Back to Journals » Drug Design, Development and Therapy » Volume 9
Stepwise design, synthesis, and in vitro antifungal screening of (Z)-substituted-propenoic acid derivatives with potent broad-spectrum antifungal activity
Authors Khedr M
Received 7 March 2015
Accepted for publication 22 April 2015
Published 10 August 2015 Volume 2015:9 Pages 4501—4513
DOI https://doi.org/10.2147/DDDT.S84178
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Professor Shu-Feng Zhou
Mohammed A Khedr
Department of Pharmaceutical Chemistry, Faculty of Pharmacy, Helwan University, Cairo, Egypt
Abstract: Fungal infections are a main reason for the high mortality rate worldwide. It is a challenge to design selective antifungal agents with broad-spectrum activity. Lanosterol 14α-demethylase is an attractive target in the design of antifungal agents. Seven compounds were selected from a number of designed compounds using a rational docking study. These compounds were synthesized and evaluated for their antifungal activity. In silico study results showed the high binding affinity to lanosterol 14α-demethylase (-24.49 and -25.83 kcal/mol) for compounds V and VII, respectively; these values were greater than those for miconazole (-18.19 kcal/mol) and fluconazole (-16.08 kcal/mol). Compound V emerged as the most potent antifungal agent among all compounds with a half maximal inhibitory concentration of 7.01, 7.59, 7.25, 31.6, and 41.6 µg/mL against Candida albicans, Candida parapsilosis, Aspergillus niger, Trichophyton rubrum, and Trichophyton mentagrophytes, respectively. The antifungal activity for most of the synthesized compounds was more potent than that of miconazole and fluconazole.
Keywords: design, broad antifungal, molecular modeling
Introduction
Fungi are eukaryotic organisms with cellular functions that resemble those of animals and plants. They can cause many diseases such as invasive candidiasis, with high mortality rates of up to 75%. Oral candidiasis is associated with many problems especially in AIDS patients.1 Other fungal infections like Aspergillosis caused by the Aspergillus species can affect the ear, brain, and lungs. The inhalation of Aspergillus spores will result in severe pulmonary disease and a high mortality rate. The incidence of life-threatening fungal infections is considered a real problem all over the world, especially in the USA. Candida species infections in hospitals are the fourth cause of infections.2 The development of a selective antifungal agent is a real challenge. Most of the available antifungal drugs have severe side effects, for example, amphotericin B, which is used for the treatment of invasive mycosis, is nephrotoxic, while miconazole has oral bioavailability problems.3 The antifungal resistance for azole-containing drugs is a real problem that occurs in AIDS patients.4 In addition, the lanosterol 14α-demethylase (L14αDM) enzyme is involved in the biosynthesis of lanosterol, which is the main component of the fungal cell membrane.5 L14αDM is a member of the CYP 450 enzymes and is encoded by the ERG11 (CYP51) gene in fungi. Azole antifungal drugs exert their antifungal effect via the inhibition of L14αDM, thus preventing the biosynthesis of ergosterol.6
The mechanism of action of azole antifungal drugs depends on the coordination of their azole moiety (imidazole and/or triazole) with the heme that is present as a cofactor in L14αDM in the form of the heme–porphyrin complex. The azole moiety resembles that of the histidine amino acid in its binding to heme.7,8 To date, the replacement of the azole heme-coordinating moiety with other heme chelator groups has not been attempted previously.
Recent advances in in silico structure and ligand-based drug design approaches have a great importance in the process of drug design, discovery, and optimization. These approaches were very successful in designing selective inhibitors of some enzymes as targets for the treatment of important diseases.9 Molecular docking is one of the widely used methods that is used in the prediction of binding modes, binding free energy, and affinity of the designed compounds.10 The main aim of this study was to design new L14αDM inhibitors that contain a heme chelator group other than the azole ring and features unique binding geometry. Finally, the in vitro antifungal activity against different fungal strains was also tested.
Materials and methods
Chemistry
Chemicals were purchased from Sigma Aldrich (St Louis, MO, USA). Monitoring of chemical reactions was done by analytical thin layer chromatography with Merck 60 F-254 silica-gel plates (Merck Millipore, Billerica, MA, USA) and visualization was carried out with AKRUSS OPTRONIC Gmbh (Hamburg, Germany) ultra violet light. Stuart SMP11 Melting Point apparatus (Bibby Scientific Limited, Staffordshire, UK) was used to check melting points. Nuclear magnetic resonance (NMR) spectra (1H and 13C) were recorded in dimethyl sulfoxide (DMSO)-d6 solvent using tetramethylsilane as internal standard, with a 400 MHz Bruker spectrometer. Chemical shifts were expressed in ppm downfield from tetramethylsilane and splitting pattern is abbreviated as s, singlet; m, multiplet; and d, doublet. Infrared (IR) spectra were recorded using Shimadzu FTIR-8400S infrared spectrophotometer (Shimadzu Corp., Kyoto, Japan). Elemental microanalysis was carried out for determination of the C, H, and N percentages. It was done in the microanalytical center, faculty of science, Cairo University.
