Back to Journals » International Journal of Nanomedicine » Volume 12
Spermidine-mediated poly(lactic-co-glycolic acid) nanoparticles containing fluorofenidone for the treatment of idiopathic pulmonary fibrosis
Authors Tang J, Li JM, Li G, Zhang HT ![]() , Wang L
, Wang L ![]() , Li D, Ding JS
, Li D, Ding JS
Received 27 April 2017
Accepted for publication 18 July 2017
Published 8 September 2017 Volume 2017:12 Pages 6687—6704
DOI https://doi.org/10.2147/IJN.S140569
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Linlin Sun
Jing Tang,1 Jianming Li,2 Guo Li,2 Haitao Zhang,2 Ling Wang,3 Dai Li,4 Jinsong Ding2
1School of Pharmaceutical Sciences, Changsha Medical University, 2Xiangya School of Pharmaceutical Sciences, Central South University, Changsha, 3Key Laboratory of Drug Targeting and Drug Delivery Systems, West China School of Pharmacy, Sichuan University, Chengdu, 4Xiangya Hospital, Central South University, Changsha, China
Abstract: Idiopathic pulmonary fibrosis is a progressive, fatal lung disease with poor survival. The advances made in deciphering this disease have led to the approval of different antifibrotic molecules, such as pirfenidone and nintedanib. An increasing number of studies with particles (liposomes, nanoparticles [NPs], microspheres, nanopolymersomes, and nanoliposomes) modified with different functional groups have demonstrated improvement in lung-targeted drug delivery. In the present study, we prepared, characterized, and evaluated spermidine (Spd)-modified poly(lactic-co-glycolic acid) (PLGA) NPs as carriers for fluorofenidone (AKF) to improve the antifibrotic efficacy of this drug in the lung. Spd-AKF-PLGA NPs were prepared and functionalized by modified solvent evaporation with Spd and polyethylene glycol (PEG)-PLGA groups. The size of Spd-AKF-PLGA NPs was 172.5±4.3 nm. AKF release from NPs was shown to fit the Higuchi model. A549 cellular uptake of an Spd–coumarin (Cou)-6-PLGA NP group was found to be almost twice as high as that of the Cou-6-PLGA NP group. Free Spd and difluoromethylornithine (DFMO) were preincubated in A549 cells to prove uptake of Spd-Cou-6-PLGA NPs via a polyamine-transport system. As a result, the uptake of Spd-Cou-6-PLGA NPs significantly decreased with increased Spd concentrations in incubation. At higher Spd concentrations of 50 and 500 µM, uptake of Spd-Cou-6-PLGA NPs reduced 0.34- and 0.49-fold from that without Spd pretreatment. After pretreatment with DFMO for 36 hours, cellular uptake of Spd-Cou-6-PLGA NPs reached 1.26-fold compared to the untreated DFMO group. In a biodistribution study, the drug-targeting index of Spd-AKF-PLGA NPs in the lung was 3.62- and 4.66-fold that of AKF-PLGA NPs and AKF solution, respectively. This suggested that Spd-AKF-PLGA NPs accumulated effectively in the lung. Lung-histopathology changes and collagen deposition were observed by H&E staining and Masson staining in an efficacy study. In the Spd-AKF-PLGA NP group, damage was further improved compared to the AKF-PLGA NP group and AKF-solution group. The results indicated that Spd-AKF-PLGA NPs are able to be effective nanocarriers for anti–pulmonary fibrosis therapy.
Keywords: idiopathic pulmonary fibrosis, fluorofenidone, spermidine, polyamine transport system, nanoparticles
Introduction
Idiopathic pulmonary fibrosis (IPF) is a chronic, progressive, irreversible, and fatal lung disease, with a median survival time of 3–5 years from diagnosis.1,2 The disease occurs primarily in older adults, predominantly males.3 IPF is characterized by usual interstitial pneumonitis and progressive interstitial fibrosis caused by excessive extracellular matrix deposition.4,5 The course of IPF is highly unpredictable, but risk factors associated with IPF may include smoking, environmental exposures, gastroesophageal reflux disease, commonly prescribed drugs, diabetes mellitus, infectious agents, and genetic factors.1,6,7 Despite IPF being classified as a rare disease, its health-care costs and social burden are significant.8
Over the last few decades, many conventional therapeutic concepts on IPF have proved to be unsuccessful. Corticosteroids and immunosuppressants are usually the main treatment for IPF, but the therapeutic benefits of these agents are limited, with poor final outcome.9 Immunomodulation by azathioprine and cyclophosphamide did not show sufficient benefit.8 Lung transplantation may be the final choice of medical therapy for refractory patients with IPF.10 Despite the majority of therapies so far having disappointing results, there is a new antifibrotic drug, pirfenidone (PD), which has shown positive effects on reducing mortality in IPF patients.3,11 PD (5-methyl-1-phenyl-2-[1H]-pyridone) was the first antifibrotic drug tested specifically for use in IPF, starting in 1999.12 It has been approved in multiple countries and regions (including Europe, Japan, Canada, and the US).13 It is presumed that pyridone agents are effective drugs for IPF.14,15
Fluorofenidone (AKF; 5-methyl-1-[3-fluorophenyl]-2-[1H]-pyridone) which has been synthesized by the Department of Pharmacochemistry, School of Pharmaceutical Sciences, Central South University (Changsha, China), has a similar chemical structure to PD.16,17 It can reduce fibroblast growth and collagen synthesis of different organs.18–21 Furthermore, AKF is able to attenuate bleomycin-induced experimental IPF by reducing inflammation, accelerating the production of caveolin 1, inhibiting the phosphorylated MAPK-signaling pathway, and limiting the fibrosis cytokine TGF-β.22 However, the curative power of AKF in the lung has been limited by its unsatisfactory pharmacokinetic properties, which include a short half-life (2 hours), as well as rapid absorption and extensive distribution.17 Therefore, improvement of AKF lung concentration is a high-priority requirement in treating IPF. One of the ideal approaches is to use nanoscale vehicles to improve the pharmacokinetic properties of AKF, and then antifibrotic capability could be extended.
At present, lung-targeted drug-delivery systems include microparticles23,24 (microspheres and microcapsules) and liposomes,25,26 and intravenous injection and pulmonary inhalation are the main administration routes.27 Encapsulating antifibrotic drugs within biodegradable polymers to form microspherical particles has been reported in a lung-targeting drug-delivery system.28 Great attention has been paid to the use of polymeric nanoparticles (NPs) as delivery vehicles for lung-disease drugs, and some studies have suggested that nanocarriers can significantly enhance antifibrotic drug efficacy.29–32 Local delivery of biodegradable PD NPs has exhibited high dispersibility and suitable distribution for inhalation therapy, with sustained lung delivery and enhanced antifibrotic efficacy.29 Nevertheless, to succeed pulmonary drug-delivery must overcome several critical and unique challenges, including toxicological evaluation of materials and mucociliary clearance.30–32 Compared with inhalation, the intravenous route is valid and efficient, which ensures the accuracy of individual doses. The nanoscale dimensions of drug-delivery systems allow entry into blood circulation directly, avoiding degradation by enzymes on the alveolar membrane.25 Enhancement of drug accumulation in lung tissue by active targeting could improve local therapeutic efficacy and reduce systemic adverse reactions.24,33–35
Among the various kinds of polymer-based NPs, poly(lactic-co-glycolic acid) (PLGA)-based formulations are considered ideal and safe vehicles for different drugs.36,37 In this regard, NPs based on amphiphilic copolymers of polyethylene glycol (PEG; as a hydrophilic) and PLGA have already been approved by the US Food and Drug Administration (FDA), and PEG-PLGA copolymers are safe and nontoxic after hydrolysis in vivo.38 In former work at our laboratory, we found that AKF-loaded PLGA microspheres improved the curative effect of AKF in treating acute lung injury via intravenous administration.39 It can be concluded that the extent of drug accumulation in the lung is closely related to therapeutic efficacy, while it is worth noting that large particles might cause pulmonary capillary embolization, which induces severe side effects.30,40 Therefore, intravenous injection of NPs is an ideal strategy to realize lung-targeted delivery of a drug and strengthen its efficacy.41–44 Based on this evidence, PLGA-NPs releasing AKF are proposed as a potential therapeutic measure for IPF. Furthermore, through the conjugation of a targeting agent, such as truncated bFGF peptide28 or aptamer45 to an NP surface, an active-targeting effect could be established to modify the pharmacokinetics of drugs and further enhance therapeutic efficiency.
As targeting ligands, polyamines (PAs), such as putrescine, spermine, and spermidine (Spd), which bind to different compounds, have been investigated on account of their therapeutic potential.46,47 PA transport is an important regulatory mechanism of intracellular PA content by cells.48 A large study showed that the lung has high capability in transporting PAs, and PAs were selectively accumulated into type II pneumocytes in reverse concentration via the polyamine-transport system (PTS).49 Owing to the unique characteristics of PAs and preferential properties of PTS as a receptor, Spd is an ideal candidate for PA-mediated targeting for NPs into type II pneumocytes in treating lung disease.50,51 Until now, PA-mediated nanocarriers for lung targeting have not been reported.
To test our hypothesis, we explored the feasibility of using Spd-modified PEG-PLGA NPs as a lung-targeted delivery system for AKF. Targeting NPs were comprehensively characterized in terms of particle size, ζ-potential, particle morphology, drug-encapsulation efficiency (EE), drug-loading capacity (LC), and in vitro drug release. Targeting ability in vitro was characterized by cellular uptake on A549 lung cancer cells. Biodistribution and antifibrotic efficacy in vivo were identified by comparing AKF solution and AKF-PLGA NPs.
Materials and methods
Materials
PLGA (average molecular weight [MW] 41,000 Da, lactic acid:glycolic acid 50:50) was obtained from Shandong Medical Instrument Research Institute (Shandong, China). NH2-PEG-COOH (MW 3,400 Da) was obtained from Seebio Biotech (Shanghai, China). The 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (EDC) and N-hydroxysuccinimide (NHS) were obtained from Qiyun Biotech (Guangzhou, China). Spd, polyvinyl alcohol (87%–90% hydrolyzed, MW 30,000–70,000 Da), 2-iminothiolane hydrochloride (ITL), 4-dimethylaminopyridine, 2-(N-morpholino) ethanesulfonic acid (MES), and MTT were obtained from Sigma-Aldrich (St Louis, MO, USA). A bicinchoninic acid (BCA) protein-assay kit was obtained from Dingguo Changsheng Biotech (Beijing, China). Dithiothreitol (DTT) and coumarin (Cou)-6 were obtained from Aladdin Biological Technology (Xi’an, China). L+-Cysteine was obtained from Xiya Reagent Chemical (Chengdu, China). AKF (MW 203.22 Da, purity >99%, lot 160704) was synthesized by the School of Pharmaceutical Sciences, Central South University (Changsha, China). PD was obtained from Meilun Biotech (Dalian, China). Paraquat (PQ) was obtained from Syngenta (Basel, Switzerland). 1,1′-dioctadecyl-3,3,3′,3′-tetramethyl indotricarbocyanine iodide (DiR) was obtained from AAT Bioquest (Sunnyvale, CA, USA). DMEM and fetal bovine serum (FBS) were obtained from Thermo Fisher Scientific (Waltham, MA, USA). Trypsin–EDTA, penicillin–streptomycin solution, and 4′, 6-diamidino-2-phenylindole (DAPI) were obtained from Solarbio Biotech (Beijing, China). Chloral hydrate was obtained from Hecang Chemical (Shanghai, China). All other reagents of analytical and chromatographic pure grade were obtained from Sinopharm Chemical Reagent (Shanghai, China). Double-deionized water was purified using a Millipore simplicity system (EMD Millipore, Billerica, MA, USA).
