × g for 15 minutes at 25°C to remove the excess TCG. The supernatant was filtered through a 0.45 μm polyvinylidene fluoride (PVDF) membrane (Millipore, Bengaluru, Karnataka, India), and the filtered solution was diluted 100 times with isopropanol. The TCG concentration was then measured by HPLC analysis.
Evaluation of emulsification and phase separation
The evaluation of emulsification was carried out for the preselected excipients to determine the most appropriate surfactant and cosurfactant. Based on the requirement for a self-forming microemulsion, a type IV composition was used, which dispersed very finely and formed small droplets.23 The SMEDDS formulations were prepared with a composition of 20% oil, 40% surfactant, and 40% cosurfactant, and 100 mg SMEDDS was diluted in 10 mL of distilled water or media at pH 1.2, pH 4.0, or pH 6.8. Subsequently, the mixture was homogenized for 30 minutes and then evaluated for emulsification and phase separation. The emulsification grade of SMEDDS was classified by the droplet size, transmittance, and phase separation (Table S1, Supplementary materials) by modifying the previously reported grading system.12 The phase separation was visually confirmed by the presence or absence of precipitation.
Pseudoternary phase diagram
The pseudoternary phase diagram was used to determine the ranges of the microemulsions. Based on the solubility studies and emulsification results for each excipient, the ternary phase diagrams were plotted by combining various percentages of Capmul MCM (oil), Cremophor EL or Tween 20 (surfactant), and Transcutol P (cosurfactant). The vertices of the triangle graph corresponded to each component. For any mixture, the total percentage of oil, surfactant, and cosurfactant was 100%. One hundred milligrams of each mixture were added to 10 mL distilled water and gently stirred using a magnetic stirrer to evaluate microemulsion-forming ability. The droplet size, transmittance, and phase separation of the microemulsion formed were measured. A mixture that formed a transparent microemulsion with droplets <300 nm (grade A and B as described in method; Evaluation of emulsification and phase separation) was considered a suitable SMEDDS formulation. All studies were repeated in triplicate.
Experimental optimization of TCG-SM formulation
The composition of the SMEDDS formulation was optimized using Scheffé’s mixture design, which is desirable for response surface methodology to optimize complex formulations.24 The experiments were designed using three factors and four responses (Table 1). Design-Expert® 11 (Stat-Ease Inc., Minneapolis, MN, USA) was used to perform the experimental design and statistical analysis.
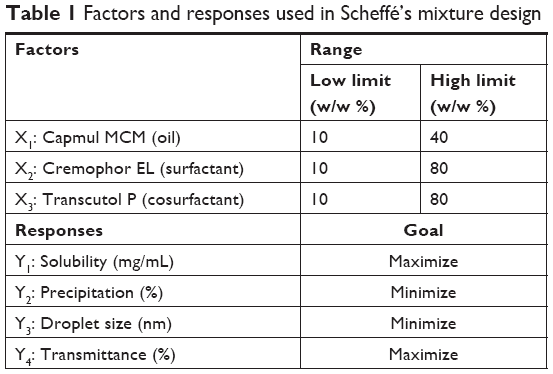 | Table 1 Factors and responses used in Scheffé’s mixture design |
The experimental design and statistical analysis were performed using Design-Expert 11 (Stat-Ease Inc.). Based on pseudoternary phase diagram, the percentages of Capmul MCM (X1, w/w %), Cremophor EL (X2, w/w %), and Transcutol P (X3, w/w %) were set within ranges of 10%–40%, 10%–80%, and 10%–80%, respectively. In all experiments, the percentages of X1, X2, and X3 added up to 100%. The solubility of TCG in SMEDDS (Y1), the precipitation of TCG (Y2), droplet size (Y3), and transmittance (Y4) were used as responses to optimize the SMEDDS composition. The 17 designed experiments were fitted to linear, cubic, quadratic, and special cubic or quadratic polynomial models. The appropriate fitting model for each response was determined by comparing various statistical parameters provided by ANOVAs, such as sequential P-values, lack of fit, squared correlation coefficient (R2), adjusted R2, and adequate precision. The desirability function was analyzed for the optimization of factors associated with desirable responses after the fitting of the statistical model.
Solubility of TCG in SMEDDS (Y1)
The solubility of TCG in SMEDDS was evaluated to prepare a formulation capable of dissolving a large amount of TCG in the minimum volume of SMEDDS formulation. Solubility measurements were conducted by modifying the method for screening excipients (see method; Preliminary screening of excipients for SMEDDS). In brief, excess TCG was added to 1 g of prepared SMEDDS and the solubility was measured after stirring for 72 hours. The samples were centrifuged at 15,000 × g for 15 minutes at 25°C, the supernatant was diluted with isopropanol, and TCG measured by HPLC.
Precipitation (Y2)
A precipitation test was performed to confirm that the microemulsion formed homogeneous droplets <200 nm in diameter. Briefly, 10 mL of distilled water was added to 100 mg of the TCG-SM, and the mixture was vortexed for 30 minutes to form a homogeneous microemulsion. The microemulsion was filtered through a 0.22 μm PVDF membrane filter, immediately diluted with isopropanol, and analyzed by HPLC. The degree of precipitation was evaluated by comparing the TCG concentration with that of TCG-SM dissolved in isopropanol. The equation used is as follows:
|
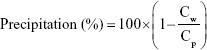
where Cw is the concentration of TCG in TCG-SM diluted with distilled water and Cp is the concentration of TCG in TCG-SM diluted in isopropanol. A precipitation percentage close to 0% indicates that the precipitation of TCG does not occur.
Droplet size (Y3)
The SMEDDS droplet size was measured after the reconstitution procedure. One hundred milligrams of SMEDDS were added to 10 mL of distilled water to prepare a reconstituted microemulsion. The microemulsion was subsequently incubated for 30 minutes at room temperature. The samples were loaded into a cuvette and then monitored with an electrophoretic laser scattering (ELS) analyzer (ELS-8000; Otsuka Electronics, Osaka, Japan). The instrument was set to measure the droplet size 50 times and calculate the average size.
Transmittance (Y4)
The transmittance of each mixture was determined by measuring the absorbance of the formulation at 620 nm using a microplate reader (Sunrise; Tecan Group Ltd., Männedorf, Switzerland) compared with distilled water as control. To measure absorbance, 100 mg of each mixture was added to 10 mL distilled water for forming the microemulsion. The transmittance was calculated using the following equation:
|
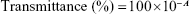
where A is the absorbance of microemulsion. A transmittance close to 100% indicates a clear and transparent microemulsion.
Characterization of TCG-SM
The optimized TCG-SM was prepared by dissolving 90 mg of TCG in 400 mg of the optimized SMEDDS. The morphology of the TCG-SM was evaluated using a transmission electron microscope (JEM 1400; JEOL Ltd, Tokyo, Japan) with an acceleration voltage of 120 kV. The TCG-SM was dispersed in distilled water. Then, 10 μL of the sample was directly mounted on a copper grid and dried at room temperature before being observed with the microscope. For droplet size measurements, 100 mg of TCG-SM was suspended by gentle mixing in 10 mL distilled water for 30 minutes. The droplet size was measured using an ELS analyzer (ELS-8000; Otsuka Electronics).
In vitro dissolution of TCG in the optimized TCG-SM was compared with that of TCG in Brilinta® 90 mg (commercial product) using the United States Pharmacopeia (USP) apparatus II method with a dissolution tester (DST-810; Labfine, Anyang, Republic of Korea). Distilled water and media at pH 1.2, pH 4.0, and pH 6.8 were prepared as dissolution media according to the USP guidelines. The volume of dissolution media was 900 mL with 37°C±0.5°C. The paddle speed was set at 50 rpm throughout the experiment. The TCG-loaded Cremophor EL (TCG-CE) and TCG-SM containing 90 mg of TCG were filled into hard gelatin capsules (capsule 00 size). The TCG-CE was prepared with the same amount of Cremophor EL as in the TCG-SM formulation to compare the effect of SMEDDS on micelle formation. At predetermined time points of 5, 10, 15, 30, 45, 60, 90, 120, and 180 minutes, samples (5 mL) were collected and filtered through a 0.45 μm membrane filter and then diluted with isopropanol. The concentration of TCG in each formulation was analyzed by HPLC.
Cell study
Cell culture
The heterogeneous human epithelial colorectal adenocarcinoma (Caco-2) cell line was purchased from Korean Cell Line Bank (Seoul, Republic of Korea). The cells were cultured in DMEM supplemented with 10% FBS and 1% penicillin/streptomycin at 37°C in a humidified 95% air and 5% CO2 atmosphere.
Cytotoxicity study
Cytotoxicity studies were performed to determine the toxicity of TCG and the SMEDDS formulation. Briefly, cells in 200 μL of culture medium were seeded into 96-well plates at a cell density of 5×104 cells/well. The cells were treated with the raw TCG solution as control, blank-CE, TCG-CE, blank-SM, and TCG-SM at equivalent concentrations of TCG and incubated for 24 hours at 37°C. The samples were diluted with culture media containing 1% DMSO (v/v) to achieve various concentrations of TCG ranging from 9 ng/mL to 90 μg/mL. After incubation for 24 hours, MTT solution (5 mg/mL, 30 μL/well) was added to the culture medium and incubated for a further 4 hours. The medium was removed, and 200 μL of DMSO was added to each well. The absorbance values of each well were measured at 560 nm using a microplate reader (Sunrise; Tecan Group Ltd.). The cell viability was calculated using the following equation:
|
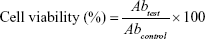
where Abtest is the absorbance value of the cells treated with samples and Abcontrol is the absorbance value of the cells incubated without samples.
Cellular uptake study
We assessed the uptake of TCG into Caco-2 cells from raw TCG solution, TCG-CE, and TCG-SM. The cells were seeded in 6-well plates at 1×106 cells/well and incubated for 24 hours. The cells were treated with raw TCG solution as control, TCG-CE, and TCG-SM at an equivalent concentration of 9 μg/mL TCG, which was previously shown to be noncytotoxic (see method; Cytotoxicity study). After incubation for 4 hours, the medium in the wells was removed and the wells were washed three times with 1 mL cold phosphate-buffered saline. Cell lysis was achieved by adding 0.5 mL of 1% (w/v) Triton X-100 solution to each well. Subsequently, 0.5 mL of acetonitrile was added to the lysed cells and shaken for 5 minutes to extract TCG from the cells. The samples were centrifuged at 15,000 × g for 10 minutes. The supernatant was filtered with a 0.45 μm membrane filter and analyzed by HPLC. The cellular uptake of TCG from different formulations was calculated by normalization to the amount of protein measured using the bicinchoninic acid assay.
