Back to Journals » International Journal of Nanomedicine » Volume 9 » Issue 1
Stable cerasomes for simultaneous drug delivery and magnetic resonance imaging
Authors Cao Z ![]() , Zhu W, Wang W, Zhang C, Xu M, Liu J, Feng S, Jiang Q, Xie X
, Zhu W, Wang W, Zhang C, Xu M, Liu J, Feng S, Jiang Q, Xie X
Received 28 April 2014
Accepted for publication 11 July 2014
Published 5 November 2014 Volume 2014:9(1) Pages 5103—5116
DOI https://doi.org/10.2147/IJN.S66919
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Zhong Cao,1,* Wenjian Zhu,1,* Wei Wang,2 Chunyang Zhang,1 Ming Xu,2 Jie Liu,1 Shi-Ting Feng,3 Qing Jiang,1 Xiaoyan Xie2
1Department of Biomedical Engineering, College of Engineering, 2Department of Medical Ultrasonics, The First Affiliated Hospital of Sun Yat-sen University, Institute of Diagnostic and Interventional Ultrasound, 3Department of Radiology, The First Affiliated Hospital, Sun Yat-sen University, Guangzhou, People’s Republic of China
*These authors contributed equally to this work
Abstract: Magnetic liposomes have been frequently used as nanocarriers for targeted drug delivery and magnetic resonance imaging in recent years. Despite great potentials, their morphological/structural instability in the physiological environment still remains an intractable challenge for clinical applications. In this study, stable hybrid liposomal cerasomes (ie, liposomes partially coated with silica) which can co-encapsulate Fe3O4 nanoparticles and the anticancer drug paclitaxel were developed using thin film hydration method. Compared with the drug loaded liposomes, the paclitaxel-loaded magnetic cerasomes (PLMCs) exhibited much higher storage stability and better sustained release behavior. Cellular uptake study showed that the utilization of an external magnetic field significantly facilitated the internalization of PLMCs into cancer cells, resulting in potentiated drug efficacy of killing tumor cells. The T2 relaxivity (r2) of our PLMCs was much higher than that of free Fe3O4 nanoparticles, suggesting increased sensitivity in T2-weighted imaging. Given its excellent biocompatibility also shown in the study, such dual functional PLMC is potentially a promising nanosystem for effective cancer diagnosis and therapy.
Keywords: MRI, paclitaxel, SPIO, superparamagnetic iron oxide
Introduction
Over the past 2 decades, various diagnostic and drug-delivery systems have been developed for cancer therapy. In the efforts to improve the accuracy of diagnosis/prognosis and to improve the therapeutic efficiency, the joint delivery of therapeutic and diagnostic agents has proven to be a very promising direction.1 The so-called “theranostic strategy” is capable of combining dual functions into one nanomedicinal system; that is, simultaneous drug therapy (eg, chemotherapy), and monitoring of pathological progress and therapeutic efficacy with medical imaging tools such as magnetic resonance imaging (MRI).2,3
MRI has emerged as one of the most powerful techniques for clinical diagnosis of vital diseases such as cancer. The high spatial resolution, noninvasiveness and excellent soft-tissue contrast are the main advantages of MRI over other imaging techniques. The use of contrast agents further enables MRI to detect solid tumors at an early stage. In recent years, superparamagnetic iron oxide (SPIO) as an MRI T2 contrast agent has attracted a great deal of attention due to its desirable properties including high MRI T2 sensitivity.4–6 On the other hand, liposomes have been intensively investigated as a main type of drug-delivery systems being clinically used at present.7 Liposomes have excellent biocompatibility and can be easily modified with facile chemical approaches (eg, PEGylation) to improve their basic properties including sustained drug release and long circulation, or to introduce new functionalities such as tumor active targeting. In addition, incorporation of MRI contrast agents like SPIO into liposomes will yield the theranostic system with combined properties/advantages of both liposomal drug delivery and MRI diagnosis. Recently, iron oxide nanoparticles (NPs)-encapsulated phospholipid vesicles, the so-called magnetic liposomes (MLs), have been reported.8–10 These MLs appeared to be a versatile delivery system possessing good biocompatibility, easy chemical functionalization, and potential to site-specific drug delivery by using magnetic targeting. MLs have also gained much attention because of their effective T2 contrast enhancement in noninvasive MRI application.11–13
Despite many advantages, a major limitation of using MLs in vivo is their low colloidal stability. MLs are prone to aggregation/fusion to form large vesicles in suspension generally, resulting in short circulation time and inclusion leakage after intravenous administration.14 Therefore, the development of more stable MLs is still an urgent need nowadays. To this end, a type of hybrid liposomal “cerasome” bearing a surface cross-linked with the molecularly thin siloxane networks has drawn much attention.15 In our previous work, we developed anticancer drugs-loaded cerasome which exhibited advanced controlled release behavior and much higher stability than the conventional liposomes.16,17 These liposomal nanohybrid cerasomes demonstrated great potential as a promising drug-delivery system, which has driven us to further develop a nanosystem combining the advantages of both cerasome carriers and SPIO magnetic NPs at the present study, aiming at obtaining magnetic-targeted drug-delivery systems with increased stability.
