Back to Journals » International Journal of Nanomedicine » Volume 15
Sorafenib-Loaded Nanoparticles Based on Biodegradable Dendritic Polymers for Enhanced Therapy of Hepatocellular Carcinoma
Authors Li Z, Ye L, Liu J, Lian D, Li X
Received 5 November 2019
Accepted for publication 21 February 2020
Published 5 March 2020 Volume 2020:15 Pages 1469—1480
DOI https://doi.org/10.2147/IJN.S237335
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Linlin Sun
Zihuang Li,1 Ling Ye,2 Jingwen Liu,1 Daizheng Lian,1 Xianming Li1
1Department of Radiation Oncology, The Second Clinical Medical College of Jinan University, Shenzhen Municipal People’s Hospital, Shenzhen 518020, People’s Republic of China; 2Department of Oncology, The First Affiliated Hospital of Jinan University, Guangzhou 510632, People’s Republic of China
Correspondence: Zihuang Li; Xianming Li
Department of Radiation Oncology, The Second Clinical Medical College of Jinan University, Shenzhen Municipal People’s Hospital, Shenzhen 518020, People’s Republic of China
Tel +86 755 22942428
Email [email protected]; [email protected]
Purpose: In spite of its enhanced efficacy and reduced side effects in clinical hepatocellular carcinoma (HCC) therapy, the therapeutic efficacy of antitumor angiogenesis inhibitor sorafenib (SFB) is still restricted due to short in vivo half-life and drug resistance. Here, a novel SFB-loaded dendritic polymeric nanoparticle (NP-TPGS-SFB) was developed for enhanced therapy of HCC.
Methods: NP-TPGS-SFB was fabricated by encapsulating SFB with biodegradable dendritic polymers poly(amidoamine)-poly(γ-benzyl-L-Glutamate)-b-D-α-tocopheryl polyethylene glycol 1000 succinate (PAM-PBLG-b-TPGS).
Results: NP-TPGS-SFB exhibited excellent stability and achieved acid-responsive release of SFB. It also exhibited much higher cellular uptake efficiency in HepG2 human liver cells than PEG-conjugated NP (NP-PEG-SFB). Furthermore, MTT assay confirmed that NP-TPGS-SFB induced higher cytotoxicity than NP-PEG-SFB and free SFB, respectively. Lastly, NP-TPGS-SFB significantly inhibited tumor growth in mice bearing HepG2 xenografts, with negligible side effects.
Conclusion: Our result suggests that NP-TPGS-SFB may be a novel approach for enhanced therapy of HCC with promising potential.
Keywords: dendritic block copolymer, sorafenib, enhanced therapy, TPGS, hepatocellular carcinoma
Introduction
Hepatocellular carcinoma (HCC), the third most lethal type of cancer worldwide,1 has been frequently diagnosed as a highly graded hemangioma.2 To combat this disease, anti-angiogenic strategies have been put forward as a potential therapy for HCC. Sorafenib (SFB), a multikinase inhibitor, is an angiogenesis inhibitor used to treat advanced liver cancer, with the potential to greatly improve the survival rate of liver cancer patients.3–5 However, there are still some problems with SFB, since it has poor water solubility and a short half-life in vivo.6 Besides, liver cancer can evade anti-angiogenic therapy and become resistant to SFB, causing a high recurrence rate.7–9 Therefore, new methods to solve these problems are urgently required.