Molecular Operating Environment (MOE) 2014.09 package license was purchased from Chemical Computing Group Inc, Montreal, QC, Canada11 and Leadit 2.1.2 software license was purchased from BioSolveIT GmbH, Sankt Augustin, Germany.12 The synthetic strategy employed to produce the targeted compounds is illustrated in Figure 1. Synthesis of all compounds was performed as per the reported procedures.13
 | Figure 1 Synthetic pathway employed to produce the target compounds. |
General procedure employed in the preparation of (Z)-2-(2,4-dichlorophenoxy)-3-substituted phenyl-2-propenoic acid derivatives
A mixture of 5 mmol of 2,4-dichlorophenoxy acetic acid, 5 mmol of appropriate substituted aromatic aldehydes, 5 mmol of triethylamine, and 4 mL acetic anhydride was heated with stirring at 105°C–110°C for 36 hours. The reaction mixture was cooled to 60°C, and 3 mL of water was added to decompose the excess acetic anhydride. The mixture was poured on ice and was extracted with ether. The ether layer was washed with water and then extracted with three portions of 5% sodium carbonate solution. The combined sodium carbonate extracts were neutralized with hydrochloric acid, heated to 70°C–80°C. The mixture was filtered, acidified, and the formed product was recrystallized from ethanol.
(Z)-2-(2,4-dichlorophenoxy)-3-phenyl-2-propenoic acid (I)
Yellowish white solid. Yield: 35%, mp 130°C–135°C. 1H NMR (DMSO-d6) δ: 7.06 (d, J =8.8 Hz, 1H, Ph), 7.35 (d, J =8.9 Hz, 1H, Ph), 7.57 (s, 1H, Ph), 7.30 (m, 5H, Ph), 7.95 (s, 1H, =CH), 12.90 (s, 1H, COOH). 13C NMR (DMSO-d6) δ: 169.47, 152.25, 140.29, 128.75, 129.52, 128.11, 128.36, 126.91, 126.02, 126.5, 125.92, 124.25, and 117.21. IR γmax cm−1 (KBr): 3,030, 2,850, 1,685, 1,650, 1,540, 750. Anal. Calcd for C15H10Cl2O3 (Mwt: 309.15): C, 58.27%; H, 3.26%. Found: C, 58.11%; H, 3.09%.
(Z)-2-(2,4-dichlorophenoxy)-3-(4-chlorophenyl)-2-propenoic acid (II)
White solid. Yield: 32%, mp 110°C–115°C. 1H NMR (DMSO-d6) δ: 7.06 (d, J =8.8 Hz, 1H, Ph), 7.35 (d, J =8.9 Hz, 1H, Ph), 7.57 (s, 1H, Ph), 6.4 (d, J =8.4 Hz, 2H), 6.90 (d, J =8.4 Hz, 2H), 7.89 (s, 1H, =CH), 12.95 (s, 1H, COOH). 13C NMR (DMSO-d6) δ: 165.11, 152.36, 142.46, 140.91, 134.82, 130.89, 129.81, 129.45, 129.37, 129.17, 127.32, 122.30, and 118.51. IR γmax cm−1 (KBr): 3,032, 2,845, 1,735, 1,660, 1,550, 750. Anal. Calcd for C15H9Cl3O3 (Mwt: 343.59): C, 52.43%; H, 2.64%. Found: C, 52.40%; H, 2.50%.
(Z)-2-(2,4-dichlorophenoxy)-3-(4-bromophenyl)-2-propenoic acid (III)
Pale brown solid. Yield: 35.3%, mp 120°C–125°C. 1H NMR (DMSO-d6) δ: 7.06 (d, J =8.8 Hz, 1H, Ph), 7.35 (d, J =8.9 Hz, 1H, Ph), 7.57 (s, 1H, Ph), 6.99 (d, J =8.5 Hz, 2H), 7.93(d, J =8.3 Hz, 2H), 7.83 (s, 1H, =CH), 13.03 (s, 1H, COOH). 13C NMR (DMSO-d6) δ: 160.66, 151.60, 142.50, 136.12, 133.25, 132.55, 129.95, 129.52, 128.95, 127.45, 126.82, 126.62, and 119.51. IR γmax cm−1 (KBr): 3,035, 2,865, 1,734, 1,660, 1,550, 750. Anal. Calcd for C15H9BrCl2O3 (Mwt: 388.04): C, 46.42%; H, 2.33%. Found C, 46.39%; H, 2.15%.