Theranostic nanomedicine, paclitaxel (PTX)-loaded magnetic cerasomes (PLMCs), prepared via a thin film hydration method were evaluated for potential applications in cancer treatments (Figure 1). The PLMCs were characterized by dynamic light scattering (DLS), transmission electron microscopy (TEM), vibrating-sample magnetometer (VSM), and relaxometry measurements. The cell-uptake and cytotoxicity of PLMCs were investigated by confocal laser scanning microscopy and the tetrazolium-based colorimetric (MTT) assay, respectively.
 | Figure 1 Schematic illustration of PTX-loaded magnetic cerasomes. |
Materials and methods
Materials
N,N-dihexadecyl-N-(3-triethoxysilyl)propylsuccinamide (ie, cerasome-forming lipid [CFL]) was synthesized by the procedure in the Supplementary materials (Figure S1 and S2). PTX was obtained from Shanghai Jinhe Bio-Technology (Shanghai, People’s Republic of China). Iron(III) acetylacetonate, triethylene glycol, and Triton-X 100 were purchased from Sigma-Aldrich Co. (St Louis, MO, USA). Dialysis membrane (molecular weight cut-off: 12,000–14,000) was obtained from Spectrum Laboratories (Los Angeles, CA, USA). All organic solvents used in the experiments were of analytic grade. Distilled and deionized water used for vesicle preparation was obtained using a Milli-Q gradient system. Unless otherwise stated, all reagents and chemicals were commercially obtained and used without further purification.
Synthesis of sodium citrate modified Fe3O4 NPs
The hydrophilic Fe3O4 NPs were synthesized as previously reported.18 Briefly, iron(III) acetylacetonate (2 mmol) and triethylene glycol (30 mL) were mixed in a reaction flask by magnetic stirring under argon. The mixture was heated to 280°C and refluxed for 30 minutes. The black mixture was cooled to room temperature, precipitated into acetic acetate, and then magnetically separated to recover a black precipitate. The above procedure was repeated three times. To get the water-soluble Fe3O4 NPs, the purified product was redispersed into 50 mL 0.1 M sodium citrate solution and ultrasonically agitated for 30 minutes. After acetone was added for flocculation, magnetic separation was applied to obtain a black precipitate which was then washed three times with acetone to remove excess sodium citrate. The product was dispersed in deionized water and stored at 4°C prior to use.
Preparation of PLMCs
Co-encapsulation of Fe3O4 NPs and PTX within cerasomes was performed using thin-film hydration.16 Briefly, CFL and PTX were co-dissolved in 5 mL chloroform. After evaporation of chloroform, the obtained thin film was dispersed in 4 mL diluted water containing sodium citrate-modified Fe3O4 at a CFL/PTX ratio of 25/1 (mol/mol), followed by sonication for 5 minutes with a probe-type 60 Sonic Dismembrator (Thermo Fisher Scientific, Waltham, MA, USA) to prepare magnetic cerasomes (MCs) or PLMCs. Fluorescent magnetic cerasomes (FMCs) were prepared by incorporating 3% 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-(7-nitro-2-1,3-benzoxadiazol-4-yl) (NBD-DOPE) into cerasomes. The nonencapsulated Fe3O4 NPs were removed by gel filtration on Sephadex™ G-50 columns.
Characterization of PLMCs
The hydrodynamic diameter and zeta potential of PLMCs were measured by DLS in aqueous medium using a Malvern Zetasizer Nano ZS90 (Malvern Instruments, Malvern, UK) at room temperature. The morphology of PLMCs was analyzed by TEM using a JEM 1,400 microscope. To display the morphology, each sample was placed on a copper grid and then negatively stained by aqueous solution of uranyl acetate. The magnetization of Fe3O4 NPs and PLMCs were measured in their solid state at room temperature using a VSM (Lake Shore 7307). Fourier transform infrared spectroscopy (FTIR) was recorded on a Bruker VERTEX 70 spectrometer in the range between 4,000 cm−1 and 500 cm−1 with a resolution of 2 cm−1. All powder samples were compressed into KBr pellets for the FTIR measurements. Matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF) mass spectrometry was performed on a Bruker ultrafleXtreme equipped with a 337 nm N2 laser in the reflector mode using a 25 kV accelerating voltage. Dithranol (97%; Sigma-Aldrich Co.) was used as matrix.
Determination of Fe3O4 and PTX loading contents and encapsulation efficiencies
To determine the PTX loading content, preweighed freeze-dried PLMCs were redissolved in methanol/water (70/30, v/v) and filtered to remove Fe3O4 NPs if applied. Concentration of PTX was determined on a high-performance liquid chromatography system (1260; Agilent Technologies, Santa Clara, CA, USA) equipped with an Agilent Eclipse XDB-C18 (4.6×150 mm; Agilent Technologies) column and the detector wavelength was set at 227 nm. Mixture of methanol and water (70/30, v/v) was used as the mobile phase at a flow rate of 1.0 mL/min. The area under the peak was calculated by numerical integration (Simpson’s rule). A standard curve was preestablished using known amounts of PTX dissolved in the same methanol/water mixture. The drug-loading content (DLC) and encapsulation efficiency (EE) of the vesicles were evaluated using the following formulas:
|
|

|
|

The SPIO loading contents of PLMCs were determined using a polarized Zeeman Atomic Absorption Spectrophotometer (Hitachi Z-2000 series, Hitachi Ltd, Tokyo, Japan). PLMCs were first weighed before being suspended in 1 M HCl solution to allow for cerasome-forming lipid degradation and complete dissolution of the PLMCs. The iron concentration was then determined at the specific Fe absorption wavelength (248.3 nm) based on a preestablished calibration curve. SPIO loading content was calculated as the ratio of iron oxide over the total weight of PLMCs.