Biodegradable polymeric nanocarriers have been widely studied to improve the solubility of hydrophobic drugs, prolong the half-life, and improve the targeted enrichment efficiency of drugs to tumors through the enhanced permeation and retention (EPR) effect.10–18 For example, the nanodrug Genexol®-PM, which has been on the market, is loaded with anti-tumor drug paclitaxel using amphiphilic biodegradable block copolymer mPEG-b-PDLLA.19 The nanodrug has been shown to enhance the efficacy and reduce the side effects of paclitaxel in some cancer cases, including breast cancer, non-small cell lung cancer and ovarian cancer. However, as with most PEGylated polymeric micelles self-assembled from block copolymers, nanodrugs face several problems. First, polymeric micelles self-assembled from amphiphilic polymers may disassociate rapidly because of the vast dilution after being injected into the body, although they are very stable above the critical micelle concentration (CMC) in vitro.20–22 Second, nanocarriers may be released prematurely during circulation, reducing delivery efficiency, since their stability also depends on the interaction between hydrophobic segments and drug. Currently, the interactions between many chemotherapy drugs and polymers are weak, leading to premature release during circulation and reduced delivery efficiency.22,23 Finally, common PEG-based nanocarriers can hardly overcome cancer drug resistance. Therefore, it is of great value to develop new nanocarriers that can improve the stability of the vector in vivo, effectively load drugs and overcome the cancer drug resistance.
Owing to their covalent nature, multi-arm dendritic block copolymers boost excellent structural stability in vivo, drawing a lot of attention.24–27 As the first commercialized dendrimer family, poly(amidoamine) (PAMAM) dendrimers with tens of terminal functional groups have been widely used as macroinitiators to synthesize dendritic polymers.28–31 For example, the biocompatible and biodegradable PAMAM-based poly(γ-benzyl-L-glutamate) (PBLG) was synthesized from directly initiating the ring-opening polymerization of γ-benzyl-L-glutamate-N-carboxyanhydride (BLG-NCA) monomers by amino-terminated PAMAM. Recently reports show that aryl group-containing drugs like paclitaxel are efficiently loaded by nanoparticles fabricated from aromatic groups-containing polymers via noncovalent pi-pi stacking interaction, with significantly improved stability in vivo.32–34 Another unique advantage of PBLG is that it can be hydrolyzed into hydrophilic PGlu in acid environment such as endosome and lysosome of cancer cells, leading to accelerated drug release. Thus, we anticipated that PAMAM-PBLG-based SFB-loaded nanoparticles would be quite stable and avert the premature drug release during circulation, and achieve accelerated drug release after cellular uptake.
The soluble vitamin E derivative D-α-tocopheryl polyethylene glycol 1000 succinate (TPGS), esterified from the acid group of vitamin E succinate and polyethylene glycol (PEG) 1000, has been listed as a safe pharmaceutical excipient by the Food and Drug Administration (FDA).35–37 It is well known that TPGS could improve the cellular uptake,38 and prevent P-glycoprotein from circumventing drug resistance by interfering with the structure and function of mitochondria.39 Mei group has reported that surface modification of PLGA nanoparticles (NPs) with TPGS prolonged the half-life of drugs in vivo and facilitated their cellular uptake.27 Thus, we confirmed that through conjugation of TPGS with PAMAM-PBLG-based nanoparticles rather than PEG, the stability of the nanoparticles is maintained, cellular drug uptake is improved and drug resistance is overcome. As far as we know, no such TPGS-conjugated PAMAM-PBLG was yet reported to prepare SFB-loaded nanoparticles.
Thus, to prepare SFB-loaded nanoparticle, we designed a novel design of TPGS-containing dendritic polymeric PAM-PBLG-b-TPGS. This nanoparticle, referred as NP-TPGS-SFB, consisted of dendritic molecule PAMAM-G3, aryl-containing segment PBLG and a TPGS polymer (Figure 1). We then tested the physicochemical properties such as size, zeta potential, drug loading content, encapsulation efficiency, in vitro stability and drug release behaviors of NP-TPGS-SFB. Furthermore, the cellular uptake and in vitro cytotoxicity of NP-TPGS-SFB were evaluated in the human liver cancer cells HepG2. Finally, the antitumor efficacy and safety of NP-TPGS-SFB in mice bearing HepG2-derived tumor xenografts were evaluated.