(Z)-2-(2,4-dichlorophenoxy)-3-(4-fluorophenyl)-2-propenoic acid (IV)
White solid. Yield: 31.25%, mp 115°C–120°C. 1H NMR (DMSO-d6) δ: 7.06 (d, J =8.8 Hz, 1H, Ph), 7.35 (d, J =8.9 Hz, 1H, Ph), 7.57 (s, 1H, Ph), 7.46 (d, J =8.4 Hz, 2H), 7.93 (d, J =8.4Hz, 2H), 7.86 (s, 1H, =CH), 12.94 (s, 1H, COOH). 13C NMR (DMSO-d6) δ: 170.66, 165.47, 160.35, 150.37, 142.42, 133.21, 132.50, 130.55, 129.25, 127.95, 127.65, 126.52, 119.60, 117.85, and 116.75. IR γmax cm−1 (KBr): 3,035, 2,865, 1,737, 1,665, 1,570, 770. Anal. Calcd for C15H9Cl2FO3 (Mwt: 327.14): C, 55.07%; H, 2.77%. Found C, 54.98%; H, 2.63%.
(Z)-2-(2,4-dichlorophenoxy)-3-(4-nitrophenyl)-2-propenoic acid (V)
Yellow solid, Yield: 37%, mp 105°C–110°C. 1H NMR (DMSO-d6) δ: 7.06 (d, J =8.8 Hz, 1H, Ph), 7.35 (d, J =8.9 Hz, 1H, Ph), 7.57 (s, 1H, Ph), 8.06 (d, J =8.5Hz, 2H, Ph), 7.30 (d, J =8.5Hz, 2H, Ph), 7.80 (s, 1H, =CH), 13.2 (s, 1H, COOH). 13C NMR (DMSO-d6) δ: 161.52, 152.11, 150.24, 142.25, 140.02, 129.95, 129.53, 129.37, 129.24, 127.85, 126.41, 124.25, and 119.42. IR γmax cm−1 (KBr): 3,040, 2,955, 1,735, 1,665, 1,570, 775. Anal. Calcd for C15H9Cl2NO5 (Mwt: 354.15): C, 50.87%; H, 2.56%, N, 3.95%. Found C, 50.64%; H, 2.51%, N, 3.72%.
(Z)-2-(2,4-dichlorophenoxy)-3-(4-cyanophenyl)-2-propenoic acid (VI)
White solid. Yield: 32%, mp 143°C–148°C. 1H NMR (DMSO-d6) δ: 7.06 (d, J =8.8 Hz, 1H, Ph), 7.35 (d, J =8.9 Hz, 1H, Ph), 7.57 (s, 1H, Ph), 7.51 (d, J =8.5 Hz, 2H, Ph), 7.65 (d, J =8.5 Hz, 2H, Ph), 7.55 (s, 1H, =CH), 13.03 (s, 1H, COOH). 13C NMR (DMSO-d6) δ: 160.23, 150.35, 142.37, 138.70, 133.36, 130.21, 130.12, 129.23, 127.73, 127.55, 126.45, 119.70, 119.55, and 115.80. IR γmax cm−1 (KBr): 3,045, 2,840, 1,730, 1,665, 1,550, 765. Anal. Calcd for C16H9Cl2NO3 (Mwt: 334.16): C, 57.51%; H, 2.71%, N, 4.19%. Found C, 57.32%; H, 2.47%, N, 4.05%.
(Z)-2-(2,4-dichlorophenoxy)-3-(2,4-dinitrophenyl)-2-propenoic acid (VII)
Yellow solid. Yield: 35.4%, mp 152°C–157°C. 1H NMR (DMSO-d6) δ: 7.06 (d, J =8.8 Hz, 1H, Ph), 7.35 (d, J =8.9 Hz, 1H, Ph), 7.57 (s, 1H, Ph), 8.05 (d, J =8.9 Hz, 1H, Ph), 8.5 (d, J =9.1 Hz, 2H, Ph), 8.3 (s, 1H, Ph), 7.85 (s, 1H, =CH), 13.2 (s, 1H, COOH). 13C NMR (DMSO-d6) δ: 160.56, 153.45, 152.35, 149.39, 142.85, 135.12, 132.73, 130.09, 129.85, 128.51, 127.82, 127.50, 126.41, 120.75, and 119.53. IR γmax cm−1 (KBr): 3,055, 2,940, 1,734, 1,655, 1,540, 740. Anal. Calcd for C15H8Cl2N2O7 (Mwt: 399.14): C, 45.13%; H, 2.02%, N, 7.01%. Found C, 45.02%; H, 1.96%, N, 6.89%.
