Back to Archived Journals » Reports in Organic Chemistry » Volume 5
Stereoselective total synthesis of lippialactone
Authors Sabitha G, Karra PR, Sudina PR
Received 17 March 2015
Accepted for publication 18 April 2015
Published 21 July 2015 Volume 2015:5 Pages 57—63
DOI https://doi.org/10.2147/ROC.S84568
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Sean Kerwin
Gowravaram Sabitha, Purushotham Reddy Karra, Purushotham Reddy Sudina
Natural Products Chemistry Division, Council of Scientific & Industrial Research-Indian Institute of Chemical Technology, Hyderabad, India
Abstract: The stereoselective synthesis of lippialactone was achieved in high overall yield from commercially available D-mannitol. Key reactions involved in the synthesis are preparation of required epoxide (5), its opening with vinyl Grignard reagent, and olefin cross-metathesis. A new route to a vinyl lactone (4) was explored.
Keywords: D-mannitol, epoxide, vinyl Grignard, cross-metathesis
Introduction
Natural products possessing the 5,6-dihydro-2-pyrone (α-pyrone) subunit as a core structure, bearing on position 6 an additional alkenyl chain, are known to display a broad spectrum of pharmacological properties,1–4 such as insect growth inhibition, and antimicrobial, cytotoxic, and antitumoral activities. In 2013, Ludere et al5 isolated a new antimalarial agent, lippialactone (1)) (Figure 1), from aerial parts of Lippia javanica. Lippialactone is active against the chloroquine-sensitive D10 strain of Plasmodium falciparum, with a half maximal inhibitory concentration (IC50) value of 9.1 μg/mL, and is known to show mild cytotoxicity. The relative stereochemistry of lippialactone was determined by molecular modeling based on the determination of the relative configuration, by quantum mechanical gauge including atomic orbitals (GIAO) 13C chemical-shift calculations. Lippialactone (1)) is structurally related to synargentolide A (2)) (Figure 1), whose structure was revised by our team.6 To date, however, only a single report has appeared on the synthesis of 1).7 In continuation of our interest in the synthesis of bioactive natural δ-lactones,8–14 we herein describe a stereoselective total synthesis of lippialactone.
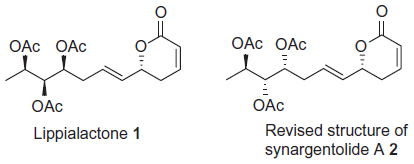 | Figure 1 Structures of lippialactone (1) and revised structure of synargentolide A (2). |
Experimental
General
Reactions were conducted under N2, in anhydrous solvents such as CH2Cl2, tetrahydrofuran (THF), and ethyl acetate (EtOAc). All reactions were monitored by thin-layer chromatography (TLC) (silica-coated plates and visualization under ultraviolet [UV] light). n-Hexane (bp 60°C−80°C) was used. Yields refer to chromatographically and spectroscopically (1H and 13C nuclear magnetic resonance [NMR]) homogeneous material. Air-sensitive reagents were transferred by syringe or double-ended needle. Evaporation of solvents was performed at reduced pressure on a BÜCHI rotary evaporator (BÜCHI Labortechnik AG, Flawil, Switzerland). 1H and 13C NMR spectra of samples in CDCl3 were recorded on Varian FT-400MHz (Varian Medical Systems Inc., Palo Alto, CA, USA), Varian FT-500MHz (Varian Medical Systems Inc.), and Bruker UXNMR FT-300MHz (Avance; Bruker Corporation, Billerica, MA, USA) spectrometers. Chemical shift δ is reported relative to tetramethylsilane (TMS) (δ=0.0) as an internal standard. Mass spectra recorded E1 conditions at 70 eV on ES-MSD (Agilent Technologies, Santa Clara, CA, USA) spectrometers. Column chromatography was performed on silica gel (60–120 mesh) supplied by Acme Chemical Co, India. TLC was performed on Merck 60 F-254 silica gel plates (Merck KGaA, Darmstadt, Germany). Optical rotations were measured with a JASCO DIP-370 Polarimeter (JASCO Corp, Tokyo, Japan).
