Back to Journals » International Journal of Nanomedicine » Volume 11
Stimuli-responsive hybrid nanocarriers developed by controllable integration of hyperbranched PEI with mesoporous silica nanoparticles for sustained intracellular siRNA delivery
Authors Prabhakar N, Zhang J, Desai D, Casals E, Gulin-Sarfraz T, Näreoja T, Westermarck J, Rosenholm JM ![]()
Received 25 August 2016
Accepted for publication 22 October 2016
Published 8 December 2016 Volume 2016:11 Pages 6591—6608
DOI https://doi.org/10.2147/IJN.S120611
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Thomas Webster
Neeraj Prabhakar,1,2 Jixi Zhang,3 Diti Desai,1 Eudald Casals,1 Tina Gulin-Sarfraz,1 Tuomas Näreoja,2,4 Jukka Westermarck,5,6 Jessica M Rosenholm1
1Pharmaceutical Sciences Laboratory, Faculty of Science and Engineering, Åbo Akademi University, 2Laboratory of Biophysics, Faculty of Medicine, University of Turku, Turku, Finland; 3College of Bioengineering, Chongqing University, Chongqing, People’s Republic of China; 4Department of Neuroscience, Karolinska Institute, Stockholm, Sweden; 5Centre for Biotechnology, University of Turku and Åbo Akademi, 6Department of Pathology, University of Turku, Turku, Finland
Abstract: Small interfering RNA (siRNA) is a highly potent drug in gene-based therapy with the challenge being to deliver it in a sustained manner. The combination of mesoporous silica nanoparticles (MSNs) and polycations in the confined pore space allows for incorporation and controlled release of therapeutic siRNA payloads. We hereby constructed MSNs with expanded mesopores and pore-surface-hyperbranched poly(ethyleneimine) (PEI) tethered with redox-cleavable linkers that could carry a high payload of siRNA (120 mg·g-1). The developed nanocarriers were efficiently taken up by cancer cells and were subsequently able to escape to the cytoplasm from the endosomes, most likely owing to the integrated PEI. Triggered by the intracellular redox conditions, the siRNA was sustainably released inside the cells over a period of several days. Functionality of siRNAs was demonstrated by using cell-killing siRNA as cargo. Despite not being the aim of the developed system, in vitro experiments using cell-killing siRNAs showed that the efficacy of siRNA transfection was comparable to the commercial in vitro transfection agent Lipofectamine. Consequently, the developed MSN-based delivery system offers a potential approach to hybrid nanocarriers for more efficient and long-term siRNA delivery and, in a longer perspective, in vivo gene silencing for RNA interference (RNAi) therapy.
Keywords: mesoporous silica nanoparticles, RNAi therapy, siRNA delivery, stimuli-responsive drug release, hybrid nanocarriers
Introduction
RNA interference (RNAi) of cellular processes by delivering small interfering RNA (siRNA) molecules to the cytoplasm, in order to knock down messenger RNA (mRNA) expression, may open an avenue for the development of highly specific therapeutic applications.1–5 In particular, a recent study demonstrating first cancer cures by systemic delivery of siRNA in patients encourages further development of siRNA delivery strategies, eg, nanoparticle-mediated delivery.6–8 Despite the availability of very efficient in vitro transfection agents, these are generally not applicable for in vivo oligonucleotide delivery. In light of the concerns that the fragile oligonucleotides utilized in RNAi suffer from, such as macromolecular form, high polarity, poor stability and low bioavailability in the physiological environment, it is critically important to develop efficient carriers that can load, retain, protect and release these oligonucleotides during the processes involving the related biomedical applications. However, well-controlled siRNA delivery remains challenging for many reasons. First, as siRNA-mediated target inhibition takes considerably longer time than direct small molecular targeting of proteins, long-term therapeutic effects will require repeated dosing if the delivery system does not meet the criteria of high payload and sustained, slow release. Second, crossing biological barriers such as the cell membrane and the endosomal compartment is difficult.2,5,7–11
Nanocarriers, especially mesoporous silica nanoparticles (MSNs), have the potential for meeting both the abovementioned challenges, as they encompass several characteristics that make them a suitable vehicle for the delivery of siRNA cargo.12–14 In cellular systems, low toxicity and high uptake of MSNs have been well established in studies stemming from the past decade.15 The ordered pore structure accompanied with the consequential high surface area and pore volume provides the nanocarriers with a high loading capacity16 and offers efficient protection for the oligonucleotide cargo before reaching the point of release.17,18 Furthermore, MSNs may facilitate slow, sustained and controlled release19 owing to their design flexibility that enables multifunctionality as well as allows for sequential release.20 Consequently, a veritable explosion of research efforts has occurred in the area of siRNA delivery by MSNs.
Notably, one of the most significant development breakthroughs for MSN-based siRNA carriers in the past decade could be the successful transition in the particle–oligonucleotide interaction designs from adsorption on the particles’ outer surface21–24 to loading into the mesopores.18,19,25,26 Apparently, the latter exhibits great advantages in the protection of siRNA from nucleases, as well as the enhancement of loading capacity by the pore space. The surface functionalization with organic cationic linkers from small aminopropyl groups6 to large polycations, such as poly(ethyleneimine) (PEI),27 poly-L-lysine (PLL)26 and poly(2-dimethylaminoethylmethacrylate) (PDMAEMA),25 was generally involved to enhance the interaction between pore surfaces and siRNA molecules by shielding the influence from the negatively charged silanol groups. Meanwhile, remarkable progress in the cationic surface functionalization was made by a change from physical polycation adsorption28 to covalent grafting,26,27 in light of the benefits in increasing the siRNA-accessible cationic groups and reducing the potential toxicity of cationic polymers.29,30
Another clear trend in research efforts of employing large-pore (>10 nm) MSNs for accommodating polycations (molecular weight [MW] less than several kilodaltons) has emerged in the aforementioned studies, most likely related to the concerns in pore diameter reduction after pore surface grafting. However, it is questionable whether large pores can generate sufficient binding affinity for the negatively charged oligonucleotide, considering that siRNA molecules possess a rigid cylindrical-shaped conformation with a length of ~5 nm and a diameter of ~2.3 nm.18 A higher mobility for shorter 20-bp oligonucleotides inside large mesopores (7–9 nm) was previously demonstrated by the method of fluorescence recovery after photobleaching.31 Hence, a side-by-side adsorption of RNA molecules may be feasible by the large size of the entrance pores. Furthermore, it is hypothesized that proper smaller pore sizes would allow for a closer intimacy between the phosphate groups of siRNA and the cationic species on the pore surfaces via sufficient binding sites from geometrical and/or curvature fitting.32,33 In this case, the pore diameter is close to the hydrodynamic radius of siRNA, and a single-file diffusion is more likely to occur.
Inspired by the concept of geometrical matching of pore size and molecular dimensions of siRNA for pursuing efficient utilization of surface cations at the expense of lower toxicity, one of our previous studies has demonstrated that MSNs with pore sizes ranging from 3.5 to 5.0 nm and a high density of amine-terminated organic linkers (one linker per 3.3–3.7 nm2) can achieve high loading capacity of a 21-bp-sized model DNA oligos (190 mg·g−1) at low N/P ratios (the element ratios of nitrogen to phosphorus in the particles loaded with DNA oligos) ranging from 0.7 to 1.0 inside the functionalized mesopores.34 Moreover, cleavable linkers bearing an amino-group end and a labile disulfide bond in the spacer arm could provide a sustained switching of the affinity between the surface functions and short oligonucleotides inside the mesopores.34 However, as a proof-of-concept study, the system did not take into account the requirement of functional entities for a practical siRNA delivery. Moreover, the density of surface amino groups was limited on the pore surface, resulting from the co-condensation approach utilized for functionalization. Therefore, incorporation of certain polycationic modalities, such as PEI, into MSNs is needed to improve their transfection capability. In this regard, our previous studies using surface-hyperbranching polymerized PEI by a “grafting from” approach35,36 has demonstrated great potential of inducing efficient cell uptake and endosomal escape.12,37 Meanwhile, the risk of potentially associated toxicity can be well circumvented by the hyperbranched structure of less than three to four generations on the external surface and one generation on the pore surface of MSNs.38
In this report, we convey the benefits of the abovementioned amine-terminated organic linkers in constructing MSN-hyperbranched PEI hybrid nanoparticles for intracellular delivery of siRNA. Organic surface linkers bearing an amino-group end and a cleavable disulfide bond in the spacer arm were grafted onto MSNs with an average pore size of 4.9 nm via surface-hyperbranching polymerized PEI (Scheme 1). To further enhance the cell uptake and endosomal escape of the nanocarriers, hyperbranched PEI with a low MW of 1,300 Da was grafted on the exterior surface of the siRNA-loaded MSN-PEI-linker particles (Scheme 2). Facilitated by the underlying hyperbranched PEI and the efficient proximity of siRNA on the pore surface, a sustained cleavage of external PEI and internal linkers was realized, leading to an enhanced intracellular uptake, endosomal escape, as well as a slow, sustained (up to 96 h) and controlled release of functional siRNA in vitro. Consequently, we found it is crucial to incorporate such convertible physicochemical properties, particularly in response to intracellular stimuli, into MSN carriers in order to achieve desired oligonucleotide delivery to ultimately generate the desired biological and therapeutic effects.