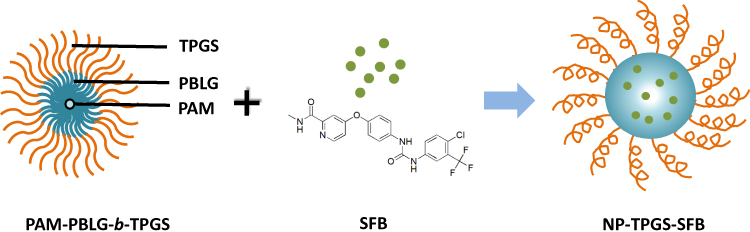 |
Figure 1 Schematic representation of the SFB-loaded polymeric nanoparticles (NP-TPGS-SFB) fabricated from SFB and PAM-PBLG-b-TPGS by nanoprecipitation method. |
Materials and Methods
Materials
N-carboxyanhydride of γ-benzyl-L-glutamate (BLG-NCA) was purchased from J&K Chemical Ltd. (Shanghai, China). PAMAM-G3-NH2 (PAM-NH2, Mw = 6900 Da) was purchased from Aladdin Industrial (Shanghai, China). N-Hydroxysuccinimide terminated PEG2k (PEG2k-NHS) and N-Hydroxysuccinimide-modified D-α-tocopheryl polyethylene glycol 1000 succinate (TPGS-NHS) were purchased from Ponsure Biotechnology (Shanghai, China). Sorafenib was obtained from Cayman Chemical Ltd. HepG2 human liver cancer cell and LO2 normal liver cell line were purchased from American Type Culture Collection (ATCC). Fetal bovine serum (FBS) was purchased from Lonza Walkersville. The Dulbecco’s Modified Eagle Medium (DMEM) growth medium and penicillin/streptomycin were both purchased from Invitrogen. Balb/C nude mice (5–6 weeks old), purchased from Guangdong Province Medical Animal Center, were maintained in an SPF (specific pathogen-free) class experimental animal room.
Synthesis of Dendritic Polymer PAM-PBLG-b-TPGS
PAM-PBLG-NH2 was synthesized through the ring-opening polymerization according to references.29 Six grams of BLG-NCA and 0.54 g of the dendrimer PAM-NH2 were dissolved in CH2Cl2 respectively and then mixed for polymerization (Figure 2) at 35°C for 2 d. The repeat units of PBLG in PAM-PBLG-NH2 were quantified by 1H-NMR spectroscopy using CDCl3 as solvent. Next, 16 N TPGS-NHS was reacted with PAM-PBLG-NH2 in CHCl3 at 25°C for 2 h to obtain the dendritic polymer PAM-PBLG-b-TPGS. PAM-PBLG-b-PEG was synthesized by a similar method as the control. The critical micelle concentration (CMC) of the polymers PAM-PBLG-b-TPGS and PAM-PBLG-b-PEG was measured according to a previously reported method using pyrene as fluorescent probe.32
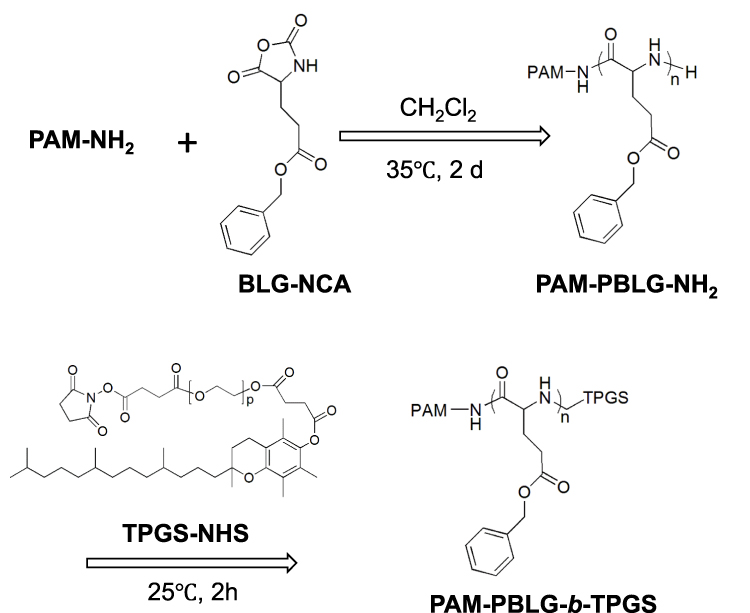 |
Figure 2 Synthesis of PAM-PBLG-b-TPGS. |
Fabrication of SFB-Loaded Polymeric Nanoparticles
The drug-loaded polymeric nanoparticles NP-TPGS-SFB were fabricated as follows. Polymer and pre-weighed amount of SFB powder were both dissolved 10 mL DMF and then added dropwise into 100 mL DI water under stirring. After that free SFB and DMF solvent in the mixture were dialyzed away through a dialysis bag in DI water (MWCO: 5 kDa). Lastly, the purified nanoparticle solution was lyophilized and preserved for further use. NP-PEG-SFB and coumarin 6-loaded NPs were both fabricated similarly and all the lyophilized NPs were redispersed in PBS before use.