(4R,4′R,5S)-2,2,2′,2′,5-pentamethyl-4,4′-bi(1,3-dioxolane) (8)
A solution of 6 (2.0 g, 8.65 mmol) in dry CH2Cl2 (15 mL), containing triethylamine (2.5 g, 24.7 mmol) and p-dimethylaminopyridine (DMAP) (0.04 g), was cooled to 0°C and treated with p-toluenesulphonyl chloride (3.30 g, 17.30 mmol). The reaction mixture was stirred at room temperature (RT) for 1 hour. It was then diluted with water and extracted with CH2Cl2. The organic layer was washed with brine and dried over anhydrous Na2SO4. Removal of solvent under reduced pressure afforded the unstable crude tosyl compound 7, which was used for further reaction. To a stirred suspension of LiAlH4 (0.49 g, 12.89 mmol), in dry THF (10 mL) at 0°C, was added dropwise a solution of compound 7 (2.5 g, 6.47 mmol) in dry THF (10 mL). The reaction mixture was refluxed for 3 hours. It was then cooled to 0°C, diluted with Et2O, and quenched by the dropwise addition of saturated aqueous Na2SO4. The solid material was filtered and washed thoroughly with hot EtOAc several times. The combined organic layers were dried over anhydrous Na2SO4. The solvent was removed under vacuo, and the residue was purified by silica gel column chromatography (SiO2, EtOAc/hexane [4:6]) to afford the compound 8 (1.20 g, 87%) as a viscous liquid. [α]D25: –39.9 (c 0.7, CHCl3); 1H NMR: (CDCl3, 500 MHz): δ 4.14–4.06 (m, 1H), 4.04–3.98 (m, 2H), 3.94 (dd, J=8.3, 5.0 Hz, 1H), 3.45 (d, J=7.9 Hz, 1H), 1.40 (s, 3H), 1.39 (s, 3H), 1.37–1.31 (m, 9H); 13C NMR: (CDCl3, 125 MHz): δ 109.7, 108.7, 82.8, 77.4, 77.3, 67.9, 27.5, 27.0, 26.9, 25.5, 18.9; infrared (IR) (neat): 2986, 2926, 2875, 1692, 1455, 1217, 1070, 771 cm–1; MS (ESI) m/z: 239 [M+Na]+.
(R)-1-((4S,5S)-2,2,5-trimethyl-1,3-dioxolan-4-yl)ethane-1,2-diol (9)
To a solution of the compound 8 (1.0 g, 4.62 mmol) in CH3CN (10 mL) was added CuCl2 · 2H2O (1.57 g, 9.20 mmol) as a solid. The reaction mixture was stirred at 0°C for 1 hour. The reaction mixture was quenched with solid NaHCO3 (5.0 g), and the resulting mixture was filtered. The filtrate was concentrated under reduced pressure, and the crude product was purified by column chromatography (1:1% EtOAc/hexane), to afford diol 9 (0.72 g, 90%) as a colorless oil; [α]D25: –38.3 (c 0.3, CHCl3); 1H NMR: (CDCl3, 300 MHz): δ 3.96–3.89 (m, 1H), 3.81–3.68 (m, 4H), 3.62–3.44 (m, 1H), 2.46 (brs, 1H), 1.38 (s, 3H), 1.36 (s, 3H). 1.27 (d, J=6.8 Hz, 3H). 13C NMR: (CDCl3, 125 MHz): δ 108.4, 82.6, 74.8, 72.1, 63.7, 27.2, 26.8, 18.9. IR (neat): 3243, 1526, 1426, 1214, 743 cm−1; MS (ESI) m/z: 177 [M+H]+.
(R)-2-hydroxy-2-((4S,5S)-2,2,5-trimethyl-1,3-dioxolan-4-yl)ethyl pivalate (10)
To a solution of the diol 9 (0.7 g, 4.02 mmol) and DMAP (catalytic) in dry CH2Cl2 (10 mL) was added Et3N (2.03 mL, 20.06 mmol) at 0°C under N2 atmosphere. This was followed by a dropwise addition of pivaloyl chloride (PivCl) (0.49 mL, 4.06 mmol) with vigorous stirring. The reaction was allowed to proceed for 4 hours with gradual warming to RT. The reaction mixture was diluted with CH2Cl2 and washed with saturated NaHCO3 solution, water, and brine. The organic layer was dried over anhydrous Na2SO4 and concentrated in vacuo. The crude product was chromatographed over silica gel to give (0.87 g, 87%) pure monopivaloate 10 as a colorless liquid. [α]D25: −18.9 (c 0.5, CHCl3); 1H NMR: (CDCl3, 500 MHz): δ 4.30 (dd, J=11.7, 3.5 Hz, 1H), 4.20–4.08 (m, 2H), 3.92–3.85 (m, 1H), 3.54 (t, J=7.6 Hz, 1H), 1.41 (s, 3H), 1.38 (s, 3H), 1.23 (s, 9H), 1.22 (d, J=6.0 Hz, 3H); 13C NMR: (CDCl3, 125 MHz): δ 179.0, 108.5, 81.6, 75.0, 71.2, 66.1, 38.8, 26.7, 27.2, 27.1, 19.0; IR (neat): 2980, 2935, 1715, 1458, 1215, 1160, 752 cm−1; high resolution mass spectroscopy (HRMS) (ESI): calc. 261.16965 C13H25O5, found 261.16963 [M-H]–.