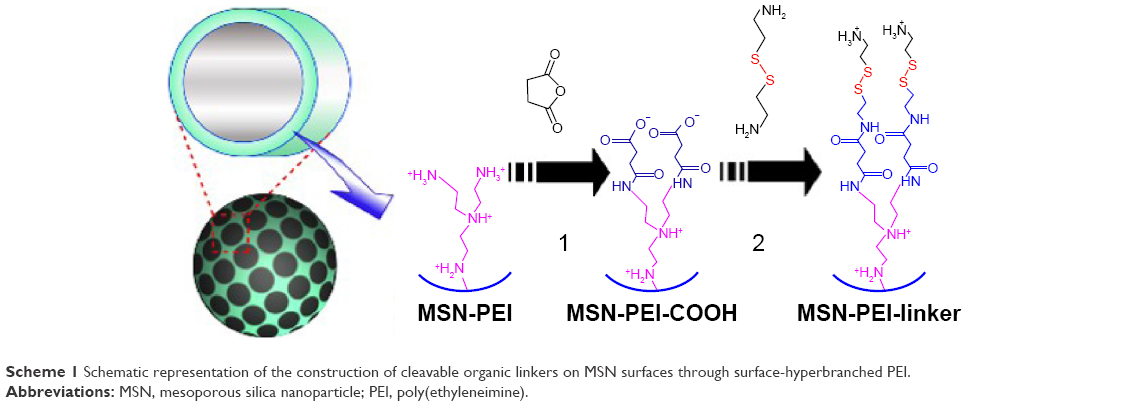 | Scheme 1 Schematic representation of the construction of cleavable organic linkers on MSN surfaces through surface-hyperbranched PEI. |
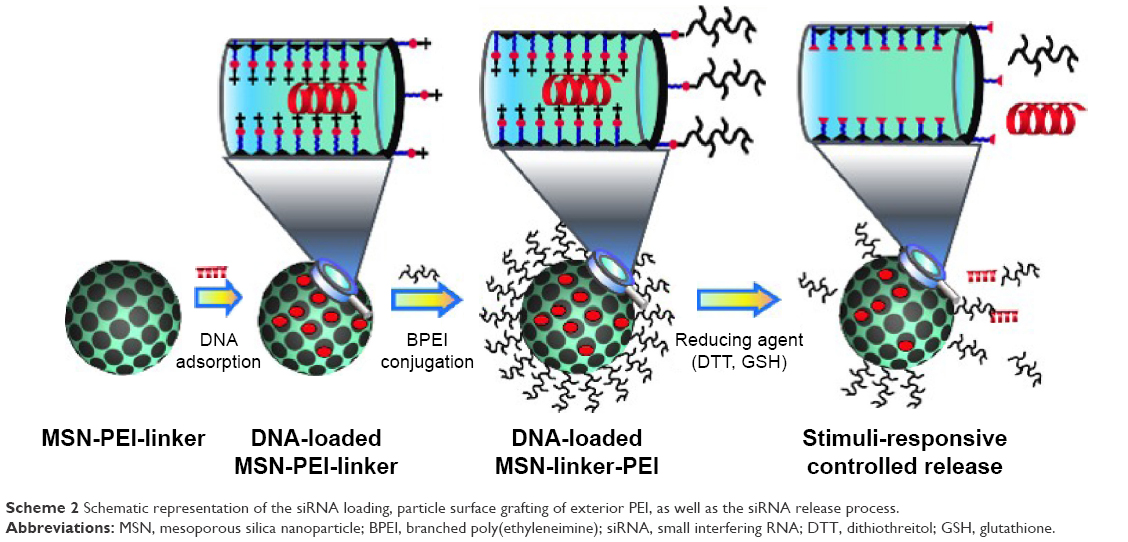 | Scheme 2 Schematic representation of the siRNA loading, particle surface grafting of exterior PEI, as well as the siRNA release process. |
Materials and methods
MSN nanocarrier synthesis and functionalization
PEI-modified MSN particles were synthesized according to a procedure previously reported by us.34 MSN particles bearing the cleavable surface linkers were prepared via succinylation of the surface amines followed by a conjugation step where disulfide-containing and amine group-terminating linkers (cystamine) were covalently coupled.34 In a typical process, 6 mg of the MSN-PEI particles were suspended in 6 mL of anhydrous ethanol and sonicated for 10 min to homogenize the suspension. Subsequently, 60 mg of succinic anhydride was added to the suspension, and the mixture was stirred at room temperature (RT) for 2 h. Finally, the carboxyl acid group-modified particles (MSN-COOH) were recovered from the suspension by centrifugation at 9,000 rpm for 15 min. The final product was washed with ethanol three times and redispersed in 2 mL MES buffer (20 mM, pH 5). Afterward, 30 μL of N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide (EDC) and 10 mg of N-hydroxysuccinimide (NHS) were added successively under sonication. After 15 min of stirring, 6 mL of cystamine dihydrochloride solution (6 mg·mL−1) was added dropwise to the suspension. The reaction was continued at RT overnight, and the product (MSN-PEI-linker) was retrieved by centrifugation followed by washing with ethanol. Material characterization was carried out according to the previous work by us.38
siRNA loading into MSN-PEI-linker particles was performed by soaking particles in a solution of siRNA in MES buffer (pH 5). For the determination of loading capacity, oligo DNA (5′-CTGTGGTTGTGTTTGCACTTT-3′ with 3′ TT-overhangs) with the same length of base pairs was used to carry out adsorption isotherm experiments. Typically, 1 mg of MSN-linker-Cys was suspended in 1 mL of MES with different initial concentrations of DNA (0.05–0.3 mg·mL−1). Subsequently, the mixture was homogenized by continuous vibration at RT for 6 h. The DNA-loaded MSN-PEI-linker particles were separated by centrifugation, and the supernatant was collected. The amount of siRNA/DNA adsorbed by MSN-PEI-linker was calculated through the concentration difference in the supernatant before and after the adsorption at a wavelength of 260 nm using a UV–Vis Spectrophotometer (NanoDrop 2000c; Wilmington, Thermo Scientific, USA).
To enhance the cell uptake and endosomal escape of the nanocarriers, hyperbranched PEI (BPEI) with an MW of 1,300 Da was post-encapsulated/conjugated on the exterior surface of the siRNA/oligo DNA-loaded MSN-PEI-linker particles. First, siRNA/oligo DNA-loaded MSN-PEI-linker particles were washed with ethanol three times and redispersed in 0.3 mL of ethanol. During the washing step, there was no release of siRNA/oligo DNA due to the high polarity of nucleic acids. Then, 18 μL of N, N′-disuccinimidyl carbonate (DSC) solution in DMF (4 mg·mL−1) was added under sonication, and the suspension was shaken for 20 min. Subsequently, 200 μL of PEI solution in ethanol (15 mg·mL−1) was added, and the suspension was mixed for 2 h. Finally, the product was retrieved by centrifugation and washed with ethanol two times.
For release studies, the oligo DNA-loaded particles were incubated at 37°C in 20 mM HEPES buffer (pH 7.4) or MES buffer (pH 5.0) at a concentration of 0.5 mg·mL−1. Particles were separated by centrifugation at different time periods for the determination of the released DNA in the media.
Cellular studies
Cell culture
MDA-MB-231 cells (human breast adenocarcinoma) were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 10% fetal bovine serum, 2 mM L-glutamine and 1% penicillin–streptomycin (v/v).
Cytotoxicity analysis
The cytotoxicity of MSN nanocarriers (without siRNA) was evaluated using WST-1 cell viability assay (Roche Diagnostics, Mannheim, Germany). A total of 10,000 MDA-MB-231 cells per well were grown in a 96-well plate in DMEM (10% fetal calf serum [FCS], 1% amino acids, 1% penicillin–streptomycin) and incubated overnight to adhere. Then, MSN nanocarriers were sonicated for 15 min and added to 1 mL of pre-warmed (37°C) growth medium at three different fivefold increasing concentrations of 2 μg, 10 μg and 50 μg. Then, the growth medium containing particles was further sonicated for 15 min. The growth media of cells in 96-well plates were replaced with media containing three different concentrations of MSN nanocarriers. A 5 μM of staurosporine (toxin) was added as a positive control, whereas negative control cells were untreated (pure cell media only). After incubating the particles for time points from 24 h to 72 h at 37°C, 5% CO2, 10 μL of WST-1 cell proliferation reagent (Roche Diagnostics) was added to the wells containing 100 μL of cell growth media, and the plate was again allowed to incubate for 3 h at 37°C, 5% CO2. After incubation period, the absorbance was read at 430 nm by Tecan Ultra microplate reader (MTX Lab Systems, Inc.). The number of viable cells was correlated with the observed absorbance from each individual sample.
Cellular uptake (by confocal microscopy)
Cellular uptake was evaluated by fixed cell confocal imaging at 24 h and 48 h time points: the microscopy setup consisted of Leica TCS SP5 STED (Leica Microsystems), LASAF software (Leica application suite), photo multiplier tube (PMT) and 100× oil objectives. The imaging was performed using sequential scanning option consisting of single-photon excitation for MSN nanocarriers chemically conjugated with fluorescein isothiocyanate (FITC), while multi-photon excitation was used for Hoechst (nuclear) dye. FITC-conjugated MSN nanocarriers were excited by argon laser at 488 nm, and emission from MSN nanocarriers was collected between 510 nm and 540 nm. Ti-sapphire femtosecond pulse excitation at 740 nm was used for Hoechst nuclear dye. The Hoechst emission was constantly collected in the blue wavelength range. MDA-MB-231 cells were grown as described earlier at 60%–70% confluent over coverslips. A 20 μg·mL−1 of MSN nanocarriers conjugated with FITC was prepared in 1 mL of cell growth media. The particles were sonicated and vortexed to allow proper de-aggregation of particles, if occurring. Then, the cell media with particles were added to cells growing over coverslips. After incubation for 24 h and 48 h, the medium was removed and cells were washed 1× with phosphate-buffered saline (PBS). Cells were then fixed with 4% paraformaldehyde (PFA) for 10 min at room temperature. After 10 min, cells were washed 3× with PBS. Hoechst nuclear dye was added to fixed cells along with mounting media to stain the nucleus.
Cellular uptake (by flow cytometry)
First, 40,000 MDA-MB-231 cells were grown in a 24-well plate for flow cytometry analysis. The quantification of 10 μg·mL−1 FITC-conjugated MSN nanocarriers uptake was performed with BD LSRII flow cytometer (BD Biosciences) using FL1-H channel (FITC compatible channel). The cellular uptake observed from MDA-MB-231 cells incubated with 10 μg·mL−1 MSN nanocarriers for 24 h and 48 h was analyzed with Flowing Software (Cell Imaging Core, BTK). MS excel software was used for the statistical analysis of the results.
Sustained siRNA release and intracellular co-localization
To monitor the intracellular trafficking and siRNA release, FITC-conjugated MSN nanocarriers were loaded with fluorescently labeled Alexa 555 siRNA (42 nM final siRNA concentration, BLOCK-iT™ Alexa Fluor® Red Fluorescent Control; Life Technologies). BLOCK-iT Alexa Fluor Red Fluorescent Control is used as a positive transfection control.39 The FITC-MSN nanocarrier and Alexa 555 siRNA complexes were incubated with MDA-MB-231 cells for live cell confocal imaging. The microscopy setup consisted of Leica TCS SP5 STED, LASAF software (Leica application suite), PMT and 63× water objectives. The cells were maintained at 37°C, 5% CO2, during the microscopy. FITC-conjugated MSN nanocarriers were excited by argon laser at 488 nm, and for Alexa 555, HeNe diode laser was used at 561 nm, and emissions were collected between 510–540 nm and 570–610 nm. Image analysis for co-localization was performed over 10 images per time point (N=10) with ImageJ, and co-localization of FITC/Alexa 555 analysis was performed with Volocity® 3D Image Analysis Software (PerkinElmer).40
siRNA transfection efficiency
The transfection efficiency of MSN nanocarriers was performed with different complementary techniques (live cell monitoring of colony growth, colony counting with crystal violet staining and WST-1 cell viability assay). Evaluation and optimization of siRNA delivery by MSN nanocarrier were performed with AllStars Hs Cell Death Control siRNA (Qiagen). AllStars Hs Cell Death Control siRNA is a blend of potent siRNAs targeting genes crucial for cell survival. The AllStars Hs Cell Death Control siRNA is used as a positive transfection.41–45 A total of 25,000 MDA-MB-231 cells were added to two 24-well plates; one 24-well plate each for long-term monitoring of colony growth by live cell imaging with IncuCyte ZOOM® (Essen Instruments, Ann Arbor, MI, USA).46,47 MSN nanocarriers were loaded with 10 wt% AllStars Hs Cell Death Control siRNA, and finally, 50 nM concentration was added to MSN nanocarrier samples. Equivalent (50 nM concentration) of AllStars Hs Cell Death Control siRNA was prepared with Lipofectamine® RNAiMAX (Invitrogen, Thermo Scientific, MA, USA) and added to respective samples as a positive control for transfection efficiency. A 5 μM concentration of staurosporine was added to positive control samples for cell death. Negative control samples were untreated. After 120 h, WST-1 was added to the first 24-well plate to quantify the surviving cell population, while the second 24-well plate was used for crystal violet staining. Cell medium was removed, washed 1× with PBS and fixed with 4% PFA for 10 min at RT. A 0.2% crystal violet solution in 10% EtOH was added to each well for 20 min to stain the cell colonies; 3× wash with PBS was used to remove unbound dye from cells. Finally, the 24-well plate was imaged and surviving cell colonies (N=4, four wells for each treatment) were counted by ImageJ plugins (Figures S1 and S2) “Cell Counter” and “ColonyArea”, which automatically quantify the colony growth area and intensity percentage.48
Results and discussion
MSN nanocarrier preparation and characterization
As a starting material, amino-functionalized MSNs (“MSN-NH2”) were prepared according to our previously published protocols.34 Scanning electron microscopy (SEM) and transmission electron microscopy (TEM) images of the MSN-NH2 particles are shown in Figure 1A and B. Uniform spherical particles were observed with an average diameter of 70 nm and radially aligned mesopores. Amino groups were modified onto the particle surface by a co-condensation approach to facilitate an efficient surface polymerization of PEI. TEM image of MSN-PEI particles stained with osmium (Figure 1C) clearly reveals the reduction in the contrast of the mesopores by the presence of a large amount of scattered black dots, implying hyperbranched polymers on both the exterior particle surface and the interior mesopore surface.