Size, Zeta Potential and Morphology
The size and zeta potential of NPs were determined using a Malvern Mastersizer 2000 and the tests were finished triplicate for calculating the average values. Scanning electron microscopic (SEM) images were obtained on a field-emission SEM.
Drug Loading Content and Encapsulation Efficiency
The drug loading content (LC) and drug encapsulation efficiency (EE) of the SFB-loaded NPs were both quantified in line with the previous work.40 After centrifuging at 15,000 rpm for 15 min, the supernatant of SFB-loaded NPs solution was collected and measured by UV–Vis spectrophotometer at 270 nm. Then, the background of NPs was subtracted by testing the supernatant of non-drug-loaded NPs. The test was performed three times, followed by calculating the LC and EE according to:
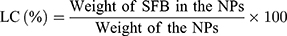
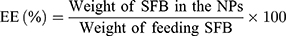
In vitro Stability and Drug Release Study
The stability of NP-TPGS-SFB in PBS and DMEM + 10% FBS over 7 days was tested by DLS. The SFB release rate from it under different pH conditions (pH 7.4 and 5.0) was studied through dialysis. Five milliliters of NPs solution (1 mg/mL) was added in a dialysis bag (MWCO: 5 kDa) and then immersed in a tube containing 50 mL of cell culture media at pH 7.4 and 5.0, respectively. The tube was vibrated in an orbital shaker water bath at 200 rpm and 37°C. At defined periods 1 mL of the solution outside the dialysis bag was fetched to determine the concentrations of SFB by UV–Vis spectrophotometer at 270 nm. Then, the cumulative release of SFB from NPs at each time point was calculated.
Cellular Uptake Study of NPs
The cellular uptake behaviors of coumarin-6 loaded NPs were studied by confocal laser scanning microscopy (CLSM). Firstly, HepG2 cells (1×105 cells/well) were seeded in 12-well culture plates and cultured in serum-containing DMEM medium overnight. Next, after washed one time with PBS, the cells were added with coumarin 6-loaded NP-PEG and NP-TPGS (coumarin 6 concentration as 2 μg/mL) and incubated for another 3 h. Then, the cells were washed three times with cold PBS and fixed by methanol for 20 min. After that, the cells were observed by CLSM (Olympus Fluoview FV-1000, Japan). Coumarin 6-loaded NPs and the DAPI-stained cell nuclei were, respectively, observed through green channel (coumarin 6) at 485 nm excitation and blue channel (DAPI) at 430 nm excitation.
For quantitative analysis, HepG2 cells (1×105 cells/well) were seeded in 12-well culture plates and cultured in serum-containing DMEM medium overnight. The cells were balanced with Hank’s buffered salt solution (HBSS) at 37°C for 1 h. Then, coumarin 6-loaded NPs at concentrations of 100, 200 and 500 μg/mL, respectively, were added into the cells and incubated for 3 h. After that, the cells were washed three times with cold PBS followed by putting 50 mL of 0.5% Triton X-100 in 0.2 N sodium hydroxide into each sample well to lyse the cells. Cellular uptake efficiency, expressed as fluorescence intensity (%), was calculated as the percentage of fluorescence in the cells versus the amount of fluorescence present in the feed medium.