In vitro permeability in Caco-2 cell line
The Caco-2 cell line was used as a model system to study gastrointestinal epithelial permeability. The cells were seeded onto polycarbonate membrane (HTS Transwell-24; Corning, Cambridge, MA, USA) with a surface area of 0.33 cm2 and a diameter of 6.5 mm at a density of 5×104 cells/well. DMEM was used as culture medium, and cells were grown for 21 days to reach a monolayer. The wells containing cell monolayers with transepithelial electrical resistance values above 300 Ω·cm2 were used for the permeability study. Raw TCG solution as control, TCG-CE, and TCG-SM at a dose equivalent to 9 μg/mL of TCG was prepared by diluting with Hanks’ balanced salt solution (HBSS, pH 7.4). Permeability studies were performed to assess apical–basolateral (A–B) transport and basolateral–apical (B–A) transport. The Caco-2 cell monolayer was washed with prewarmed HBSS before the permeability study. To measure A–B transport, 250 μL of the test solution was added to the apical chamber and 800 μL HBSS to the basolateral chamber. To measure B–A transport, 800 μL of test solution was added to the basolateral chamber and 250 μL HBSS to the apical chamber. Samples of 70 μL were then taken from the apical or basolateral chamber at predetermined time intervals (30, 60, 120, and 180 minutes) and immediately replaced with the same volume of HBSS. The TCG concentration in the buffer was determined by liquid chromatography with tandem time-of-flight mass spectroscopy (LC-TOF MS/MS) analysis (see method; LC-TOF MS/MS analysis of TCG). The apparent permeability (Papp) across the Caco-2 cell monolayer was calculated using the following equation:
|
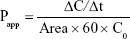
where Papp is the apparent permeability coefficient (cm/s), ΔC/Δt is the rate of drug appearance in the receiving chamber (nmol/s), Area is the surface area of the Transwell membrane (0.33 cm2), and C0 is the initial concentration of transported TCG (μM). The efflux ratio (ER) was then calculated using the equation:
|
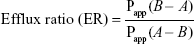
to determine the ratio of B–A transport (Papp (B–A)) to A–B transport (Papp (A–B)).
Pharmacokinetic studies
Evaluation of in vivo oral bioavailability
All animal experiments were conducted in accordance with the “Guiding Principles in the Use of Animals in Toxicology” adopted by the Society of Toxicology (Reston, VA, USA), and the experimental protocols were approved by Chungnam National University Institutional Animal Care Committee (Protocol no CNU-00911, Daejeon, Republic of Korea). Male Sprague Dawley rats aged 6–7 weeks with a body weight of 200–250 g were obtained from Samtaco Bio Korea Inc. (Osan, Republic of Korea) and housed at 22°C in a dark–light cycle of 12 hours where food and water could be freely accessed. Animals were adapted for 2 weeks before starting the study. The rats were randomly divided into three groups of eleven rats. The raw TCG suspension as control, TCG-CE, and TCG-SM at an equivalent dose of TCG was orally administered to rats at a dose of 10 mL/kg. The raw TCG suspension was prepared by suspending TCG in 0.5% (w/v) carboxymethylcellulose solution with low viscosity. The TCG-CE and TCG-SM were diluted with distilled water. Approximately 0.3 mL of blood samples was collected from the retro-orbital plexus using heparinized tubes at 0, 0.17, 0.33, 0.66, 1.0, 1.5, 2, 4, 6, 8, 12, and 24 hours and centrifuged immediately at 17,000 × g for 15 minutes at 4°C. The plasma samples were kept frozen at −80°C until LC-TOF MS/MS analysis.
LC-TOF MS/MS analysis of TCG
The concentrations of TCG were determined using an LC-TOF MS/MS system consisting of two Shimadzu LC-20AD pumps, a Shimadzu CBM-20A HPLC pump controller (Shimadzu Corp., Kyoto, Japan), a CTC HTS PAL autosampler (CEAP Technologies, Carrboro, NC, USA) and a quadrupole time-of-flight TripleTOF™ 5600 mass spectrometer (SCIEX, Foster City, CA, USA). The chromatographic column used for this assay was a Kinetex® XB-C18 column (50×2.1 mm, 2.6 μm; Phenomenex, Torrance, CA, USA). The mobile phase for TCG consisted of solvent A (0.1% formic acid in distilled water) and solvent B (0.1% formic acid in acetonitrile), with a gradient elution (0–0.5 minute, 10% solvent B; 0.5–1.0 minute, 10%–95% solvent B; 1.0–1.5 minutes, 95% solvent B; 1.5–1.6 minutes, 95%–10% solvent B; 1.6–3.0 minutes, 10% solvent B). The flow rate was 0.4 mL/min, and the injection volume was 10 μL. The scan mass spectra of TOF MS/MS and TOF MS were recorded using the positive ion mode. To analyze TCG and an internal standard (ISTD, verapamil), m/z 100–600 with 0.15 second accumulation time was used for TOF MS/MS. For quantification, the TCG and ISTD (m/z 523.1 and 455.3) and their respective product ions (m/z 153.0 and 165.1) were selected for quantitative analysis. The temperature of ion source was set at 500°C with a curtain gas flow of 33 L/min and 4.5 kV of the ion spray. For TCG and ISTD, the declustering potentials were 100 and 125 V and the collision energies were 40 and 30 V, respectively. The calibration curves for TCG was prepared at concentrations of 3–2,200 ng/mL and showed a good linearity with an R2 value >0.99.
Pharmacokinetic data analysis
WinNonlin software (8.0; Pharsight Co., Sunnyvale, CA, USA) was used to perform noncompartmental analysis. The maximum plasma concentration (Cmax), the time to reach the maximum plasma concentration (Tmax), and the half-life (T1/2) were observed from the plasma concentration–time profiles. The area under the concentration–time curve (AUC0–∞) was calculated by the linear trapezoidal method. The relative bioavailability (RBA) was calculated using the following equation:
|
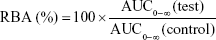
where AUC0–∞ (test) was the AUC0–∞ of TCG-CE or TCG-SM, and AUC0–∞ (control) was the AUC0–∞ of raw TCG suspension.
Pharmacodynamic study
Preparation of platelet-rich plasma (PRP) and platelet-poor plasma (PPP)
Light transmission aggregometry (LTA) was used to evaluate the antiplatelet activities of TCG. PRP and PPP were prepared prior to the experiment. Rat blood was collected via the jugular vein or postorbital plexus and drawn into a tube containing 3.2% sodium citrate solution (1/9 vol of blood). The blood was centrifuged at 200 × g at room temperature for 15 minutes to obtain PRP. The remaining blood was centrifuged at 2,000 × g at room temperature for 10 minutes to obtain PPP.
Antiplatelet activity test
The in vitro and ex vivo antiplatelet activities of raw TCG suspension as control and TCG-SM were evaluated using an automated aggregometer (Chrono-Log Model 700, Chrono-Log Corporation, Havertown, PA, USA). The platelet number of PRP was measured using a hematology analyzer (Advia 2120i; Siemens Healthineers, Forchheim, Germany) and adjusted to 5×107 platelets/mL against a PPP blank.
For in vitro studies, rat blood for preparation of PRP was collected via the jugular vein under anesthesia with pentobarbital (50 mg/kg, intraperitoneal injection). The obtained PRP was incubated with the raw TCG suspension or TCG-SM for 30 minutes at room temperature before aggregation was induced. Then, 240 μL PRP was stirred and incubated for 1 minute at 37°C before 10 μL ADP (20 μM) was added to induce platelet aggregation. Platelet aggregation was measured 10 minutes after addition of ADP.
For ex vivo studies, rats were randomly assigned to two treatment groups (n=9). Blood was collected from the jugular vein or postorbital plexus at 1, 2, 5, 10, and 24 hours after a single oral administration of raw TCG suspension or TCG-SM at an equivalent dose of TCG (2, 5, and 10 mg/kg). Approximately 1.8 mL blood samples were collected into a tube containing 0.2 mL 3.2% sodium citrate solution (pH 7.4). The blood was centrifuged (200 × g for 15 minutes at room temperature) to obtain PRP. After obtaining PRP, the remaining blood was centrifuged (2,000 × g for 10 minutes at room temperature) to obtain PPP. Then, the antiplatelet activities of raw TCG suspension and TCG-SM were evaluated as described above.
Pharmacodynamic data analysis
The values for in vitro 50% effective concentration (EC50) were calculated from nonlinear regression for the log concentration–response relationship for inhibition of platelet aggregation. To evaluate the ex vivo antiplatelet activity, the values of the AUC were calculated from the aggregometer and were used to calculate the percentage inhibition of platelet aggregation (IPA%) using the following equation:
|
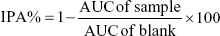
where the AUC of sample and AUC of blank were the AUC of sample PRP and blank PPP, respectively. The IPA% values of samples at predicted time-points were normalized to that of PRP at 0 hour. The values for the ex vivo 50% effective dose (ED50) were calculated from the linear regression for log dose–response (95% CI of X when Y=50). The area under the inhibitory curves (AUIC0–24) was calculated from the response–time relationship. GraphPad Prism 7 (GraphPad Software Inc., La Jolla, CA, USA) was used to calculate the EC50, ED50, and AUIC0–24 values.
Statistical analysis
Data are expressed as mean ± SD, except for the antiplatelet activity results, which are presented as mean ± standard error of the mean (SEM). Student’s t-test was used to assess the significance of differences. For the pharmacokinetic studies, one-way ANOVA was used and performed using Prism 7 (GraphPad Software Inc.). Statistical significance was defined as P<0.05.