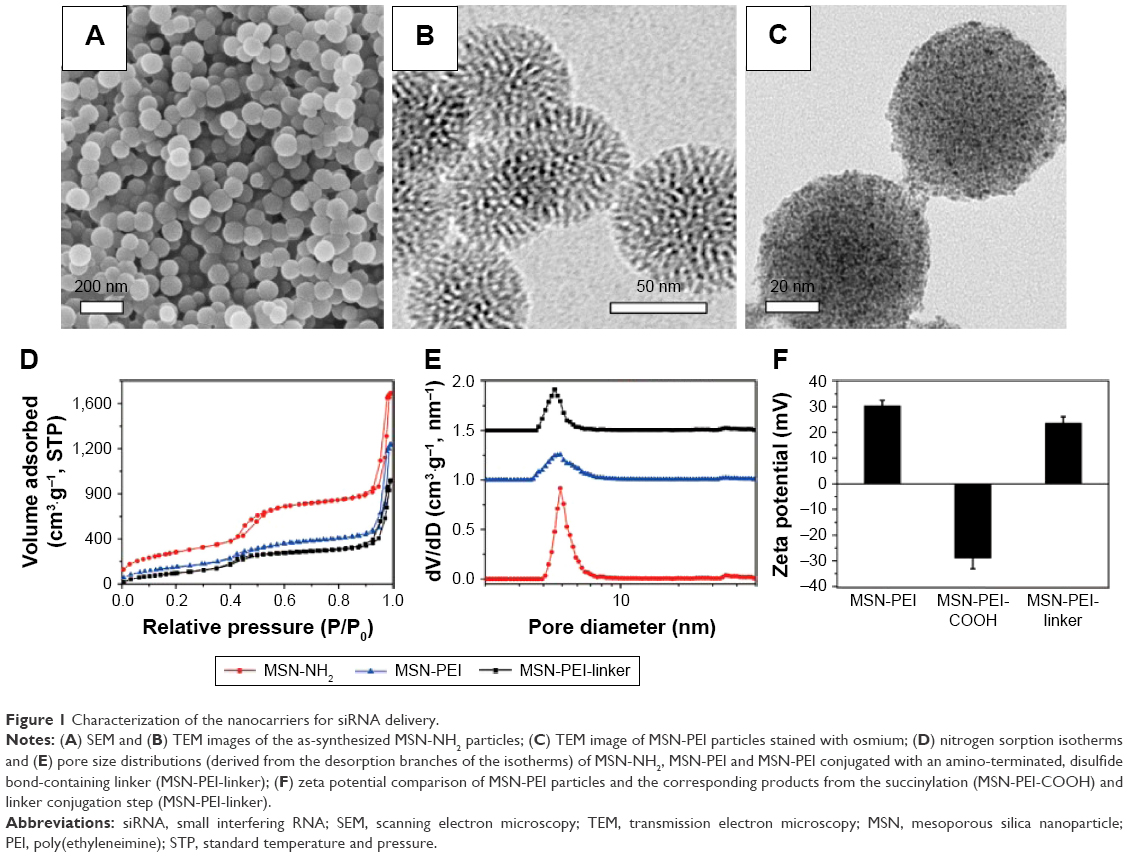 | Figure 1 Characterization of the nanocarriers for siRNA delivery. |
The construction of MSN particles bearing cleavable surface linkers was conceived on the basis of the abundant primary amines obtained in MSN-PEI (Supplementary materials). In order to confirm that the spatial dimension after these modifications still meets the requirements of siRNA loading, the textual properties of the nanocarriers were examined by nitrogen sorption. The typical nitrogen adsorption–desorption isotherms for MSN-NH2, MSN-PEI and MSN-PEI-linker confirmed the porous nature of the particles (Figure 1D). Compared with MSN-NH2, the isotherms of MSN-PEI and MSN-PEI-linker display an apparently decreased specific surface area (from 1,043 m2·g−1 to 566 m2·g−1 and 523 m2·g−1) and pore volume (from 0.85 cm3·g−1 to 0.58 cm3·g−1 and 0.46 cm3·g−1), implying a successful polymer and linker modification. The mesopore diameters remained almost the same after PEI polymerization (4.8 nm; Figure 1E) but decreased slightly after linker modification (4.5 nm; Figure 1E). The pore size of MSN-PEI-linker is identical to MSN-NH2 with the same linker modification that was used for loading oligonucleotides in our previous study.34 The mechanism behind the phenomenon is that the spatially restricted mesopore environment involves pronounced restrictions by the surface-hyperbranching polymerization method in solvent, and in turn, the hyperbranching inside the nanopores stops after one generation.25,36 This only contributes to the increment of aminoethyl group density on the pore surface (from 0.5 mmol·g−1 in the case of MSN-NH2 to 1.5 mmol·g−1 in the case of MSN-PEI),34 instead of protruding thick polymer layers that might cause a decrease in the pore size. Consequently, the surface-hyperbranching polymerization holds the biggest advantage for anchoring high-density amino groups inside small pores,36 as compared with previous “graft to” strategy where polycations can only be functionalized on large-pore MSNs.26,27
Organic surface linkers bearing amino-group ends and a cleavable disulfide bond in the spacer arm were thus functionalized onto the MSN surfaces by successive covalent modification steps. As shown in Figure 1F, MSN-PEI particles possess a zeta potential of +30 mV that remarkably decreased to −31 mV after succinylation, because of the derivatized carboxyl acid groups. However, after the conjugation of disulfide-containing cystamine molecules, the surface charge of the particles converted back to positive (+25 mV), resulting from the terminal amino groups. These amino groups on the surfaces are the main binding sites for siRNA, considering that siRNA adsorption in our system involved a direct electrostatic interaction between the cationic amino end groups and the oppositely charged phosphate groups on oligonucleotide molecules.
Using oligo DNA with a length of 21 bp (5′-CTGTGGTTGTGTTTGCACTTT-3′ with 3′ TT-overhangs) as a model before siRNA evaluations, the loading capacity of this oligo DNA in MSN-PEI was evaluated by measuring the adsorbed oligo DNA amount in MES buffer (pH 5). Figure 2A depicts the oligo DNA-loading comparison of MSN-PEI-linker material at different initial concentrations. The loading amount of oligo DNA in the particles reached two plateaus with loading amounts of 120 mg·g−1 and 280 mg·g−1, when the initial concentrations in the absorption solution were >0.15 mg·mL−1. Two similar plateaus (−16 mV, −30 mV) were also found in the zeta potential comparison of the particles from the loading solution, as shown in Figure 2B. The onset of the first plateau can be interpreted as the saturated adsorption of oligo DNA in the mesopores, according to our previous studies.34 However, the second plateau might be related with the strong complexing and packing ability of oligo DNA by the secondary and quaternary amines of hyperbranched PEI (three to four generations) located on the exterior surface of MSN-PEI-linker. Considering the stronger protection of siRNA from degradation in mesopores, a lower initial concentration of oligo DNA/siRNA (0.15 mg/mL) was used in the subsequent studies. To enhance the cell uptake and endosomal escape of the nanocarriers, hyperbranched PEI with a low MW of 1,300 kDa was grafted on the exterior surface of the oligo DNA-loaded MSN-PEI-linker particles. By increasing the employed amount of PEI 1,300 (0.3–4.8 mmol·g−1 with respect to the mass of the MSN-PEI-linker particles), the zeta potential value of the particles shifted from negative (−16 mV) to strongly positive (27 mV) as depicted in Figure 2C.