In vitro Cytotoxicity of SFB-Loaded Polymeric Nanoparticles
The 50% growth inhibitory concentrations (IC50) of SFB, NP-PEG-SFB and NP-TPGS-SFB on the HepG2 cells were determined through MTT assay. HepG2 cells (1×104 cells/well) were seeded in 96-well culture plates and incubated for 24 h. Then, SFB, NP-PEG-SFB and NP-TPGS-SFB with equivalent drug concentrations ranging from 0.1 to 12.5 μg/mL or Drug-free NP-TPGS with the same polymer concentrations were added into the wells and incubated for another 24 and 48 h, respectively. The cell viability was quantified by formazan absorbance at 490 nm using a Bio-Rad 680 microplate reader. The viability study of different drug formulations on LO2 cells was carried out as controls in similar methods.
Hemolysis Assay
In the hemolysis assay, red blood cells (RBCs) were separated from plasma by centrifugation at 1600 rpm for 5 min, washed with saline, and resuspended at a 2% (v/v) cell concentration. Then, SFB-loaded NPs (5–50 μg/mL) were added into the RBC suspension and the samples were incubated at 37°C for 3 h. Deionized water was added as a positive control, and saline solution was added for the negative control group. Then, the mixtures were centrifuged at 1600 rpm for 5 mins and the supernatants were collected. After incubating for 30 min at room temperature to allow haemoglobin oxidation, oxyhemoglobin absorbance was measured by a UV–Visible spectrophotometer at 576 nm as an indication of RBC lysis. The hemolysis ratio (%) was calculated using the following formula:
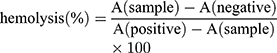
here, A(sample), A(negative), and A(positive) are the absorbances of the samples, negative control, and positive control, respectively.
In vivo Antitumor Efficacy
The protocols for animal assays were approved by the Administrative Committee on Animal Research in Shenzhen Municipal People’s Hospital. All of in vivo experiments complied with the guidelines of the institutional animal ethics committee. The mice were divided into five groups randomly (n = 5) followed by establishing the HepG2 xenograft model by the subcutaneous injection of 5*106 HepG2 cells (100 μL) into the right flank of each mouse. The tumors were all allowed to grow to 50 mm3 in volume before treatment. Then, the mice were injected intravenously with Saline, SFB, NP-PEG-SFB and NP-TPGS-SFB (5 mg/kg as the dose of SFB) via the tail vein three times on days 0, 4 and 8. After 14 days of treatment, mice were sacrificed by cervical decapitation. To evaluate the antitumor activity, tumor volume (V) was calculated using the following equation:
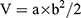
where a and b are the length and width of the tumor, respectively, which were measured by a caliper. The mice were weighted simultaneously to evaluate the systemic toxicity.
Histopathology Evaluation
The histopathology of tumor and major organs was evaluated by optical microscopy following treatments with hematoxylin and eosin (H&E). On the 14th day, after the mice were sacrificed, their tumor, heart, lung, liver, spleen and kidney were collected, dehydrated in PBS with 10% formaldehyde overnight. Next, paraffin was used for the tissues embedding followed by cutting them into 5 μm slices. Then, the slices of tissues were observed by optical microscopy and the photographs were taken.
Statistical Analysis
All the experiments were finished at least triplicate. Unless noted otherwise, the Data are stated as mean ± SD and analyzed for significance using Student’s t-test. Statistically significant was indicated when probability value (P) <0.05. *P < 0.05; **P < 0.01; ***P < 0.001.
Results and Discussion
Synthesis and Characterization of Dendritic Block Copolymer
The synthesis of dendritic block copolymers PAM-PBLG-b-TPGS and PAM-PBLG-b-PEG was performed according to previous work (Figure 2).29 The chemical structure of copolymer can be verified by 1H-NMR. Figure 3A shows the 1H-NMR spectrum of PAM-PBLG-NH2, where a is the characteristic peak of PAMAM methylene, b, c, d and e are the characteristic peaks of -CHCH2CH2C(O)-, -CHCH2CH2C(O)-, -C(O)CH(CH2)NH- and C6H5CH2- in PBLG, respectively. By calculating the peak area of a, c, d and e, it can be concluded that the unit number of BLG in each dendritic molecule is 240. As PAMAM-G3 contains 32 primary amines, the average PBLG segment repeat unit initiated by each primary amine is 7.5. Peak g in Figure 3B corresponds to -CH2CH2O- of TPGS, which indicating the successful synthesis of PAM-PBLG-b-TPGS. 12 TPGS was calculated to be conjugated on each dendritic molecule. According to previous reported method,32 CMC data of PAM-PBLG-b-TPGS and PAM-PBLG-b-PEG were measured as 0.56 and 0.85 μg/mL, respectively. These CMCs were substantially lower than conventional block copolymers,33 which is likely attributed to dendritic structure of polymers, strong π-π stacking and hydrophobic interactions between the polymer chains.
