Back to Journals » Clinical Interventions in Aging » Volume 12
Spectrum of noncerebrovascular rapidly progressive cognitive deterioration: a 2-year retrospective study
Authors Zhang Y, Gao T ![]() , Tao Q
, Tao Q ![]()
Received 25 June 2017
Accepted for publication 5 September 2017
Published 9 October 2017 Volume 2017:12 Pages 1655—1659
DOI https://doi.org/10.2147/CIA.S144821
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Zhi-Ying Wu
Yun Zhang, Ting Gao, Qing-Qing Tao
Department of Neurology and Research Center of Neurology in Second Affiliated Hospital, Zhejiang University School of Medicine, Hangzhou, China
Purpose: The number of cognitive deterioration patients has been steadily increasing as the population ages in China. Patients with cognitive deterioration demonstrated diverse patterns, often making the diagnosis difficult, especially in rapidly progressive cognitive deterioration (RPCD) patients. The purpose of this study was to exhibit the disease spectrum and frequency of noncerebrovascular RPCD in patients from a medical college hospital of southeastern China.
Materials and methods: We performed a 2-year retrospective cohort study including 310 RPCD patients who had been admitted to the Department of Neurology, Second Affiliated Hospital, Zhejiang University School of Medicine, from January 1, 2015 to December 31, 2016. RPCD patients’ information on epidemiologic data and clinical aspects were collected. All the data were analyzed using SPSS.
Results: Of a total of 310 patients hospitalized for RPCD diagnosis, mean age of onset was 55.92±18.89 years. The most common cause of RPCD was viral encephalitis, accounting for 21.9% (68) of the cases, followed by Alzheimer’s disease and autoimmune encephalitis, accounting for 14.5% (45) and 9.0% (28) of the cases, respectively. Creutzfeldt–Jakob disease accounted for 7.1% (22) of the cases. Patients in the secondary RPCD group tended to be younger than those in the primary RPCD group and experienced a more rapid progression course.
Conclusion: Our study suggests that the most common causes of RPCD are secondary neurological diseases and most of them are potentially reversible under appropriate treatment of the underlying disease. The spectrum and frequency of RPCD in our cohort is comparable with a previous study performed in the European population.
Keywords: cognitive deterioration, spectrum, primary causes, secondary causes
Introduction
Although no precise definition for rapidly progressive cognitive deterioration (RPCD) or dementia (RPD) exists, RPCD or RPD usually refers to conditions that progress from the onset of first symptom to cognitive deterioration or dementia in less than 1–2 years, most occur over weeks to months.1,2 The causes of RPCD are diverse, spanning the spectrum of primary neurological diseases such as prion diseases, neurodegenerative disorders,3 autoimmune encephalopathies, and secondary neurological diseases such as cerebral vascular disease, infectious encephalopathies, and neoplasm.4–8 As these diseases exhibit rapid progression course and some of them are caused by reversible or curable conditions, urgent evaluation and diagnosis for patients with RPCD is necessary.9
The potential causes of RPCD are diverse, often making the diagnosis difficult. From this perspective, data about the spectrum and relative frequency of diseases demonstrating with RPCD could be informative and helpful for the decisions regarding the proper investigations. Studies focusing on the relative frequency of the underlying diseases in cases of RPCD or RPD are few. To our knowledge, currently only two studies performed in the European population examined the causes of RPCD,10,11 and several studies conducted in the United States have examined the causes among patients suspected for Creutzfeldt–Jakob disease (CJD).2,12,13 However, no evidence in this aspect has been reported in Chinese Han population or other Asian population.
Here, we performed a 2-year retrospective study to examine the spectrum and relative frequency of the causes of RPCD in patients from a medical college hospital of southeastern China. A total of 310 RPCD patients’ information on epidemiologic data and clinical aspects were collected and analyzed. To the best of our knowledge, this is the first study on the relative frequency of underlying diseases in cases of RPCD patients from China.
Materials and methods
Subjects
Patients included in this study were referred to the Department of Neurology, Second Affiliated Hospital, Zhejiang University School of Medicine, during a 2-year period (from January 1, 2015 to December 21, 2016). The institutional review board of the Second Affiliated Hospital, Zhejiang University School of Medicine, approved this study, and informed consent was waived because of the retrospective nature of the study. All of the patients’ data were kept confidential. A total of 310 patients hospitalized for RPCD diagnosis were included in our study according to the inclusion criteria which included: 1) patients should have cognitive impairment and 2) cognitive deterioration should have evolved within 2 years.