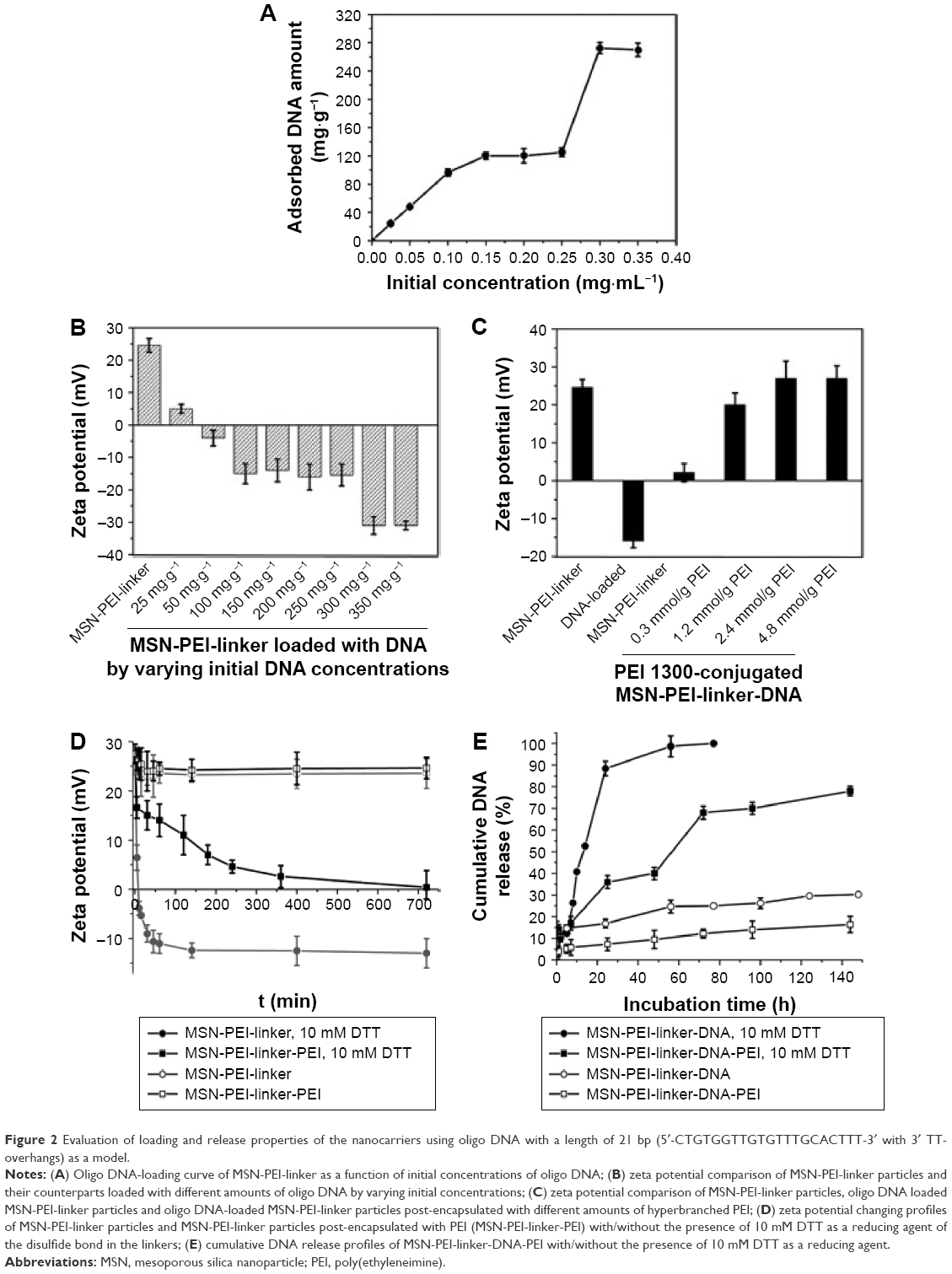 | Figure 2 Evaluation of loading and release properties of the nanocarriers using oligo DNA with a length of 21 bp (5′-CTGTGGTTGTGTTTGCACTTT-3′ with 3′ TT-overhangs) as a model. |
Zeta potential can be regarded as a physical indicator of charging on both the external particle surface and the internal pore surface with the uniform distribution of functional groups. To test the ability of the designed linkers to be cleaved from the surface of MSN-Linker-Cys particles in a redox-responsive manner, the measurements of the zeta potential as a function of time were carried out in the presence of a reducing agent (DTT, Dithiothreitol). As revealed in Figure 2D, the zeta potential was only slightly changed in the absence of the reducing agents for MSN-PEI-linker particles and MSN-PEI-linker particles post-grafted with PEI (MSN-PEI-linker-PEI), demonstrating high stability of the surface functions when particles were suspended in an aqueous solution. In the presence of 10 mM DTT, a rapid decrease of the zeta potential from +25 mV to −12 mV within 120 min was observed for MSN-PEI-linker. However, it took >720 min for MSN-PEI-linker-PEI to reach a reduced zeta potential approaching zero. This indicates an effect of the particle surface-grafted PEI on a more sustained breakage of the disulfide bond in the organic linkers. Once the surface linkers of the MSN-PEI-linker particles were cleaved by the reducing agent, the thiol group terminated surface functions repelled DNA and, consequently, resulted in DNA release.
Cumulative DNA release profiles in HEPES buffer (pH 7.2) were obtained and are displayed in Figure 2E. A slight burst release of DNA (5%) was observed for DNA-loaded MSN-PEI-linker-PEI particles, and slow cumulative release was found up to 144 h. Interestingly, the introduction of the reducing agent led to an appreciably faster and more efficient time-dependent release of oligo DNA over the whole release period. Over 15% of the loaded oligo DNA was released in the beginning in the presence of 10 mM DTT, and the release ratio went up to ~80% after 144 h of incubation. The amount of burst release was suppressed by adding a capping layer of PEI, as was also observed in our earlier study; albeit in which case, the capping layer consisted of PEG chains, which do not promote endosomal escape of cargo.34
Cytotoxicity evaluation of MSN nanocarriers
Cell studies were performed under standard conditions using MDA-MB-231 cells. MSNs have been previously shown to be biologically compatible with human cells in multiple studies.15,44,45 In polycation-based polymer nanocarriers, the molar ratio of amines in the polymer backbone to phosphates in the nucleic acid (N/P ratio) determines the maximum payload of cargo. A high N/P ratio of 4.5–135 may thus be associated with a risk of toxicity induced by the amines. In MSN-based delivery systems, it was shown that a low N/P ratio ranging from 0.7 to 1.0 inside the functionalized mesopores is sufficient to retain the siRNA molecules due to better bond coordination.34 Thus, we investigated the viability of cells transfected with MSNs functionalized with PEI-linker-PEI polymers over the time course.
Different concentrations of MSN nanocarriers (2–50 μg·mL−1) were incubated with MDA-MB-231 cells up to 72 h in the presence of a positive control (toxin: staurosporine) and negative controls. The cell viability of MSN nanocarrier-incubated cells remained comparative to negative cells, and MSN nanocarriers (MSN-PEI-linker-PEI) did not contribute to any significant toxicity.
Cell viability test was performed with a toxin, staurosporine, as a positive control.49 The cell viability compared to the toxin demonstrated the cytocompatability of MSN nanocarriers at different concentrations (2 μg·mL−1, 10 μg·mL−1 and 50 μg·mL−1) over 72 h (Figure 3). At lower nanocarrier concentrations (2 μg·mL−1 and 10 μg·mL−1), the viability was similar to that of negative control cells, while low or minimal cytotoxicity was observed with higher dose of nanocarriers (50 μg·mL−1). This confirms that the functionalized MSNs have low impact on cell viability by themselves. The higher concentration of nanocarriers is proportional to higher PEI dose, and PEI at higher doses has shown cellular toxicity (Figure S1).50,51 Consequently, for all the following cellular experiments, we used particle concentrations equal to 20 μg·mL−1 or lower.
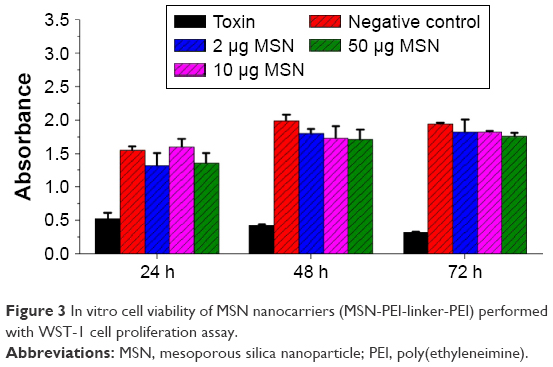 | Figure 3 In vitro cell viability of MSN nanocarriers (MSN-PEI-linker-PEI) performed with WST-1 cell proliferation assay. |
Cellular uptake of MSN nanocarriers
The cellular uptake of nanomaterials remains a primary concern and prerequisite for any efficient imaging, drug or gene delivery system. PEI functionalization has been used due to its positive charge as an effective transfection agent to deliver DNA/siRNA (Figure S2).52–54 To establish the uptake efficacy of our system, we used a concentration of 20 μg·mL−1 for MSN-PEI-linker-PEI nanocarriers labeled with FITC (without siRNA). MSN nanocarriers were added to MDA-MB-231 cells, and cellular uptake was evaluated after 24 h and 48 h of incubation using qualitative analysis (confocal microscopy). Flow cytometry was also used to quantify the relative uptake. Confocal microscopy revealed that most of the cells had endocytosed MSN nanocarriers (Figure 4A–E) after 24 h. The fluorescence signal from MDA-MB-231 cells also shows that no MSN nanocarriers were observed inside the nucleus. In general, MSN nanocarriers remained widely spread throughout the cytoplasm and some aggregation was also observed (Figure 4D–E) that might implicate their endosomal localization in the cells.
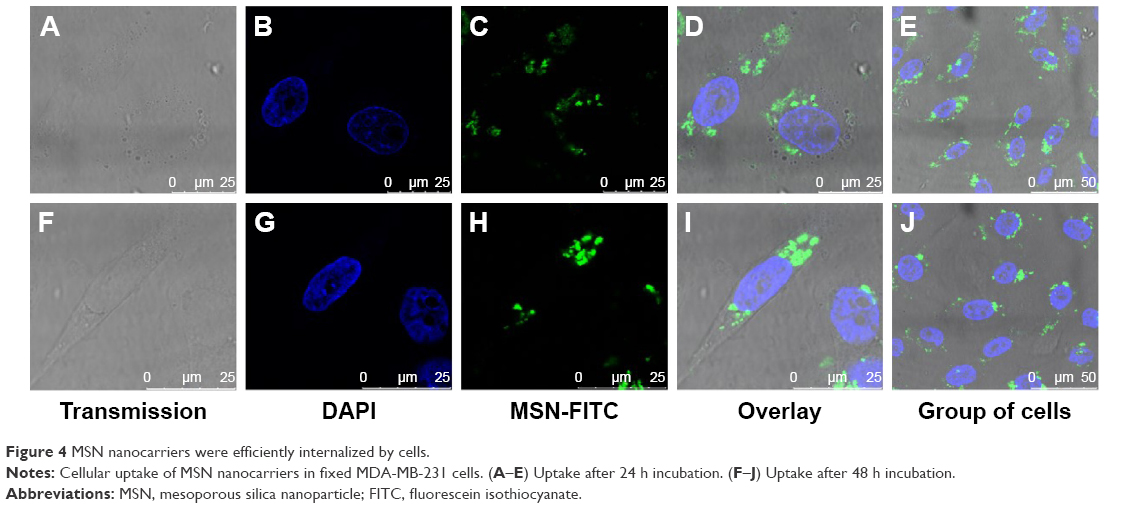 | Figure 4 MSN nanocarriers were efficiently internalized by cells. |
At 48 h incubation time, there was less overall distribution of MSN nanocarriers. At this time point, most MSN nanocarriers were aggregated or observed to be strongly confined in the cytoplasmic space (Figure 4F–J). This would suggest endosomal localization of the MSN nanocarriers prior to decomposing. There was no significant increase in cellular uptake after 48 h, as judged by the microscopical analysis (Figure 4). Taken together, in the MDA-MB-231 cells, the uptake of MSN nanocarriers was efficient and took place primarily during the first 24 h. Subsequently, the MSNs were clustered together in endosomal compartments.
Flow cytometry was performed to quantify the observed uptake utilizing a larger population of MDA-MB-231 cells and to compare the uptake efficiency with respect to time. The analysis was performed on a population of live cells (Figure 5A) with fluorescence-assisted cell sorting (FACS). The uptake of MSN nanocarriers is shown as a peak shift of the average fluorescence signal (Figure 5B–E). The cells incubated with MSNs can be clearly distinguished from the unlabeled control cells (Figure 5E). These results validate efficient cellular uptake of MSN nanocarriers after 24 h incubation and further show that almost all the cells had taken up MSN nanocarriers (24 h and 48 h). The PEI-induced positive charge has been reported previously as boosting cellular uptake. Therefore, the small MSN nanocarriers (~70 nm) along with additional high positive charge were capable of efficiently permeating across the cell membrane.