Molecular modeling
Sequence similarity search
The PDB (protein data bank) search module provided in MOE 2014.09 was used for searching for the best template. Gap start was used as default (−12), gap extent (−2), E-value cutoff (10) E-value accept (1e-012), Z iterations (100), and Z-cutoff (6).
Molecular alignment
Molecular alignment was done using Clustalo tool provided in Uniprot server. The alignment was done between the lanosterol 14α-demethylase of Candida albicans FASTA sequence (uniprot ID: P10613), strain (SC5314/ATCC MYA-2876), and Saccharomyces cerevisiae FASTA sequence (uniprot ID: P10614). Both sequences were from the same ERG11 gene. The length of C. albicans was 528 residues, while that of S. cerevisiae was 530 residues.
Homology modeling
The crystal structure of S. cerevisiae L14αDM (PDB entry code 4LXJ) was used as a template with 62.82% sequence identity.14 S. cerevisiae L14αDM was used as template chain A, and C. albicans L14αDM sequence was used as query. AMBER99 was used as a forcefield, and all settings were kept as default in which strain cutoff 1.5 and distance cutoff 1.2 were used. Heme–porphyrin complex was kept in the new model.
Molecular docking studies with MOE 2014.09
All the compounds were built and saved as moe files. Rigid receptor was used as a docking protocol. Both receptor-solvent were kept as a “receptor”. Triangle matcher was used as a placement method. Two rescorings were computed, rescoring 1 was selected as London dG and rescoring 2 was selected as affinity. Force field was used as a refinement.
Molecular docking studies with Leadit 2.1.2
All compounds were built and saved as mol2 files. The homology model of C. albicans L14α-DM was used for docking. The protein was loaded into Leadit 2.1.2 and the receptor components were chosen by selection of chain A as a main chain, which is complexed with heme–porphyrin and azole inhibitor. Binding site was defined by choosing azole inhibitor as a reference ligand to which all coordinates were computed. Amino acids within a radius of 6.5 Å were selected in the binding site. All chemical ambiguities of residues were left as default. Ligand binding was driven by enthalpy (classic Triangle matching). For scoring, all default settings were restored. Intraligand clashes were computed by using clash factor =0.6. Maximum number of solutions per iteration =200. Maximum number of solution per fragmentation =200. The base placement method was used as a docking strategy. Docking results are tabulated in Figure 2.
 | Figure 2 Possible binding modes of the designed compounds within the Candida albicans L14αDM homology model. |
Antifungal screening
The antifungal screening, minimum inhibitory concentrations (MICs), and IC50 (half maximal inhibitory concentration) calculation for the synthesized compounds was done at the Regional Center for Mycology and Biotechnology, Al-Azhar University, Cairo, Egypt. Eight synthesized compounds and three references standards (miconazole, fluconazole, and amphotericin B) were evaluated for their in vitro antifungal activity against five fungal strains; C. albicans (RCMB 05036), Candida parapsilosis (RCMB 05064), Aspergillus niger (RCMB 02568), Trichophyton rubrum (RCMB 010162), and Trichophyton mentagrophytes (RCMB 010173). MTT micro dilution assay was used for the evaluation of their antifungal activity.15 MIC was determined by using 0.49, 0.98, 1.95, 3.9, 7.81, 15.63, 31.25, 62.5, and 125 μg/mL concentrations.
Results and discussion
The first discovery of azole drugs was after the in vitro screening of 1-phenethyl imidazole derivatives (Figure 3A) against a large number of fungal species. They revealed broad-spectrum antifungal activity. Since then, this scaffold was used to develop more potent antifungal agents.16 Further modifications of the 1-phenethyl imidazole scaffold resulted in the common structure of azole antifungal drugs (Figure 3B). They share common structural features like the azole heme-coordinating group, a halo-substituted aromatic ring that is separated by two carbon atoms from the azole moiety, and a hydrophobic side chain that may be a halo-substituted aromatic ring as well. Most of the well-known antifungal drugs share these common structural features (Figure 4).
 | Figure 3 (A) Chemical structure of the first 1-phenethyl imidazole scaffold. (B) General structure of azole antifungal drugs. |
 | Figure 4 Structures of some azole antifungal drugs. |
In this work, a stepwise design approach was used. Five steps were used to obtain the final proposed design as described in the following sections.