Results and discussion
Preparation and characterization of optimized TCG-SM
Screening of excipients for TCG-SM
To develop a SMEDDS to dissolve the unit dose of TCG (90 mg) using a minimum volume of formulation, the first step was to select the appropriate excipients to achieve high solubility of TCG. The solubility of TCG for several excipients was shown in Figure 1. As shown in Figure 1A, Capmul MCM produced significantly higher solubility (101.19 mg/mL) than other oils (0.03–42.97 mg/mL). The solubility of TCG in the oil components of SMEDDS is crucial because oils are liable to solubilize hydrophobic drugs and can further improve the lipophilicity of the dissolved drug.25 Therefore, Capmul MCM with the highest solubility was used as the oil of the SMEDDS formulation. Of the screened surfactants, Tween 20, Tween 80, Labrasol, Cremophor EL, and Pluronic L64 all provided good solubility for TCG (>110 mg/mL). Generally, surfactants lower the surface energy and allow the oil to disperse into small droplet sizes in the aqueous phase.26 Among the cosurfactants screened, tetraglycol and Transcutol P produced the highest solubility for TCG (420.34 and 369.51 mg/mL, respectively) (Figure 1B). Cosurfactants can help increase the solubility of the drug in micelles, form micelles with the surfactant in the aqueous phase, prevent drug precipitation, and maintain a more stable micelle.27
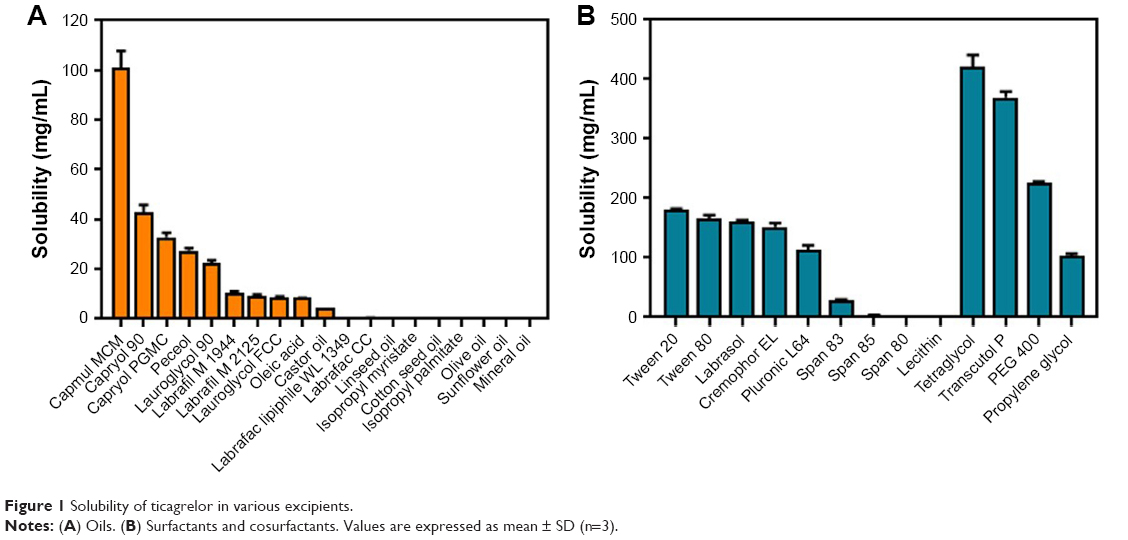 | Figure 1 Solubility of ticagrelor in various excipients. |
The droplet size, transmittance, and phase separation were evaluated to confirm the self-emulsifying ability of the preselected surfactants and cosurfactants. They were graded as shown in Table S1 (supplementary materials). The results of emulsification studies related to various combinations of surfactants and cosurfactants were listed in Table 2. Compared with other surfactants, Cremophor EL exhibited grade A emulsifying ability and showed no phase separation. Tween 20 exhibited excellent emulsifying ability (grade A or B) and showed no phase separation in distilled water and pH 6.8 media. Although Tween 20 showed phase separation in pH 1.2 and pH 4.0 media, an emulsion was formed when the samples were remixed. For the cosurfactants, the emulsifying ability of Transcutol P was higher than that of tetraglycol, especially in combination with Cremophor EL or Tween 20. Based on the above results, small droplet size (<300 nm), high transmittance (>90%, grades A and B), and a lack of phase separation were observed in SMEDDS formulations that combined Tween 20 or Cremophor EL as surfactant, Transcutol P as cosurfactant, and Capmul MCM as oil.
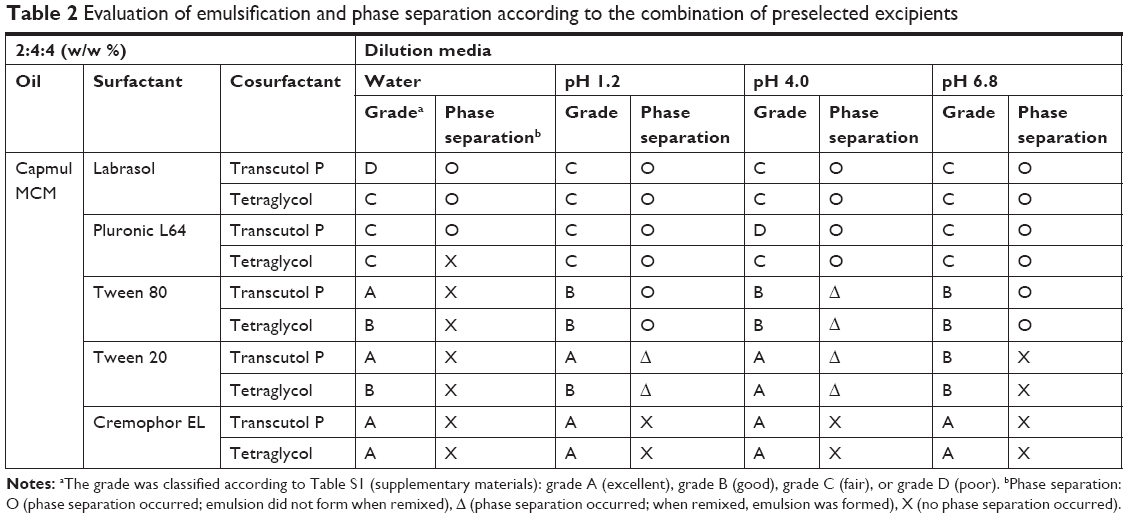 | Table 2 Evaluation of emulsification and phase separation according to the combination of preselected excipients |
Next, pseudoternary phase diagrams were prepared for both surfactants to determine the final components and set the range to be used in the experiments. In Figure 2A and B, the green line represents a microemulsion with a clear and/or transparent appearance (grade A), the red line represents microemulsions with a slightly less clear appearance (grade B), and the blue line indicates grade C emulsions with a bluish-white appearance. The grade A and B regions of SMEDDS formulations including Cremophor EL as surfactant were wider than those for SMEDDS formulation using Tween 20 as surfactant. This indicated that SMEDDS formulations including Cremophor EL as surfactant had better self-microemulsification properties than those containing Tween 20. A small droplet size results in an increased interfacial surface area, which reduces the micelle surface energy, enables faster drug release, and improves drug absorption.28 Thus, the final composition selected for SMEDDS was a combination of Capmul MCM/Cremophor EL/Transcutol P. When the concentration of oil exceeded 40%, unstable emulsions were formed, and it was verified that the droplet size increased as the oil content increased. In addition, the SMEDDS formulation could form droplets of <300 nm when the surfactant and cosurfactant were present in the mixture in the range of 10%–80%. Therefore, SMEDDS formulations were developed with composition of 10%–40% oil and 10%–80% surfactant/cosurfactant to produce transparent and homogeneous microemulsions (grade A, B) and to avoid the range in which unstable microemulsions were formed.
 | Figure 2 Pseudoternary phase diagram. |
Design of experiments for optimizing TCG-SM
Scheffé’s mixture design was applied to optimize the TCG-SM. The results of responses were fitted to Design-Expert 11 software and statistical analysis suggested different fitting models and their relationship to the variables. The solubility of TCG in SMEDDS (Y1), precipitation (Y2), droplet size (Y3), and transmittance (Y4) were important variables in the preparation of SMEDDS that maintained a stable formulation and improved the oral absorption of insoluble drugs. A high solubility of TCG in SMEDDS allows the SMEDDS to include a high drug content in a minimum volume of a stable formulation without precipitation. When the SMEDDS formulation is dispersed in an aqueous phase, some of the drugs are not encapsulated in micelles and are present as free drug, which can lead to precipitation.29 Therefore, the relationship between precipitation and variables was confirmed. A small droplet size increases the surface area of micelles, which enhances drug absorption by intestinal membrane contact and enables rapid drug dissolution.30 Transmittance was chosen to monitor the goal of forming a clear homogeneous microemulsion when dispersed in an aqueous phase.
As shown in Table 3, the statistical models of Y1, Y2, Y3, and Y4 responses were fitted to linear, quadratic, cubic, and special quadratic models, respectively. Various statistical parameters for each model such as sequential P-value, lack of fit, R2, and adjusted R2 value were considered when fitting the statistical models. The sequential P-values of all suggested models were <0.05, which indicated that the model parameters were significant at the 95% confidence level.31 The lack-of-fit P-values of the models were >0.05, suggesting that the fitted model was appropriate.32 The R2 and adjusted R2 were analyzed to determine how much the variability of experimental data was reflected by the models.33 All R2 and adjusted R2 values for Y1, Y3, and Y4 were >0.9, indicating that the data were close to the fitted values.34 Although the R2 value of Y2 was 0.7732, the precision of Y2 was adequate at 8.0892, which indicates that the Y2 model could be used to navigate the design space (adequate precision >4).35 All R2 and adjusted R2 values of responses were similar (difference between R2 and adjusted R2 <0.2). A similar value for R2 and adjusted R2 has been reported to be an appropriate goodness-of-fit indicator.36
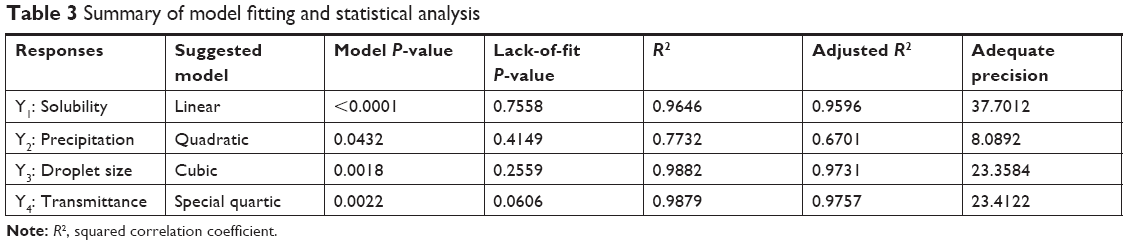 | Table 3 Summary of model fitting and statistical analysis |
The relationship between the factors was schematically illustrated in three-dimensional response surface plots and coefficient equations (Figure 3 and Table S2, supplementary materials). As shown in Figure 3 and Table S3 (supplementary materials), the solubility of TCG in SMEDDS (Y1) ranged from 126.51 to 330.59 mg/mL and the precipitation of TCG (Y2) ranged from 0.2% to 67.7% (Figure 3A and B). For droplet size (Y3) and transmittance (Y4), the ranges of Y3 and Y4 were from 54.7 to 1,023.6 nm and 49.5% to 99.2%, respectively (Figure 3C and D). The solubility of TCG in SMEDDS (Y1) and precipitation (Y2) increased as X3 increased. There was no significant interaction effect between X1, X2, and X3 in the solubility of TCG. In the case of droplet size (Y3), the effect of the interaction between X2 and X3 was significant, and the Y3 value tended to decrease as X2 increased and X3 decreased. The transmittance (Y4) was influenced by the interaction of X2 and X3 except for the main effect, and the value of Y4 tended to increase as X2 increased and X3 decreased.