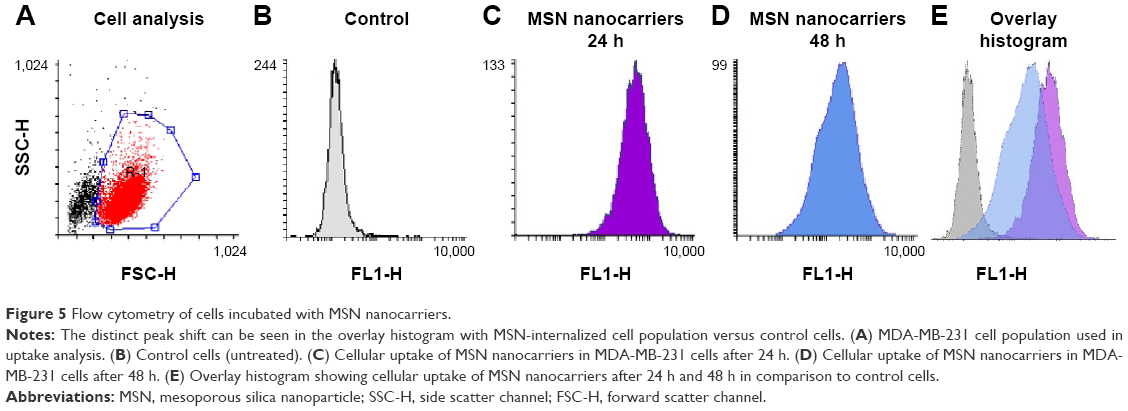 | Figure 5 Flow cytometry of cells incubated with MSN nanocarriers. |
Sustained siRNA release and endosomal escape
Parameters such as targeted delivery, long circulation time, slow sustained release and surviving the low-pH, acid-activated enzymes in the endosome are key limiting steps in intracellular siRNA delivery and need to be addressed to realize the full potential of RNAi in therapeutic applications.55–57 Understanding the events taking place after endocytosis of MSNs and before release of siRNAs to the cytoplasm and overcoming these barriers are vital for the performance of MSN nanocarriers58 and overall gene silencing efficiency.59–62 We chose to study the siRNA release from MSN nanocarriers and its endosomal release mechanism at an individual cell level using live cell confocal microscopy. We labeled MSN nanocarriers with FITC (green) and loaded them with fluorescent red-labeled (Alexa 555) siRNA as a positive control for transfection and tracking of release from MSN nanocarriers. Furthermore, quantitative co-localization of the fluorophore colors was performed (green and red). Live cell imaging started immediately after the addition of green-labeled MSN nanocarriers loaded with red-labeled siRNA to MDA-MB-231 cells. Cellular internalization of MSN nanocarriers was observed during the first 0–2 h of transfection, showing the MSN nanocarriers were rapidly internalized. We observed yellow fluorescent signals from the particles loaded with siRNA, both internalized as well as outside the cells (Figure 6A). This indicates that no significant level of premature release or leaching of siRNA was observed from MSN nanocarriers, neither intracellularly nor extracellularly. High co-localization was observed at 0–2 h (Figures 6A and 7), which also indicates that MSN nanocarriers were efficiently loaded with siRNA. The disulfide-containing cystamine linkers can be cleaved in a redox-responsive manner. The visualization of cells at 24 h in Figure 6B suggested that most of the loaded siRNA were largely retained within MSN nanocarriers. With the localization of the siRNA molecules within the mesopores, it is implied that the siRNA would be protected against the intracellular degradation mechanisms. Nearly all cells imaged during the 24 h time period displayed fluorescent yellow dots throughout the imaging period, with some occurrences showing separation of red pixels from the green ones (Figure 6B). However, these dots were characteristically different from co-localized yellow dots and remained few in number. The partial release was mostly observed during live cell imaging at 48 h (Figure 6C), yet most of the observed cells contained MSN nanocarrier–siRNA composites intact in the endosomes.
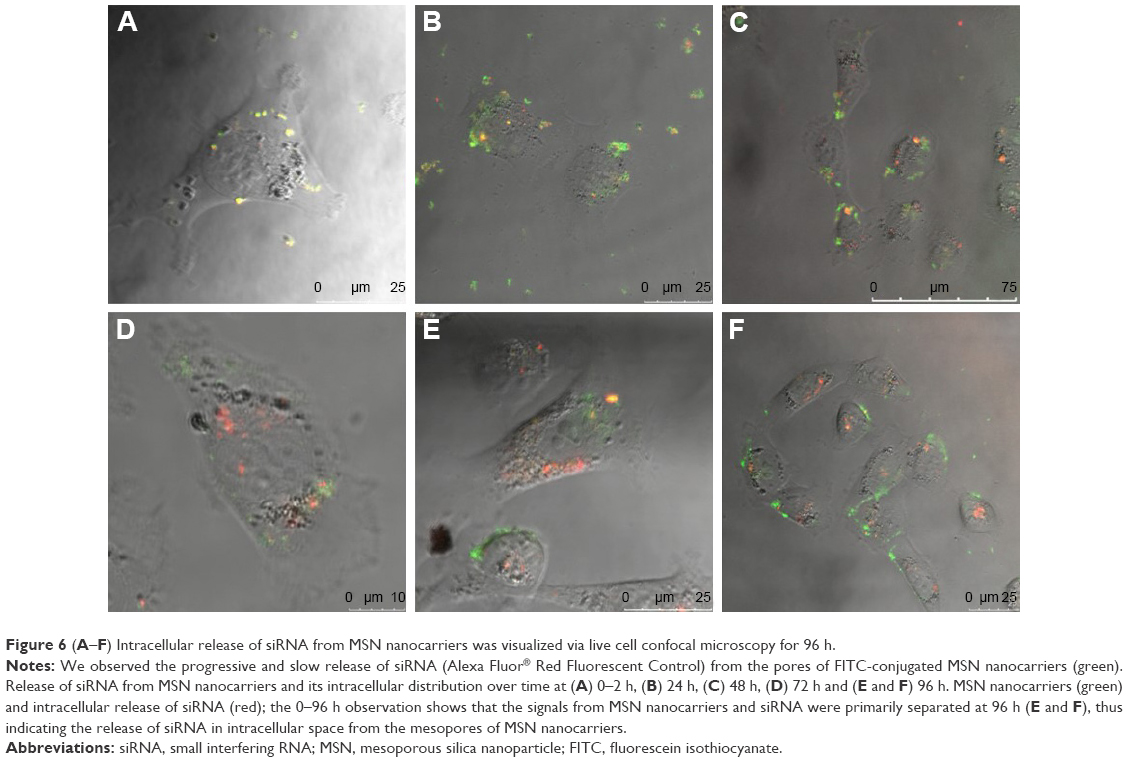 | Figure 6 (A–F) Intracellular release of siRNA from MSN nanocarriers was visualized via live cell confocal microscopy for 96 h. |
The co-localization of MSN nanocarriers and siRNA at 24 h suggests that internalized particles had not yet released their cargo, which was also consistent with our earlier result (Figure 2E). The degree of co-localization was determined to be 0.25 for 48 h live cell imaging (Figure 7), which indicates release of fluorescently labeled siRNA from the MSN nanocarriers. We observed the progressive increase in distinct red dots (Alexa 555 siRNA) and diffuse red staining in the cytoplasm, and furthermore, we observed a significant number of intense green dots (MSN nanocarriers) at separate locations inside the cells (Figures 6 and 7). Both of these observations can be interpreted as partial release of siRNA from MSN nanocarriers. A majority of the MSN nanocarriers in cells can be seen entrapped in endosome-like structures after release of the cargo (Figure 6C–F). However, the presence of co-localized MSN nanocarriers and siRNA in the cells, despite slow release, persisted until the end of the observation at 72 h live cell imaging, suggesting that all the siRNA content had not been released at this time point. This implies that long-term sustained release and protection of loaded siRNA can be achieved by MSN nanocarriers at least up to 72 h after transfection.
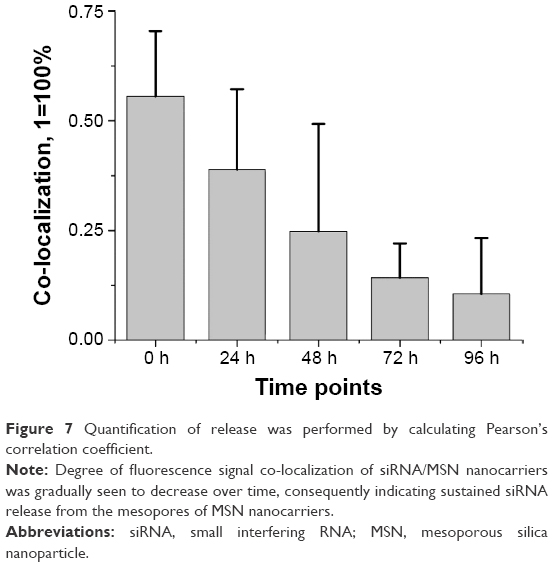 | Figure 7 Quantification of release was performed by calculating Pearson’s correlation coefficient. |
Moreover, at 96 h (Figure 6E–F), it was apparent that all cells or all MSNs do not exhibit similar synchronized release of siRNA visualized by localization of red fluorescence. Some cells exhibit more efficient release, while in other cells, less release of siRNA was observed. Neither do all MSNs within the same cell release their cargo at any specific time point. We observed the red staining to be more diffuse as compared to green, suggesting siRNA localization in cytoplasm or nucleus after the release (Figure 6C–F). This observation can be correlated with our finding with the release behavior of MSN nanocarriers in HEPES buffer (pH 7.2) (Figure 2E) and suggested that the MSN nanocarriers were capable of slow and sustained release of siRNA inside cells.