In vitro drug release
In vitro release of PTX from PLMCs was analyzed using a dialysis method at 37°C and was compared with PTX-loaded magnetic liposomes (PLMLs). An aliquot of PLMCs dispersion was put in a dialysis bag, then immersed in 50 mL phosphate-buffered saline (PBS) (pH 7.4) containing 0.1% (v/v) Tween 80 to maintain a good sink condition for poorly water-soluble drugs. The release medium was slowly stirred magnetically. At predetermined time intervals, buffered solution (2 mL each) outside the dialysis bag was removed for analysis and replaced with fresh buffer solution. The release experiment lasted for 100 hours. PTX concentration was analyzed by high-performance liquid chromatography.
Confocal laser scanning microscopy study
The assay was carried out using NBD-DOPE labeled magnetic cerasomes, FMCs. HeLa cells were incubated with various concentrations of FMCs with or without the presence of an external cylindrical sintered N-35 Nd-Fe-B magnet from Ningbo permanent magnetics Co., Ltd., (Ningbo, People’s Republic of China) (Dimension: length =30 mm, height =30 mm, width =30 mm; field strength: ~0.42 T; positioned beneath the cell culture dishes). The cells were stained with 2-(4-amidinophenyl)-6-indolecarbamidine dihydrochloride for 1 minute, and the glass slides were rinsed three times with PBS before assessment on a confocal laser scanning microscope (LSM 510 META; Carl Zeiss Meditec AG, Jena, Germany) using argon laser at an excitation wavelength of 488 nm and an emission wavelength of 543 nm.
In vitro MRI
MRI measurements were performed on a clinical 1.5 T MRI scanner (Magnetom Vision; Siemens, Erlangen, Germany) at room temperature. An 11 cm circular coil (C3; Siemens Healthcare Sector, Erlangen, Germany) was used for all the in vitro MRI studies. Fast spin-echo T1-weighted images were acquired using the following parameters: repetition time (TR)/echo time (TE), 500/15 ms; field of view (FOV), 150 mm × 150 mm; section thickness, 1.5 mm; matrix, 256×256. T2-weighted images were acquired using the following parameters: TR/TE, 2,600/100 ms; FOV, 150 mm; matrix, 256×256; section thickness, 1.5 mm. T2 relaxation data were acquired by using a single-section mixed inversion-recovery spin-echo sequence that was initiated with an inversion recovery pulse (TR/TE, 4,000/160 ms, and inversion time 400 ms) followed by a spin-echo pulse (TR/TE 3,500/20 ms) and a single-section multi-spin-echo (TR/TE 2,000/160 ms). Both sequences were performed with the following parameters: stepped echo time, 20–160 ms for eight steps; echo spacing, 20 ms; FOV, 70 mm; matrix, 256×256; section thickness, 2 mm. In both cases, a circular region of interest was selected in each sample and the values of T2 relaxation times were obtained. The increase in r2 relaxation rates (1/T) with increasing Fe concentration was analyzed by linear least squares regression analysis. T2 relaxivity was calculated from the slope of the linear plots of r2 relaxation rates versus Fe concentration.
Hemolysis assay
Blood compatibility of PLMCs was evaluated by hemolysis assay. Fresh human blood anticoagulated with heparin was donated from a volunteer. The human red blood cells (HRBCs) were collected by centrifugation at 1,500 rpm for 10 minutes and washed several times with PBS until the supernatant became colorless. After the last wash, the HRBCs were diluted five times with PBS. The diluted HRBC suspension (400 μL) was added to 1.2 mL of deionized water as a positive control, 1.2 mL of PBS solution as a negative control, and 1.2 mL of a cerasomal microcapsule solution at various concentrations. The samples were incubated with moderate shaking for 3 hours at 37°C and then centrifuged. The supernatants were measured at 541 nm on a UV-DU730 absorption spectrophotometer to analyze the release of hemoglobin. The percent hemolysis of each sample was calculated by the following formula:
|
|
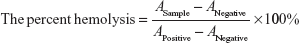
where A is the ultraviolet absorbance of hemoglobin.