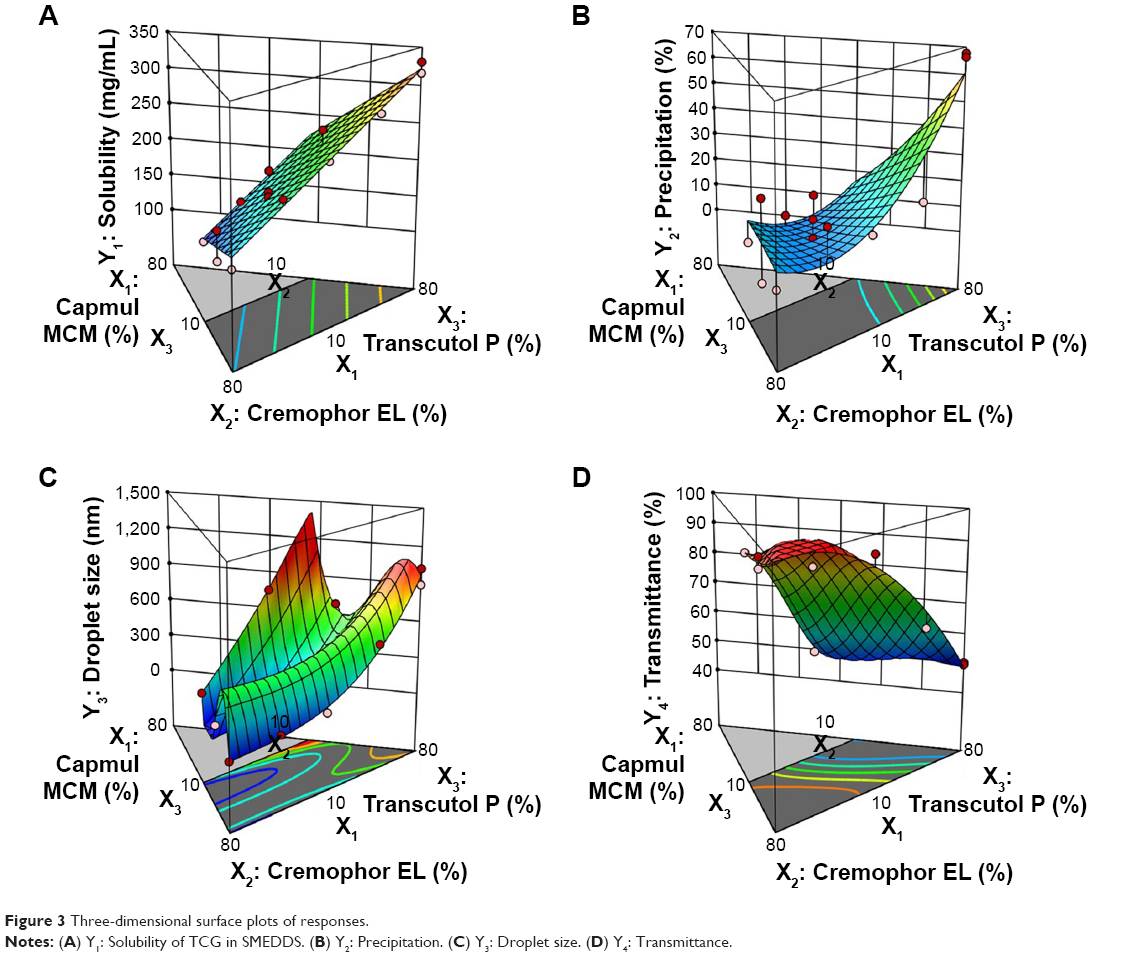 | Figure 3 Three-dimensional surface plots of responses. |
The factors were optimized using the desirability function by considering all the responses. The goals of Y1 and Y4 were set to be maximized and those of Y2 and Y3 were set to be minimized. Figure 4A shows the desirability plot that reflected the effect of the different variables on the four responses. The optimized values of X1, X2, and X3 were 10.0%, 53.8%, and 36.2%, respectively. The desirability value of the corresponding point was 0.766. The optimized factors and the values of predicted responses are listed in Table 4. To confirm the prediction accuracy, the difference between the predicted and actual values for each response was calculated as a percentage. Although the percentage error associated with Y1 (6.25%) and Y3 (5.61%) was slightly high, the errors between predicted values and actual values for Y2 (2.50%) and Y4 (1.36%) were very low. The fact that the errors were <10% indicated that the optimization of TCG-SM was successful.
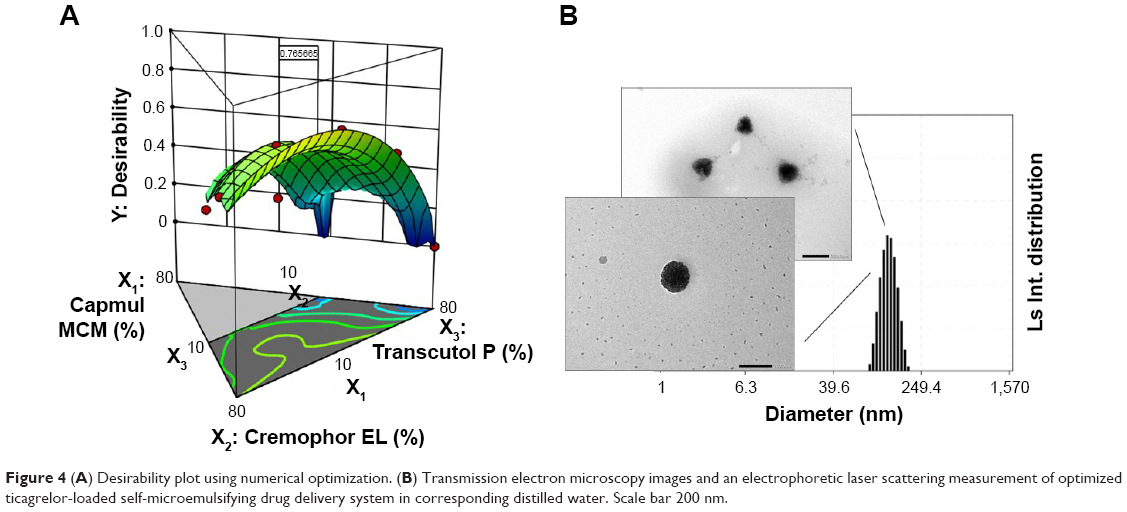 | Figure 4 (A) Desirability plot using numerical optimization. (B) Transmission electron microscopy images and an electrophoretic laser scattering measurement of optimized ticagrelor-loaded self-microemulsifying drug delivery system in corresponding distilled water. Scale bar 200 nm. |
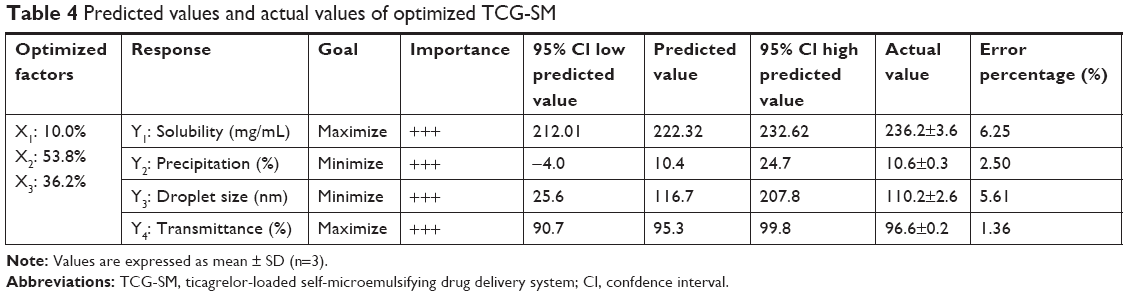 | Table 4 Predicted values and actual values of optimized TCG-SM |
The transmission electron microscopy data of optimized TCG-SM showed that the microemulsion with spherical droplet shapes in the nanometer range were observed and of a size similar to that obtained by ELS measurement (116.4±5.7 nm), as shown in Figure 4B.
It has been reported that self-forming microemulsions exhibit enhanced dissolution patterns.37,38 In this study, we assumed that the dissolution rates of TCG were significantly improved by the optimized TCG-SM formulation. For that reason, the dissolution profiles of Brilinta as control (commercial product), TCG-CE, and TCG-SM were determined in the media at pH 1.2, pH 4.0, and pH 6.8 and in distilled water (Figure 5). During the 3-hour experimental period, Brilinta showed very low dissolution of <10% in all media. The TCG-CE formulation slightly increased the dissolution of TCG within 3 hours in all media, with >40% dissolution in pH 1.2 media, pH 4.0 media, and distilled water (46.3%, 41.3%, and 43.1%, respectively), but not pH 6.8 media (34.6%). The optimized TCG-SM significantly enhanced the dissolution of TCG within 30 minutes in all media, with >85% dissolution (89.4%, 90.8%, 86.5%, and 97.1% in pH 1.2, pH 4.0, pH 6.8 media, and distilled water, respectively).