All patients were evaluated by neurologic examinations and underwent blood tests, including a complete blood count; hematocrit; platelets; serum electrolytes; blood sugar; acute C-reactive protein; liver, kidney and thyroid function tests; serum cholesterol and lipid levels; as well as treponemal serology. Magnetic resonance imaging of the brain including T1, T2, and fluid-attenuated inversion-recovery sequences were performed in all patients. Besides, most of the patients underwent lumbar puncture except for those who had contraindications, and routine cytological and biochemical examination of the cerebrospinal fluid was performed. For patients suspected for mitochondrial disease, additional lactic acid test was performed.
Diagnosis
In the current study, causes of acute cognitive impairment due to infectious, toxic, or metabolic reasons were also included. Dementia was diagnosed according to the Diagnostic and Statistical Manual of Mental Disorders-Fifth Edition.14 Patients with dementia were classified according to the most consistent etiologic diagnosis. Specifically, patients suspected for probable Alzheimer’s disease (AD) were diagnosed according to National Institute on Aging and Alzheimer’s Association (NIA-AA) criteria as described previously.15 Dementia with Lewy body (DLB) and Parkinson disease dementia were defined according to the consensus guidelines as described previously.16 Diagnosis of other subtypes of dementia such as frontotemporal lobar degeneration (FTLD),17 progressive supranuclear palsy,18 CJD,19 and multiple system atrophy (MSA) were done according to the International Consensus Criteria.20
Statistical analysis
Statistical analysis was conducted using the SPSS 16.0 package (IBM Corporation, Armonk, NY, USA). One-way ANOVA was performed to evaluate the differences in age at onset and time of progression to cognitive deterioration. Categorical data, such as gender, were evaluated by chi-square tests.
Results
Characteristics of subjects with different causes of RPCD
Demographic data of the diagnostic categories by etiology for the 310 patients with RPCD are shown in Table 1. A total of 310 patients were hospitalized with RPCD diagnosis, and mean age of onset was 55.92±18.89 years. The most common cause of RPCD was viral encephalitis, accounting for 21.9% (68) of the cases, followed by probable AD and autoimmune encephalitis, accounting for 14.5% (45) and 9.0% (28) of the cases, respectively. CJD accounted for 7.1% (22) of the cases. The age of patients at presentation ranged from 13 to 87 years, with a mean age of 56.15±18.94 years.
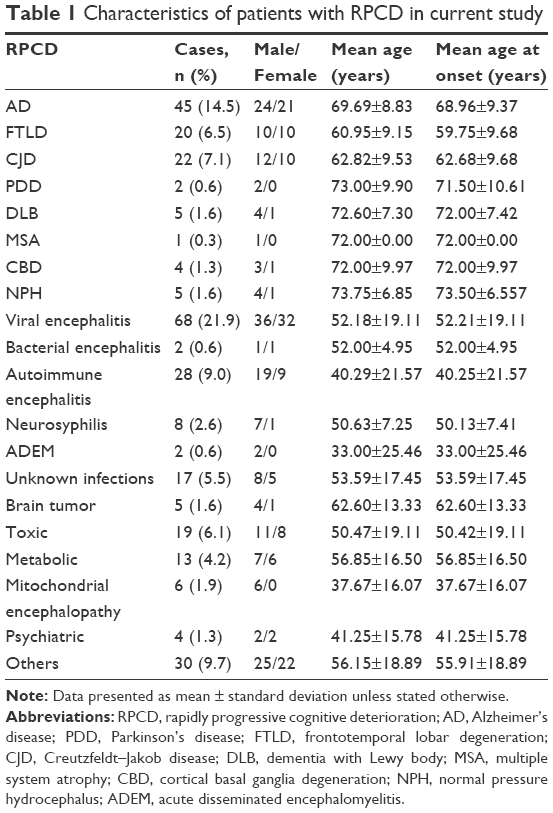 | Table 1 Characteristics of patients with RPCD in current study |
Frequency of primary neurological diseases and secondary neurological diseases that cause RPCD
The frequency of primary neurological diseases that cause RPCD is presented in Table 2. The most common primary neurological disease that causes RPCD was AD, accounting for 45.5% (45) of the cases, followed by CJD and FTLD, accounting for 22.2% (22) and 20.2% (20) of the cases. The frequency of secondary neurological diseases that cause RPCD is given in Table 3. The most common secondary neurological disease that causes RPCD was viral encephalitis, accounting for 41.5% (68) of the cases, followed by autoimmune encephalitis, accounting for 17.2% (28) of the cases. The causes of RPCD in our study are shown in Figure 1. Patients in the secondary cognitive deterioration group tended to be younger than those in the primary cognitive deterioration group and experienced a more rapid progression course.