Selection of the heme-coordinating group
L14αDM contains a heme–porphyrin complex in its active site. The azole ring of the antifungal drugs was selected due to its similarity to the imidazole ring of histidine;17 the binding of azole antifungal drugs to L14αDM was reported to be achieved through coordination with the heme–porphyrin complex, such as the binding of econazole (Figure 5).18
 | Figure 5 Econazole–heme coordination. |
It was reported that the carboxylic group could form bidentate coordination with metals like iron. The carboxylic acid group acts as iron chelator, with the following three features: the coordinating atom is an oxygen atom, which is very selective for iron chelation; the bidentate iron-complex is a neutral complex with a favored partition coefficient; and finally, it does not form any polymeric complexes, which can cause problems in crossing cell membranes.19,20 The first aim of this work was to replace the azole moiety in the general structure of the azole antifungal agent (Figure 3B) with another group. Here, the carboxylic group was selected for that purpose due to the features previously mentioned.
Selection of the side chain
The diversity in the structure of the side chain in all azole antifungal drugs encouraged the investigation of the effect of this side chain. Most of the azole drugs have a halo-substituted benzyloxy side chain, such as econazole, isoconazole, and miconazole. The para-substituted halo phenoxy acetic acid moiety was successful in the case of omoconazole. Recently, the 2,4-dichloro phenoxy moiety was reported to have antifungal properties.21 The 2,4-dichloro phenoxy side chain is considered to be a new moiety with promising antifungal activity and it was selected to be in the side chain of the proposed design.
Selective geometry of the inhibitor
Most of the azole antifungal drugs were found to be a (R and S) racemic mixture. Heme coordination is achieved using the (R) isomer.22 By observing the binding of the (R) isomer, we can conclude that both substituted aromatic rings must be oriented in one direction, away from the azole moiety. Some trials were performed to obtain a rigid conformation in which one isomer with the required geometry was present. One of these trials used an oxime group in the structure of miconazole to obtain oxiconazole (Figure 4) with broad-spectrum antifungal activity.23 It is worth noting that the (Z)-isomer of oxiconazole was superior to the (E)-isomer in terms of antifungal activity.
According to these findings from the literature, a proposed structure for an L14αDM inhibitor was designed in which a carboxylic group was used for heme-coordination; the 2,4-dichloro phenoxy moiety was selected as a main side chain, in addition to a substituted aromatic ring. The final selection of the substituted aromatic rings was achieved after a rational docking study for selection of the top ranked compounds for synthesis. The geometry was fixed to the Z-configuration by adding C=C in the two carbon linker between the heme- chelator group and the substituted aromatic ring (Figure 6).
 | Figure 6 Proposed design for antifungal agents with carboxylic heme chelator. |
Chemistry discussion
The synthesis of the proposed structures was achieved using Perkins reaction, which is a reaction between phenylacetic acids or phenoxy acetic acids and appropriate aldehydes.13,24 Here, the 2,4-dichloro phenoxy acetic acid and different aromatic substituted aldehydes were refluxed together in the presence of triethyl amine and acetic anhydride (Figure 1). The reaction resulted in the formation of C=C with respect to the two aryl groups from both the 2,4-dichloro phenoxy acetic acid and the substituted aldehydes in a low yield. The configuration of the product was confirmed by many X-ray crystallographic studies and was reported to be in the Z-configuration.25,26 All compounds showed IR spectra with absorbance bands at 1,685–1,737 for carbonyl group, 1,550–1,600 for C=C phenyl. The most characteristic feature in the 1H-NMR spectra was the C=CH proton as a singlet at δ 7.8–7.95.
Homology modeling
Unfortunately, the crystal structure of L14αDM for C. albicans and most of the fungal species are not available. Many studies reported the homology model of C. albicans L14αDM.27 However, in all previously reported models, the template that was used was from Mycobacterium tuberculosis, and it shared a low percentage of identity with the query sequence. Recently, the crystal structure of S. cerevisiae L14αDM (PDB entry code 4LXJ) was resolved;28 the sequence alignment of both C. albicans and S. cerevisiae L14αDM appeared to share 62.82% identity (Figure 7). In this work, a homology model of C. albicans L14αDM was built in order to predict the orientation and binding of the designed compounds to heme within the active site, as compared to miconazole and fluconazole.