(4S,5R)-2,2,4-trimethyl-5-((S)-oxiran-2-yl)-1,3-dioxolane (5)
To a cooled (−80°C) solution of the pivaloate compound 10 (0.8 g, 3.05 mmol) and triethylamine (1.32 mL, 13.04 mmol) in dry CH2Cl2 (10 mL) was added methanesulfonyl chloride (0.69 mL, 6.05 mmol), dropwise. The reaction mixture was stirred for 15 minutes at the same temperature and kept in a freezer (−20°C) overnight. The reaction mixture was diluted with more CH2Cl2 and washed with water and brine. The CH2Cl2 layer was dried over anhydrous Na2SO4 and concentrated on a rotary evaporator. The crude yellow solid was dissolved in MeOH, 0.9 g (2.67 mmol, 2.5 equiv based on theoretical yield) of finely powdered K2CO3 was added, and the mixture was stirred vigorously at RT for 2 hours. Methanol was evaporated on a rotary evaporator. The residue was taken in water and extracted with diethyl ether, and the combined organic layers were washed with brine and dried over anhydrous Na2SO4 before concentration. The crude product was chromatographed over silica gel to give (0.35 g, 85%) epoxide 5 as a colorless liquid. [α]D25: –42.1 (c 0.8, CHCl3); 1H NMR: (CDCl3, 300 MHz): δ 4.14–3.99 (m, 1H), 3.35 (dd, J=8.3, 5.2 Hz, 1H), 3.02–2.95 (m, 1H), 2.80 (t, J=5.2 Hz, 1H), 2.68 (dd, J=5.2, 3.0 Hz, 1H), 1.39 (s, 3H), 1.37 (s, 3H), 1.31 (d, J=6.0 Hz, 3H). 13C NMR: (CDCl3, 125 MHz): δ 109.0, 82.6, 73.5, 50.8, 43.5, 27.2, 26.5, 17.7. IR (neat): 3019, 1550, 1473, 1212, 742 cm−1; MS (ESI) m/z: 181 [M+Na]+.
(S)-1-((4S,5S)-2,2,5-trimethyl-1,3-dioxolan-4-yl)but-3-en-1-ol (11)
Freshly prepared vinylmagnesium bromide (2.46 mL, 2.4 mmol) in THF (1 m solution) was added dropwise to a solution of CuI (0.036 g, 0.15 mmol) in THF (15 mL) at −20°C. The mixture was stirred for 30 minutes, and chiral epoxide 5 (0.30 g, 1.89 mmol) in THF (5 mL) was added dropwise. After completion of the reaction, a saturated solution of NH4Cl (10 mL) was added and diluted with Et2O (5 mL). The two layers were separated, and the aqueous layer was extracted with Et2O (3×5 mL). The combined organic layers were washed with brine (2×5 mL), dried with anhydrous Na2SO4, and concentrated under reduced pressure, and gave alcohol 11 (0.30 g, 88%) as a colorless oil; [α]D25: +2.0 (c 0.5, CHCl3); 1H NMR: (CDCl3, 500 MHz): δ 5.90–5.78 (m, 1H), 5.16–5.07 (m, 2H), 4.09–4.01 (m, 1H), 3.54 (brs, 1H), 3.50–3.45 (m, 1H), 2.28 (brs, 2H), 2.21 (brs, 1H), 1.39 (s, 3H), 1.37 (s, 3H), 1.26 (d, J=5.9 Hz, 3H). 13C NMR: (CDCl3, 125 MHz): δ 134.2, 117.7, 108.4, 84.3, 73.0, 69.2, 39.2, 27.4, 26.8, 17.8. IR (neat): 3019, 2926, 1693, 1517, 1213, 746 cm−1; HRMS (ESI): calc. 187.13288 C10H18O3, found 186.13290 [M-H]-.
(S)-1-((4R,5S)-2,2,5-trimethyl-1,3-dioxolan-4-yl)but-3-en-1-yl acetate (3)
To a solution of the compound 11 (0.2 g, 1.07 mmol) in dry CH2Cl2 (20 mL) were added Et3N (0.46 mL, 4.54 mmol) followed by acetic anhydride (0.21 mL, 2.05 mmol) and catalytic amount of DMAP at 0°C. The reaction mixture was continued to stir for 1 hour and then diluted with CH2Cl2 (5 mL). The organic layer was washed with 5% NaHCO3 solution (2×5 mL), then brine (2×5 mL), and dried over anhydrous Na2SO4. The solvent was removed under reduced pressure, and the crude residue was purified on silica gel column chromatography (30% EtOAc/hexane) to provide the compound 3 (0.22 g, 92%) as a colorless oil. [α]D25: +9.0 (c 0.8, CHCl3); 1H NMR: (CDCl3, 500 MHz): δ 5.84–5.65 (m, 1H), 5.18–4.95 (m, 2H), 3.93–3.80 (m, 1H), 3.61 (dd, J=8.3, 3.0 Hz, 1H), 2.43 (t, J=6.9 Hz, 2H), 2.07 (s, 3H), 1.40 (s, 3H), 1.38 (s, 3H), 1.28 (d, J=6.0 Hz, 3H). 13C NMR: (CDCl3, 125 MHz): δ 170.5, 133.2, 118.2, 108.4, 82.4, 72.6, 70.2, 35.7, 27.3, 26.5, 20.9, 17.7. IR (neat): 2934, 1739, 1514, 1373, 1216, 748 cm−1; MS (ESI) m/z: 305 [M+Na]+.