 | Table 2 Spectrum and frequency of primary cognitive deterioration in current study |
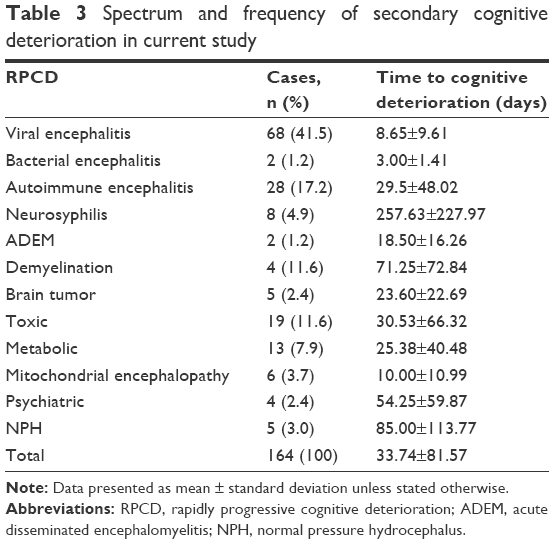 | Table 3 Spectrum and frequency of secondary cognitive deterioration in current study |
 | Figure 1 Causes of rapidly progressive cognitive deterioration. |
Discussion
Compared with the common chronic causes of cognitive impairment and dementia, RPCD usually has a relatively fast rate of progression course and some of them are caused by reversible conditions. For a neurologist, attention must be directed to identify potentially curable causes of RPCD. A systemic history, clinical assessment, and investigations should be prioritized in an effort to identify the potentially reversible causes of RPCD.21 The diverse causes of RPCD were a great challenge for the selection of a proper investigation protocol. Data regarding the relative frequency of diseases presenting with RPCD may be informative for the diagnosis.
In this study, we performed a 2-year retrospective cohort study including 310 RPCD patients to determine the spectrum and frequency of the causes of RPCD in the southeastern Chinese population. In our study, the most common cause of RPCD was secondary neurological disease, which was in line with the data reported in a previous study performed in the Greek cohort. Viral encephalitis was the most common cause among the secondary neurological diseases, accounting for 41.5%, followed by autoimmune encephalitis, accounting for 17.2% of the cases. In another cohort including 640 cases from the University of California at San Francisco (UCSF), CJD was the most frequent cause of RPCD. This result was not surprising considering that UCSF is a major referral center for prion diseases. However, secondary neurological disease was also the most frequent cause among the remaining non-CJD cases, indicating that high proportion of RPCD cases were caused by secondary neurological diseases which could be potentially reversible under appropriate treatment of the underlying disease.
In our study, the most common primary neurological disease that caused RPCD was AD, accounting for 45.5%, and it was also the second most common cause of RPCD, accounting for 14.5% of the cases. Neurodegenerative dementias such as AD, FTLD, MSA, and DLB typically show insidious onset with relatively slowly progressive course. A previous study performed in the Mayo Clinic demonstrated that AD, FTLD, and DLB can also cause a RPCD.22 Similarly, AD was the second most common cause of RPD in a Greek cohort, accounting for 17.6% of the cases, suggesting the frequency and spectrum of RPCD in our cohort is comparable with that of the European population.
There are several limitations in our study that need to be discussed. First, all the patients were selected from one single hospital, which may lead to inherent selection bias making some causes of RPCD to be overrepresented or underrepresented. Second, the diagnoses in our study lacked pathologic data for confirmation and most diagnoses were derived from single adjudicator. In addition, the sample size in the current study was relatively small, hence future multicenter independent studies with larger sample size are needed to replicate our results.
Conclusion
In conclusion, our study suggests the following: 1) the most common causes of RPCD are secondary neurological diseases and many of them are potentially reversible under appropriate treatment of the underlying disease and 2) the frequency and spectrum of RPCD in our cohort is comparable with a previous study performed in the European population.
Disclosure
The authors report no conflicts of interest in this work.