 | Figure 7 The similar residues are in grey color, hydrophobic residues in blue color, and the metal binding site at position 470 of in template and query. |
Model validation
Ramachandran plot
The Ramachandran plot of the built model was generated and analyzed. Residues in the phi-psi core were 90%. Residues in the phi-psi enabled region were found to be 7%. Residues in omega core were found to be 96%, and in the omega enabled region, they were 3%. Conversely, residues in the omega generous region were 0%. The free energy of protein folding was computed and found to be −508.68 (Figure 8).
 | Figure 8 Ramachandran plot for the homology model of Candida albicans L14αDM. |
Molecular docking
Upon building of the model of C. albicans L14αDM, the heme–porphyrin site was identified. All surrounding residues were found to be hydrophobic, such as Tyr 126, Tyr 140, Phe 134, Phe 241, Phe 236, and Thr 130 (Figure 9).
 | Figure 9 Identification of the hydrophobic binding site for docking. |
The binding site of the azole-containing antifungal drugs is a hydrophobic site, which contains a number of hydrophobic residues such as Phe 134, Phe 236, Tyr 126, Tyr 140, and Phe 241. All well-known L14αDM inhibitors (Figure 4) have two halo-substituted aromatic rings. The main reasons for using these halo-substituted aromatic rings is to produce an electron poor ring that will be stabilized by π–π interactions with the hydrophobic residues in the binding pocket. These hydrophobic interactions will facilitate the fixation of the inhibitor within the pocket and enhance its coordinating with heme–porphyrin complex.
The molecular docking study was performed for a variety of compounds with different substitutions of aromatic ring B as shown in Figure 6. Both electron donating groups and electron withdrawing groups were used. The aim was to study the binding modes of the designed compounds and to compute the binding affinity, binding free energy, lipophilic contribution score, clash penalty score, and the ligand entropy conformation score for all compounds. The docking study enabled the prediction of those compounds with high scoring when compared to miconazole and fluconazole, to select the top-ranked compounds, and to correlate that to the observed activity (Table 1).
 | Table 1 Docking results using Leadit 2.1.2 |
For example, the docking results revealed some interesting insights and interactions that could be used in the prediction of binding modes and biological activity. Compounds VIII, IX, X, XI, XII, and XIII, with all electron donating group substitutions (4-OCH3, 3,5-dimethoxy, 3,4,5-trimethoxy, 4-(N, N-dimethyl)-, 4-hydroxy, 4-methyl, respectively), showed high ligand entropy conformation values than those with halo substitution and electron withdrawing substitution (Compounds I–VII). This score was the main reason in decreasing the docking scores of compounds VIII–XIII that ranged from −18 to −17.01 kcal/mol (Table 1).
On the other hand, the calculated affinity score was very high for compound VII (−25.83 kcal/mol) and compound V (−24.49 kcal/mol). The affinity values for compounds I–VII were higher than those of miconazole (−18.19 kcal/mol) and fluconazole (−16.08 kcal/mol). Compounds I–VII showed the same ligand entropy conformation score (2.80), which was much lower than that of miconazole (7.00) and fluconazole (8.40), which indicates favored binding, as shown in Table 1.
The higher value of the ligand entropy conformation score in both miconazole and fluconazole was inversely proportional to the binding affinity. The best value for the lipophilic contribution was found to be −12.60 and −12.44 for the most active compounds (VII and V, respectively). By analysis of the interactions, we can conclude that the Z-configuration of the synthesized compounds was successful in enabling the carboxylic group to chelate with the heme in the heme–porphyrin complex. The 2,4-dichloro phenoxy moiety in all compounds, in addition to the halo and/or an electron-withdrawing aromatic substituted rings, resulted in an electron-poor phenyl ring, and that was the cause of π–π interactions with neighboring hydrophobic residues such as Tyr 126, Tyr 140, Phe 134, Phe 241, and Phe 236. The presence of the para nitro group in compounds V and VII showed an extra hydrogen bond with Thr 130 (Figure 10). The possible binding mode for the top selected compounds is summarized in Figure 2.
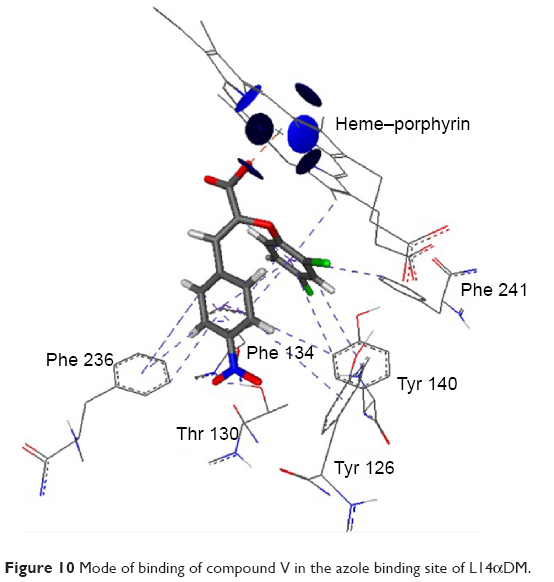 | Figure 10 Mode of binding of compound V in the azole binding site of L14αDM. |
By comparing the binding of compound V and miconazole, we can conclude that miconazole does not form any hydrogen bonding with Thr 130. Also, it has weak π–π interactions with all hydrophobic residues at the site, as compared to compound V. The orientation of the carboxylic group was at exactly the same position as C=N of the imidazole ring, which means that they are competing for heme coordination (Figure 11).