(R)-2-(((4-methoxybenzyl)oxy)methyl)oxirane (12)
To a stirred suspension of NaH (1.0 g, 41.66 mmol) in THF (10 mL) at 0°C was added para-methoxybenzyl alcohol (PMBOH) (3.0 g, 21.73 mmol) in THF (5 mL) dropwise. After being stirred under nitrogen for 30 minutes at 0°C, (Bu)4NI catalytic amount and (±) epichlorohydrin × (2.0 g, 21.73 mmol) in THF (15 mL) were added as well. The mixture was stirred for 16 hours, quenched with saturated NH4Cl solution, and extracted into EtOAc (3×10 mL). The combined organic layer was washed with water, then brine, dried over anhydrous Na2SO4, and the volatiles were evaporated. The crude residue was purified by column chromatography (hexane/EtOAc 8:2) to afford 12 (3.6 g, 87%) as a viscous liquid.
A mixture of (S,S)-bis(3,5-di-tert-butylsalicylide)-1,2-cyclohexanediamino-Co(II) complex (0.031 g, 0.51 mmol), toluene (2 mL), and acetic acid (0.062 g, 1.02 mmol) were stirred in open air for 1 hour at RT. The solvent was evaporated under reduced pressure, and compound 12 (1.0 g, 5.15 mmol) and water (1.0 mL, 55.55 mmol) were added to the brown residue at bath temperature below 15°C, and the reaction mixture was stirred at RT for 36 hours. The crude reaction mixture was purified by silica gel column chromatography (hexane/EtOAc [8:2]) to afford the chiral epoxide 12 (0.88 g, 42%). [α]25D =-10.3 (c=0.5, CHCl3). 1H-NMR(CDCl3, 500 MHz): δ 7.28 (d, J=8.3 Hz, 2H), 6.88 (d, J=9.0 Hz, 2H), 4.52 (ABq, J=18.8, 11.3 Hz, 2H), 3.81 (s, 3H), 3.74 (dd, J=11.3, 3.0 Hz, 1H), 3.41 (dd, J=11.3, 6.0 Hz, 1H), 3.22–3.15 (m, 1H), 2.80 (t, J=4.5 Hz, 1H), 2.61 (dd, J=5.2,3.0 Hz, 1H). 13C-NMR (CDCl3, 125 MHz): δ 159.0, 131.6, 129.7, 129.2, 113.6, 72.7, 70.3, 55.0, 50.7, 44.1. IR (neat): 1609, 1513, 1249, 1093 cm−1; ESI-MS: m/z 217 [M+Na]+.
(R)-6-((4-methoxybenzyl)oxy)hex-2-yne-1,5-diol (13)
Under nitrogen, lithium bis(trimethylsilyl)amide (1 M solution in hexane, 5.1 mL, 5.14 mmol) was added to a solution of propargylic alcohol (0.2 mL, 3.85 mmol) in THF (10 mL) at −78°C, and the mixture was stirred for 1 hour. Then, BF3 · OEt2 (0.39 mL, 3.08 mmol) was added to the solution, and the stirring was continued for 15 minutes at −78°C. Finally, a solution of chiral epoxide 12 (0.5 g, 2.57 mmol) in dry THF (10 mL) was added, and after the reaction mixture was stirred for 3 hours at −78°C, it was quenched by adding a saturated aqueous NH4Cl solution (20 mL). The resulting mixture was extracted with EtOAc (2×20 mL), and the combined organic layers were dried with anhydrous Na2SO4 Evaporation of the solvent, followed by column chromatography (50% EtOAc/hexane) afforded pure alcohol 13 (0.547 g, 85%) as a colorless oil; [α]25D =–3.3 (c=0.2, CHCl3). 1H-NMR (CDCl3, 500 MHz): δ 7.25 (d, J=8.4 Hz, 2H), 6.88 (d, J=8.4 Hz, 2H), 4.48 (s, 2H), 4.19 (brs, 2H), 3.98–3.89 (m, 1H), 3.80 (s, 3H), 3.56–3.41 (m, 2H), 3.32–2.78 (brs, 2H), 2.52–2.36 (m, 2H). 13C-NMR (CDCl3, 125 MHz)):δ 159.2, 129.7, 129.4, 113.7, 81.9, 80.6, 73.0, 72.6, 68.7, 55.2, 50.9, 23.7. IR (neat): 3383, 1610, 1512, 1248, 1028 cm−1; ESI-MS: m/z 273 [M+Na]+.
(R,Z)-6-((4-methoxybenzyl)oxy)hex-2-ene-1,5-diol (14)
A suspension of compound 13 (0.45 g, 1.80 mmol), Lindlar catalyst (0.026 gr, 5 wt%), and quinoline (catalytic amount) in EtOAc (2 mL) was stirred at RT under hydrogen atmosphere (1 atm) until partially reduced product appeared on TLC. The reaction mixture was filtered through a pad of Celite® with EtOAc (5 mL). The filtrate was concentrated under reduced pressure, and the crude product was purified by silica gel column chromatography (50% EtOAc/hexane) to afford compound 14 as a colorless liquid (0.40 g, 90%). [α]25D =+4.8 (c=0.3, CHCl3). 1H-NMR (CDCl3, 500 MHz): δ 7.25 (d, J=6.0 Hz, 2H), 6.9 (d, J=7.9 Hz, 2H), 5.95–5.75 (m, 1H), 5.68–5.51 (m, 1H), 4.48 (s, 2H), 4.22–3.99 (m, 2H), 3.81 (s, 3H), 3.53–3.29 (m, 2H), 2.40–2.20 (brs, 2H). 13C-NMR (CDCl3, 125 MHz)):δ159.2, 131.3, 129.7, 129.4, 128.4, 113.8, 73.5, 73.0, 69.2, 57.4, 55.2, 31.0. IR (neat): 3394, 1611, 1513, 1248, 1032 cm−1; ESI-MS: m/z 275 [M+Na]+.