References
Geschwind MD. Rapidly progressive dementia. Continuum (Minneap Minn). 2016;22(2 Dementia):510–537. | ||
Geschwind MD, Haman A, Miller BL. Rapidly progressive dementia. Neurol Clin. 2007;25(3):783–807. | ||
Grau-Rivera O, Gelpi E, Carballido-Lopez E, Sanchez-Valle R, Lopez-Villegas MD. Rapidly progressive dementia with psychotic onset in a patient with the C9ORF72 mutation. Clin Neuropathol. 2015;34(5):294–297. | ||
Mead S, Rudge P. CJD mimics and chameleons. Pract Neurol. 2017;17(2):113–121. | ||
Bucelli RC, Ances BM. Diagnosis and evaluation of a patient with rapidly progressive dementia. Mo Med. 2013;110(5):422–428. | ||
Weiss D, Brockmann K, Nagele T, Gasser T, Kruger R. Rapid emergence of temporal and pulvinar lesions in MELAS mimicking Creutzfeldt-Jakob disease. Neurology. 2011;77(9):914. | ||
Briani C, Baracchini C, Zanette G, Zanusso G, Carollo C, Monaco S. Rapidly progressive dementia in hypereosinophilic syndrome. Eur J Neurol. 2001;8(3):279–280. | ||
Schielke E, Nolte C, Muller W, Bruck W. Sarcoidosis presenting as rapidly progressive dementia: clinical and neuropathological evaluation. J Neurol. 2001;248(6):522–524. | ||
Degnan AJ, Levy LM. Neuroimaging of rapidly progressive dementias, part 1: neurodegenerative etiologies. AJNR Am J Neuroradiol. 2014;35(3):418–423. | ||
Papageorgiou SG, Kontaxis T, Bonakis A, Karahalios G, Kalfakis N, Vassilopoulos D. Rapidly progressive dementia: causes found in a Greek tertiary referral center in Athens. Alzheimer Dis Assoc Disord. 2009;23(4):337–346. | ||
Sala I, Marquie M, Sanchez-Saudinos MB, et al. Rapidly progressive dementia: experience in a tertiary care medical center. Alzheimer Dis Assoc Disord. 2012;26(3):267–271. | ||
Geschwind MD, Shu H, Haman A, Sejvar JJ, Miller BL. Rapidly progressive dementia. Ann Neurol. 2008;64(1):97–108. | ||
Day GS, Tang-Wai DF. When dementia progresses quickly: a practical approach to the diagnosis and management of rapidly progressive dementia. Neurodegener Dis Manag. 2014;4(1):41–56. | ||
Widiger TA, Samuel DB. Diagnostic categories or dimensions? A question for the Diagnostic And Statistical Manual Of Mental Disorders – fifth edition. J Abnorm Psychol. 2005;114(4):494–504. | ||
Jack CR Jr, Albert MS, Knopman DS, et al. Introduction to the recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Demen. 2011;7(3):257–262. | ||
McKeith IG. Consensus guidelines for the clinical and pathologic diagnosis of dementia with Lewy bodies (DLB): report of the Consortium on DLB International Workshop. J Alzheimers Dis. 2006;9(3 Suppl):417–423. | ||
Neary D, Snowden JS, Gustafson L, et al. Frontotemporal lobar degeneration: a consensus on clinical diagnostic criteria. Neurology. 1998;51(6):1546–1554. | ||
Litvan I, Agid Y, Calne D, et al. Clinical research criteria for the diagnosis of progressive supranuclear palsy (Steele-Richardson-Olszewski syndrome): report of the NINDS-SPSP international workshop. Neurology. 1996;47(1):1–9. | ||
Manix M, Kalakoti P, Henry M, et al. Creutzfeldt-Jakob disease: updated diagnostic criteria, treatment algorithm, and the utility of brain biopsy. Neurosurg Focus. 2015;39(5):E2. | ||
Laurens B, Vergnet S, Lopez MC, et al. Multiple system atrophy – state of the art. Curr Neurol Neurosci Rep. 2017;17(5):41. | ||
Rosenbloom MH, Atri A. The evaluation of rapidly progressive dementia. Neurologist. 2011;17(2):67–74. | ||
Josephs KA, Ahlskog JE, Parisi JE, et al. Rapidly progressive neurodegenerative dementias. Arch Neurol. 2009;66(2):201–207. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.