Cell cultures and animals
A549 cells were provided by Xiangya Central Experiment Laboratory (Changsha, China). Cells were maintained in DMEM supplemented with 10% (v:v) heat-inactivated FBS at 37°C in a humidified atmosphere (95%) containing 5% CO2. Cell-experiment protocols were approved by the Faculty Committee, Xiangya School of Pharmaceutical Sciences, Central South University.
Male Sprague Dawley rats (body weight 180–220 g) and male athymic nude mice (nu/nu, BALB/c, 4–6 weeks old, body weight 18–22 g) were provided by SJA Laboratory Animals (Changsha, China). Sprague Dawley rats were kept in a pathogen-free environment and fed ad libitum. All animal experiments were approved by and carried out in accordance with the protocols of Central South University Animal Care and Use Committee.
Preparation of spermidine-modified NPs
Synthesis of PLGA-PEG block copolymer
Carboxylate-functionalized copolymer PLGA-PEG-COOH was synthesized by conjugating COOH-PEG-NH2 to PLGA-COOH. Briefly, PLGA–COOH (5 g, 0.12 mmol) dissolved in chloroform (10 mL) was converted to PLGA-NHS with excess NHS (135 mg, 1.1 mmol) and EDC (230 mg, 1.2 mmol). Then, PLGA-NHS was precipitated and washed repeatedly with ice-cold methanol to remove residual NHS. After vacuum-drying, PLGA-NHS (1 g, 0.024 mmol) was dissolved in chloroform (4 mL), followed by addition of N, N-diisopropylethylamine (28 mg, 0.22 mmol) and NH2-PEG-COOH (250 mg, 0.074 mmol). This polymer was stirred for 12 hours, precipitated, and washed with ice-cold methanol to remove unreacted PEG. After vacuum-drying, the copolymer was dissolved in CDCl3 and characterized by 1H nuclear magnetic resonance (NMR; Mercury Plus 400; Varian, Palo Alto, CA, USA) for confirming the conjugation percentage of PEG to PLGA.
Preparation of PLGA NPs
Emulsification-solvent evaporation was employed for the preparation of AKF NPs. Briefly, PLGA-PEG copolymer (20 mg) and AKF (5 mg) were dissolved in 1 mL dichloromethane and added to 2% polyvinyl alcohol aqueous solution (4 mL). The mixture was sonicated by a microtip-probe sonicator for 2 minutes at 80 W energy output over an ice bath. The emulsion was stirred at 30°C for 3 hours to evaporate the organic solvent (Figure 1A). Blank NPs were prepared without the addition of AKF. Cou6- or DiR-labeled NPs were prepared in the same way by adding Cou6 (0.05% w:w) or DiR (0.4% w:w) for encapsulation. Next, a centrifuge (23,000× g, 4°C for 20 minutes) separated the NPs from the solvent, before they were washed three times with double-deionized water and dried.
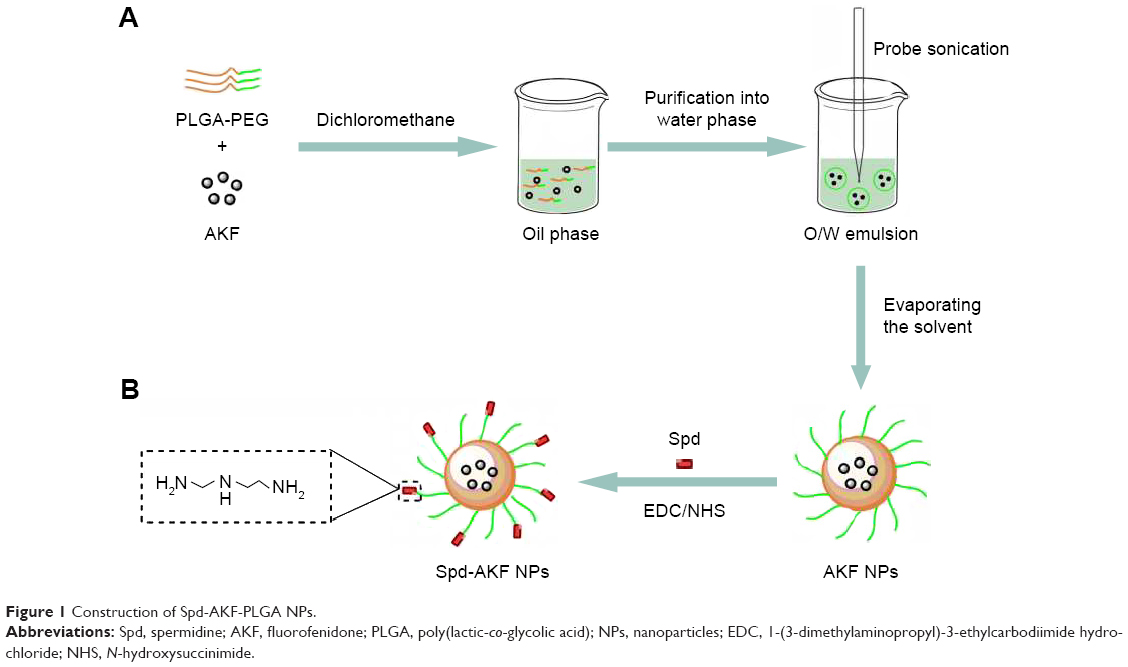 | Figure 1 Construction of Spd-AKF-PLGA NPs. |
Binding of Spd to PLGA NPs
Spd was modified on the surface of PLGA NPs using an EDC/NHS technique. PLGA NPs were suspended in MES (0.1 M, pH 6, 2 mL), then added with excess NHS (100 mM) and EDC (250 mM) at 25°C for 30 minutes under magnetic stirring (Figure 1B). The solution was transferred to a centrifuge tube and centrifuged at 20,000× g, 4°C for 20 minutes to remove the residual reactants. Activated NPs were then dispersed in PBS (0.1 M, pH 8, 10 mL) to react with Spd (10 mM, 100 μL) for 1 hour under magnetic stirring. Link-coupled Spd-PLGA NPs were washed three times with PBS (0.01 M, pH 7.4) and freeze-dried.
Characterization of PLGA NPs
Particle size, ζ-potential, and surface morphology
Particle size and ζ-potential of Spd-AKF-PLGA NPs and AKF-PLGA NPs were determined by dynamic light-scattering analysis using a ζ-potential/particle sizer (Nano ZS; Malvern Instruments, Malvern, UK). The shape and surface characteristics of NPs were investigated by transmission electron microscopy (TEM; Titan G2 60-300; FEI, Hillsboro, OR, USA) after samples had been suspended in water and then dropped into Formvar-coated copper grids.
Drug-entrapment efficiency and -loading capacity
In order to determine the EE and LC of NPs, a predetermined amount of NPs were ultrasonicated in methanol for 20 minutes to degrade and release AKF. AKF level was determined with high-performance liquid chromatography (HPLC; LC 2010C; Shimadzu, Kyoto, Japan) using a reverse-phase C18 octadecylsilyl column (200×4.6 mm, 5 μm; Diamonsil, Beijing, China) with methanol and water (60/40, v/v) as the mobile phase at flow rate of 1 mL/min. The column temperature was 40°C and sample injection volume 20 μL. The detection wavelength was 316 nm. EE and LC were calculated using the formula:
|

|
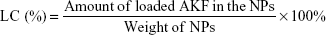
Determination of Spd conjugated on PLGA NPs
The amount of Spd conjugated on PLGA NP surfaces was determined by BCA. Briefly, 2 mL Spd-AKF-PLGA NPs (2.5 mg/mL) were pretreated with 400 μL of ITL (20 mM) for 1 hour at 37°C. After being washed with DTT (1 mM) and PBS (0.1 M, pH 7.4), NPs were resuspended and reacted with 200 μL BCA for 30 minutes at 37°C, then samples were analyzed by a microplate reader (Tecan Infinite M200) at 256 nm. The same approach without NPs was used for the control.
Drug release of PLGA NPs
The drug-release properties of NPs were studied by the dialysis-bag method. Firstly, free AKF solution, AKF-PLGA NPs, or Spd-AKF-PLGA NPs (0.5 mL) were put into a dialysis bag (MW cutoff 3,400 Da). Then, the dialysis bag was hung in a conical flask containing PBS (50 mL, pH 7.4) and shaken horizontally (100 rpm, 37°C). Finally, dissolution medium (1 mL) was taken out at designated time points (1, 2, 3, 4, 6, 12, 24, 48, 72, 96, 120, 144, 168, and 192 hours) for HPLC analysis and fresh PBS (1 mL) added immediately.
Biocompatibility and cellular uptake of PLGA NPs
Cou6 as the fluorescent probe was incorporated into the NPs. Intracellular accumulation of Cou6-PLGA NPs in A549 cells was quantitatively assayed with flow cytometry (FACSCalibur; BD Biosciences, San Jose, CA, USA). The quantitative uptake of Spd-Cou6-PLGA NPs in A549 cells was determined by measurement of fluorescence on a cell-imaging station (inverted fluorescence microscope; Olympus, Tokyo Japan). A549 cells were seeded at a concentration of 5×105 cells per well in a six-well plate and cultured for 24 hours. After cells had been rinsed with PBS, Spd-Cou6-PLGA NPs and Cou6-PLGA NPs were diluted separately with culture medium to 400 μg/mL (PLGA content), and these NPs (2 mL) were added to each well and coincubated with the cells for 1 hour. After that, A549 cells were digested by trypsin enzymes and washed with PBS (pH 7.4) twice, then fixed with 70% ethanol and counterstained with DAPI. Next, centrifugation separated the A549 cells from PBS, concentrating them into Eppendorf tubes. Finally, cells were resuspended in PBS (pH 7.4, 2 mL) with FBS (2%, v/v), and intracellular fluorescence intensity of NPs was measured.
In vivo fluorescence imaging
DiR, a lipophilic near-infrared probe, was encapsulated into the NPs for in vivo imaging. The nude mice (aged 46 weeks, 18–22 g, male) were injected with DiR-PLGA NPs or Spd-DiR-PLGA NPs (0.2 mL) separately at a dose of DiR 0.1 mg/kg via tail vein, and then anesthetized with isoflurane/oxygen (1.5%). Subsequently, the mice under anesthesia were put into a chamber of an imaging system (FMT 4000 fluorescence tomography system; PerkinElmer, Waltham, MA, USA). The fluorescence images were captured at 1, 3, 6, 24, and 48 hours after injection. Distribution information for DiR-PLGA NPs or Spd-DiR-PLGA NPs in mice was acquired through analyzing fluorescence intensity, which was processed and analyzed using TrueQuant system software.
In vivo biodistribution
Male Sprague Dawley rats were used in the biodistribution study of NPs, and received a single intravenous injection of Spd-AKF-PLGA NPs, AKF-PLGA NPs, or free AKF solution at an equivalent AKF dose of 30 mg/kg. After administration, blood, heart, liver, spleen, lung, and kidney were collected from rats scheduled to be killed at 1, 3, 6, 12, 24, and 48 hours. For determination of AKF in vivo, tissue samples (0.5 g) were homogenized in 0.5 mL physiological saline and extracted with 1 mL acetonitrile using ultrasonic extraction for 1 hour. The mixture was centrifuged (12,000 rpm, 5 minutes) and the supernatant analyzed by HPLC, which employed PD as the internal standard.