 | Figure 11 Comparison between miconazole (blue color) and compound V (element color) in their binding modes. |
When measuring the distance between N3 of imidazole and the first carbon in the C–C linker, it was found to be 3.55 Å. Conversely, in the case of compound V, the distance between the oxygen atom and the C=C linker was 2.4 Å. This finding can introduce a new structural feature for the design of a potent antifungal drug (Figure 12).
 | Figure 12 Comparison between the distance of heme-chelator group and the first carbon in C–C linker in both miconazole and compound V. |
Antifungal in vitro screening
In this study, two azole antifungal drugs (miconazole and fluconazole) were used as lanosterol 14α-demethylase inhibitor standards to which all the synthesized compounds were compared. In addition, the gold antifungal drug amphotericin B, which is a well-known, broad-spectrum antifungal agent, was also used as a third standard to be used for comparing the spectrum of these synthesized compounds. According to the results of the in vitro antifungal screening, most of the tested compounds showed potent and broad-spectrum antifungal activity against five different fungal strains. For activity against C. albicans, compounds II, V, and VII were the most active, with IC50 values of 8, 7.01, and 7.91 μg/mL, respectively, which were more potent than miconazole, fluconazole, and amphotericin B. Compound V was the most active against C. parapsilosis with an IC50 value of 7.59 μg/mL, and it was also more potent than the reference drugs.
Compounds II, V, and VII showed the highest activity against A. niger with IC50 values of 21.4, 7.25, and 16.6 μg/mL, respectively. On the other hand, antifungal activity against T. rubrum was expressed by compound V (IC50: 31.6 μg/mL) and compound VII (IC50: 33.3 μg/mL). All of these compounds were more potent in their activity against T. rubrum than miconazole (IC50: 36.4 μg/mL), fluconazole (IC50: 72.7 μg/mL), and amphotericin B (IC50: 44.6 μg/mL). Only compound V showed the highest activity against T. mentagrophytes (IC50: 41.6 μg/mL), which was almost the same as that of miconazole but more potent than fluconazole (Table 2).
 | Table 2 In vitro antifungal screening results of the synthesized compounds against different fungal strains |
Conclusion
Seven compounds with the (Z)-substituted propenoic acid scaffold were designed by a rational design approach. The compounds were synthesized, and tested for their in vitro antifungal activity. All compounds showed potent antifungal activity in the submicromolar range against five fungal strains, including dermatophytes. Compound V was the most active with IC50s of 7.01, 7.59, 7.25, 31.6, and 41.6 μg/mL against C. albicans, C. parapsilosis, A. niger, T. rubrum, and T. mentagrophytes, respectively. The use of the carboxylic group was successful in obtaining potent broad-spectrum activity. The use of the Z-configuration was site specific in terms of achieving a unique binding geometry. These agents may be a good start for further biological studies in vivo.
Disclosure
The authors report no conflicts of interest in this work.