In vitro cytotoxicity
After HeLa cells were incubated with PTX, PTX-loaded cerasomes (PLCs), and PLMCs, the cytotoxicity was evaluated by the MTT assay. In the case of PLMCs, the effect of the Nd-Fe-B magnet was also evaluated. The cell culture plate was positioned under external an magnetic field to impose magnetic influence for 2 hours, and then the cells were incubated for another 2 hours without the presence of the magnetic field. In control experiments, the cells were incubated for 4 hours without the presence of a magnetic field. After discarding the supernatants, cells were washed twice with PBS and incubated for another 20 hours in 200 μL of fresh RPMI 1,640 culture medium. Then, 20 μL MTT solution (5 mg/mL in PBS) was added to each well, followed by 4 hours’ incubation. The precipitate was dissolved in 150 μL dimethyl sulfoxide and analyzed on a microplate reader (BIO-680; Bio-Rad Laboratories Inc., Hercules, CA, USA) at 490 nm.
Results and discussion
Preparation and characterization of PLMCs
PLMCs were prepared using the thin-film hydration method. In our previous study, we demonstrated that cerasomes with a liposome-like structure can be developed based upon hydrolyzing the triethoxysilyl groups of CFLs.16 Herein, we further incorporated the sodium citrate modified Fe3O4 NPs and PTX into the hydrophilic inner core and hydrophobic membrane of the cerasomes. TEM analysis shows that the hydrophilic SPIO NPs are monodispersed with uniform morphology and size (Figure 2A). Figure 2B shows the morphology of blank cerasomes, which were negatively stained with uranyl acetate. Apparently, spherical hollow cerasomes with a thin bilayer membrane have been obtained in our study. TEM observation of PLMCs further evidenced the encapsulation of clustered Fe3O4 NPs inside the inner aqueous core of the vesicles (Figure 2C). Since the samples were not stained, only the Fe3O4 NP clusters were shown and thus the size of PLMCs seems to decrease in the TEM image. The lipophilic membrane was clearly shown in the image when PLMCs were negatively stained (Figure 2C insert). It could be clearly seen that the encapsulated Fe3O4 NPs still maintained their initial morphology and size (about 9 nm) (Figure 2). The size and size distribution of blank cerasomes (ie, PTX and blank cerasomes without PTX nor Fe3O4 NP) and PLMCs were analyzed by DLS. The hydrodynamic diameters of blank cerasomes and PLMCs were 135.9±5.6 nm (mean ± standard deviation) and 148.6±7.2 nm (n=3), respectively. TEM and DLS detected the sample in dry state and inside aqueous solution, respectively, which may also lead to the deviation in the detected sizes by two methods. NPs should neither be too small (kidney excretion of particles) nor should they be too big (increased uptake of particles through reticuloendothelial system). These sizes are suitable for in vivo application, since NPs between 10 nm and 150 nm in diameter have been proved to effectively escape fast clearance or uptake by the reticuloendothelial system.19
 | Figure 2 TEM images. |
Different PTX/lipid molar ratios were tried to prepare PLMCs in our experiment and a 1:25 ratio yielded the highest drug EE and drug-loading content up to 86.6% and 4.45%, respectively, which is comparable to other published data.20,21 The loading contents of Fe3O4 NPs in the vesicles was 8.2%. The zeta potential of MLs was −7.8±1.7 mV (mean ± standard deviation, n=3) since the lipid molecules used do not bear a charge. In contrast, MCs possessed a zeta potential of −23.8±2.2 mV (mean ± standard deviation, n=3), due to the negatively charged hydroxyl groups of the polysiloxane networks formed on the surface. It is noteworthy that the surface charge density of vesicles is an important parameter determining their colloidal stability and a mechanism for cellular interactions. For instance, due to their lack of surface charges, neutral liposomes are more prone to aggregation and thus less stable in aqueous medium.
The magnetic responsiveness of PLMCs in solution was visualized by a simple experiment in which a 0.42 T magnet was placed near the glass vials (Figure 3A and B). PLMCs in water deposited quickly on the wall adjacent to the magnet. This observation provides direct evidence that PLMCs possess prompt responsiveness to the external magnetic field. The magnetic properties of the Fe3O4 NPs and PLMCs were examined using a VSM. The two saturation magnetization curves showed no hysteresis loop (Figure 3C), which means that encapsulating Fe3O4 NPs into PLMCs did not change their superparamagnetism. As contrast agents should rapidly relax magnetic moment vectors to generate ideal contrast in MRI signal when the external magnetic field is removed, superparamagnetism is very important for the PLMCs to be applied as MRI probes.22 The saturation magnetizations were 68.4 Fe emu·g−1 for Fe3O4 NPs and 57.8 Fe emu·g−1 for PLMCs. Loss of saturation magnetization is due to the presence of the cerasome bilayer surrounding the magnetic cores. Although the saturation magnetization value decreased about 10%, the magnetization is still strong enough for the bioseparation and MRI test. Both the superparamagnetism and relatively high magnetization make the PLMCs suitable for magnetic-targeted or MRI-visible drug delivery. The FTIR spectrum of cerasome shows a strong peak at 1,095 cm−1 ascribing to the characteristic stretching vibration of the Si–O–Si bond, directly evidencing the formation of the siloxane frameworks (Figure 4). PTX shows the characteristic bands of the acylamino groups at 1,632 cm−1. Furthermore, the peaks at 1,723 cm−1 and 711 cm−1 were assigned to the vibration absorption bands of the carbonyl groups and aromatic ring of PTX, respectively. After PTX and Fe3O4 NPs were incorporated into cerasomes, the peaks at 578 cm−1 and 1,723 cm−1 appeared due to the characteristic vibrations of Fe–O and C=O bonds, indicating the presence of Fe3O4 NPs and PTX. The above FTIR analyses strongly demonstrated the successful co-encapsulation of PTX and Fe3O4 within cerasomes.