Drug-efficacy assessment
The anti-pulmonary fibrosis effect of PLGA NPs was assessed in the PQ-poisoning rat model, and efficacy verified with pathological sections. A total of 150 male rats were randomly assigned to the control group (normal saline), experimental groups, and the positive-control group (PQ). Four experimental groups were injected with AKF solution, PD solution (FDA-approved therapeutic for treatment of IPF), AKF-PLGA NPs and Spd-AKF-PLGA NPs at a dosage of 30 mg/kg (AKF or PD every other day) for 28 days after 30 minutes of intraperitoneal injection of PQ (24 mg/kg). During the course of therapy, lung tissue was collected at 0, 7, 14, 21, and 28 days and washed rapidly with saline. Changes in interstitial tissue were observed with H&E and Masson’s trichrome staining. Images were obtained on an Olympus BX41 microscope at a final magnification of 400×.
Statistical analyses
All statistical analyses were carried out using one-way analysis of variance, and P<0.05 was considered statistically significant. Results are expressed as means ± SD.
Results
Characterization of PLGA-PEG block copolymer
PLGA-PEG copolymer was synthesized via an EDC/NHS technique.52,53 PLGA-COOH was transformed into PLGA-NHS and then reacted with NH2-PEG-COOH to obtain PLGA-PEG (Figure 1). The structure and chemical composition of PLGA-PEG-COOH copolymer were identified by 1H NMR spectra (Figure 2). The characteristic peak at 3.6 ppm (Figure 2A) belonged to the methene proton of the PEG chain, and the peaks at 1.5, 4.8, and 5.2 ppm (Figure 2B) belonged to the methyl, methene, and methine protons of the PLGA segment, respectively. Through a combination of PLGA and PEG, these peaks (Figure 2C) all appeared in the PLGA-PEG copolymer NMR spectra. The conjugation efficiency of NH2-PEG-COOH to PLGA-COOH was estimated to be 25% after calculation of relative MWs of characteristic peaks at 5.2 and 3.6 ppm.
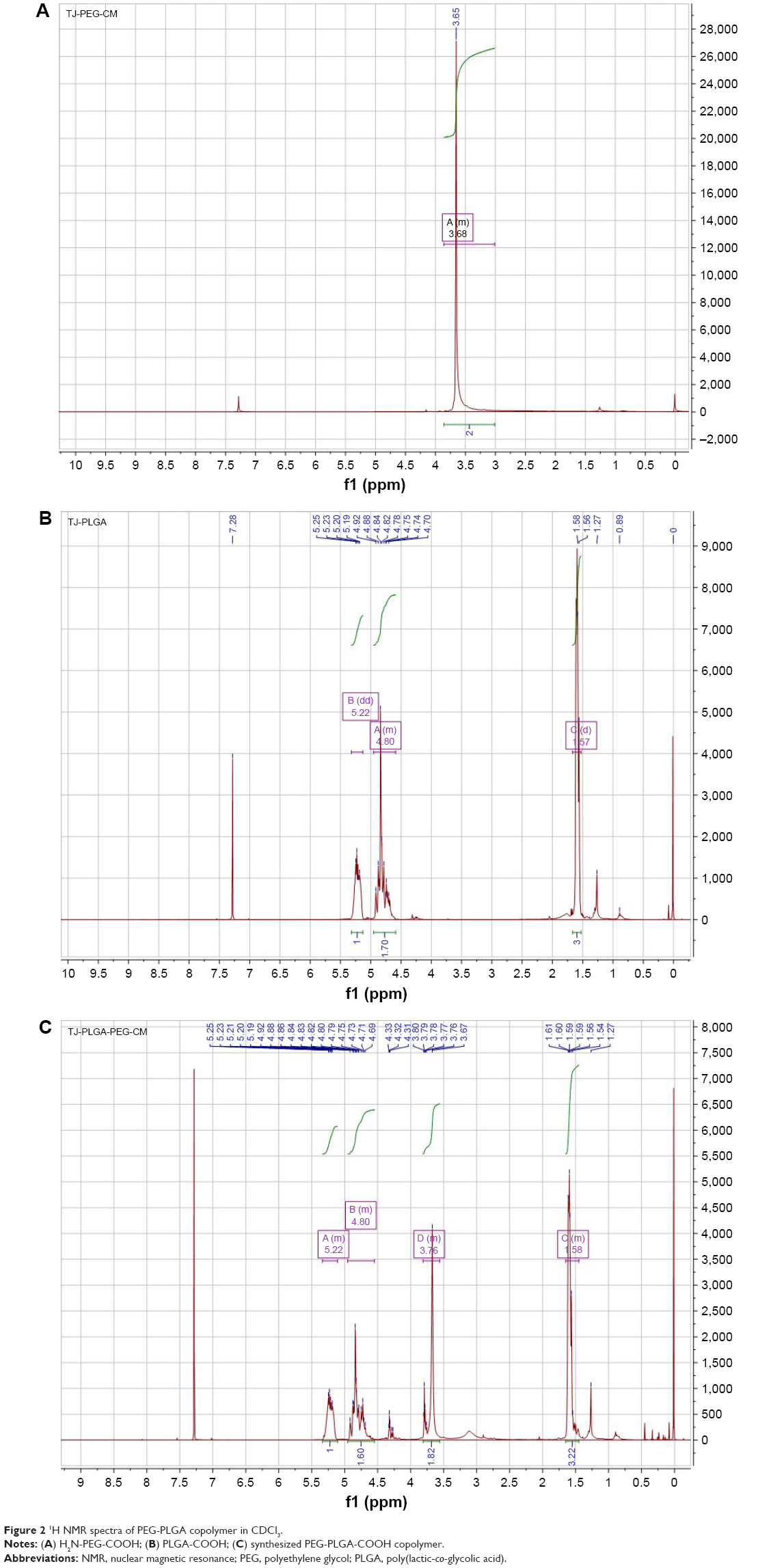 | Figure 2 1H NMR spectra of PEG-PLGA copolymer in CDCl3. |
Characterization of PLGA NPs
Particle size, ζ-potential, and surface morphology
AKF-PLGA NPs were prepared by solvent evaporation and conjugated by Spd. Dynamic light-scattering results showed that the average diameter of AKF-PLGA NPs was around 171.5 nm, with a polydispersity index (PDI) of 0.052 (Table 1). After conjugation by Spd, the diameter (172.5 nm) and PDI (0.034) remained broadly stable, but the ζ-potential increased from −11 mV to −2.54 mV. The results of size and ζ-potential were due to the low MW and more positive charges of Spd. TEM images showed that AKF-PLGA NPs and Spd-AKF-PLGA NPs were spherical with uniform size and relatively smooth surfaces (Figure 3).
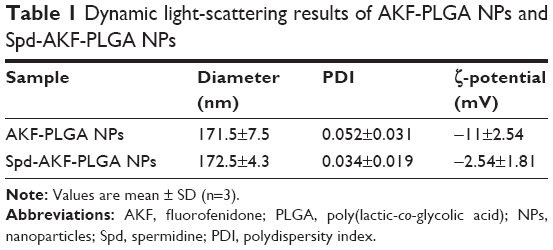 | Table 1 Dynamic light-scattering results of AKF-PLGA NPs and Spd-AKF-PLGA NPs |
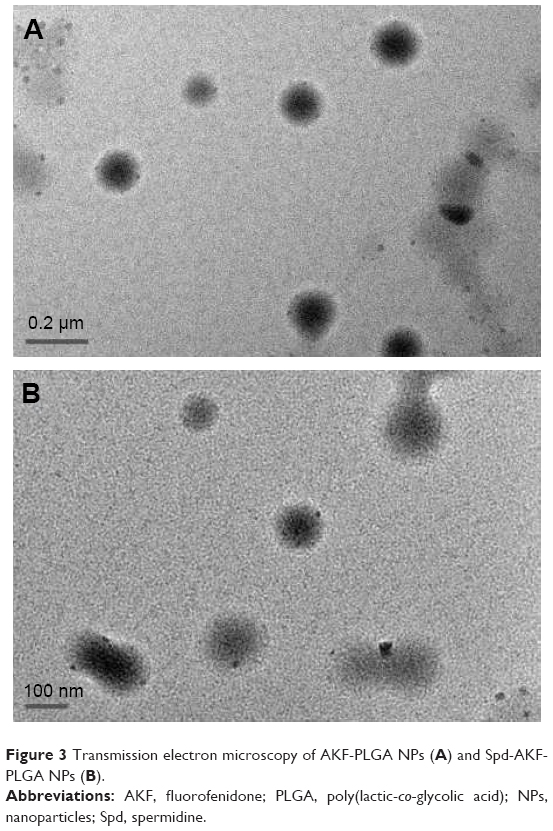 | Figure 3 Transmission electron microscopy of AKF-PLGA NPs (A) and Spd-AKF-PLGA NPs (B). |
Drug-encapsulation efficiency and -loading capacity
In our study, AKF-PLGA NPs had EE of 50.1%±3.7% and LC of 95±0.4 μg/g (n=3). Due to the existence of Spd on the particle surface and repeated washing of NPs, AKF-PLGA NP EE and LC for targeted particles decreased slightly: 48.3%±2.6% and 93±0.6 μg/g (n=3), respectively.
Surface-modification rate of Spd on PLGA NPs
The quantitative analysis method for surfaces modified with Spd on NPs was the BCA protein-assay kit.54,55 Briefly, terminal primary amine groups of Spd reacted with ITL reagent to generate thiol groups. Next, the sulfhydryl reduced cupric into cuprous in an alkaline environment, which generated purple complexes with BCA reagent and had strong absorbance at 256 nm. The amount of Spd modified on NPs was 9.2±0.3 mM/g.
Drug release of PLGA NPs
The drug release of AKF solution was approximately 92% of total quantity within 4 hours. AKF released from AKF-PLGA NPs or Spd-AKF-PLGA NPs presented a burst-release stage at the first stage (4 hours) and a sustained-release stage in the subsequent 192 hours (Figure 4). The burst-release percentage of AKF-PLGA NPs and Spd-AKF-PLGA NPs was 39%±4.9% and 31.2%±5.2%, respectively. Various mathematical kinetic models are used to describe drug release.56,57 As shown in Table 2, the release model of AKF-PLGA NPs was the same as with free AKF (both fit the first-order model), which was probably caused by a large amount of AKF adsorbed on the surface of NPs. Spd-AKF-PLGA NPs fit the Higuchi model, and the initial release reduction might have been related to the wash times before conjugation of Spd, while the sustained release was due to slow degradation of polymer-based carriers.58
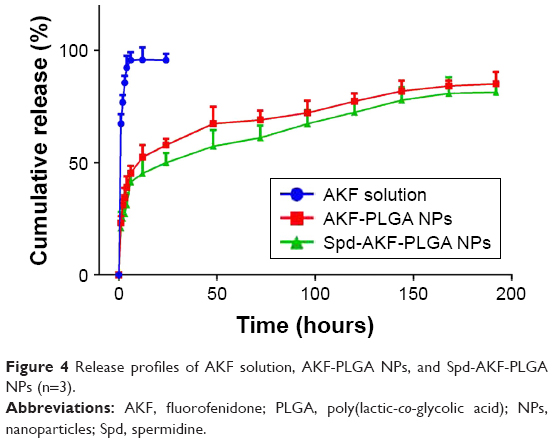 | Figure 4 Release profiles of AKF solution, AKF-PLGA NPs, and Spd-AKF-PLGA NPs (n=3). |
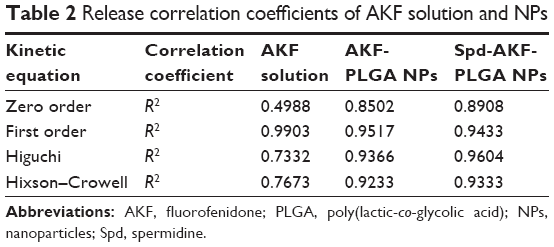 | Table 2 Release correlation coefficients of AKF solution and NPs |
Cellular uptake of NPs
A549 cells were used for the cellular uptake of NPs’ model, due to their similar features and functions to alveolar epithelial cells. Meanwhile, Cou6 was used as a probe encapsulated into NPs for the evaluation of cellular uptake of Cou6-PLGA NPs and Spd-Cou6-PLGA NPs.59–61 Fluorescence images and flow-cytometry analysis were carried out to compare the cellular uptake of drug (Figures 5–7). After incubation of the A549 cells with Cou6-PLGA NPs or Spd-Cou6-PLGA NPs for 1 hour, stronger Cou6 fluorescence intensity in A549 cells indicated that the cells internalized more Spd-Cou6-PLGA NPs. The cellular uptake of the Spd-Cou6-PLGA NPs was found to be almost twice as high as the Cou6-PLGA NPs. This suggested that the uptake of Spd-modified NPs may be specifically recognized by lung epithelial cells via PTS.