(R)-6-(((4-methoxybenzyl)oxy)methyl)-5,6-dihydro-2H-pyran-2-one (15)
Bis(acetoxy)iodobenzene (BAIB) (0.53 g, 1.65 mmol) was added to a solution of alcohol 14 (0.35 g, 1.38 mmol) and 2,2,6,6-tetramethyl-1-piperidinyloxy (TEMPO) (0.021 g, 0.13 mmol) in 10 mL of CH2Cl2. The reaction mixture was stirred until the alcohol was no longer detectable (TLC), and then it was diluted with CH2Cl2 (20 mL). The mixture was washed with a saturated aqueous solution of Na2S2O3 (20 mL) and extracted with CH2Cl2 (4×20 mL). The combined organic extracts were washed with aqueous NaHCO3 (30 mL) and brine (30 mL), dried (Na2SO4), and concentrated under reduced pressure and purified by silica gel column chromatography (hexane/EtOAc [7:3]) to afford 15 as a colorless oil (0.31 g, 90%). [α]25D =–20.3 (c=0.06, CHCl3). 1H-NMR (CDCl3, 500 MHz): δ 7.26 (d, J=8.5 Hz, 2H), 6.92–6.87 (m, 3H), 6.03–6.00 (m, 1H), 4.62–4.55 (m, 1H), 4.53 (d, J=3.5 Hz, 2H), 3.81 (s, 3H), 3.66 (d, J=5.6 Hz, 2H), 2.59–2.51 (m, 1H), 2.42–2.35 (m, 1H). 13C-NMR (CDCl3, 125 MHz)):δ 163.7, 159.3, 144.9, 129.3, 129.6, 121.1, 113.8, 76.5, 73.2, 70.4, 55.2, 26.1. IR (neat): 1722, 1512, 1247 cm−1; ESI-MS: m/z 271 [M+Na]+.
(R)-6-(hydroxymethyl)-5,6-dihydro-2H-pyran-2-one (16)
To a cooled (0°C) solution of 15 (0.25 g, 1.00 mmol) in CH2Cl2 (4.5 mL) and H2O (0.5 mL) was added 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) (0.34 g, 1.50 mmol) and stirred at RT for 2 hours. After completion of the reaction, saturated NaHCO3 solution was added, and the aqueous layer was extracted with CH2Cl2 (2 × 10 mL). The combined organic extract was dried over anhydrous Na2SO4 and concentrated in vacuo. The crude residue was purified by column chromatography using hexanes/EtOAc (7:3) as an eluent to give alcohol 16 (0.11 g, 92%) as a colorless liquid. [α]25D =–29.6 (c=0.2, CHCl3). 1H-NMR (CDCl3, 300 MHz): δ7.00–6.91 (m, 1H), 6.04 (dd, J=9.8, 3.0 Hz, 1H), 4.62–4.52 (m, 1H), 3.89 (dd, J=12.0, 3.0 Hz, 1H), 3.76 (dd, J=12.8, 5.2 Hz, 1H), 2.69–2.55 (m, 1H), 2.39–2.28 (m 1H). 13C-NMR (CDCl3, 125 MHz)):δ 163.9, 145.3, 120.8, 78.3, 63.7, 25.2. IR (neat): 3401, 2923, 1712, 1514, 1373, 1216, 748 cm−1; ESI-MS: m/z 151 [M+Na]+.
(R)-6-vinyl-5,6-dihydro-2H-pyran-2-one (4)
To an ice-cooled solution of 2-(iodooxy) benzoic acid (0.32 g, 1.17 mmol) in anhydrous CH3CN (7 mL) was added a solution of alcohol 16 (0.100 g, 0.78 mmol). The mixture was refluxed for 1 hour and then allowed to cool to RT. The solvent was removed under reduced pressure, and the unstable crude aldehyde product was used directly for the next step without further purification by column chromatography.
In a reaction flask, a 2.5 M solution of n-BuLi in hexane (0.9 mL, 2.28 mmol) was added under N2 atmosphere to a stirred suspension of methyltriphenylphosphonium iodide (0.54 g, 1.51 mmol) in dry THF (15 mL) at −78°C. The mixture was allowed to warm to RT, stirred for 1 hour, and cooled to −78°C again. To this mixture, a solution of above crude aldehyde in dry THF (10 mL) was added dropwise, and the resulting mixture was stirred at RT for 2 hours, quenched with aqueous NH4Cl, and extracted with EtOAc (2×10 mL). The combined organic extracts were dried over anhydrous Na2SO4 and concentrated in vacuo. The residue was purified by column chromatography (hexane/EtOAc 8:2) to give compound 4 as a colorless oil (0.07 g, 75%). [α]25D =-10.2 (c=0.2, CHCl3). 1H NMR: (CDCl3, 500 MHz): δ 6.93–6.87 (m, 1H), 6.08–6.04 (m, 1H), 6.0–5.92 (m, 1H), 5.41 (dd, J=17.2, 0.9 Hz, 1H), 5.30 (dd, J=10.5, 0.7 Hz, 1H), 2.53–2.39 (m, 2H). 13C NMR: (CDCl3, 125 MHz): δ 163.7, 144.4, 134.7, 121.5, 117.8, 77.7, 29.3. IR (neat): 2930, 1720, 1520, 1314, 1201, 752 cm−1; ESI-MS: m/z 147 [M+Na]+.