 | Figure 3 Magnetic properties of PLMCs. |
 | Figure 4 FTIR spectra of PLMCs. |
Storage stability and drug-release studies
Cerasomes have an inorganic silicate framework with a high degree of polymerization, which make them much more stable than liposomes in aqueous solution. This notion was demonstrated by determining the size change and drug loss over a prolonged storage time of 1 month at room temperature. As shown in Figure 5A, the size of PLMCs detected by DLS remained fairly constant during this period. On the contrary, the diameter of PLMLs increased significantly from less than 200 nm to 2.26 μm, most likely due to the aggregation or the fusion of liposomes.17 This enhanced stability in aqueous medium is believed to be a highly desirable property for the potential in vivo application of cerasomes, since it is well known that the size of nanocarriers is one of the key parameters affecting the dosage, targeting, and rate of clearance from the body.
 | Figure 5 Physical stability tests and in vitro drug release. |
Resistance of cerasomes to surfactant-induced membrane dissolution was evaluated by DLS. Triton X-100 (TX-100) was added to the suspension at various surfactant/lipid molar ratios, and then the hydrodynamic diameters of the cerasomes were quantified (Figure 5B). The DLS analysis revealed almost constant size of PLMCs against 40 equivalents of TX-100. In contrast, liposomes that were not well integrated into the polysiloxane networks were removed from the vesicles by the addition of a small amount of TX-100. The excellent morphological stability of cerasomes was attributed to the formation of siloxane networks on their surfaces. Consistent results indicating better stability of cerasomes were also obtained in examining drug loss in sample storage. As shown in Figure S3, PLMLs in aqueous solution only retained 21.26% of their initial drug content after 1 month. In contrast, PLMCs appeared much more potent in preventing drug loss in the hydrated state. That is, only 24.26% PTX leakage in 1 month’s storage. Furthermore, the MALDI-TOF mass spectrometry (Figure S4) of PLMCs showed a wide range of lipid oligomers from monomer to nonamer. As listed in Table S1, all the detectable lipid oligomer species of PLMCs have molecular weights consistent with the calculated ones. These results further directly evidence that the formation of siloxane network on the vesicular surface of PLMCs has contributed to their high stability which was not affected by Fe3O4 NP embedment. The marked difference in morphological stability between the MCs and the conventional MLs is expectably beneficial to their practical applications.
Subsequently, kinetics of the drug release from MCs was assessed in vitro by dialysis in physiological solution (PBS) and PTX concentration in the dialysate was measured (Figure 5C). As a control study, release of free PTX out of the dialysis bag completed in 10 hours. PTX release from PLMLs is very fast. Approximately 92.32% of PTX was released within 22 hours. In comparison, only 61.26% and 44.57% of the free PTX was released out of the dialysis bag over 100 hours. PTX release was sustained, not completed in 100 hours. Approximately 83.9% and 66.2% PTX was released from PLMCs and PLCs, respectively, within 200 hours. PLMCs released PTX slightly faster than PLCs. The decelerated release of PTX from the two cerasomes can be attributed to their increased stability compared to PLMLs. Moreover, the siloxane networks may have partially blocked the drug release channels. Nevertheless, the siloxane network is not so highly developed on the cerasome surface. The MALDI-TOF mass spectrometry (Figure S4) of PLMCs proved that a majority of lipid oligomers were monomers. This effect perturbs the membrane structure and may induce the formation of pores that are sufficiently large to allow for the leakage of the PTX contents. Sustained drug release in cancer therapy is important not only for achieving prolonged anticancer efficacy but also for preventing cancer from relapsing and developing drug resistance.23 The co-loading of Fe3O4 NPs and sustained PTX release profile make the PLMCs a promising functional nanomaterial for magnetic-controllable cancer therapy.
In vitro MRI
The MRI contrast-enhancing capability of PLMCs with various iron concentrations was evaluated and compared with that of the sodium citrate modified Fe3O4 NPs with the same iron concentrations using T2-weighted MRI. As shown in Figure 6A, the signal intensity of the MR imaging decreased with the increase of the iron concentration for both samples. Figure 6B shows that the T2 relaxation time decreases as the iron concentration increases, and the trend is fitted well by a linear line for both samples. The transverse-relaxivity (r2) value for the PLMCs reached 53.45 mM Fe−1s−1, which was remarkably higher than that for the sodium citrate-modified Fe3O4 NPs (9.83 mM Fe−1s−1). The high transverse relaxivity which indicates good MRI T2 detection sensitivity is attributed to the clustering of Fe3O4 NPs inside the inner core of cerasomes (Figure 2C), which raises the cross-sectional area.