Functional assay for the delivery of biologically active siRNA by the constructed MSNs
We next evaluated the RNAi in a functional experiment using a cell-killing siRNA (AllStars Hs Cell Death Control siRNA). As investigated earlier, the co-localization analysis suggested that the optimal release of siRNA from MSN nanocarriers can be achieved during 72–96 h. Therefore, an ideal timeframe for achieving efficient gene knockdown would be 100–120 h, or even longer. The employed cell-killing siRNA has been used previously as a positive control for transfection.41–45 The aim of the experiment was to investigate transfection efficiency of MSN nanocarriers in MDA-MB-231 cells; therefore, we used a very general siRNA transfection approach rather than selecting some specific cellular target. The transfection efficacy was benchmarked against a commercial transfection agent, Lipofectamine RNAiMax, as a positive (cell death) control. It is a widely used in vitro transfection agent, and it has excellent transfection efficiency at in vitro level. The cells were transfected with 50 nM of cell-killing siRNA with both Lipofectamine and MSN nanocarriers, as well as 5 μM concentration of staurosporine used as toxin. The results from both different experimental analyses were used for MSN nanocarriers RNAi efficiency evaluation. We conducted a 120 h live cell imaging experiment to establish the time-dependent transfection efficacy (Figure 8A). The cells treated with staurosporine were dead shortly after addition of the toxin, while non-treated negative control cells reached almost 100% confluence (Figure 8A–C). Transfection with MSN nanocarriers and Lipofectamine, both loaded with 50 nM cell-killing siRNA, demonstrated a similar time-dependent reduction in cell viability (Figure 8A). Furthermore, we conducted two end point cell viability assays at 120 h; crystal violet staining (Figure 8B) and WST-1 assay (Figure 8C). In the WST-1 assay, the MSN nanocarriers and Lipofectamine control had nearly equal efficacies, and in the crystal violet staining, the difference was nonsignificant. This could establish the versatility of MSN nanocarriers toward a general in vivo transfection agent, which can be further applied with a broad spectrum of other types of siRNAs. The experiments were designed to provide both qualitative and quantitative transfection efficiency of MSN nanocarriers. Therefore, we included different sets of experiments (live cell growth monitoring, WST-1 cell viability assay and crystal violet staining for direct visualization of colony growth comparison) to properly investigate the knockdown efficiency (Figure 8).
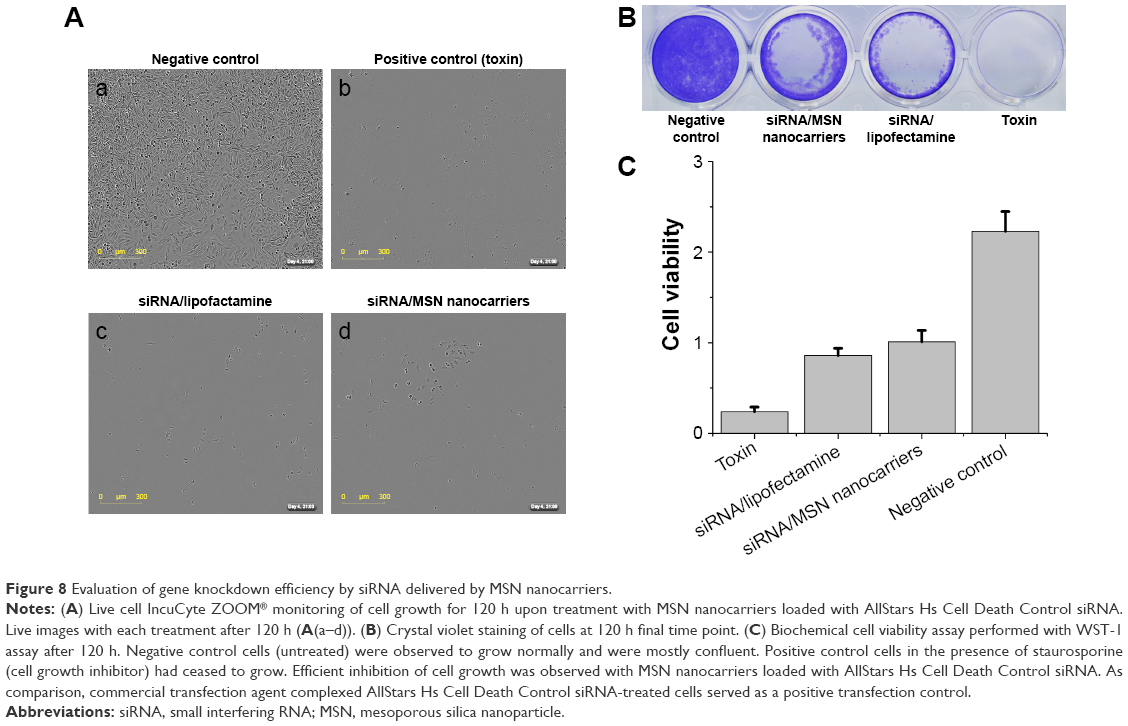 | Figure 8 Evaluation of gene knockdown efficiency by siRNA delivered by MSN nanocarriers. |
Taken together, the live cell imaging, WST-1 and crystal violet staining results demonstrated that the MSN nanocarriers were efficient delivery vehicles for RNAi (Figure 8A–C). The measurements by different techniques provide a broader understanding of the knockdown efficiency of MSN nanocarriers. However, intensity percent calculations were performed with the same crystal violet data, showing that differences in the intensity of MSN nanocarrier-treated cells were remarkably close to the commercial transfection agent (Figures S3 and S4). Consequently, based on these results, obtained with all different techniques, the gene knockdown trend remained the same under the in vitro conditions investigated. The next step for the MSN system is in vivo delivery, which is not applicable for Lipofectamine and other in vitro transfection agents. All in all, it can be summarized that the developed MSN nanocarriers were efficient for cellular uptake, non-toxic, provided a slow sustained intracellular release and an overall promising knockdown efficiency, all of which will prospectively enable more efficient in vivo gene silencing for improved RNAi therapy.
Conclusion
By integrating the advantage of MSN and hyperbranched PEI in siRNA delivery, a novel organic–inorganic hybrid nanocarrier system with a sub-100 nm diameter was successfully developed for RNAi. Facilitated by surface-hyperbranching polymerization, the surfaces of MSNs were grafted with a high density of amino groups, which resulted in an appropriate pore size of 4.5 nm, as well as easier construction of cleavable organic linkers for the payload of siRNA molecules with high affinity (120 mg·g−1) in the mesopore space. Subsequent conjugation of low MW PEI on the exterior surface as a capping layer led to more stable encapsulation of siRNA, as well as enhanced intracellular uptake and endosomal escape of siRNA-loaded particles. A slow, sustained (up to 96 h) and controlled release of functional siRNA was revealed intracellularly by tracking the co-localization of the nanocarriers and the cargo in live cells. Promising long-term gene knockdown efficiency was demonstrated by intracellular delivery of cell-killing siRNA to MDA-MB-231 cells. Given the efficient retention of siRNA within the protective environment provided by the mesoporous space at extracellular conditions, followed by endosomal escape and a sustained intracellularly triggered release of siRNA, the developed system incorporates the desired properties suitable for in vivo siRNA delivery. In summary, this study may bring new understanding and insights of host–guest interactions into the application of porous nanocarriers for long-term siRNA delivery.
Acknowledgments
The authors would like to acknowledge the technical assistance provided by Taina Kalevo-Mattila, Dr Anni Laine and Amanpreet Kaur from Cancer Cell Signalling group from Turku Centre of Biotechnology, University of Turku, and Åbo Akademi University, Finland. The authors are grateful for IncuCyte Imaging assistance by Tiina Kähkönen and Dr Yu Lan from Cell Biology and Anatomy, Faculty of Medicine, University of Turku. Dr Takahiro Deguchi, Laboratory of Biophysics, University of Turku, is acknowledged for data analysis of the co-localization experiment. The financial contributions from Jane and Aatos Erkko Foundation (DD, EC, JW, JMR), Sigrid Juselius Foundation (JW), Foundation for Finnish Cancer Institute (JW), Academy of Finland (projects #260599, 284542) (JMR), Suomen Kulttuurirahasto (DD), Forskarskolan vid Åbo Akademi (TGS), National Natural Science Foundation of China (NSFC, Grant No 51502027), Basic Advanced Research Project of Chongqing 572 (Grant No cstc2015jcyjA10051) (JZ) and the Doctoral Education Network in Materials Research at Åbo Akademi University (NP) are greatly acknowledged. EC thanks Agency for Management of University and Research Grants from Generalitat de Catalunya (AGAUR) for financial support through Beatriu de Pinos program (2014 BP_A 00132).
Author contributions
NP designed and performed the cellular studies and wrote the manuscript. JZ designed, synthesized and characterized the initial MSN nanocarriers and contributed in manuscript writing. DD, EC and TGS prepared MSN nanocarriers and performed siRNA loading at different stages of the experiments. TN helped with technical details with cellular studies and experimental design. JW and JMR conceived, designed and coordinated the study and assisted in manuscript writing. All authors read, commented and approved the final manuscript. All authors contributed toward data analysis, drafting and critically revising the paper and agree to be accountable for all aspects of the work.
Disclosure
The author reports no conflicts of interest in this work.