(S,E)-4-((R)-6-oxo-3,6-dihydro-2H-pyran-2-yl)-1-((4R,5S)-2,2,5-trimethyl-1,3-dioxolan-4-yl)but-3-en-1-yl acetate (17)
To a solution of the compound 3 (0.1 g, 0.43 mmol) and 4 (0.13 g, 1.27 mmol) in CH2Cl2 (3 mL), 10 mol-% Grubbs catalyst II (0.03 g, 0.04 mmol) was added and stirred at RT for 24 hours under N2. Most of the solvent was then distilled off, and the concentrated solution was left to be stirred at RT for 2 hours. The mixture was evaporated to dryness to give a brown residue, which was purified by column chromatography (SiO2; 6:4% EtOAc/hexane) to give 17 (0.11 g, 82%) as a colorless liquid. [α]D25: +13.4 (c 0.2, CHCl3); 1H NMR: (CDCl3, 500 MHz): δ 6.90–6.85 (m, 1H), 6.05 (tt, J=9.7, 2.1, 1.5 Hz, 1H), 5.81–5.68 (m, 2H), 5.04–4.98 (m, 1H), 4.91–4.85 (m, 1H), 3.91–3.84 (m, 1H), 3.61 (dd, J=8.2, 3.0 Hz, 1H), 2.50–2.45 (m, 2H), 2.44–2.37 (m, 2H), 2.10 (s, 3H), 1.41 (s, 3H), 1.39 (s, 3H), 1.29 (d, J=6.1 Hz, 3H). 1C NMR: (CDCl3, 75 MHz): δ 170.5, 163.8, 144.4, 130.4, 129.4, 121.5, 108.5, 82.3, 77.5, 72.5, 69.9, 34.1, 29.5, 20.8, 17.7. IR (neat): 1734, 1514, 1378, 1213, 745 cm−1; HRMS (ESI): calc. 342.19111 C17H28O6N, found 342.19097 [M+NH4]+.
(4S,5S,6R,E)-5,6-dihydroxy-1-((R)-6-oxo-3,6-dihydro-2H-pyran-2-yl)hept-1-en-4-yl acetate (18)
To a stirred solution of compound 17 (0.05 g, 0.15 mmol) in anhydrous CH2Cl2 (7 mL), TiCl4 (0.008 mL, 0.05 mmol) was added at 0°C, and the reaction mixture was stirred at the same temperature for 1 hour. The reaction mixture was quenched with solid NaHCO3 and filtered. The solvent was removed under reduced pressure. The residue was purified by silica gel column chromatography (1:1% EtOAc/hexane) to afford 18 (0.03 g, 85%) as a colorless oil. [α]D25: +18.9 (c 0.4, CHCl3); 1H NMR: (CDCl3, 500 MHz): δ 6.89 (ddd, J=9.7, 4.8, 3.3 Hz, 1H), 6.08–6.03 (m, 1H), 5.93–5.84 (m, 1H), 5.76–5.67 (m, 1H), 5.10–5.00 (m, 1H), 4.96–4.85 (m, 1H), 3.77–3.64 (m, 1H), 3.41 (t, J=5.0 Hz, 1H), 2.48–2.29 (m, 4H), 2.09 (s, 3H), 1.30 (d, J=6.4 Hz, 3H). 13C NMR: (CDCl3, 125 MHz): δ 170.1, 164.3, 145.0, 133.5, 129.3, 123.0, 80.0, 78.4, 73.7, 70.2, 67.2, 64.5, 31.9, 29.6, 21.0, 18.9. IR (neat): 3018, 2925, 1693, 1516, 1417, 1212, 747 cm−1; HRMS (ESI): calc. 302.15981 C14H24O6N, found 302.15985 [M+NH4]+.