 | Figure 6 Magnetic resonance imaging properties of PLMCs and Fe3O4 NPs. |
Cellular uptake study
The intracellular delivery of cerasomes was assessed by monitoring cell-uptake of the fluorescently (NBD-DOPE) labeled MCs. HeLa cells incubated with the nonmagnetic fluorescent cerasomes (FCs) exhibited negligible green fluorescence even after 4 hours’ incubation, indicating very low level of cell-uptake of FCs (Figure 7). This result may be attributed to the fact that the negatively charged FCs could not effectively bind to the likewise negatively charged cell membrane. In comparison, at the same incubation time, the fluorescence of cells treated with FMCs appeared slightly stronger. The surface charges of FCs and FMCs were −26.6±4.2 mV and −21.8±2.6 mV, respectively. This change in zeta potential upon encapsulation of Fe3O4 NPs may contribute to the cell-uptake difference between FCs and FMCs. Gravity-driven adhesion to cell membrane is assumed to be another contributor to the cell-uptake, which somewhat facilitated endocytosis. Excitingly, the presence of an external magnetic field (~0.42 T) greatly potentiated the internalization of FMCs into HeLa cells. In this case, very strong green fluorescence was observed in cytoplasm at 2 hours’ incubation time. Cell-uptake of FMCs was further increased notably when the cell incubation with FMCs was prolonged to 4 hours. The above data reveal that the PLMC represents a promising candidate for magnetic-targeted drug delivery into the tumorous sites and cells, which may enable more effective cancer therapy.
 | Figure 7 CLSM images of HeLa cells treated with NBD-DOPE-labeled MCs for 2 hours and 4 hours. |
Hemolysis assay and cell growth inhibition studies
The hemocompatibility and cytotoxicity of MCs are critical to their in vivo application as a novel drug carrier. We investigated the hemolytic behavior of PLMCs at various concentrations of HRBCs. Our study set zero and 100% hemolysis when incubating the HRBCs with PBS and deionized water, respectively. The hemolytic effect on HRBCs was determined by measuring the absorbance of supernatant at 541 nm by ultraviolet–visible spectroscopy. As expected, the hemolysis percentage of HRBCs is positively related to the concentration of PLCs and PLMCs over a broad range of 10–400 μM (Figure 8). The PLMCs did not exhibit appreciable hemolysis up to 400 μM. At this concentration, only 2.2% hemolysis was exhibited on exposure for 3 hours.
 | Figure 8 Hemolysis assay for magnetic cerasomes at different concentrations. |
The cytotoxicity of MCs was assessed using the MTT assay. No appreciable cell growth inhibition was observed when HeLa cells were exposed to drug-free MCs at various lipid concentrations up to 400 μM no matter whether the external magnetic field existed or not (Figure 9A), indicating excellent biocompatibility of the carrier itself. Since potency of the delivered PXT was evaluated at MC concentrations less than 16 μM (CFL/PTX ratio of 25/1 mol/mol), cytotoxicity caused by the carrier itself is thus negligible in HeLa cells. The four PTX-containing formulations exhibited obvious cytotoxicity in a PTX-concentration-dependent manner in HeLa cells (Figure 9B). Free PTX and PLCs (ie, PTX-loaded but SPIO-free cerasomes) induced relatively low cytotoxicity. At the highest PTX concentration (1.0 μM), the cell viabilities were 52.67% and 63.89% for free PLMCs and PLCs, respectively. In comparison, PTX showed slightly higher cytotoxicity than PLCs and PLMCs (44.67% cell viability at 1.0 μM PTX). Notably, a significant increase in cytotoxicity was observed for PLMCs when an external magnetic field (~0.42 T) was applied during the cell culture. The cell viability was decreased to as low as 18.28% at the PTX concentration of 1.0 μM. This result clearly evidenced the magnetic-targeted delivery event, which essentially enhanced the endocytosis of PLMCs and led to the highly effective growth inhibition of cancer cells. The above cytotoxicity data are consistent with the results obtained in the cell-uptake studies. In this magnetic-targeting strategy, the external magnetic field is expected to guide the accumulation of PLMCs in tumor tissues and facilitate their internalization into tumor cells.24
 | Figure 9 Cytotoxicity study of PTX-free cerasomes and PTX-loaded cerasomes compared to PTX in human HeLa cells. |
Conclusion
PLMCs with high stability in aqueous solution, sustained drug release profile, and MRI capability have been successfully developed by loading PTX and Fe3O4 NPs into the cerasomal lipophilic membrane and aqueous inner core, respectively. Magnetic field-guided delivery of the obtained PLMCs enabled much more efficient uptake of cerasomes by tumor cells, significantly enhancing the cell-killing effect of the delivered anticancer drug PTX. Moreover, the PLMCs displayed much higher MRI T 2 sensitivity than free Fe3O4 NPs, which makes them a very promising multifunctional platform for simultaneous magnetic-targeted chemotherapy and MRI diagnosis of cancer. Further in vivo studies to explore this potential are currently under way in our laboratory.
Acknowledgments
This work was supported by National Natural Science Foundation of China (81101142, 81301238, and 51103183), Natural Science Foundation (S2011040005922), and the Key Laboratory of Sensing Technology and Biomedical Instruments (2011A060901013) of Guangdong province.
Disclosure
The authors report no conflicts of interest in this work.