References
Fire A, Xu S, Montgomery MK, Kostas SA, Driver SE, Mello CC. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature. 1998;391(6669):806–811. | ||
Wu SY, Lopez-Berestein G, Calin GA, Sood AK. RNAi therapies: drugging the undruggable. Sci Transl Med. 2014;6(240):240ps7. | ||
Tabernero J, Shapiro GI, LoRusso PM, et al. First-in-humans trial of an RNA interference therapeutic targeting VEGF and KSP in cancer patients with liver involvement. Cancer Discov. 2013;3(4):406–417. | ||
Shih Y-M, Sun C-P, Chou H-H, et al. Combinatorial RNA interference therapy prevents selection of pre-existing HBV variants in human liver chimeric mice. Sci Rep. 2015;5:15259. | ||
Xu X, Ho W, Zhang X, Bertrand N, Farokhzad O. Cancer nanomedicine: from targeted delivery to combination therapy. Trends Mol Med. 2015;21(4):223–232. | ||
Schiffelers RM, Ansari A, Xu J, et al. Cancer siRNA therapy by tumor selective delivery with ligand-targeted sterically stabilized nanoparticle. Nucleic Acids Res. 2004;32(19):e149. | ||
Miele E, Spinelli GP, Miele E, et al. Nanoparticle-based delivery of small interfering RNA: challenges for cancer therapy. Int J Nanomedicine. 2012;7:3637–3657. | ||
Guo S, Huang L, Guo S, Huang L. Nanoparticles escaping RES and endosome: challenges for siRNA delivery for cancer therapy, nanoparticles escaping RES and endosome: challenges for siRNA delivery for cancer therapy. J Nanomater. 2011;2011:12. | ||
Whitehead KA, Langer R, Anderson DG. Knocking down barriers: advances in siRNA delivery. Nat Rev Drug Discov. 2009;8(2):129–138. | ||
Takahashi Y, Nishikawa M, Takakura Y. Nonviral vector-mediated RNA interference: its gene silencing characteristics and important factors to achieve RNAi-based gene therapy. Adv Drug Deliv Rev. 2009;61(9):760–766. | ||
Grimm D. Small silencing RNAs: state-of-the-art. Adv Drug Deliv Rev. 2009;61(9):672–703. | ||
Trewyn BG, Slowing II, Giri S, Chen H-T, Lin VS-Y. Synthesis and functionalization of a mesoporous silica nanoparticle based on the sol–gel process and applications in controlled release. Acc Chem Res. 2007;40(9):846–853. | ||
Argyo C, Weiss V, Bräuchle C, Bein T. Multifunctional mesoporous silica nanoparticles as a universal platform for drug delivery. Chem Mater. 2014;26(1):435–451. | ||
Tang F, Li L, Chen D. Mesoporous silica nanoparticles: synthesis, biocompatibility and drug delivery. Adv Mater. 2012;24(12):1504–1534. | ||
Rosenholm JM, Sahlgren C, Lindén M. Towards multifunctional, targeted drug delivery systems using mesoporous silica nanoparticles – opportunities & challenges. Nanoscale. 2010;2(10):1870–1883. | ||
Hong S, Shen S, Tan DC, et al. High drug load, stable, manufacturable and bioavailable fenofibrate formulations in mesoporous silica: a comparison of spray drying versus solvent impregnation methods. Drug Deliv. 2016;23(1):316–327. | ||
Chen T, Yang N, Fu J. Controlled release of cargo molecules from hollow mesoporous silica nanoparticles based on acid and base dual-responsive cucurbit[7]uril pseudorotaxanes. Chem Commun. 2013;49(58):6555–6557. | ||
Li X, Xie QR, Zhang J, Xia W, Gu H. The packaging of siRNA within the mesoporous structure of silica nanoparticles. Biomaterials. 2011;32(35):9546–9556. | ||
Na H-K, Kim M-H, Park K, et al. Efficient functional delivery of siRNA using mesoporous silica nanoparticles with ultralarge pores. Small. 2012;8(11):1752–1761. | ||
Aznar E, Oroval M, Pascual L, Murguía JR, Martínez-Máñez R, Sancenón F. Gated materials for on-command release of guest molecules. Chem Rev. 2016;116(2):561–718. | ||
Radu DR, Lai C-Y, Jeftinija K, Rowe EW, Jeftinija S, Lin VS-Y. A polyamidoamine dendrimer-capped mesoporous silica nanosphere-based gene transfection reagent. J Am Chem Soc. 2004;126(41):13216–13217. | ||
Hom C, Lu J, Liong M, et al. Mesoporous silica nanoparticles facilitate delivery of siRNA to shutdown signaling pathways in mammalian cells. Small. 2010;6(11):1185–1190. | ||
Xia T, Kovochich M, Liong M, et al. Polyethyleneimine coating enhances the cellular uptake of mesoporous silica nanoparticles and allows safe delivery of siRNA and DNA constructs. ACS Nano. 2009;3(10):3273–3286. | ||
Finlay J, Roberts CM, Dong J, Zink JI, Tamanoi F, Glackin CA. Mesoporous silica nanoparticle delivery of chemically modified siRNA against TWIST1 leads to reduced tumor burden. Nanomedicine. 2015;11(7):1657–1666. | ||
Lin D, Cheng Q, Jiang Q, et al. Intracellular cleavable poly(2-dimethylaminoethyl methacrylate) functionalized mesoporous silica nanoparticles for efficient siRNA delivery in vitro and in vivo. Nanoscale. 2013;5(10):4291–4301. | ||
Hartono SB, Gu W, Kleitz F, et al. Poly-l-lysine functionalized large pore cubic mesostructured silica nanoparticles as biocompatible carriers for gene delivery. ACS Nano. 2012;6(3):2104–2117. | ||
Hartono SB, Yu M, Gu W, et al. Synthesis of multi-functional large pore mesoporous silica nanoparticles as gene carriers. Nanotechnology. 2014;25(5):55701. | ||
Li X, Chen Y, Wang M, Ma Y, Xia W, Gu H. A mesoporous silica nanoparticle – PEI – Fusogenic peptide system for siRNA delivery in cancer therapy. Biomaterials. 2013;34(4):1391–1401. | ||
Lee S-Y, Huh MS, Lee S, et al. Stability and cellular uptake of polymerized siRNA (poly-siRNA)/polyethylenimine (PEI) complexes for efficient gene silencing. J Control Release. 2010;141(3):339–346. | ||
Patil ML, Zhang M, Betigeri S, Taratula O, He H, Minko T. Surface-modified and internally cationic polyamidoamine dendrimers for efficient siRNA delivery. Bioconjug Chem. 2008;19(7):1396–1403. | ||
Lebold T, Schlossbauer A, Schneider K, et al. Controlling the mobility of oligonucleotides in the nanochannels of mesoporous silica. Adv Funct Mater. 2012;22(1):106–112. | ||
Li X, Zhang J, Gu H. Study on the adsorption mechanism of DNA with mesoporous silica nanoparticles in aqueous solution. Langmuir. 2012;28(5):2827–2834. | ||
Möller K, Müller K, Engelke H, Bräuchle C, Wagner E, Bein T. Highly efficient siRNA delivery from core–shell mesoporous silica nanoparticles with multifunctional polymer caps. Nanoscale. 2016;8(7):4007–4019. | ||
Zhang J, Niemelä M, Westermarck J, Rosenholm JM. Mesoporous silica nanoparticles with redox-responsive surface linkers for charge-reversible loading and release of short oligonucleotides. Dalton Trans. 2014;43(10):4115–4126. | ||
Rosenholm JM, Penninkangas A, Lindén M. Amino-functionalization of large-pore mesoscopically ordered silica by a one-step hyperbranching polymerization of a surface-grown polyethyleneimine. Chem Commun. 2006;(37):3909–3911. | ||
Rosenholm JM, Lindén M. Wet-chemical analysis of surface concentration of accessible groups on different amino-functionalized mesoporous SBA-15 silicas. Chem Mater. 2007;19(20):5023–5034. | ||
Rosenholm JM, Peuhu E, Eriksson JE, Sahlgren C, Lindén M. Targeted intracellular delivery of hydrophobic agents using mesoporous hybrid silica nanoparticles as carrier systems. Nano Lett. 2009;9(9):3308–3311. | ||
Zhang J, Desai D, Rosenholm JM. Tethered lipid bilayer gates: toward extended retention of hydrophilic cargo in porous nanocarriers. Adv Funct Mater. 2014;24(16):2352–2360. | ||
Yonekawa T, Yang S, Counter CM. PinX1 localizes to telomeres and stabilizes TRF1 at mitosis. Mol Cell Biol. 2012;32(8):1387–1395. | ||
Dunn KW, Kamocka MM, McDonald JH. A practical guide to evaluating colocalization in biological microscopy. Am J Physiol Cell Physiol. 2011;300(4):C723–C742. | ||
Lalevée S, Lapaire O, Bühler M. miR455 is linked to hypoxia signaling and is deregulated in preeclampsia. Cell Death Dis. 2014;5(9):e1408. | ||
Harradine KA, Kassner M, Chow D, et al. Functional genomics reveals diverse cellular processes that modulate tumor cell response to oxaliplatin. Mol Cancer Res. 2011;9(2):173–182. | ||
Kitchens CA, McDonald PR, Shun TY, Pollack IF, Lazo JS. Identification of chemosensitivity nodes for vinblastine through small interfering RNA high-throughput screens. J Pharmacol Exp Ther. 2011;339(3):851–858. | ||
Kukkonen-Macchi A, Sicora O, Kaczynska K, et al. Loss of p38γ MAPK induces pleiotropic mitotic defects and massive cell death. J Cell Sci. 2011;124(2):216–227. | ||
Ovaska K, Laakso M, Haapa-Paananen S, et al. Large-scale data integration framework provides a comprehensive view on glioblastoma multiforme. Genome Med. 2010;2(9):65. | ||
McCloy RA, Rogers S, Caldon CE, Lorca T, Castro A, Burgess A. Partial inhibition of Cdk1 in G2 phase overrides the SAC and decouples mitotic events. Cell Cycle. 2014;13(9):1400–1412. | ||
Wang J, Yang J, Gu P, Klassen H. Effects of glial cell line-derived neurotrophic factor on cultured murine retinal progenitor cells. Mol Vis. 2010;16:2850–2866. | ||
Guzmán C, Bagga M, Kaur A, Westermarck J, Abankwa D. ColonyArea: an ImageJ plugin to automatically quantify colony formation in clonogenic assays. PLoS One. 2014;9(3):e92444. | ||
Antonsson A, Persson JL. Induction of apoptosis by staurosporine involves the inhibition of expression of the major cell cycle proteins at the G2/M checkpoint accompanied by alterations in Erk and Akt kinase activities. Anticancer Res. 2009;29(8):2893–2898. | ||
Breunig M, Lungwitz U, Liebl R, Goepferich A. Breaking up the correlation between efficacy and toxicity for nonviral gene delivery. Proc Natl Acad Sci. 2007;104(36):14454–14459. | ||
Florea BI, Meaney C, Junginger HE, Borchard G. Transfection efficiency and toxicity of polyethylenimine in differentiated Calu-3 and nondifferentiated COS-1 cell cultures. AAPS PharmSci. 2002;4(3):E12. | ||
Godbey WT, Wu KK, Mikos AG. Poly(ethylenimine) and its role in gene delivery. J Control Release. 1999;60(2–3):149–160. | ||
Godbey WT, Wu KK, Mikos AG. Tracking the intracellular path of poly(ethylenimine)/DNA complexes for gene delivery. Proc Natl Acad Sci U S A. 1999;96(9):5177–5181. | ||
Lungwitz U, Breunig M, Blunk T, Göpferich A. Polyethylenimine-based non-viral gene delivery systems. Eur J Pharm Biopharm. 2005;60(2):247–266. | ||
Aagaard L, Rossi JJ. RNAi therapeutics: principles, prospects and challenges. Adv Drug Deliv Rev. 2007;59(2–3):75–86. | ||
Akinc A, Querbes W, De S, et al. Targeted delivery of RNAi therapeutics with endogenous and exogenous ligand-based mechanisms. Mol Ther. 2010;18(7):1357–1364. | ||
Bumcrot D, Manoharan M, Koteliansky V, Sah DWY. RNAi therapeutics: a potential new class of pharmaceutical drugs. Nat Chem Biol. 2006;2(12):711–719. | ||
Sakurai Y, Hatakeyama H, Sato Y, et al. Endosomal escape and the knockdown efficiency of liposomal-siRNA by the fusogenic peptide shGALA. Biomaterials. 2011;32(24):5733–5742. | ||
Urban-Klein B, Werth S, Abuharbeid S, Czubayko F, Aigner A. RNAi-mediated gene-targeting through systemic application of polyethylenimine (PEI)-complexed siRNA in vivo. Gene Ther. 2005;12(5):461–466. | ||
Nguyen J, Szoka FC. Nucleic acid delivery: the missing pieces of the puzzle? Acc Chem Res. 2012;45(7):1153–1162. | ||
Kanasty R, Dorkin JR, Vegas A, Anderson D. Delivery materials for siRNA therapeutics. Nat Mater. 2013;12(11):967–977. | ||
Dominska M, Dykxhoorn DM. Breaking down the barriers: siRNA delivery and endosome escape. J Cell Sci. 2010;123(8):1183–1189. |
Supplementary materials
Calculation of amine group amount and nitrogen to phosphorus (N/P) ratio
The total amount of accessible primary amines on MSN-PEI was 1.467 mmol·g−1 as determined by a ninhydrin test.1 The loading amount of oligo DNA in the particles reached 120 mg·g−1 (0.0086 mmol·g−1) containing 0.36 mmol·g−1 phosphate groups of DNA. The oligonucleotides were loaded primarily in the mesopores by interaction with the amine groups. Consequently, the N/P ratio was calculated to be ~4.1.