(2R,3S,4S,E)-7-((R)-6-oxo-3,6-dihydro-2H-pyran-2-yl)hept-6-ene-2,3,4-triyl triacetate (lippialactone (1))
To a stirred solution of the lactone 18 (10 mg, 0.03 mmol) in dry CH2Cl2 (5 mL) were added Et3N (0.03 mL, 0.29 mmol), followed by acetic anhydride (0.01 mL, 0.09 mmol) and a catalytic amount of DMAP, at 0°C. The reaction mixture was continuously stirred for 1 hour and then diluted with CH2Cl2 (3 mL), The organic layer was washed with 5% NaHCO3 solution (2×2 mL), then brine (2×2 mL), and dried over anhydrous Na2SO4. The solvent was removed under reduced pressure, and the crude residue was purified on silica gel column chromatography (30% EtOAc/hexane) to provide the lactone 1 (9 mg, 90%) as a colorless oil. [α]D25: +22.1 (c 0.3, CHCl3); 1H NMR: (CDCl3, 500 MHz): δ 6.86 (ddd, J=9.6. 5.0, 3.3 Hz, 1H), 6.03 (dt, J=9.7, 3.5 Hz, 1H), 5.72 (dd, J=15.4, 6.8 Hz, 1H), 5.65 (dd, J=15.4, 5.9 Hz, 1H), 5.14–5.09 (m, 1H), 5.08–5.03 (m, 2H), 4.88–4.82 (m, 1H), 2.45–2.37 (m, 2H), 2.36–2.26 (m, 2H), 2.11 (s, 3H), 2.07 (s, 3H), 2.03 (s, 3H), 1.19 (d, J=5.7 Hz, 3H). 13C NMR: (CDCl3, 125 MHz): δ 170.1, 163.7, 144.4, 131.1, 128.3, 121.5, 77.5, 74.1, 70.4, 68.7, 33.9, 29.5, 21.0, 20.8, 20.6, 16.4. IR (neat): 3018, 2924, 1747, 1709, 1645, 1550, 1531, 1213, 746 cm−1; ESI-MS: m/z 386 [M+NH4]+. HRMS (ESI): calc. 386.18094 C18H28O8N, found 386.18098 [M+NH4]+.
Results and discussion
The retrosynthetic analysis of lippialactone (1) is summarized in Figure 2. Lippialactone (1) could be accomplished from homoallyl alcohol 3 and vinyl lactone 4 via an olefin cross-metathesis reaction. In turn, the construction of three contiguous stereogenic hydroxyl groups in compound 3 could be achieved from commercially available D-mannitol, and vinyl lactone 4 could be prepared from a known chiral epoxide 1215 by a new synthetic route.
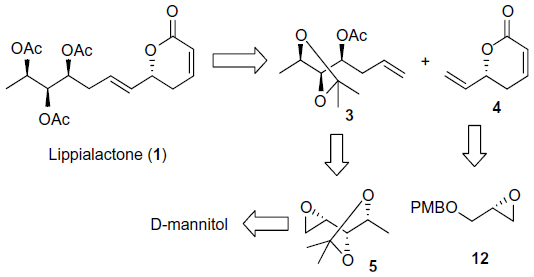 | Figure 2 Retrosynthetic analysis. |
The synthesis of fragment 3 (Figure 3) commenced from a primary alcohol 6, which was prepared from D-mannitol16,17 according to the reported procedure. Tosylation of the primary hydroxyl group in compound 6, with TsCl, Et3N, and DMAP in CH2Cl2, gave compound 7, which on treatment with lithium aluminum hydride (LAH) gave a terminal methyl compound 8. Selective deprotection of the terminal acetonide with CuCl2.2H2O in CH3CN afforded diol 9. Selective protection of the primary hydroxy group in 9, as the pivaloyl ester, with PivCl, Et3N, and DMAP in dichloromethane (DCM) gave compound 10. Mesylation of the secondary hydroxyl group in compound 10, with MsCl, Et3N, and DMAP in CH2Cl2, gave secondary mesylate. This was followed by the treatment with anhydrous K2CO3 in anhydrous MeOH, at RT, which afforded epoxide 5 with the required stereocenter. Opening of epoxide 5 with vinylmagnesium bromide in the presence of CuI afforded homoallyl alcohol 11. The resulting free secondary hydroxyl group was acetylated to give a monoacetate compound 3.
 | Figure 3 Synthesis of fragment 3. |
The other fragment, vinyl lactone 4, was prepared from a known epoxide 1215 as shown in Figure 4. The epoxide was subjected to regioselective ring opening with propargyl alcohol using LiHMDS, then BF3OEt2 at −78°C to furnish alcohol 13, in 85% yield. The triple bond in 13 was reduced to a Z-double bond using Lindlar catalyst to afford 14, in 90% yield. Oxidative cyclization of 1,5-diol 14 with TEMPO and [bis(acetoxy)iodo]benzene [(PhI(OAc)2) (BAIB)]18 produced the desired δ-lactone 15, in 90% yield.
 | Figure 4 Synthesis of vinyl lactone 4. |
Oxidative removal of the 4-methoxybenzyl ether (PMB) group (DDQ/CH2Cl2:H2O, 9:1, 0°C-rt, 92%) and oxidation of the resulting alcohol 16 to the corresponding aldehyde, and subsequent treatment with one carbon Wittig reagent furnished vinyl lactone 4 ((a) IBX/CH3CN, b) PPh3=CH2, THF, 0°C-rt, 75% over two steps).
With the two key fragments in hand, the CM reaction was planned. Olefin cross-metathesis19–21 between fragment 3 and vinyl lactone 4 was promoted smoothly by the second-generation Grubb’s catalyst in CH2Cl2 under reflux, to yield the desired lactone 17 (82%) exclusively (Figure 5).
 | Figure 5 Synthesis of lippialactone (1). |
Finally deprotection of acetonide group in compound 17, followed by acetylation of the resulting diol 18 produced the target molecule, lippialactone (1). The spectroscopic and analytical data of the synthetic compound are in good agreement with those reported for the natural product.