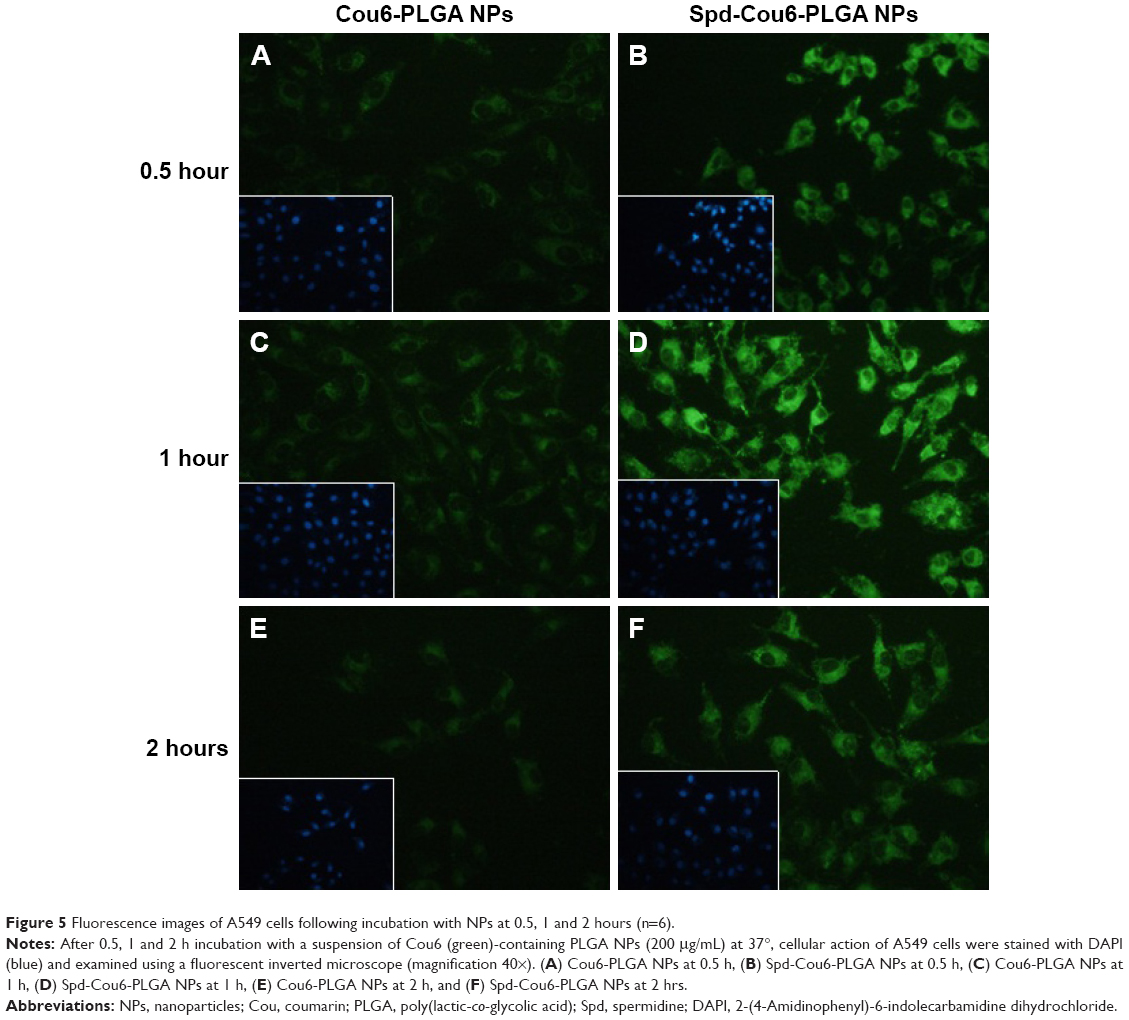 | Figure 5 Fluorescence images of A549 cells following incubation with NPs at 0.5, 1 and 2 hours (n=6). |
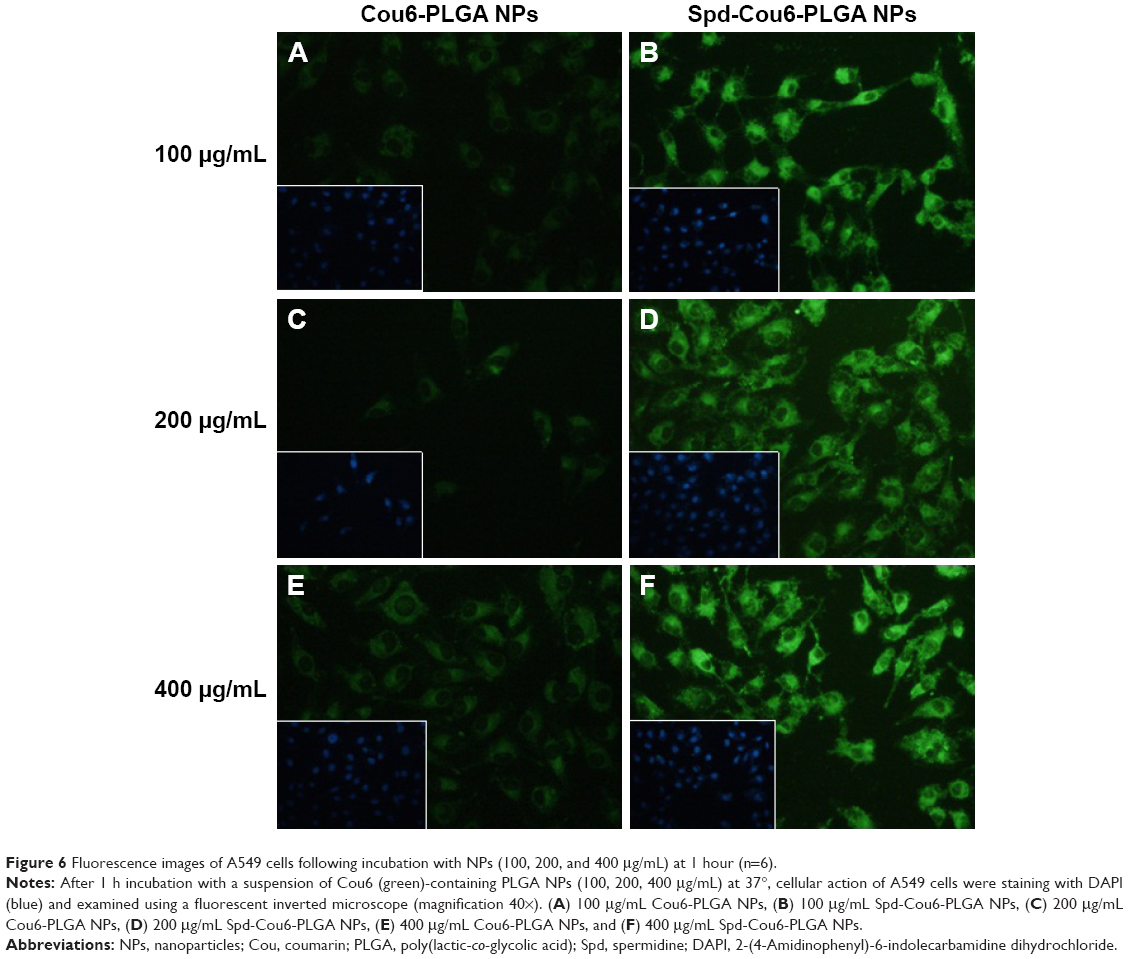 | Figure 6 Fluorescence images of A549 cells following incubation with NPs (100, 200, and 400 μg/mL) at 1 hour (n=6). |
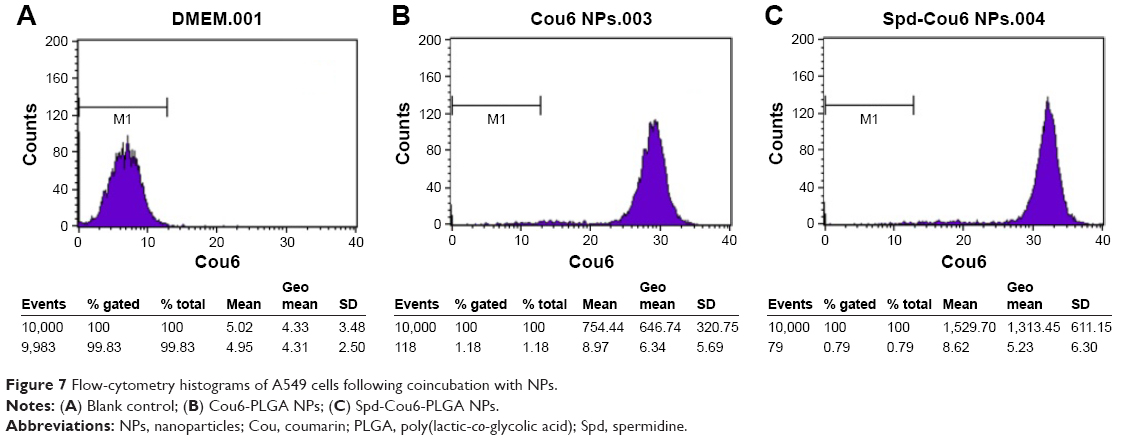 | Figure 7 Flow-cytometry histograms of A549 cells following coincubation with NPs. |
Treatment with Spd or DFMO
The full picture of the PTS is still not clear, and a gene for a PA-specific transporter has not yet been isolated. As such, it is difficult to directly demonstrate how PA-modified NPs enter into PTS-overexpressed cells. Moreover, a specific PTS inhibitor is lacking. In this experiment, DFMO, a PA biosynthetic enzyme inhibitor, was employed to reduce intracellular concentration of PAs. In order to maintain PA content, cells need an increase in PA intake. Therefore, adding DFMO to A549 cells can indirectly increase PAs, including PA-modified NPs.
In order to confirm Spd-functionalized NP transfer was mediated mainly by the PTS, DFMO was employed to coincubate with NPs to improve the intake of PA conjugates, such as Spd-PLGA NPs. In addition, free Spd was employed to coincubate with Spd-PLGA NPs to demonstrate there was competition between PAs and PA conjugates. The process of cell culture was the same as described earlier. After culturing for 24 hours, cells were treated with 2 mL free Spd (5, 50, 500 μmol/mL) for 24 hours or DMFO (5 mM) for 36 hours, separately. After being rinsed with PBS, Spd-Cou6-PLGA NPs and Cou6-PLGA NPs (400 μg/mL) were added, and the incubation continued for another hour. Then, cells were analyzed by FACS.
Free Spd and DFMO were preincubated in A549 cells further, to prove the uptake of Spd-Cou6-PLGA NPs via the PTS.62 This was due to free Spd compete with Spd modified NPs for the polyamine binding site on A549 cells. As shown in Figures 8 and 9, the uptake of Spd-Cou6-PLGA NPs significantly decreased with increased Spd concentrations in incubation. At higher Spd concentrations of 50 and 500 μM, the uptake of Spd-Cou6-PLGA NPs reduced 0.34- and 0.49-fold more than that without Spd pretreatment (P<0.05; P<0.001).