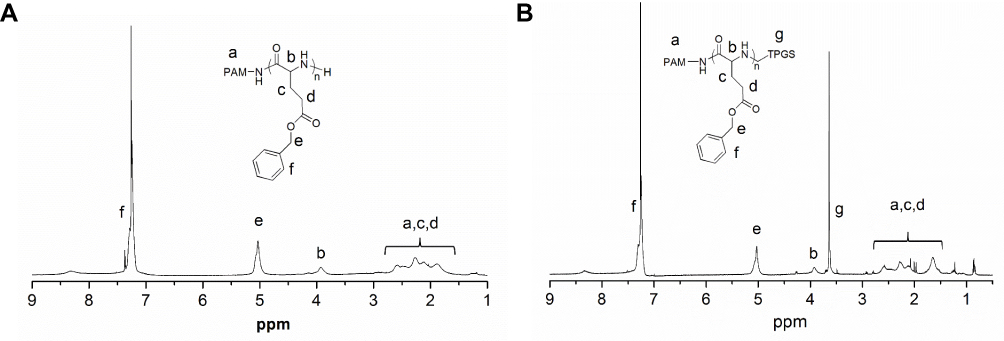 |
Figure 3 1H-NMR spectra of PAM-PBLG-NH2 (A) and PAM-PBLG-b-TPGS (B) in CDCl3. |
Characterization of SFB-Loaded Nanoparticles (NPs)
After PAM-PBLG-b-TPGS and PAM-PBLG-b-PEG were obtained, we used them to prepare the SFB-loaded and drug-free polymeric nanoparticles. DLS results showed that NP-TPGS-SFB was narrow and monodispersed and the average hydrodynamic diameter was about 118.3 nm (Figure 4A), similar to the surface PEG-modified NP-PEG-SFB (Table 1). The size of both NPs is slightly larger than drug-free NPs. SEM results showed that NP-TPGS-SFB, NP-PEG-SFB and drug-free NP-TPGS were all uniformly distributed spherical NPs with particle size around 100 nm (Figure 4B, C and D), consistent with DLS results. NP-TPGS-SFB and NP-PEG-SFB had 15.5% and 13.8% LC and more than 80% EE, respectively, indicating that dendritic polymers based on PBLG could efficiently load SFB. These results were consistent with other previous reports.33
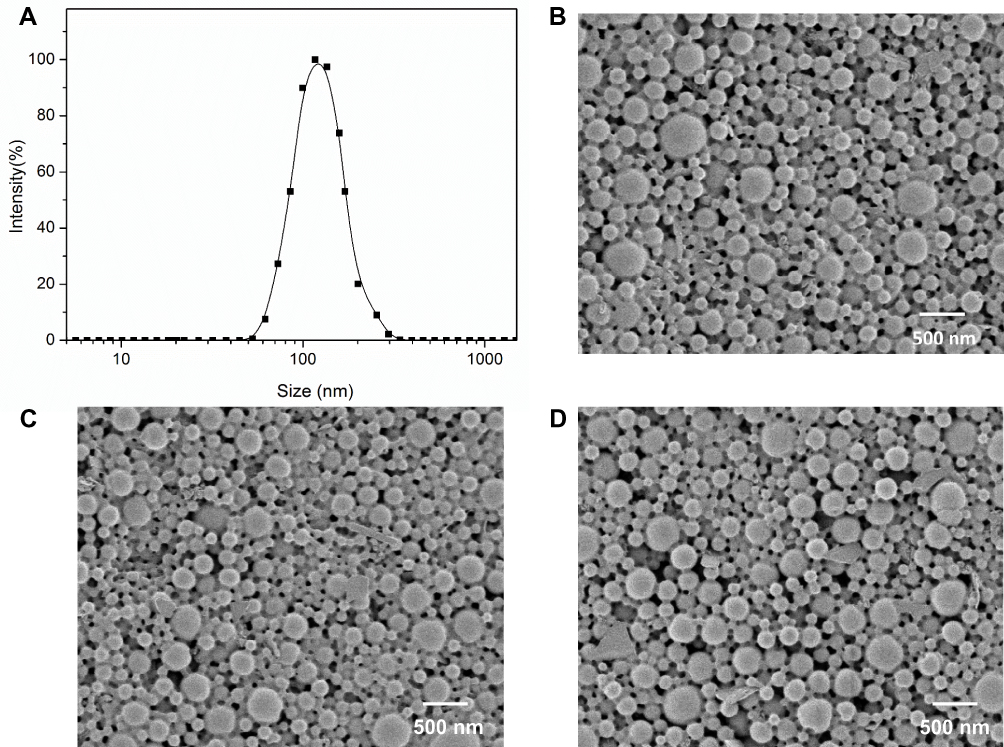 |
Figure 4 Characterization of NP-TPGS-SFB. (A) DLS size distribution of NP-TPGS-SFB. SEM images of NP-TPGS-SFB (B), NP-PEG-SFB (C) and drug-free NP-TPGS (D). |