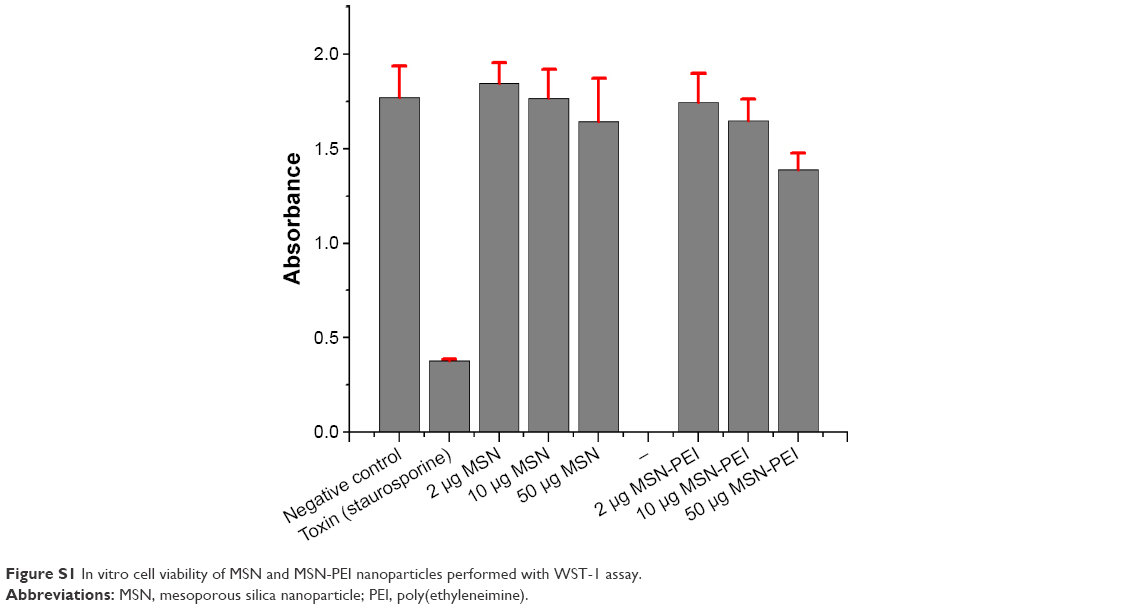 | Figure S1 In vitro cell viability of MSN and MSN-PEI nanoparticles performed with WST-1 assay. |
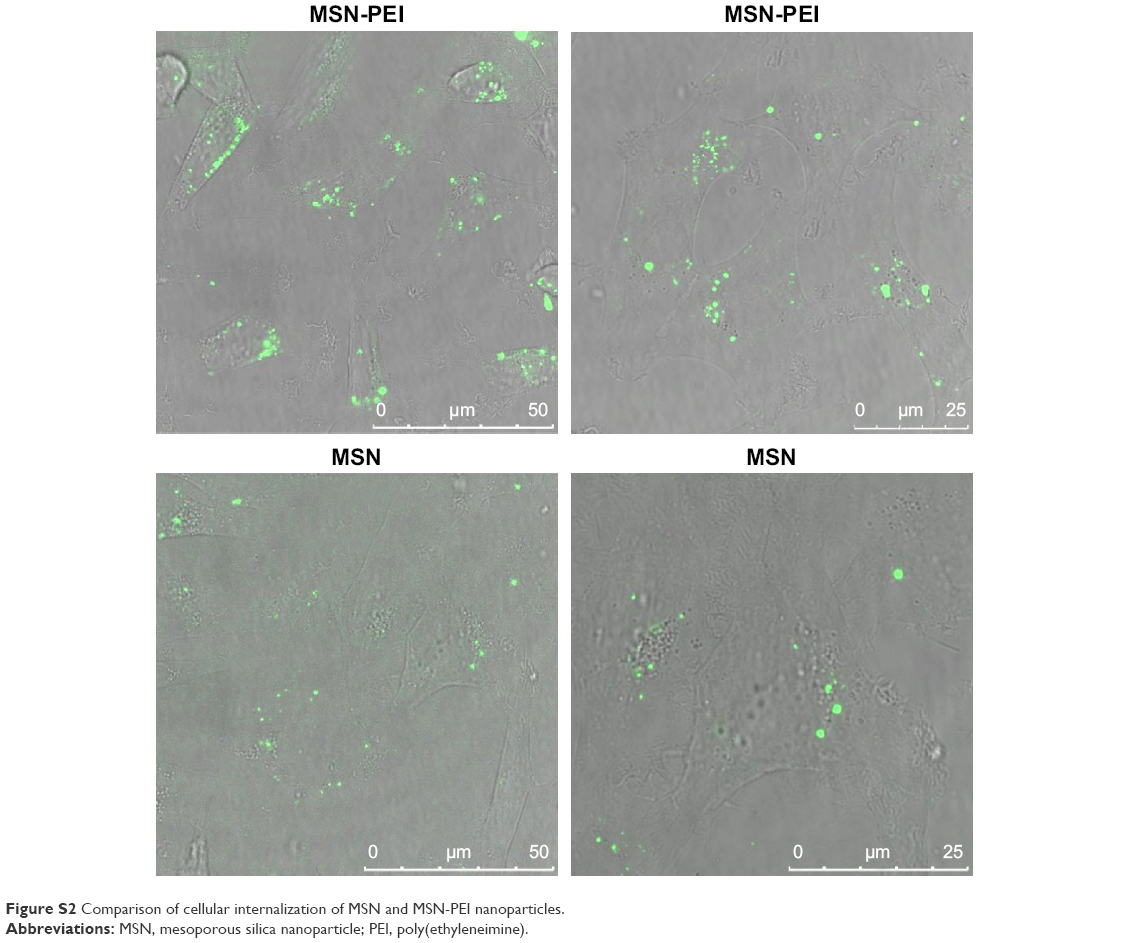 | Figure S2 Comparison of cellular internalization of MSN and MSN-PEI nanoparticles. |
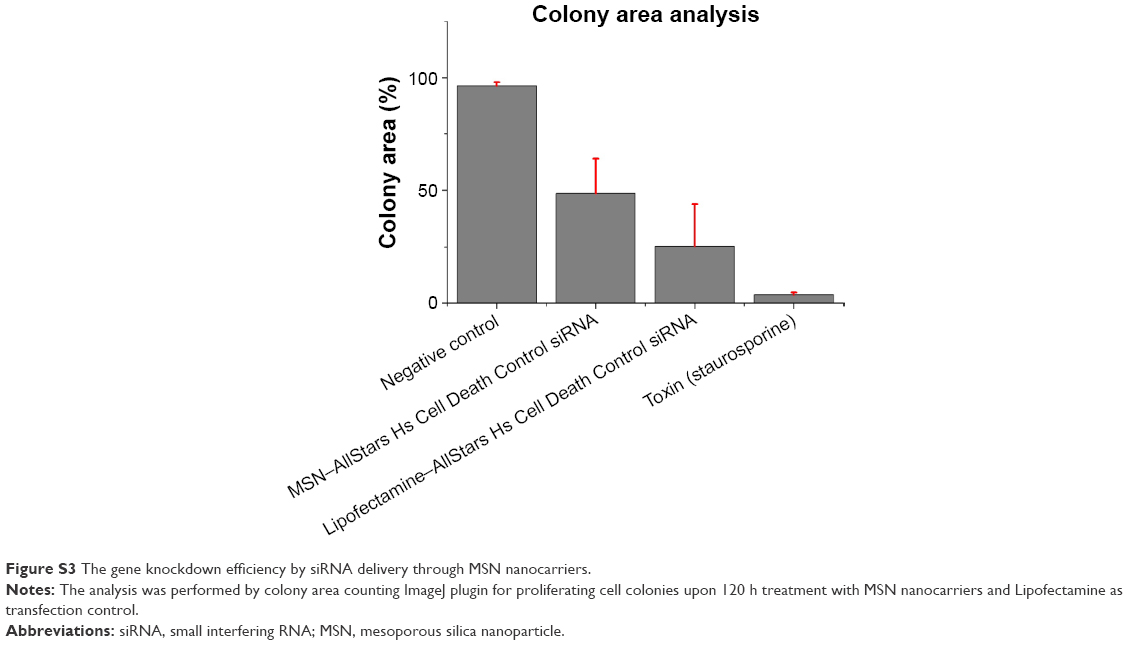 | Figure S3 The gene knockdown efficiency by siRNA delivery through MSN nanocarriers. |
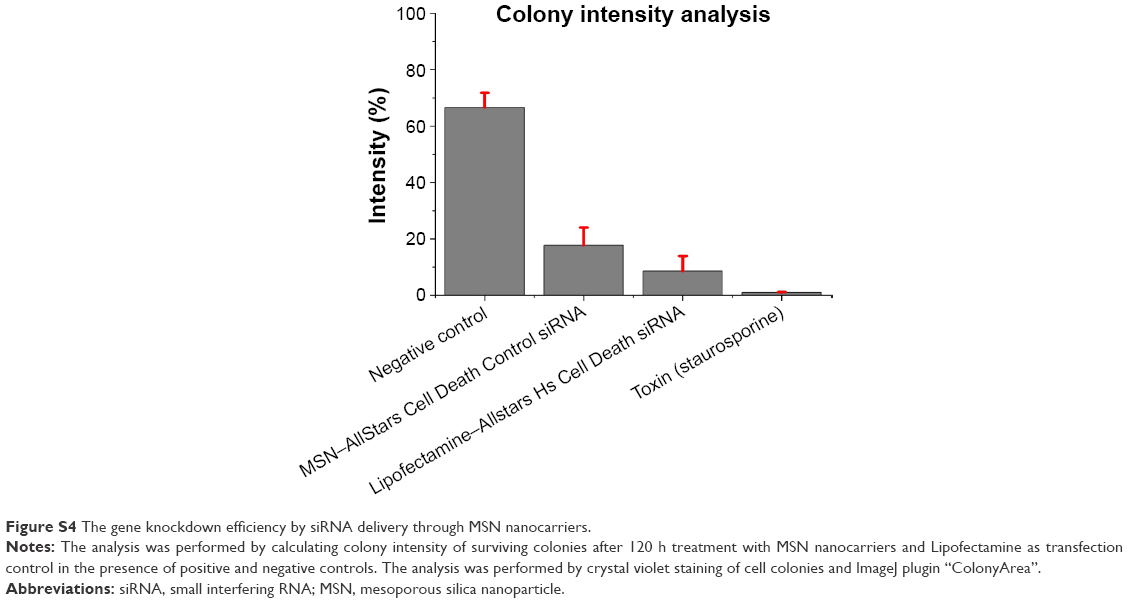 | Figure S4 The gene knockdown efficiency by siRNA delivery through MSN nanocarriers. |
Reference
Zhang J, Desai D, Rosenholm JM. Tethered lipid bilayer gates: toward extended retention of hydrophilic cargo in porous nanocarriers. Adv Funct Mater. 2014;24(16):2352–2360. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.