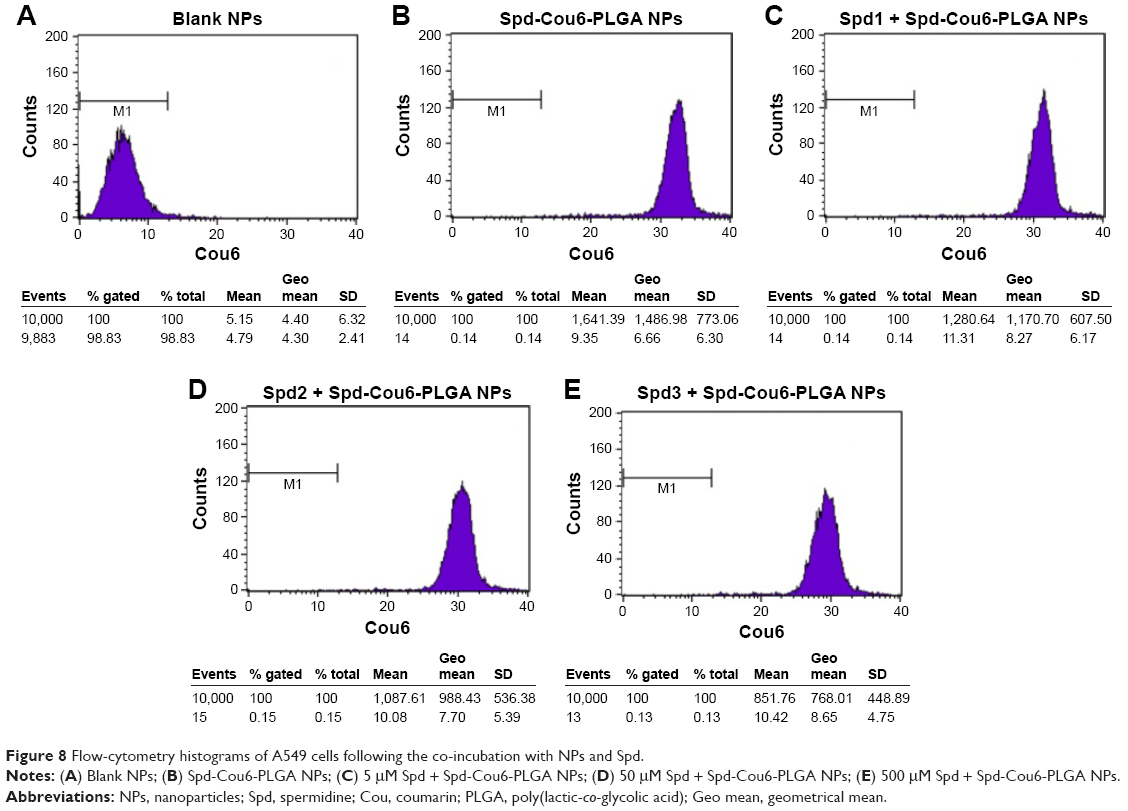 | Figure 8 Flow-cytometry histograms of A549 cells following the co-incubation with NPs and Spd. |
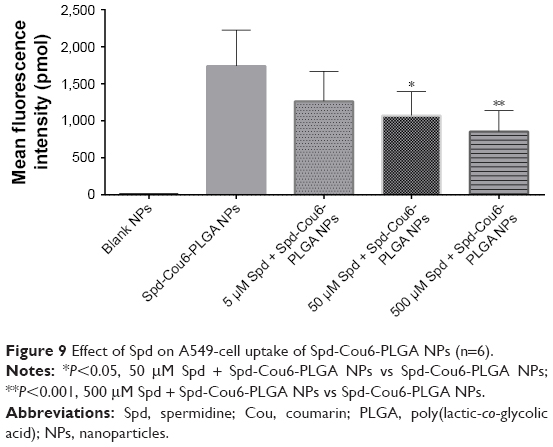 | Figure 9 Effect of Spd on A549-cell uptake of Spd-Cou6-PLGA NPs (n=6). |
After pretreatment with DFMO for 36 hours, cellular uptake of Spd-Cou6-PLGA NPs reached 1.26-fold that of the no-DFMO-pretreatment group (P<0.05) (Figures 10 and 11). However, the uptake of Cou6-PLGA NPs was obviously not enhanced after preincubation with DMFO. DFMO is a PA biosynthesis inhibitor, which can upregulate the PTS to increase the membrane uptake of exogenous PA by blocking intracellular PA synthesis.63 Therefore, the enhanced activity of the PTS for PA improved the cell internalization of Spd-Cou6-PLGA NPs. In contrast, the role of DFMO had no significant effect on the uptake of Cou6-PLGA NPs, where endocytosis was not via the PTS. These results of pretreatment with Spd and DFMO indicated that Spd-Cou6-PLGA NPs were taken up through the PTS.
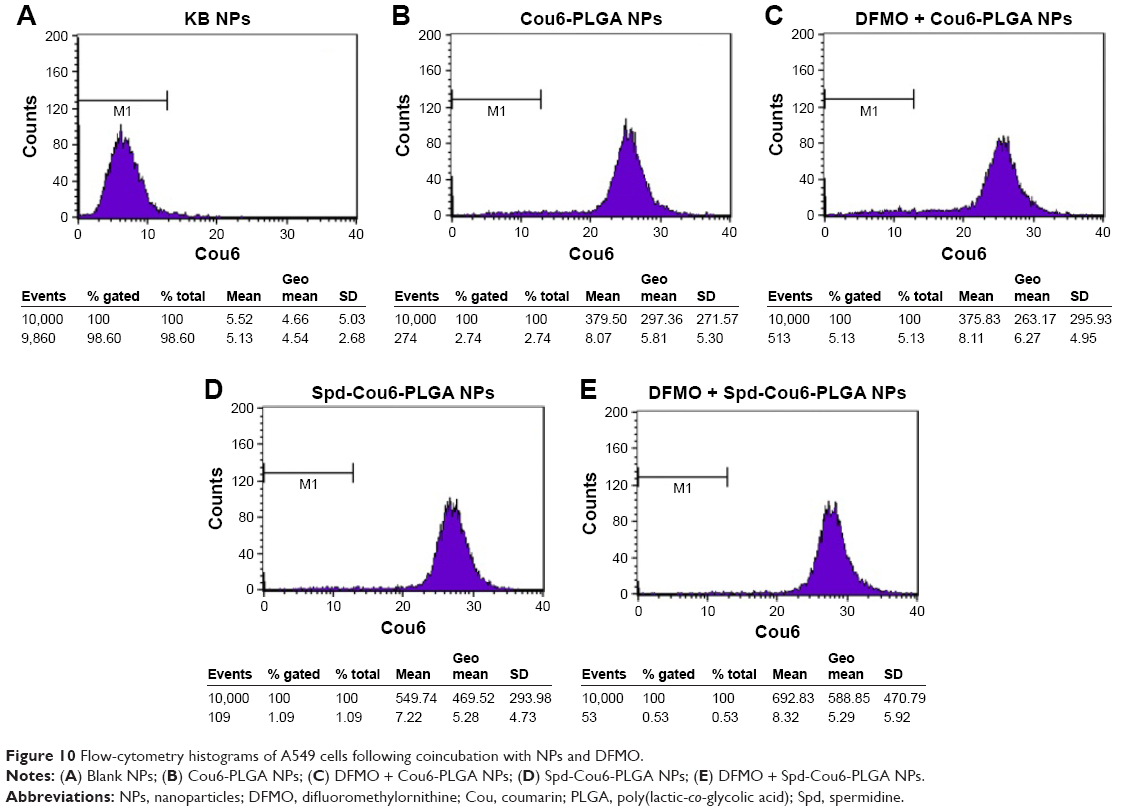 | Figure 10 Flow-cytometry histograms of A549 cells following coincubation with NPs and DFMO. |
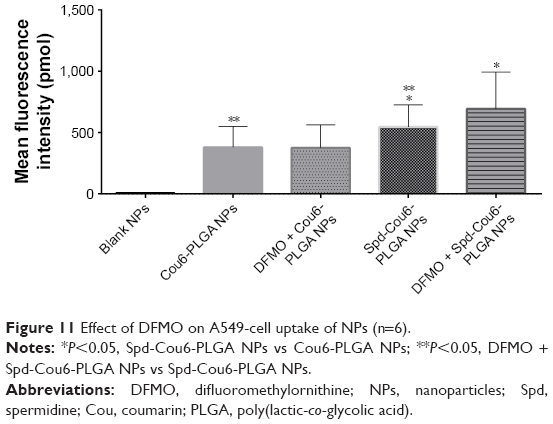 | Figure 11 Effect of DFMO on A549-cell uptake of NPs (n=6). |
In vivo fluorescence imaging
DiR is an indocarbocyanine dye with poor water solubility.64 It is often used for in vivo distribution studies because its emission wavelength is above 700 nm, which effectively eliminates emission interference from the animals used.65 It has been reported that the labeling of lipophilic nanocarriers using the indocarbocyanine-dye family provides a reliable means of near-infrared reflection imaging.66 Figure 12A and B show the lung fluorescence images captured at 1, 3, 6, 24, and 48 hours after injection of DiR-PLGA NPs and Spd-DiR-PLGA NPs in nude mice. Fluorescence accumulation in the lung zone was observed in both groups, but Spd-DiR-PLGA NPs displayed more intensive signals at each time point. Relative fluorescence intensity, calculated from quantitative analysis of the fluorescence distribution, is shown in Figure 13. This revealed again that Spd-modified DiR-PLGA NPs accumulated more in lung tissue than DiR-PLGA NPs.