References
Akbarzadeh A, Mikaeili H, Zarghami N, Mohammad R, Barkhordari A, Davaran S. Preparation and in vitro evaluation of doxorubicin-loaded Fe3O4 magnetic nanoparticles modified with biocompatible copolymers. Int J Nanomedicine. 2012;7:511–526. | ||
Liu T, Qian Y, Hu X, Ge Z, Liu S. Mixed polymeric micelles as multifunctional scaffold for combined magnetic resonance imaging contrast enhancement and targeted chemotherapeutic drug delivery. J Mater Chem. 2012;22:5020–5030. | ||
Ma Y, Liang X, Tong S, Bao G, Ren Q, Dai ZF. Gold nanoshell nanomicelles for potential magnetic resonance imaging, light-triggered drug release, and photothermal therapy. Adv Funct Mater. 2013;23(7): 815–822. | ||
Ren T, Liu Q, Lu H, Liu H, Zhang X, Du J. Multifunctional polymer vesicles for ultrasensitive magnetic resonance imaging and drug delivery. J Mater Chem. 2012;22:12329–12338. | ||
Cheng D, Hong G, Wang W, et al. Nonclustered magnetite nanoparticle encapsulated biodegradable polymeric micelles with enhanced properties for in vivo tumor imaging. J Mater Chem. 2011;21:4796–4804. | ||
Wahajuddin, Arora S. Superparamagnetic iron oxide nanoparticles: magnetic nanoplatforms as drug carriers. Int J Nanomedicine. 2012;7: 3445–3471. | ||
Torchilin VP. Recent advances with liposomes as pharmaceutical carriers. Nat Rev Drug Discov. 2005;4(2):145–160. | ||
Frascione D, Diwoky C, Almer G, et al. Ultrasmall superparamagnetic iron oxide (USPIO)-based liposomes as magnetic resonance imaging probes. Int J Nanomedicine. 2012;7:2349–2359. | ||
Skouras A, Mourtas S, Markoutsa E, et al. Magnetoliposomes with high USPIO entrapping efficiency, stability and magnetic properties. Nanomedicine. 2011;7(5):572–579. | ||
Amstad E, Kohlbrecher J, Müller E, Schweizer T, Textor M, Reimhult E. Triggered release from liposomes through magnetic actuation of iron oxide nanoparticle containing membranes. Nano Lett. 2011;11(4): 1664–1670. | ||
Mitchell N, Kalber TL, Cooper MS, et al. Incorporation of paramagnetic, fluorescent and PET/SPECT contrast agents into liposomes for multimodal imaging. Biomaterials. 2013;34(4):1179–1192. | ||
Faria MR, Cruz MM, Gonçalves MC, Carvalho A, Feio G, Martins MB. Synthesis and characterization of magnetoliposomes for MRI contrast enhancement. Int J Pharm. 2013;446(1–2):183–190. | ||
Mikhaylov G, Mikac U, Magaeva AA, et al. Ferri-liposomes as an MRI-visible drug-delivery system for targeting tumours and their microenvironment. Nat Nanotechnol. 2011;6(9):594–602. | ||
Wang B, Zhang L, Bae SC, Granick S. Nanoparticle-induced surface reconstruction of phospholipid membranes. Proc Natl Acad Sci USA. 2008;105(47):18171–18175. | ||
Katagiri K, Hashizume M, Ariga K, Terashima T, Kikuchi J. Preparation and characterization of a novel organic-inorganic nanohybrid “cerasome” formed with a liposomal membrane and silicate surface. Chemistry. 2007;13(18):5272–5281. | ||
Cao Z, Ma Y, Yue X, Li S, Dai Z, Kikuchi J. Stabilized liposomal nanohybrid cerasomes for drug delivery applications. Chem Commun (Camb). 2010;46(29):5265–5267. | ||
Jin Y, Yue X, Zhang Q, Wu X, Cao Z, Dai Z. Cerasomal doxorubicin with long-term storage stability and controllable sustained release. Acta Biomater. 2012;8(9):3372–3380. | ||
Wan J, Cai W, Meng X, Liu E. Monodisperse water-soluble magnetite nanoparticles prepared by polyol process for high-performance magnetic resonance imaging. Chem Comm (Camb). 2007;47:5004–5006. | ||
Davis ME, Chen Z, Shin DM. Nanoparticle therapeutics: an emerging treatment modality for cancer. Nat Rev Drug Discov. 2008;7(9): 771–782. | ||
Cai L, Wang X, Wang W, et al. Peptide ligand and PEG-mediated long-circulating liposome targeted to FGFR overexpressing tumor in vivo. Int J Nanomedicine. 2012;7:4499–4510. | ||
Yang T, Cui FD, Choi MK, et al. Enhanced solubility and stability of PEGylated liposomal paclitaxel: in vitro and in vivo evaluation. Int J Pharm. 2007;338(1–2):317–326. | ||
Sahoo Y, Goodarzi A, Swihart MT, et al. Aqueous ferrofluid of magnetite nanoparticles: fluorescence labeling and magnetophoretic control. J Phys Chem B. 2005;109(9):3879–3885. | ||
Nooter K, Stoter G. Molecular mechanisms of multidrug resistance in cancer chemotherapy. Pathol Res Pract. 1996;192(7):768–780. | ||
Kim J, Lee JE, Lee SH, et al. Designed fabrication of a multifunctional polymer nanomedical platform for simultaneous cancer-targeted imaging and magnetically guided drug delivery. Adv Mater. 2008; 20(3):478–483. |
Supplementary materials
Stable cerasomes for simultaneous drug delivery and magnetic resonance imaging
Synthesis of dihexadecylamine
1-Bromohexadecane (19.8 g, 64 mM) and potassium carbonate (27.3 g, 198 mM) were added to a stirred solution of n-hexadecylamine (30.7 g, 130 mM) in ethanol (300 mL). After being refluxed and stirred for 180 hours, the hot mixture was filtered and the filtrate was cooled down to room temperature. The precipitate was then purified via recrystallization from ethanol and hexane, which yielded 14.85 g (50.2%). 1H nuclear magnetic resonance (NMR) (400 MHz, CDCl3, 25°C, tetramethylsilane [TMS]): d=0.88 (t, J=6.6 Hz, 6H, NCH2CH2(CH2)13CH3), 1.25 (m, 52H, NCH2CH2 (CH2)13CH3), 1.59 (m, 4H, NCH2CH2(CH2)13CH3), 2.64 (t, 4H, NHCH2CH2(CH2)13CH3), 3.64 (m, 1H, NH).