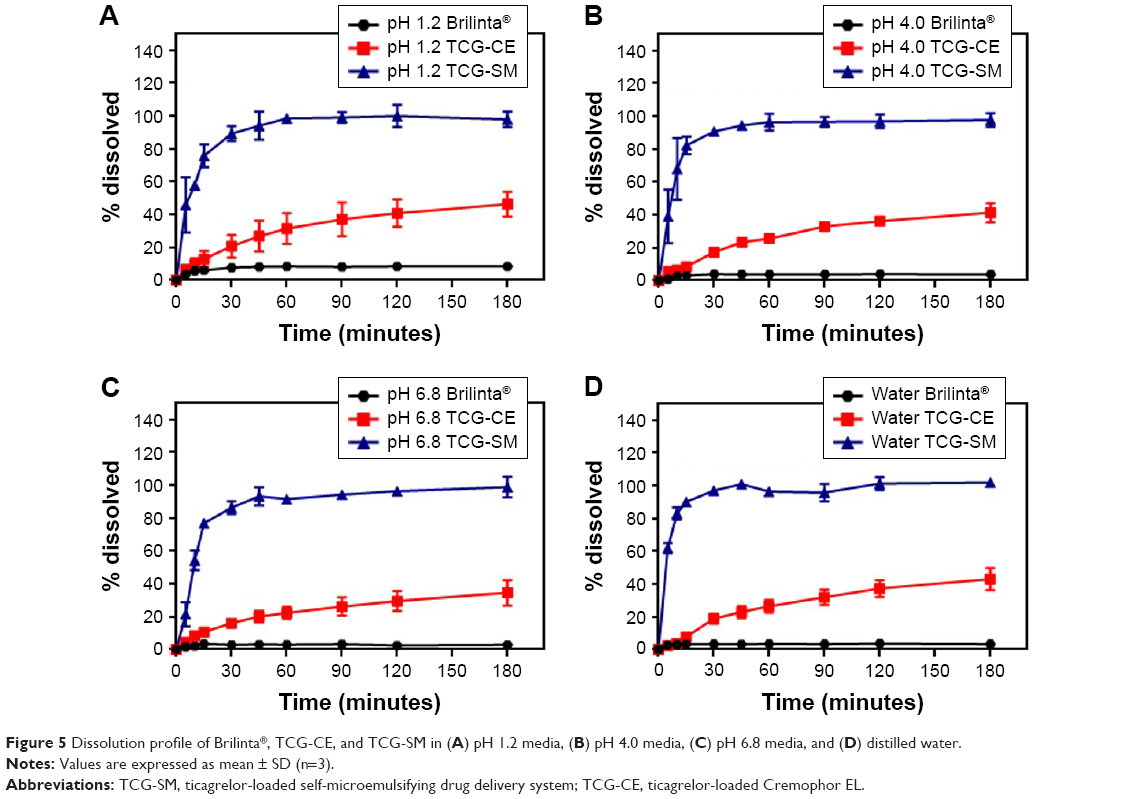 | Figure 5 Dissolution profile of Brilinta®, TCG-CE, and TCG-SM in (A) pH 1.2 media, (B) pH 4.0 media, (C) pH 6.8 media, and (D) distilled water. |
The SMEDDS formulation exhibited an excellent solvent capacity for hydrophobic drugs and formed a self-microemulsion after the penetration of water.39 The oil in the SMEDDS can improve the emulsification ability and improve the interfacial fluidity of surfactants in micelles when the formulation is diluted with aqueous medium in the presence of surfactants with a high hydrophilic–lipophilic balance.40 These results demonstrated that TCG-SM showed a higher dissolution than TCG-CE, which did not form oil-dispersed micelles because it contained only surfactant. These characteristics of the optimized TCG-SM indicate that the formulation not only provides a large surface area by forming nano-sized droplets but also improves the solubility and dissolution of TCG.
Cell study
In vitro cytotoxicity study
The cytotoxicity of the raw TCG solution, blank-CE, blank-SM, TCG-CE, and TCG-SM for Caco-2 cells was studied using a standard MTT assay. Figure 6A shows the viability of cells incubated with five different formulations for 24 hours. TCG-CE and TCG-SM exhibited a similar dose-dependent inhibitory effect to raw TCG solution on proliferation of Caco-2 cells. However, formulations without TCG (blank-CE and blank-SM) showed no cytotoxicity on Caco-2 cells. These results indicated that the oil, surfactant, and cosurfactant used in SMEDDS were not toxic for Caco-2 cells at the concentration used in the SMEDDS (400 μg/mL, calculated from the corresponding TCG content), while the raw TCG solution, TCG-CE, and TCG-SM exhibited significant cytotoxicity at concentrations of TCG above 9 μg/mL. Interestingly, the cytotoxicity appeared to be associated with the TCG concentration, but not with the formulations. This confirmed that the formulations are highly biocompatible and can be used as drug carriers for oral administration, while TCG-SM displays similar cytotoxic activity in vitro to the commercial TCG formulation (Brilinta) based on the dose of drug. Therefore, further cell studies were performed with formulations containing 9 μg/mL of TCG, a lower concentration than the half-maximal inhibitory concentration of raw TCG solution (13.1±2.3 μg/mL) calculated from the nonlinear fitted curve.
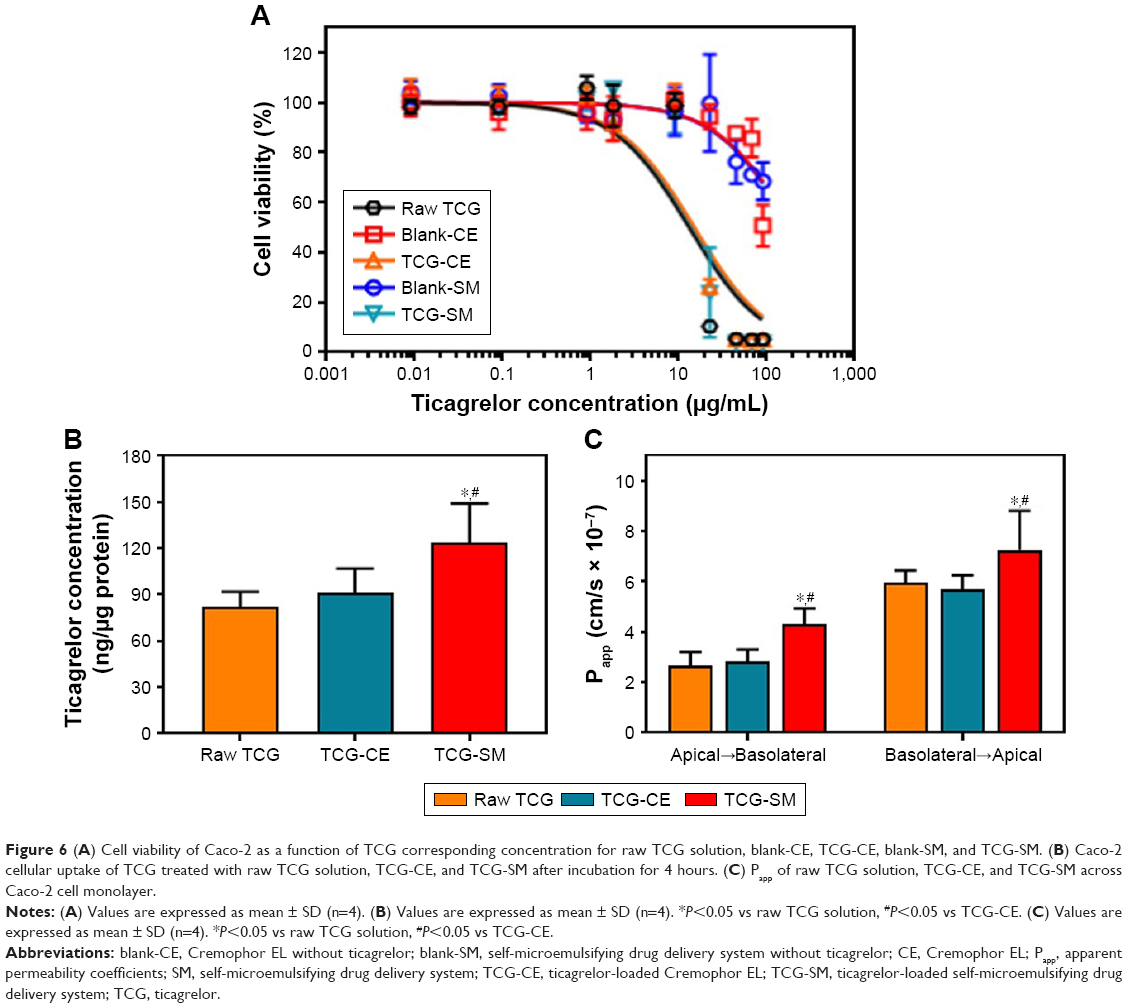 | Figure 6 (A) Cell viability of Caco-2 as a function of TCG corresponding concentration for raw TCG solution, blank-CE, TCG-CE, blank-SM, and TCG-SM. (B) Caco-2 cellular uptake of TCG treated with raw TCG solution, TCG-CE, and TCG-SM after incubation for 4 hours. (C) Papp of raw TCG solution, TCG-CE, and TCG-SM across Caco-2 cell monolayer. |
Cellular uptake study
To confirm whether the SMEDDS formulation could enhance the uptake of loaded TCG, Caco-2 cells were incubated with raw TCG solution, TCG-CE, or TCG-SM, and their intracellular TCG content was quantified by HPLC analysis. As shown in Figure 6B, Caco-2 cell uptake of TCG from TCG-SM was 1.50 and 1.35 times higher than that from raw TCG solution and TCG-CE, respectively, after incubation for 4 hours (P<0.05). In the case of TCG-CE, the uptake was 1.11 times higher than that of raw TCG solution, but these were not significantly different (P>0.05). Based on these results, we concluded that cell uptake of TCG was improved by TCG-SM.