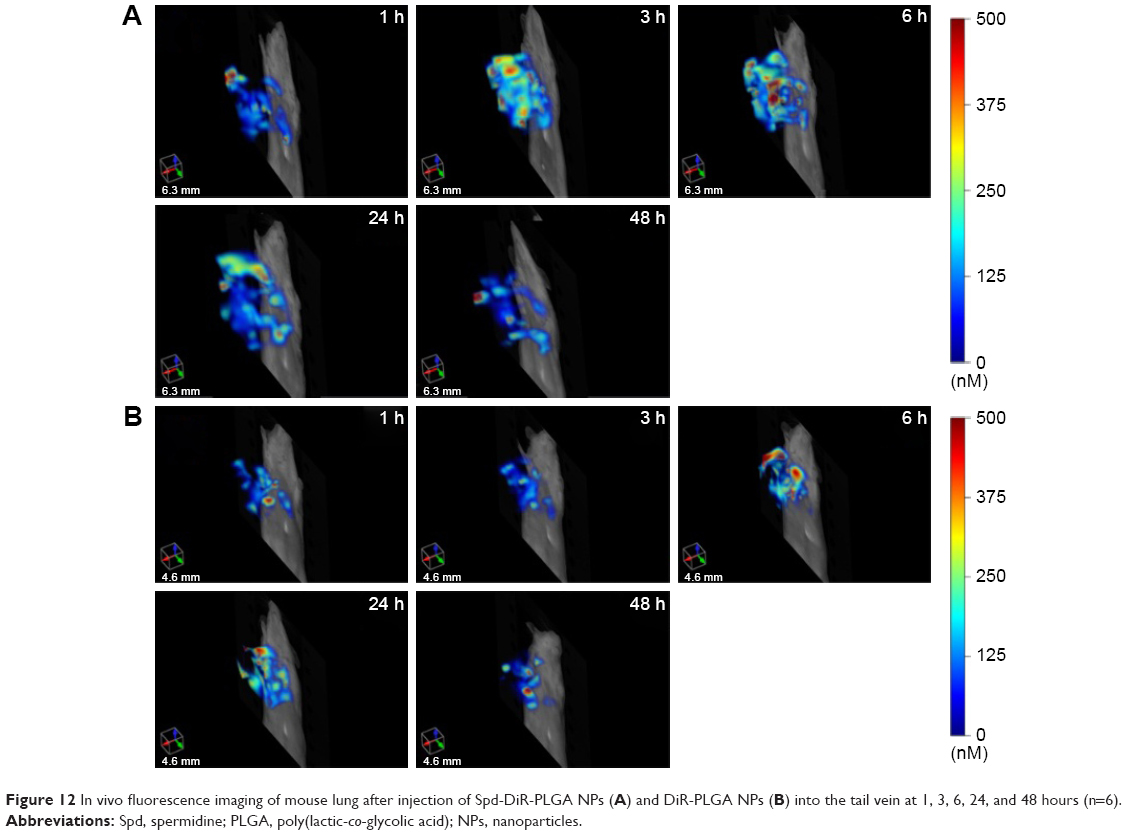 | Figure 12 In vivo fluorescence imaging of mouse lung after injection of Spd-DiR-PLGA NPs (A) and DiR-PLGA NPs (B) into the tail vein at 1, 3, 6, 24, and 48 hours (n=6). |
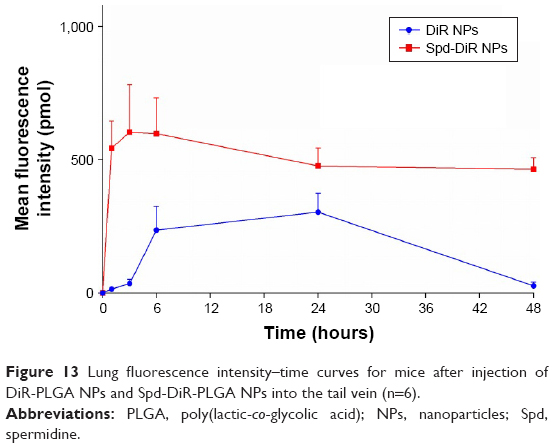 | Figure 13 Lung fluorescence intensity–time curves for mice after injection of DiR-PLGA NPs and Spd-DiR-PLGA NPs into the tail vein (n=6). |
Tissue distribution
Levels of AKF in plasma and tissues were determined by HPLC. The initial study in vivo verified the biodistribution and lung-target characteristics of the NPs.67,68 As shown in Figure 14A, at 1–3 hours after administration of AKF solution, plasma drug concentration reached maximum. After that, the drug concentration was gradually eliminated and showed no selective accumulation in any tissue. As to AKF-PLGA NPs, the distribution of the drug in different tissues were significantly different, especially in liver (Figure 14B). Spd-AKF-PLGA NPs increased drug concentration in lung within 24 hours (Figure 14C). Drug-concentration curves in lung of AKF solution, AKF-PLGA NPs, and Spd-AKF-PLGA NPs are shown in Figure 14D, and clearly indicate that the drug concentrations of Spd-AKF-PLGA NPs in lung were significantly higher than those of AKF-PLGA NPs and AKF solution at different time points. Meanwhile, the drug-targeting index of Spd-AKF-PLGA NPs in lung was 3.62 and 4.66 times higher than that of AKF-PLGA NPs and AKF solution, respectively. It is suggested that the Spd-AKF-PLGA NPs accumulated effectively in the lung.
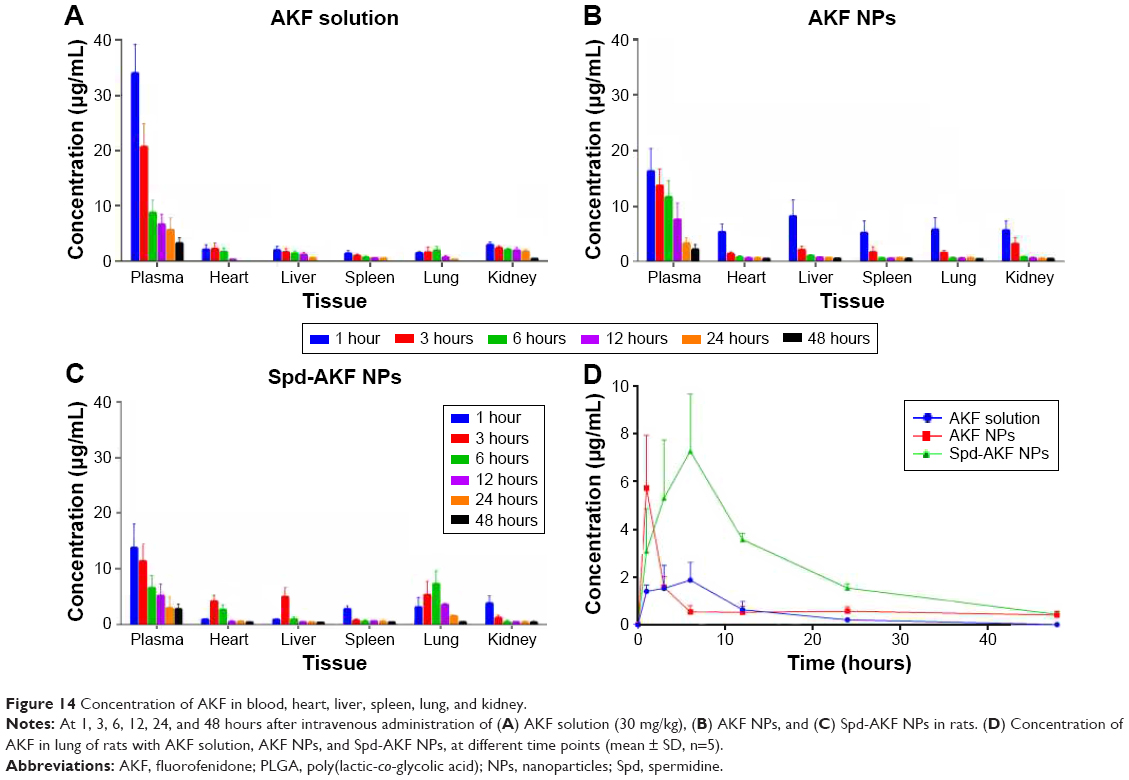 | Figure 14 Concentration of AKF in blood, heart, liver, spleen, lung, and kidney. |
Drug-efficacy assessment
Lung-histopathology changes and collagen deposition were observed by H&E and Masson staining. Microscopy of the lung sections after 28 days’ treatment is shown in Figure 15. In the control group, the alveolar walls were complete, with no bleeding or neutrophil infiltration. Due to the PQ poisoning, the positive-control group displayed significant pulmonary interstitial damage, while alveolar walls were thickened with pink collagen.69–71 Compared to the positive-control group, the PD-solution and AKF-solution groups were characterized by a slight improvement of interstitial edema. The alveolar space had decreased by 21 days in the AKF-PLGA NP groups. Particularly in the Spd-AKF-PLGA NP group, the damage was further improved compared to that of the AKF-PLGA NP group. The results showed that the degree of fibrosis was alleviated and the area of destroyed alveoli decreased (red boxes in Figure 15). Based on these results, it could be concluded that the Spd-AKF-PLGA NPs were effective nanocarriers for anti-IPF therapies.
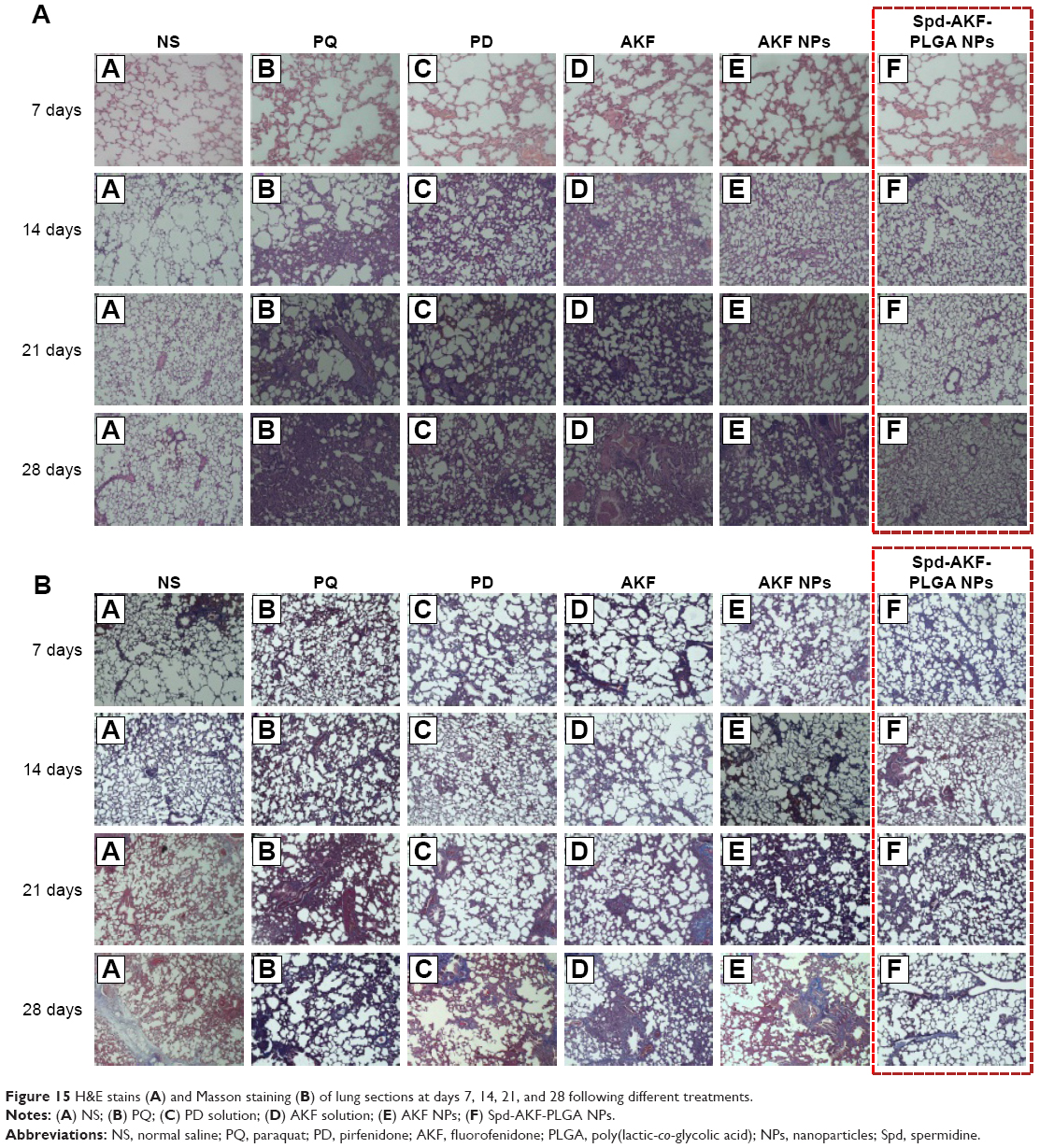 | Figure 15 H&E stains (A) and Masson staining (B) of lung sections at days 7, 14, 21, and 28 following different treatments. |
Discussion
In recent years, targeted drug-delivery systems are urgently needed for maximization of drug efficacy and minimization of side effects. Biocompatible and biodegradable polymers, such as PLGA and PEG, are frequently used to encapsulate a variety of therapeutic compounds, drugs, siRNA, DNA, and molecular/dye probes inside the matrix shield.24,45,72 PLGA and PEG have many unique functional features, not only encapsulating hydrophilic and hydrophobic drugs but also allowing therapeutic targeting. We have previously described in detail the application of PLGA microspheres for controlled and sustained drug delivery of AKF to control PQ-induced inflammation in Sprague Dawley rat lungs, though these microspheres failed to address active targeting of type II pneumocytes and relied on particle size for lung targeting.39 PEGylated Spd-AKF-PLGA NPs used in this study are an improved viable formulation of PLGA-PEG that can not only bypass the elimination by the reticuloendothelial system but can also target and selectively deliver antifibrotic drug (AKF) to lung epithelial cells for maximum therapeutic efficacy.36,73,74
IPF is a progressive disease of excessive accumulation of fibrotic tissue in the lungs. Despite years of advances in the understanding of its underlying mechanisms, IPF is still incurable. PD is one of the antifibrotic agents with proven efficacy in mild–moderate IPF. However, the need for new therapeutic options remains high. AKF, the analog of PD, exerts better anti-inflammatory and anti-fibrotic effects in tissues.75,76 Therefore, it may be used as a promising antifibrotic drug in the treatment of IPF. A large number of polymer-material vehicles are designed to help delivery of drugs to target organs and improve drug delivery into target cells. With this in mind, a nanoscale drug carrier has been developed in an attempt to deliver AKF to lung cells and provide a new therapeutic option for IPF therapy.