Conclusion
In conclusion, the stereoselective synthesis of lippialactone from D-mannitol has been achieved. Vinyl lactone used in the present synthesis was prepared by a new synthetic route. LAH reduction, epoxide opening, and olefin cross-metathesis reactions were used as key steps.
Acknowledgments
KPR thanks Council of Scientific & Industrial Research and SPR thanks the University Grants Commission, New Delhi, India for the financial support in the form of fellowships.
Disclosure
The authors report no conflicts of interest in this work.
References
Yasui K, Tamura Y, Nakatani T, Kawada K, Ohtani M. Total synthesis of (-)-PA-48153C, a novel immunosuppressive 2-pyranone derivative. J Org Chem. 1995;60(23):7567–7574. | |
Fang XP, Anderson JE, Chang CJ, Mclaughlin JL, Fanwick PE. Two new styryl lactones, 9-deoxygoniopypyrone and 7-epi-goniofufurone, from Goniothalamus giganteus. J Nat Prod. 1991:54(4):1034–1043. | |
Argoudelis AD, Zieserl JF. The structure of U-13,933, a new antibiotic. Tetrahedron Lett. 1966;7(18):1969–1973. | |
Davies-Coleman MT, Rivett DE. Naturally occurring 6-substituted 5,6-dihydro-alpha-pyrones. Fortschr Chem Org Naturst. 1989;55:1–35. | |
Ludere MT, van Ree T, Vleggaar R. Isolation and relative stereochemistry of lippialactone, a new antimalarial compound from Lippia javanica. Fitoterapia. 2013;86:188–192. | |
Sabitha G, Gopal P, Reddy CN, Yadav JS. First stereoselective synthesis of synargentolide A and revision of absolute stereochemistry. Tetrahedron Lett. 2009;50(46):6298–6302. | |
Radha Krishna P, Nomula R, Kunde R. First total synthesis of lippialactone and its C9 epime. Synthesis. 2014;46(03):307–312. | |
Sabitha G, Bhaskar V, Reddy SSS, Yadav JS. Stereoselective total synthesis of (+)-(6R,2’S)-cryptocaryalactone and (–)-(6S,2′S)-epi cryptocaryalactone. Tetrahedron. 2008;64(44):10207–10213. | |
Sabitha G, Gopal P, Reddy CN, Yadav JS. A concise and efficient synthesis of (5R, 7S)-kurzilactone and its (5S, 7R)-enantiomer by the Mukaiyama aldol reaction. Synthesis. 2009;2009(19):3301–3304. | |
Sabitha G, Prasad MN, Shankaraiah K, Jhillu S. Stereoselective total synthesis of obolactone via prins cyclization. Synthesis. 2010;2010(7):1171–1175. | |
Sabitha G, Reddy SSS, Yadav JS. Total synthesis of cryptopyranmoscatone B1 from 3,4,6-tri-O-acetyl-d-glucal. Tetrahedron Lett. 2010; 51(48):6259–6261. | |
Sabitha G, Reddy CN, Raju A, Yadav JS. Stereoselective total synthesis of the cytotoxic lactone (−)-spicigerolide. Tetrahedron: Asymmetry. 2011;22(4):493–498. | |
Sabitha G, Rao AS, Yadav JS. Stereoselective synthesis of (–)-synparvolide B. Tetrahedron: Asymmetry. 2011;22(8):866–871. | |
Sabitha G, Reddy DV, Reddy SSS, Yadav JS, Ganesh Kumar C, Sujitha P. Total synthesis of desacetylumuravumbolide, umuravumbolide and their biological evaluation. RSC Advances. 2012;2(18):7241–7247. | |
Tsuboi S, Takeda S, Yamasaki Y, et al. A convenient synthesis of platelet- activating factors and their analogues from chiral epichlorohydrin. Chemistry Lett. 1992;21(8):1417–1420. | |
Wu W, Wu Y. Chemoselective hydrolysis of terminal isopropylidene acetals and subsequent glycol cleavage by periodic acid in one pot. J Org Chem. 1993;58(13):3586–3588. | |
Ghosh S, Rao RV. Total synthesis of aspinolide B: a ring-closing metathesis approach. Tetrahedron Lett. 2007;48(39):6937–6940. | |
Hansen TM, Florence GJ, Lugo-Mas P, Chen J, Abrams JN, Forsyth CJ. Highly chemoselective oxidation of 1,5-diols to δ-lactones with TEMPO/BAIB. Tetrahedron Lett. 2003;44(1):57–59. | |
Torssell S, Somfai P. A practical synthesis of D-erythro-sphingosine using a cross-metathesis approach. Org Biomol Chem. 2004;2(11):1643–1646. | |
Dwyer CL. Metathesis of olefins. In: Chiusoli GP, Maitlis. Metal-Catalysis in Industrial Organic Processes. Cambridge: RSC Publishing; 2006:201–217. | |
Chiusoli GP. In: Weissermel K, Arpe HJ. Industrial Organic Chemistry. 4th ed. Wiley-VCH Verlag GmbH & Co. 2008;3:59–89. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.