Permeability test
Caco-2 cell monolayer models were also established to evaluate the permeability of raw TCG solution, TCG-CE, and TCG-SM. Figure S1 (Supplementary materials) illustrates that the cumulative amount of transported TCG was time-dependent for all formulations. TCG-SM exhibited better transepithelial transport than the other formulations, which was consistent with the analysis of Caco-2 cellular uptake of TCG discussed above. As shown in Figure 6C, the observed mean Papp (A–B) values of raw TCG solution, TCG-CE, and TCG-SM were 2.64×10−7 cm/s, 2.81×10−7 cm/s, and 4.30×10−7 cm/s, respectively. The mean Papp (A–B) of TCG in TCG-SM was 1.63 times higher than that of raw TCG solution (P<0.05). Similarly, mean Papp (B–A) values of 5.96×10−7 cm/s, 5.67×10−7 cm/s, and 7.24×10−7 cm/s were obtained for raw TCG solution, TCG-CE, and TCG-SM, respectively. The mean Papp (B–A) of TCG in TCG-SM was 1.23 times higher than that of the raw TCG solution (P<0.05). This result is probably due to an increase in the drug transported (A–B), thus increasing the level of B–A transport. Observed ERs were 2.26, 2.02, and 1.68 for raw TCG solution, TCG-CE, and TCG-SM, respectively. The ER of TCG-SM was decreased by 25.7% compared with that of raw TCG solution. These results support the improved permeability of TCG by a SMEDDS formulation. The enhancement in the Papp (A–B) of TCG-SM compared with raw TCG solution and TCG-CE could be due to smaller droplet size and higher lipophilicity for the reconstituted TCG-SM.41 These reduced-size droplets containing TCG solubilized by SMEDDS formulation easily permeated across the barrier membrane, which indicates improvement in the apparent permeability, flux, and relative permeability of TCG. In addition, the enhancement of the lipophilicity of TCG by microemulsion increased its adhesion to intestinal membrane, which could be associated with improved drug transport and optimizing intestinal absorption and permeation.42 Based on these results, we could confirm the enhancement of the transepithelial permeability of TCG by the SMEDDS formulation.
Pharmacokinetic and pharmacodynamic studies
Pharmacokinetic study
To evaluate the pharmacokinetics of the formulations, samples of raw TCG suspension, TCG-CE, and TCG-SM were orally administered to rats at a dose of 10 mg/kg TCG. The relevant pharmacokinetic parameters of each formulation are shown in Table 5. Cmax and AUC0–∞ of TCG after oral administration differed significantly in the following order: TCG-SM> TCG-CE> raw TCG suspension (Figure 7). As expected, raw TCG suspension exhibited poor Cmax (68.94±25.67 ng/mL) and AUC0–∞ (443.26±147.80 ng·h/mL). The TCG-CE exhibited a slightly increased Cmax (85.10±35.57 ng/mL), but the AUC0–∞ (427.25±153.96 ng·h/mL) was similar to that of the raw TCG suspension. However, the Cmax (439.18±172.01 ng/mL) and AUC0–∞ (2,525.29±390.27 ng·h/mL) of TCG-SM were significantly higher than those of raw TCG suspension and TCG-CE (P<0.05). TCG-CE and TCG-SM formulations showed slightly reduced Tmax compared with raw TCG suspension, but the difference was not significant (P>0.05). Therefore, the TCG-SM formulation improved the oral bioavailability of TCG by 637.1% compared with the raw TCG suspension. The improved oral bioavailability of TCG-SM was correlated with the result of the in vitro dissolution test and cell studies, which showed that TCG-SM was advantageous for achieving increased drug release, higher cellular uptake, and enhanced permeability across the intestinal epithelium.
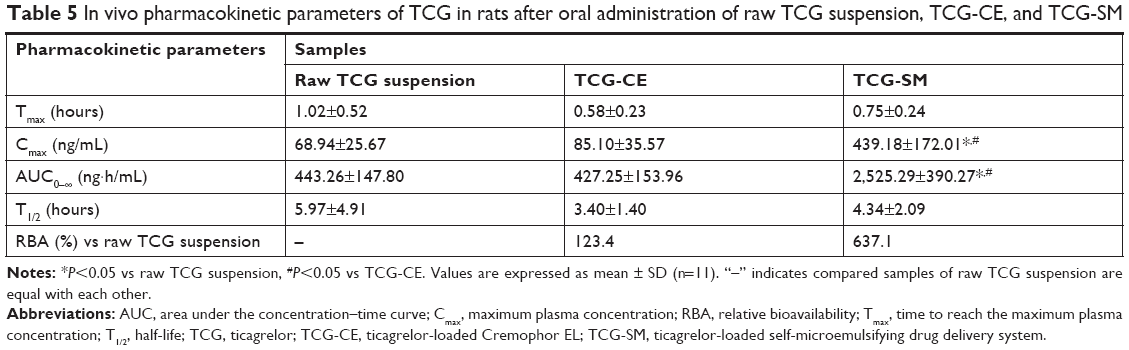 | Table 5 In vivo pharmacokinetic parameters of TCG in rats after oral administration of raw TCG suspension, TCG-CE, and TCG-SM |
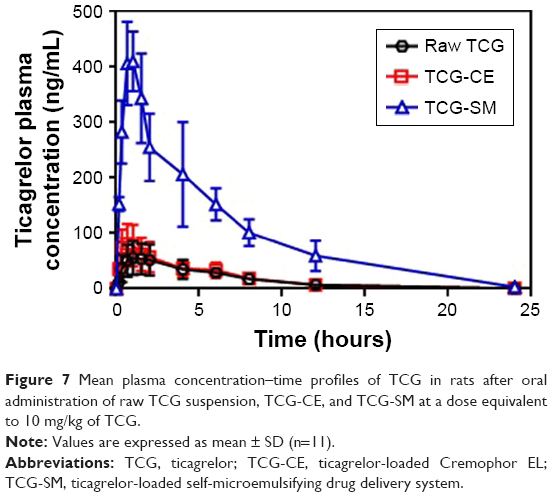 | Figure 7 Mean plasma concentration–time profiles of TCG in rats after oral administration of raw TCG suspension, TCG-CE, and TCG-SM at a dose equivalent to 10 mg/kg of TCG. |
Pharmacodynamic study
In vitro antiplatelet activity test
To confirm the antiplatelet activity of TCG and formulations, in vitro antiplatelet activity tests were carried out. The platelet aggregation in the presence of different concentrations of TCG (5.2, 15.7, 31.4, 261.3, 522.6, 1,567.7, 3,135.4, and 5,225.7 ng/mL) in raw TCG suspension and TCG-SM diluted with distilled water were measured using an LTA method. Both raw TCG suspension and TCG-SM inhibited platelet aggregation in a dose-dependent manner (Figure 8). In the blank formulations (blank-solution and blank-SM), corresponding to the negative control, mean platelet aggregation induced by 20 μM ADP ranged from 56% to 67% (data not shown). The responses to different formulations were normalized to platelet aggregation with blank PRP. The mean EC50 values for raw TCG suspension and TCG-SM were 39.45 ng/mL and 58.39 ng/mL, respectively; ie, the EC50 value of TCG-SM was higher than that of raw TCG suspension. This result was suggested to be because of the influence on platelet aggregation of the excipients used in SMEDDS. Although the excipients used in TCG-SM have not been studied in detail, Poloxamer 188, which is frequently used as a surfactant, inhibits platelet aggregation.43 In vitro, it was difficult to confirm the synergistic effect on antiplatelet activity of the SMEDDS formulation because the drug-loaded formulations were added directly to PRP for the assessment of in vitro antiplatelet activity. These results did not allow for the enhancement of absorption of the drug caused by the formulation. Therefore, there was no significant difference in the efficacy of the two formulations. However, further ex vivo studies were performed to assess whether TCG-SM has more potent antiplatelet activity than raw TCG suspension in rats.
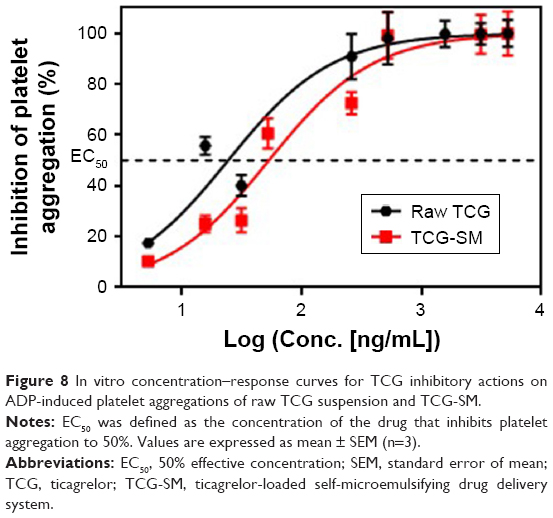 | Figure 8 In vitro concentration–response curves for TCG inhibitory actions on ADP-induced platelet aggregations of raw TCG suspension and TCG-SM. |
Ex vivo antiplatelet activity test
Ex vivo platelet aggregation was used to evaluate the antiplatelet activity of TCG in different formulations and was measured as the degree of platelet aggregation induced by 20 μM ADP in blood samples taken at 1, 2, 5, 10, and 24 hours after oral administration. A dose-related inhibitory effect on platelet aggregation was demonstrated after a single oral administration of raw TCG suspension and TCG-SM (2, 5, and 10 mg/kg) (Figure 9A). As indicated in Table 6, the AUIC0–24 data showed that the level of antiplatelet activity of TCG-SM was greater than that of raw TCG suspension at the same drug dose. The AUIC0–24 of the rats treated with 2, 5, and 10 mg/kg of TCG-SM was 270.4%±113.3%·hours, 907.8%±200.5%·hours, and 1,254.0%±233.9%·hours, respectively, which was much higher than in rats treated with raw TCG suspension (2, 5, and 10 mg/kg; 117.0%±88.08%·hours, 444.3%±205.7%·hours, and 907.0%±408.8%·hours, respectively), demonstrating the significantly improved antiplatelet activity of orally delivered TCG-SM in Sprague Dawley rats. In addition, the raw TCG suspension (10 mg/kg) exhibited a similar AUIC0–24 (907.0%±408.8%·hours) to that of the 5 mg/kg dose of TCG-SM (907.8%±200.5%·hours). These results indicated that the application of the SMEDDS formulation could achieve higher drug efficacy even with a lower dose of drug. We also calculated the ED50 of raw TCG suspension and TCG-SM at various time-points after their administration (Figure 9B and Table 6). The ED50 values for raw TCG suspension at 2, 5, and 10 hours after administration were 5.1, 4.5, and 8.4 mg/kg, respectively, while those of TCG-SM were 3.5, 2.5, and 4.4 mg/kg, respectively. The ED50 values of both formulations were lowest at the 5-hour time-point, indicating the time of peak antiplatelet activity. The ED50 values of TCG-SM were lower than those of raw TCG suspension at all time-points. These results indicated that TCG-SM not only improved the bioavailability of TCG but also increased its antiplatelet activity by enhancing absorption of the drug.