References
Donald JA, Manfred EW. Burger’s medicinal chemistry drug discovery. In: Therapeutic Agents. Vol. 2. 5th ed. New York, NY: John Wiley and Sons, Inc; 1996:6309. | ||
Richardson M, Lass-Florl C. Changing epidemiology of systematic fungal infections. Clin Microbiol Infect. 2008;14:5–24. | ||
Heel RC, Brogden RN, Pakes GE, et al. Miconazole: a preliminary review of its therapeutic efficacy in systematic fungal infections. Drugs. 1980;19:7–30. | ||
Marichal P, Koymans L, Willemsens S, et al. Contribution of mutations in the cytochrome P450 14α-demethylase (Erg11p, Cyp 51p) to azole resistance in Candida albicans. Microbiology. 1999;145:2701–2713. | ||
Hof H. A new broad-spectrum azole antifungal: posaconazole-mechanism of action and resistance, spectrum of activity. Mycoses. 2006;49:2–6. | ||
Vanden BH, Marichal P. Mode of action of anti-candida drugs: focus on terconazole and other Ergosterol biosynthesis inhibitors. Am J Obstet Gyrecol. 1991;165:1193–1199. | ||
Podust LM, Poulos TL, Waterman MR. Crystal structure of cytochrome P450 14 alpha sterol demethylase (Cyp 51) from Mycobacterium tuberculosis in complex with azole inhibitors. Proc Natl Acad Sci U S A. 2001;98(6):3068–3073. | ||
Strushkerich N, Usanov SA, Park HV. Structural basis for human cyp51 inhibition by antifungal azoles. J Mol Biol. 2010;397(4):1067–1078. | ||
Gita SR, Manoj VR, Bajaj JS. In silico structure-based design of potent and selective small peptide inhibitor of PTP1B. J Biomol Struct Dyn. 2006;23(4):377–384. | ||
Kenneth MM, Dagmar R, Charles HR. Drug Design: Structure and Ligand-Based Approaches. 1st ed. Cambridge, New York: Cambridge University Press. 2010:181–197. | ||
Molecular Operating Environment (MOE). 2014.09. Montreal, QC, Canada: Chemical Computing Group Inc. | ||
Rarey M, Kramer B, Lengauer T, Klebe G. A fast flexible docking method using an incremental construction algorithm. J Mol Biol. 1996;261(3):470–489. | ||
Domenick P, Erwin S. α-Aryloxy and α-Aryl thiol cinnamic acids. J Am Chem Soc. 1994;69(2):3022–3023. | ||
Brian CM, Thomas MT, Mikhail VK, et al. Architecture of a single membrane spanning cytochrome P450 suggests constraints that orient the catalytic domain relative to a bilayer. Proc Natl Am Sci U S A. 2014;111(10):3865–3870. | ||
Marwa MA. Synergism between clotrimazole and cinnamon oil: an effective (weapon) drug in vitro and in vivo against some multidrug resistance dermatophytes. Egypt Acad J Biol Sci. 2012;4(1):69–83. | ||
Godefroi EF, Heeres J, Van CJ, et al. The preparation and antimycotic properties of derivatives of 1-phenethyl imidazole. J Med Chem. 1969;12:784–791. | ||
Zory VT. Organic Mechanochemistry and Its Practical Applications. Boca Raton, FL: CRC press, Taylor & Francis group; 2006:78. | ||
Protein Data Bank (PDB). Available from: http://www.rcsb.org/pdb/explore/explore.do?structureId=3jus. Accessed June 17, 2015. | ||
Hossein H, Siamak M, Khashayar K. Advances in iron chelation: an update. Expert Opin Ther Pat. 2011;21(6):819–856. | ||
Hider RC, Liu ZD. Emerging understanding of the advantages of small molecules such as hydroxypyridinones in the treatment of iron overload. Curr Med Chem. 2003;10:1051–1064. | ||
Sun NB, Shi YX, Liu XH, et al. Design, synthesis, antifungal activities and 3D-QSAR of new N,N’-diacylhydrazines containing 2,4-dichlorophenoxy moiety. Int J Mol Sci. 2013;14:21741–21756. | ||
Mclean KJ, Lafite P, Levy C, et al. The structure of Mycobacterium tuberculosis CYP125: molecular basis for cholesterol binding in a P450 needed for host infection. J Biol Chem. 2009;284(51):35524–35533. | ||
Mixich G, Thiele K, Ein BZ. Stereospezifischen synthese von antimykotisch wirkasaman imidazolyl oximathern. Arzneim-Forsch/Drug Res. 1997;29:1510–1513. German. | ||
Bagavant GM, Patwardhan BH. Synthesis of α-aryloxy cinnamic acids. Proc Indian Acad Sci. 1975;82(6):224–229. | ||
Cushman MS, Layfayette W, Hamel E. Stilbene derivatives as anticancer agents. United States patent US 5,430,062. 1995. | ||
Guy S, Yacine PJ, Jeam M. A re-investigation of reservatrol synthesis by Perkins reaction. Application to the synthesis of aryl cinnamic acids. Tetrahedron. 2003;59:3315–3321. | ||
Maryam I, Asghar D. Homology modeling of lanosterol 14α-demethylase of Candida albicans and insights into azole binding. Med Chem Res. 2014;23:2890–2899. | ||
Monk BC, Tomasiak TM, Keniya MV, et al. Architecture of a single membrane cytochrome P450 suggests constraints that orient the catalytic domain relative to bilayer. Proc Natl Acad Sci U S A. 2014;111(10):3865–3870. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.