In the present study, we designed an Spd-functionalized and PTS-based lung-targeted drug-delivery system to enhance AKF accumulation in lung. Spd, one of the PAs, is a low-MW aliphatic alkylamine essential for cell growth whose concentration is controlled by biosynthesis and cellular uptake. An extensive body of research has demonstrated that active transport of PAs into cells is mediated by a specific transporter – the PTS.77 The lung and more specifically alveolar epithelial cells appear to be endowed with much higher PTS than any other major organ.78 In the lung, the accumulation of natural PAs in the epithelium has been studied in various mammalian species, including rat, rabbit, hamster, and human.79–82 This suggests that Spd or PA conjugates might be substrates for the PTS, although further research is required.
The import of PAs has been demonstrated and characterized in cells and tissues, but the full structure of the PTS is not clear. DMFO, an irreversible inhibitor of ornithine decarboxylase, inhibits PA synthesis in cells.62 However, cells can escape PA-synthesis blockade by importing PAs from the environment. Based on this, we investigated the uptake of NPs on A549 cells alone or in combination with DFMO. Interestingly, culture with DFMO for 36 hours inhibited endogenous PA synthesis and led to increase in cellular uptake of Spd-NPs.
Moreover, Spd-AKF-PLGA NPs presented therapeutic efficacy in PQ-induced rat lung disease, which simulated IPF. The PQ-induced IPF rat is a well-established animal model, and can be used to evaluate antifibrotic effects.83 PA and PQ compete for the same uptake system in the lung, and PAs have better affinity for the PTS.84,85 Therefore, PA-functionalized NPs may provide further protection against IPF from PQ toxicity.
Conclusion
In this study, we developed novel Spd-AKF-PLGA NPs with specific lung targeting for IPF. Our main goal was to increase the intracellular concentration of AKF in the lung cells via a targeted approach. The NPs were of appropriate size, maintained high stability, and showed satisfactory loading and release capacity for AKF. Fluorescence analysis showed that the Spd-AKF-PLGA NPs had high affinity for A549 cells and facilitated endocytosis. After pretreatment with Spd or DFMO, results further demonstrated enhanced cell uptake for targeted PLGA NPs via PTS and Spd interaction. Both Spd-AKF-PLGA NPs and AKF-PLGA NPs were distributed to lung after intravenous injection. However, Spd-AKF-PLGA NPs were able to distribute into the lung tissue more efficiently than AKF-PLGA NPs, due to long circulation in blood and Spd-mediated intracellular uptake. On histopathological analysis, fibroplasia, alveolar collapse, neutrophil infiltration, and lung-interval damage were significantly reduced by Spd-AKF-PLGA NP treatment. Overall, Spd-AKF-PLGA NPs could be used as a potential targeted drug-delivery system for the treatment of IPF.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (81603061 and 81573374), Research Foundation of Education Bureau of Hunan Province (16B032), Key Laboratory Breeding Base of Hunan Oriented Fundamental and Applied Research in Innovative Pharmaceutics (2016TP1029), and Hunan Engineering Research Center for Optimization of Drug Formulation and Early Clinical Evaluation (2015TP2005).
Disclosure
The authors report no conflicts of interest in this work.
References
Sgalla G, Cocconcelli E, Tonelli R, Richeldi L. Novel drug targets for idiopathic pulmonary fibrosis. Expert Rev Respir Med. Epub 2016 Feb 26. | ||
Xaubet A, Molina-Molina M, Acosta O, et al. Guidelines for the medical treatment of idiopathic pulmonary fibrosis. Arch Bronconeumol. 2017;53(5):263–269. | ||
Spagnolo P, Wells AU, Collard HR. Pharmacological treatment of idiopathic pulmonary fibrosis: an update. Drug Discov Today. 2015;20(5):514–524. | ||
Homer RJ, Elias JA, Lee CG, Herzog E. Modern concepts on the role of inflammation in pulmonary fibrosis. Arch Pathol Lab Med. 2011;135(6):780–788. | ||
Wilson MS, Wynn TA. Pulmonary fibrosis: pathogenesis, etiology and regulation. Mucosal Immunol. 2009;2(2):103–121. | ||
Zisman DA, Keane MP, Belperio JA, Strieter RM, Lynch JP 3rd. Pulmonary fibrosis. Methods Mol Med. 2005;117:3–44. | ||
Raghu G, Collard HR, Egan JJ, et al. An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med. 2011;183(6):788–824. | ||
Sgalla G, Biffi A, Richeldi L. Idiopathic pulmonary fibrosis: diagnosis, epidemiology and natural history. Respirology. 2016;21(3):427–437. | ||
Oku H, Shimizu T, Kawabata T, et al. Antifibrotic action of pirfenidone and prednisolone: different effects on pulmonary cytokines and growth factors in bleomycin-induced murine pulmonary fibrosis. Eur J Pharmacol. 2008;590(1–3):400–408. | ||
Kreuter M, Bonella F, Wijsenbeek M, Maher TM, Spagnolo P. Pharmacological treatment of idiopathic pulmonary fibrosis: current approaches, unsolved issues, and future perspectives. Biomed Res Int. 2015;2015:329481. | ||
Hunninghake GM. A new hope for idiopathic pulmonary fibrosis. N Engl J Med. 2014;370(22):2142–2143. | ||
Raghu G, Johnson WC, Lockhart D, Mageto Y. Treatment of idiopathic pulmonary fibrosis with a new antifibrotic agent, pirfenidone: results of a prospective, open-label phase II study. Am J Respir Crit Care Med. 1999;159(4 Pt 1):1061–1069. | ||
Valeyre D, Albera C, Bradford WZ, et al. Comprehensive assessment of the long-term safety of pirfenidone in patients with idiopathic pulmonary fibrosis. Respirology. 2014;19(5):740–747. | ||
Antoniu SA. Pirfenidone for the treatment of idiopathic pulmonary fibrosis: therapeutic potential prompts further investigation. Expert Opin Investig Drugs. 2005;14(11):1443–1447. | ||
Takeda Y, Tsujino K, Kijima T, Kumanogoh A. Efficacy and safety of pirfenidone for idiopathic pulmonary fibrosis. Patient Prefer Adherence. 2014;8:361–370. | ||
Ning W, Hu G, Peng Z, et al. Fluorofenidone inhibits Ang II-induced apoptosis of renal tubular cells through blockage of the Fas/FasL pathway. Int Immunopharmacol. 2011;11(9):1327–1332. | ||
Cao W, He L, Liu W, Huang Z, Cheng Z. Determination of AKF-PD in whole blood of rat by HPLC-UV. J Chromatogr B Analyt Technol Biomed Life Sci. 2006;833(2):257–259. | ||
Meng J, Zou Y, Hu C, et al. Fluorofenidone attenuates bleomycin-induced pulmonary inflammation and fibrosis in mice via restoring caveolin 1 expression and inhibiting mitogen-activated protein kinase signaling pathway. Shock. 2012;38(5):567–573. | ||
Peng ZZ, Hu GY, Shen H, et al. Fluorofenidone attenuates collagen I and transforming growth factor-β1 expression through a nicotinamide adenine dinucleotide phosphate oxidase-dependent way in NRK-52E cells. Nephrology (Carlton). 2009;14(6):565–572. | ||
Peng Y, Yang H, Wang N, et al. Fluorofenidone attenuates hepatic fibrosis by suppressing the proliferation and activation of hepatic stellate cells. Am J Physiol Gastrointest Liver Physiol. 2014;306(3):G253–G263. | ||
Wang L, Hu GY, Shen H, Peng ZZ, Ning WB, Tao LJ. Fluorofenidone inhibits TGF-β1 induced CTGF via MAPK pathways in mouse mesangial cells. Pharmazie. 2009;64(10):680–684. | ||
Liu J, Song C, Xiao Q, Hu G, Tao L, Meng J. Fluorofenidone attenuates TGF-β1-induced lung fibroblast activation via restoring the expression of caveolin-1. Shock. 2015;43(2):201–207. | ||
Saigal A, Ng WK, Tan RB, Chan SY. Development of controlled release inhalable polymeric microspheres for treatment of pulmonary hypertension. Int J Pharm. 2013;450(1–2):114–122. | ||
Muralidharan R, Babu A, Amreddy N, et al. Folate receptor-targeted nanoparticle delivery of HuR-RNAi suppresses lung cancer cell proliferation and migration. J Nanobiotechnology. 2016;14(1):47. | ||
Cao H, Dan Z, He X, et al. Liposomes coated with isolated macrophage membrane can target lung metastasis of breast cancer. ACS Nano. 2016;10(8):7738–7748. | ||
Vij N. Nano-based rescue of dysfunctional autophagy in chronic obstructive lung diseases. Expert Opin Drug Deliv. 2017;14(4):483–489. | ||
Wei Y, Zhao L. Passive lung-targeted drug delivery systems via intravenous administration. Pharm Dev Technol. 2014;19(2):129–136. | ||
Togami K, Miyao A, Miyakoshi K, Kanehira Y, Tada H, Chono S. Efficient delivery to human lung fibroblasts (WI-38) of pirfenidone incorporated into liposomes modified with truncated basic fibroblast growth factor and its inhibitory effect on collagen synthesis in idiopathic pulmonary fibrosis. Biol Pharm Bull. 2015;38(2):270–276. | ||
Trivedi R, Redente EF, Thakur A, Riches DW, Kompella UB. Local delivery of biodegradable pirfenidone nanoparticles ameliorates bleomycin-induced pulmonary fibrosis in mice. Nanotechnology. 2012;23(50):505101. | ||
Ungaro F, d’Angelo I, Miro A, La Rotonda MI, Quaglia F. Engineered PLGA nano- and micro-carriers for pulmonary delivery: challenges and promises. J Pharm Pharmacol. 2012;64(9):1217–1235. | ||
Hidalgo A, Cruz A, Pérez-Gil J. Pulmonary surfactant and nanocarriers: toxicity versus combined nanomedical applications. Biochim Biophys Acta. 2017;2736(17):30135–30139. | ||
Murugadoss S, Lison D, Godderis L, et al. Toxicology of silica nanoparticles: an update. Arch Toxicol. Epub 2017 Jun 1. | ||
Wang Y, Huang HY, Yang L, Zhang Z, Ji H. Cetuximab-modified mesoporous silica nano-medicine specifically targets EGFR-mutant lung cancer and overcomes drug resistance. Sci Rep. 2016;6:25468. | ||
Wang RT, Zhi XY, Yao SY, Zhang Y. LFC131 peptide-conjugated polymeric nanoparticles for the effective delivery of docetaxel in CXCR4 overexpressed lung cancer cells. Colloids Surf B Biointerfaces. 2015;133:43–50. | ||