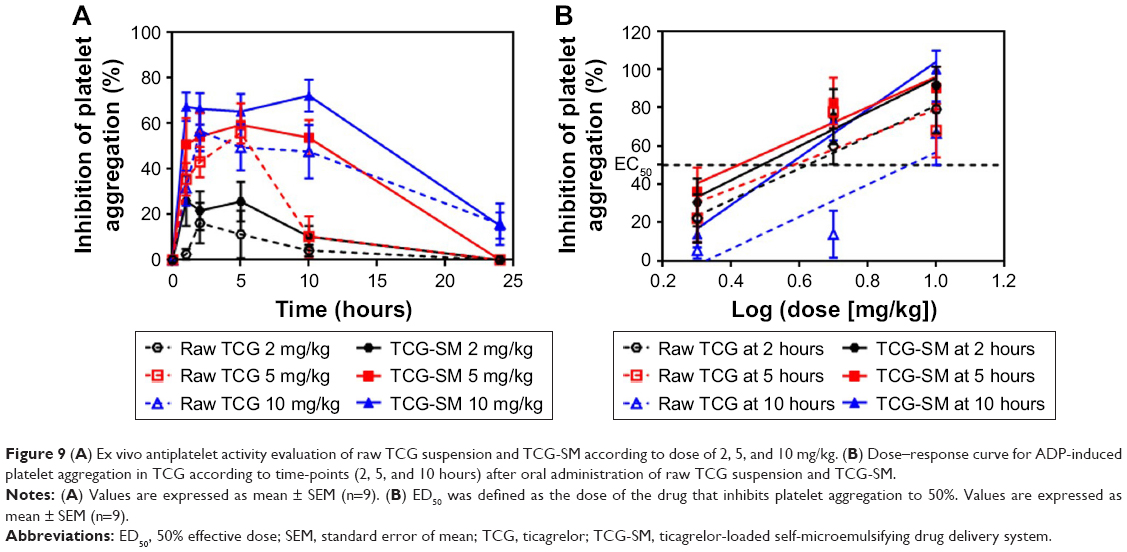 | Figure 9 (A) Ex vivo antiplatelet activity evaluation of raw TCG suspension and TCG-SM according to dose of 2, 5, and 10 mg/kg. (B) Dose–response curve for ADP-induced platelet aggregation in TCG according to time-points (2, 5, and 10 hours) after oral administration of raw TCG suspension and TCG-SM. |
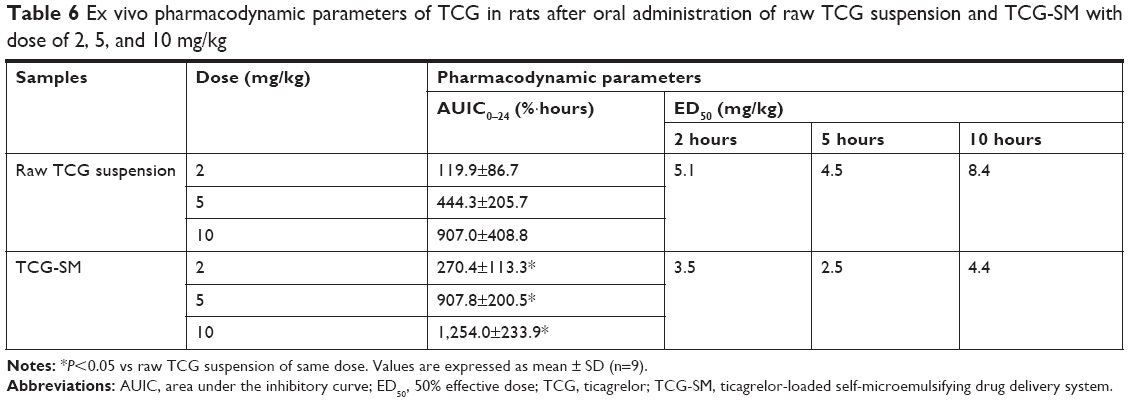 | Table 6 Ex vivo pharmacodynamic parameters of TCG in rats after oral administration of raw TCG suspension and TCG-SM with dose of 2, 5, and 10 mg/kg |
In summary, the optimized TCG-SM showed high dissolution, low cytotoxicity, and high permeability. These results led to increased bioavailability and antiplatelet activity of TCG (Figure 10).
 | Figure 10 Main design and result for the development of TCG-SM. |
Conclusion
The optimized TCG-SM contained 10% Capmul MCM (oil, X1), 53.8% Cremophor EL (surfactant, X2), and 36.2% Transcutol P (cosurfactant, X3). The SMEDDS formulation produced high solubility of TCG, small droplet size, low precipitation, and high transmittance. In addition, the formulation showed higher dissolution profile, improved cellular uptake, and permeability of TCG on Caco-2 cells. The pharmacokinetic study demonstrated the enhanced bioavailability of TCG in the TCG-SM formulation, and the ex vivo pharmacodynamic study showed that TCG-SM increased the absorption of TCG and showed greater inhibition of platelet aggregation than the same dose of raw TCG suspension. In conclusion, the optimized TCG-SM could be potentially used for improving the antiplatelet activity of TCG at a reduced dose. SMEDDS as a liquid formulation should be placed in a soft gelatin capsule for the development of drug product, which may lead to problems such as capsule aging and high manufacturing costs. So, we will proceed with the solidification study of SMEDDS to make a solid formulation such as a tablet. In addition, we will investigate the integrity of SMEDDS in body and in vitro–in vivo correlation of SMEDDS using GastroPlus program.
Acknowledgments
This work was supported by the Basic Science Research Program (2016R1A2B4011294) through the National Research Foundation of Korea (NRF) funded by the Ministry of Education, Science and Technology. The sample excipients of Gattefossé Co. and ABITEC Corporations were from the kind support of Masung & Co., Ltd. We would like to thank DW Yeom for fruitful discussions about the design of experiments study.
Disclosure
Mr Gi-Ho Son and Mr Ki-Hyun Bang are employed by Korea United Pharmaceutical Co. Ltd., Sejong, Republic of Korea. Mr Sung-Hoon Jeon is employed by SamA Pharmaceutical Co. Ltd., Suwon, Republic of Korea. The authors did not receive grants/funds from these affiliations for this study. The authors report no other conflicts of interest in this work.
References
Husted S, Emanuelsson H, Heptinstall S, Sandset PM, Wickens M, Peters G. Pharmacodynamics, pharmacokinetics, and safety of the oral reversible P2Y12 antagonist AZD6140 with aspirin in patients with atherosclerosis: a double-blind comparison to clopidogrel with aspirin. Eur Heart J. 2006;27(9):1038–1047. | ||
Teng R, Oliver S, Hayes MA, Butler K. Absorption, distribution, metabolism, and excretion of ticagrelor in healthy subjects. Drug Metab Dispos. 2010;38(9):1514–1521. | ||
van Giezen JJ, Humphries RG. Preclinical and clinical studies with selective reversible direct P2Y12 antagonists. Semin Thromb Hemost. 2005;31(02):195–204. | ||
Husted S, van Giezen JJJ. Ticagrelor: the first reversibly binding oral P2Y12 receptor antagonist. Cardiovas Ther. 2009;27(4):259–274. | ||
Wijeyeratne YD, Joshi R, Heptinstall S. Ticagrelor: a P2Y12 antagonist for use in acute coronary syndromes. Expert Rev Clin Pharmacol. 2012;5(3):257–269. | ||
Wallentin L, Becker RC, Budaj A, et al. Ticagrelor versus clopidogrel in patients with acute coronary syndromes. N Engl J Med. 2009;361(11):1045–1057. | ||
BRILINTA (ticagrelor) [package insert]. Wilmington, DE: AstraZeneca Pharmaceuticals LP; 2016. | ||
Ramesh K, Shekar C, Khadgapathi P. Enhancement of solubility and rate of in vitro drug release profiles of poorly soluble ticagrelor by solvent evaporation solid dispersion technique. Indo Am J Pharm Res. 2015;5(12):3850–3858. | ||
Pai G, Sathyanarayana MB. Fabrication and solid state characterization of ticagrelor co-crystals with improved solubility and dissolution. Int J Pharm Qual Assur. 2017;8(1):1–8. | ||
Chen ZQ, Liu Y, Zhao JH, Wang L, Feng NP. Improved oral bioavailability of poorly water-soluble indirubin by a supersaturatable self-microemulsifying drug delivery system. Int J Nanomedicine. 2012;7:1115–1125. | ||
Kamboj S, Sharma R, Singh K, Rana V. Aprepitant loaded solid preconcentrated microemulsion for enhanced bioavailability: a comparison with micronized aprepitant. Eur J Pharm Sci. 2015;78:90–102. | ||
Yeom DW, Song YS, Kim SR, et al. Development and optimization of a self-microemulsifying drug delivery system for atorvastatin calcium by using D-optimal mixture design. Int J Nanomedicine. 2015;10:3865–3878. | ||
Yeom DW, Chae BR, Son HY, et al. Enhanced oral bioavailability of valsartan using a polymer-based supersaturable self-microemulsifying drug delivery system. Int J Nanomedicine. 2017;12:3533–3545. | ||
Patel G, Shelat P, Lalwani A. Statistical modeling, optimization and characterization of solid self-nanoemulsifying drug delivery system of lopinavir using design of experiment. Drug Deliv. 2016;23(8):3027–3042. | ||