Synthesis of N,N-dihexadecylsuccinamic acid
Dihexadecylamine (6.44 g, 3.03 mM) and succinic anhydride (1.29 g, 12.9 mM) were added to dry tetrahydrofuran (50 mL) and heating was applied to facilitate the dissolving process. The solution was stirred for 24 hours at room temperature. The solvent was evaporated in vacuo and the crude product was dissolved in dichloromethane (50 mL). The solution was then washed with 10% aqueous citric acid and then saturated aqueous sodium chloride. After removing residual water using phase separation filter paper, the solvent was evaporated in vacuo. Subsequent recrystallization from acetonitrile yielded white powder (3.48 g, 95.6%). 1H NMR (400 MHz, CDCl3, 25°C, TMS): d=0.88 (t, J=6.6 Hz, 6H, NCH2CH2(CH2)13CH3), 1.26 (m, 52H, NCH2CH2(CH2)13CH3), 1.54 (m, 4H, NCH2CH2(CH2)13CH3), 2.69 (m, 4H, HOCO(CH2)2CON), 3.23 (t, 2H, J=7.8 Hz, NCH2CH2(CH2)13CH3), 3.32 ppm (t, 2H, J=7.8 Hz, NCH2CH2 (CH2)13CH3).
Synthesis of N,N-dihexadecyl-N-(3-triethoxysilyl)propylsuccinamide (cerasome-forming lipid)
1-(3-Dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (EDC) (0.71 g, 3.7 mM) was added at 0°C to a solution of N,N-dihexadecylsuccinamic acid (1.75 g, 3.09 mM) in dry dichloromethane (50 mL). After 15 minutes of stirring, 3-aminopropyltriethoxysilane (0.72 g, 4.01 mM) was added to the solution. The mixture was stirred for 6 hours at 0°C and subsequently for another 12 hours at room temperature. The solvent was then evaporated in vacuo and the crude product was purified by column chromatography. Impurities were eluted with ethyl acetate/chloroform 1:9 and then with 100% ethyl acetate, which resulted in a colorless oil (1.56 g, 65.68%). 1H NMR (400 MHz, CDCl3, 25°C, MS): d=0.59 (t, J=8.4 Hz, 2H, SiCH2), 0.85 (t, J=6.6 Hz, 6H, NCH2CH2(CH2)13CH3), 1.20 (t, J=7.3 Hz, 9H, SiOCH2CH3), 1.22–1.25 (m, 52H, NCH2CH2 (CH2)13CH3), 1.60 (br, 6H, NCH2CH2 (CH2)13CH3, SiCH2CH2CH2NH), 2.51 (t, J=6.6 Hz, 2H, NCOCH2), 2.52 (t, J=6.6 Hz, 2H, NCOCH2), 3.19–3.29 (m, 6H, NCH2CH2 (CH2)13CH3, SiCH2CH2CH2NH), 3.80 (q, J=7.3 Hz, 6H, SiOCH2CH3), 6.40 ppm (br,1H, NHCO); 13C NMR (100 MHz, CDCl3, 25°C, TMS): d=172.51,171.33, 58.38, 47.96, 46.25, 41.95, 31.91, 31.8, 29.69, 29.65, 29.61, 29.56, 29.43, 29.35, 28.93, 27.79, 27.08, 26.94, 22.91, 22.68, 18.28, 14.14, 7.76 ppm.
 | Table S1 Detectable species of lipid oligomers for PLMCs as evaluated by MALDI-TOF MS |
 | Figure S1 Synthetic procedures of the cerasome-forming lipid. |
 | Figure S2 Characterization of cerasome-forming lipid. |
 | Figure S3 PTX retention of PLMCs and PLMLs after storage for 1 month. |
 | Figure S4 MALDI-TOF MS spectrum of PLMCs. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.