Shen S, Mao C, Yang X, et al. Cationic lipid-assisted polymeric nanoparticle mediated GATA2 siRNA delivery for synthetic lethal therapy of KRAS mutant non-small-cell lung carcinoma. Mol Pharm. 2014;11(8):2612–2622. | ||
Zhang K, Tang X, Zhang J, et al. PEG-PLGA copolymers: their structure and structure-influenced drug delivery applications. J Control Release. 2014;183:77–86. | ||
Mirakabad FS, Nejati-Koshki K, Akbarzadeh A, et al. PLGA-based nanoparticles as cancer drug delivery systems. Asian Pac J Cancer Prev. 2014;15(2):517–535. | ||
Betancourt T, Byrne JD, Sunaryo N, et al. PEGylation strategies for active targeting of PLA/PLGA nanoparticles. J Biomed Mater Res A. 2009;91(1):263–276. | ||
Jing Tang J, Liu ZB, Zhang Y. Fluorofenidone-loaded PLGA microspheres for targeted treatment of paraquat-induced acute lung injury in rats. RSC Adv. 2015;5(38):30153–30159. | ||
Jiang Y, Wang F, Xu H, Liu H, Meng Q, Liu W. Development of andrographolide loaded PLGA microspheres: optimization, characterization and in vitro-in vivo correlation. Int J Pharm. 2014;475(1–2): 475–484. | ||
Kumari A, Yadav SK, Yadav SC. Biodegradable polymeric nanoparticles based drug delivery systems. Colloids Surf B Biointerfaces. 2010;75(1):1–18. | ||
Danhier F, Ansorena E, Silva JM, Coco R, Le Breton A, Préat V. PLGA-based nanoparticles: an overview of biomedical applications. J Control Release. 2012;161(2):505–522. | ||
Dandekar P, Venkataraman C, Mehra A. Pulmonary targeting of nanoparticle drug matrices. J Aerosol Med Pulm Drug Deliv. 2010;23(6):343–353. | ||
Kamaly N, Xiao Z, Valencia PM, Radovic-Moreno AF, Farokhzad OC. Targeted polymeric therapeutic nanoparticles: design, development and clinical translation. Chem Soc Rev. 2012;41(7):2971–3010. | ||
Alibolandi M, Ramezani M, Abnous K, Hadizadeh F. AS1411 aptamer-decorated biodegradable polyethylene glycol-poly(lactic-co-glycolic acid) nanopolymersomes for the targeted delivery of gemcitabine to non-small cell lung cancer in vitro. J Pharm Sci. 2016;105(5):1741–1750. | ||
Leblond P, Boulet E, Bal-Mahieu C, et al. Activity of the polyamine-vectorized anti-cancer drug F14512 against pediatric glioma and neuroblastoma cell lines. Invest New Drugs. 2014;32(5):883–892. | ||
Thibault B, Clement E, Zorza G, et al. F14512, a polyamine-vectorized inhibitor of topoisomerase II, exhibits a marked anti-tumor activity in ovarian cancer. Cancer Lett. 2016;370(1):10–18. | ||
Minois N, Carmona-Gutierrez D, Madeo F. Polyamines in aging and disease. Aging (Albany NY). 2011;3(8):716–732. | ||
Saunders NA, Ilett KF, Minchin RF. Pulmonary alveolar macrophages express a polyamine transport system. J Cell Physiol. 1989;139(3):624–631. | ||
Reguera RM, Tekwani BL, Balaña-Fouce R. Polyamine transport in parasites: a potential target for new antiparasitic drug development. Comp Biochem Physiol C Toxicol Pharmacol. 2005;140(2):151–164. | ||
Mitchell JL, Thane TK, Sequeira JM, Thokala R. Unusual aspects of the polyamine transport system affect the design of strategies for use of polyamine analogues in chemotherapy. Biochem Soc Trans. 2007;35(Pt 2):318–321. | ||
Guo J, Gao X, Su L, et al. Aptamer-functionalized PEG-PLGA nanoparticles for enhanced anti-glioma drug delivery. Biomaterials. 2011;32(31):8010–8020. | ||
Alibolandi M, Ramezani M, Sadeghi F, Abnous K, Hadizadeh F. Epithelial cell adhesion molecule aptamer conjugated PEG-PLGA nanopolymersomes for targeted delivery of doxorubicin to human breast adenocarcinoma cell line in vitro. Int J Pharm. 2015;479(1):241–251. | ||
Kakabakos SE, Tyllianakis PE, Evangelatos GP, Ithakissios DS. Colorimetric determination of reactive solid-supported primary and secondary amino groups. Biomaterials. 1994;15(4):289–297. | ||
Ghasemi M, Minier M, Tatoulian M, Arefi-Khonsari F. Determination of amine and aldehyde surface densities: application to the study of aged plasma treated polyethylene films. Langmuir. 2007;23(23):11554–11561. | ||
Arifin DY, Lee LY, Wang C. Mathematical modeling and simulation of drug release from microspheres: Implications to drug delivery systems. Adv Drug Deliv Rev. 2006;58(12–13):1274–1325. | ||
Siepmann J, Göpferich A. Mathematical modeling of bioerodible, polymeric drug delivery systems. Adv Drug Deliv Rev. 2001;48(2–3):229–247. | ||
Siepmann J, Peppas NA. Modeling of drug release from delivery systems based on hydroxypropyl methylcellulose (HPMC). Adv Drug Deliv Rev. 2001;48(2–3):139–157. | ||
Li J, Feng L, Fan L, et al. Targeting the brain with PEG-PLGA nanoparticles modified with phage-displayed peptides. Biomaterials. 2011;32(21):4943–4950. | ||
Xu A, Yao M, Xu G, et al. A physical model for the size-dependent cellular uptake of nanoparticles modified with cationic surfactants. Int J Nanomedicine. 2012;7:3547–3554. | ||
Trapani A, Mandracchia D, Di Franco C, et al. In vitro characterization of 6-coumarin loaded solid lipid nanoparticles and their uptake by immunocompetent fish cells. Colloids Surf B Biointerfaces. 2015;127:79–88. | ||
Grossi M, Phanstiel O, Rippe C, et al. Inhibition of polyamine uptake potentiates the anti-proliferative effect of polyamine synthesis inhibition and preserves the contractile phenotype of vascular smooth muscle cells. J Cell Physiol. 2016;231(6):1334–1342. | ||
Mohammed A, Janakiram NB, Madka V, et al. Eflornithine (DFMO) prevents progression of pancreatic cancer by modulating ornithine decarboxylase signaling. Cancer Prev Res (Phila). 2014;7(12): 1198–1209. | ||
El-Gogary RI, Rubio N, Wang JT, et al. Polyethylene glycol conjugated polymeric nanocapsules for targeted delivery of quercetin to folate-expressing cancer cells in vitro and in vivo. ACS Nano. 2014;8(2):1384–1401. | ||
Cheng X, Li H, Chen Y, et al. Ultrasound-triggered phase transition sensitive magnetic fluorescent nanodroplets as a multimodal imaging contrast agent in rat and mouse model. PLoS One. 2013;8(12): e85003. | ||
Wang G, Yu B, Wu Y, Huang B, Yuan Y, Liu CS. Controlled preparation and antitumor efficacy of vitamin E TPGS-functionalized PLGA nanoparticles for delivery of paclitaxel. Int J Pharm. 2013;446(1–2):24–33. | ||
Niu M, Naguib YW, Aldayel AM, et al. Biodistribution and in vivo activities of tumor-associated macrophage-targeting nanoparticles incorporated with doxorubicin. Mol Pharm. 2014;11(12):4425–4436. | ||
Karra N, Nassar T, Ripin AN, Schwob O, Borlak J, Benita S. Antibody conjugated PLGA nanoparticles for targeted delivery of paclitaxel palmitate: efficacy and biofate in a lung cancer mouse model. Small. 2013;9(24):4221–4236. | ||
Smith LL. Mechanism of paraquat toxicity in lung and its relevance to treatment. Hum Toxicol. 1987;6(1):31–36. | ||
van der Wal NA, van Oirschot JF, van Dijk A, Verhoef J, van Asbeck BS. Mechanism of protection of alveolar type II cells against paraquat-induced cytotoxicity by deferoxamine. Biochem Pharmacol. 1990;39(11):1665–1671. | ||
Fukushima T, Tanaka K, Lim H, Moriyama M. Mechanism of cytotoxicity of paraquat. Environ Health Prev Med. 2002;7(3):89–94. | ||
Yuan X, Ji WX, Chen S, et al. A novel paclitaxel-loaded poly(D,L-lactide-co-glycolide)-Tween 80 copolymer nanoparticle overcoming multidrug resistance for lung cancer treatment. Int J Nanomedicine. 2016;11:2119–2131. | ||
Khalil NM, do Nascimento TC, Casa DM, et al. Pharmacokinetics of curcumin-loaded PLGA and PLGA-PEG blend nanoparticles after oral administration in rats. Colloids Surf B Biointerfaces. 2013;101:353–360. | ||
Pamujula S, Hazari S, Bolden G, et al. Cellular delivery of PEGylated PLGA nanoparticles. J Pharm Pharmacol. 2012;64(1):61–67. | ||
Wu YH, Li XW, Li WQ, et al. Fluorofenidone attenuates bleomycin-induced pulmonary fibrosis by inhibiting eukaryotic translation initiation factor 3a (eIF3a) in rats. Eur J Pharmacol. 2016;773:42–50. | ||
Song C, He L, Zhang J, et al. Fluorofenidone attenuates pulmonary inflammation and fibrosis via inhibiting the activation of NALP3 inflammasome and IL-1β/IL-1R1/MyD88/NF-κB pathway. J Cell Mol Med. 2016;20(11):2064–2077. | ||
Abdulhussein AA, Wallace HM. Polyamines and membrane transporters. Amino Acids. 2014;46(3):655–660. | ||
Hoet PH, Nemery B. Polyamines in the lung: polyamine uptake and polyamine-linked pathological or toxicological conditions. Am J Physiol Lung Cell Mol Physiol. 2000;278(3):417–433. | ||
Dredge K, Kink JA, Johnson RM, Bytheway I, Marton LJ. The polyamine analog PG11047 potentiates the antitumor activity of cisplatin and bevacizumab in preclinical models of lung and prostate cancer. Cancer Chemother Pharmacol. 2009;65(1):191–195. | ||
Tomasi S, Renault J, Martin B, et al. Targeting the polyamine transport system with benzazepine- and azepine-polyamine conjugates. J Med Chem. 2010;53(21):7647–7663. | ||
García-Fernández AJ, Rodríguez RA, Pérez-Pertejo Y, Balaña-Fouce R. Characterization of putrescine uptake in hamster amelanocytic melanoma AMEL-3 cells. Mol Cells. 2005;20(1):127–135. | ||
Reguera RM, Tekwani BL, Balaña-Fouce R. Polyamine transport in parasites: a potential target for new antiparasitic drug development. Comp Biochem Physiol C Toxicol Pharmacol. 2005;140(2):151–164. | ||
Pourgholamhossein F, Sharififar F, Rasooli R, et al. Thymoquinone effectively alleviates lung fibrosis induced by paraquat herbicide through down-regulation of pro-fibrotic genes and inhibition of oxidative stress. Environ Toxicol Pharmacol. 2016;45:340–345. | ||
Karl PI, Friedman PA. Competition between paraquat and putrescine for accumulation by rat lung slices. Toxicology. 1983;26(3–4):317–323. | ||
Gaudreault P, Karl PI, Friedman PA. Paraquat and putrescine uptake by lung slices of fetal and newborn rats. Drug Metab Dispos. 1984;12(5):550–552. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.