Kamboj S, Rana V. Quality-by-design based development of a self-microemulsifying drug delivery system to reduce the effect of food on nelfinavir mesylate. Int J Pharm. 2016;501(1–2):311–325. | ||
Elnaggar YS, El-Massik MA, Abdallah OY. Self-nanoemulsifying drug delivery systems of tamoxifen citrate: design and optimization. Int J Pharm. 2009;380(1–2):133–141. | ||
Porter CJ, Pouton CW, Cuine JF, Charman WN. Enhancing intestinal drug solubilisation using lipid-based delivery systems. Adv Drug Deliv Rev. 2008;60(6):673–691. | ||
Porter CJH, Charman WN. In vitro assessment of oral lipid based formulations. Adv Drug Deliv Rev. 2001;50(Suppl 1):S127–S147. | ||
Balakrishnan P, Lee BJ, Oh DH, et al. Enhanced oral bioavailability of coenzyme Q10 by self-emulsifying drug delivery systems. Int J Pharm. 2009;374(1–2):66–72. | ||
Dixit AR, Rajput SJ, Patel SG. Preparation and bioavailability assessment of SMEDDS containing valsartan. AAPS Pharm Sci Tech. 2010;11(1):314–321. | ||
Krishnamoorthy B, Habibur Rahman SM, Tamil Selvan N, et al. Design, formulation, in vitro, in vivo, and pharmacokinetic evaluation of nisoldipine-loaded self-nanoemulsifying drug delivery system. J Nanopart Res. 2015;17(1):34. | ||
Bueno LM, Manoel JW, Giordani CF, et al. HPLC method for simultaneous analysis of ticagrelor and its organic impurities and identification of two major photodegradation products. Eur J Pharm Sci. 2017;97:22–29. | ||
Pouton CW. Formulation of poorly water-soluble drugs for oral administration: physicochemical and physiological issues and the lipid formulation classification system. Eur J Pharm Sci. 2006;29(3–4):278–287. | ||
Scheffé H. Experiments with mixtures. J R Stat Soc Series B. 1958;20(2):344–360. | ||
Gurram AK, Deshpande PB, Kar SS, Nayak UY, Udupa N, Reddy MS. Role of components in the formation of self-microemulsifying drug delivery systems. Indian J Pharm Sci. 2015;77(3):249–257. | ||
Hall S, Pacek AW, Kowalski AJ, Cooke M, Rothman D. The effect of scale and interfacial tension on liquid–liquid dispersion in in-line Silverson rotor–stator mixers. Chem Eng Res Des. 2013;91(11):2156–2168. | ||
Lu Y, Park K. Polymeric micelles and alternative nanonized delivery vehicles for poorly soluble drugs. Int J Pharm. 2013;453(1):198–214. | ||
Jaiswal P, Aggarwal G, Harikumar SL, Singh K. Development of self-microemulsifying drug delivery system and solid-self-microemulsifying drug delivery system of telmisartan. Int J Pharm Investig. 2014;4(4):195–206. | ||
Akula S, Gurram AK, Devireddy SR. Self-microemulsifying drug delivery systems: an attractive strategy for enhanced therapeutic profile. Int Sch Res Notices. 2014;2014:964051. | ||
Xu W, Ling P, Zhang T. Polymeric micelles, a promising drug delivery system to enhance bioavailability of poorly water-soluble drugs. J Drug Deliv. 2013;2013:340315. | ||
Mcshane BB, Gal D. Statistical significance and the dichotomization of evidence. J Am Stat Assoc. 2017;112(519):885–895. | ||
Mourabet M, El Rhilassi A, El Boujaady H, Bennani-Ziatni M, Taitai A. Use of response surface methodology for optimization of fluoride adsorption in an aqueous solution by Brushite. Arab J Chem. 2017;10:S3292–S3302. | ||
Schneider AA, Hommel G, Blettner M. Linear regression analysis: part 14 of a series on evaluation of scientific publications. Dtsch Ärztebl Int. 2010;107(44):776–782. | ||
Bewick V, Cheek L, Ball J. Statistics review 7: correlation and regression. Crit Care. 2003;7(6):451–459. | ||
Patel DV. Preparation and evaluation of extended release pellets of chiral molecules of s-metoprolol succinate by different technology. Asian J Pharm. 2017;11(3):210–223. | ||
Figueiredo Filho DB, Silva Júnior JA, Rocha EC. What is R2 all about? Leviathan. 2011;3(3):60–68. | ||
Balata GF, Essa EA, Shamardl HA, Zaidan SH, Abourehab MA. Self-emulsifying drug delivery systems as a tool to improve solubility and bioavailability of resveratrol. Drug Des Devel Ther. 2016;10:117–128. | ||
Zhou H, Wan J, Wu L, et al. A new strategy for enhancing the oral bioavailability of drugs with poor water-solubility and low liposolubility based on phospholipid complex and supersaturated SEDDS. PLoS One. 2013;8(12):e84530. | ||
Patel D, Sawant K. Self micro-emulsifying drug delivery system: formulation development and biopharmaceutical evaluation of lipophilic drugs. Curr Drug Deliv. 2009;6(4):419–424. | ||
Taha EI, Al-Saidan S, Samy AM, Khan MA. Preparation and in vitro characterization of self-nanoemulsified drug delivery system (SNEDDS) of all-trans-retinol acetate. Int J Pharm. 2004;285(1–2):109–119. | ||
Nekkanti V, Karatgi P, Prabhu R, Pillai R. Solid self-microemulsifying formulation for candesartan cilexetil. AAPS PharmSciTech. 2010;11(1):9–17. | ||
Memvanga PB, Eloy P, Gaigneaux EM, Préat V. In vitro lipolysis and intestinal transport of β-arteether-loaded lipid-based drug delivery systems. Pharm Res. 2013;30(10):2694–2705. | ||
Edwards CM, May JA, Heptinstall S, Lowe KC. Effects of pluronic F-68 (poloxamer 188) on platelet aggregation in human whole blood. Thromb Res. 1996;81(4):511–512. |
Supplementary materials
 | Table S1 The classification standards of emulsification grade |
 | Table S2 Coefficient equations of responses according to the level of factors |
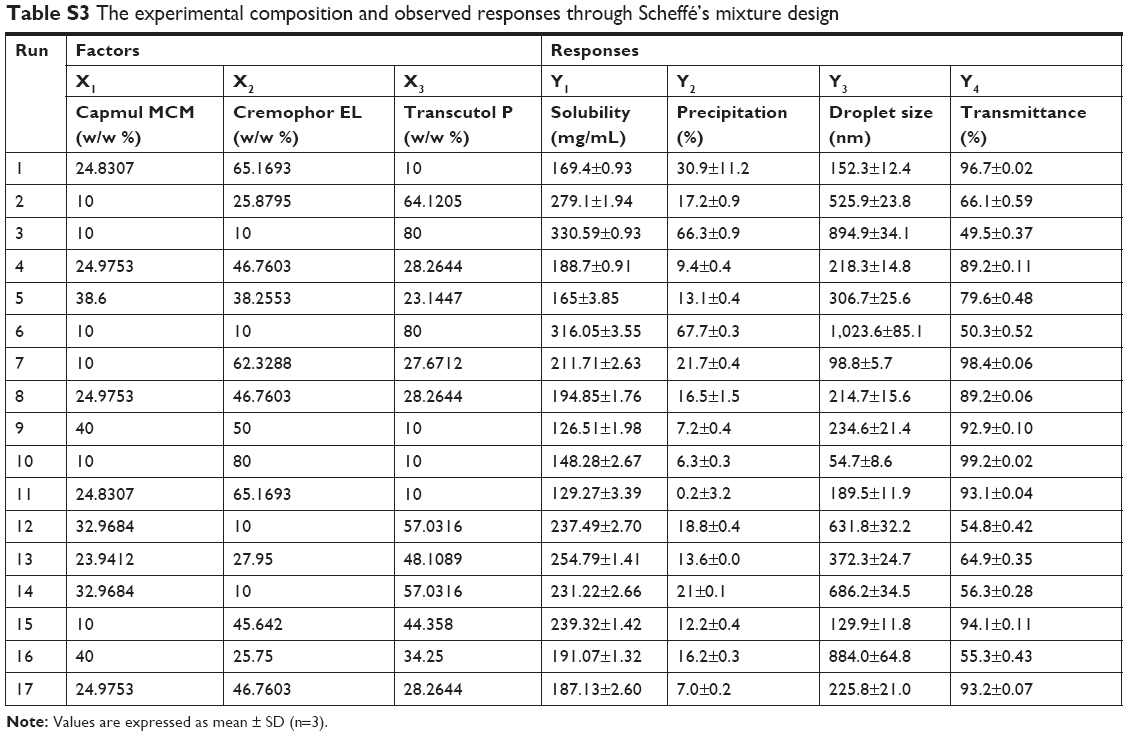 | Table S3 The experimental composition and observed responses through Scheffé’s mixture design |
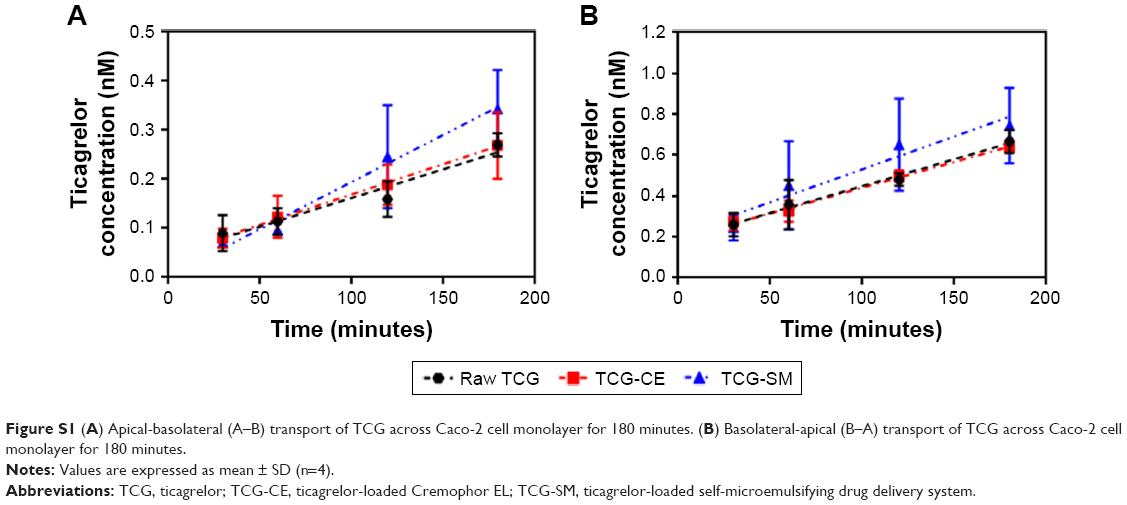 | Figure S1 (A) Apical-basolateral (A–B) transport of TCG across Caco-2 cell monolayer for 180 minutes. (B) Basolateral-apical (B–A) transport of TCG across Caco-2 cell monolayer for 180 minutes. |
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.