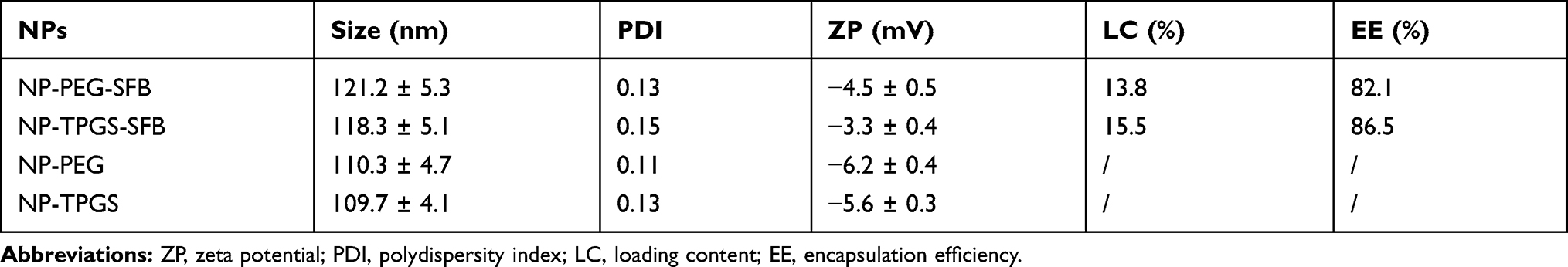 |
Table 1 Characterization of SFB-Loaded Polymeric Nanoparticles |
In vitro Stability and Drug Release Profiles
After the preparation of NP-TPGS-SFB, we tested its stability in vitro. As shown in Figure 5A, the particle size of NP-TPGS-SFB in PBS remained stable for 7 consecutive days, which was in line with our hypothesis. We then tested the drug release of NP-TPGS-SFB in vitro. We compared the release of NP-TPGS-SFB in cell culture medium at pH 7.4 and 5.0. As shown in Figure 5B, only a small amount of SFB was released at pH 7.4, with 20.3% after 12 days. This is consistent with the results reported in the literature and indicates that NP-TPGS-SFB is stable under physiological conditions, effectively retaining SFB.33 However, SFB was released faster from the NP at pH 5.0 and more than 47% after 12 days. This is due to the gradual hydrolysis of PBLG under acidic conditions, thus accelerating the release of SFB.41 These results indicate that, under physiological conditions, NP-TPGS-SFB impairs the premature release of SFB and promotes the fast and efficient release of SFB in cancer cells.
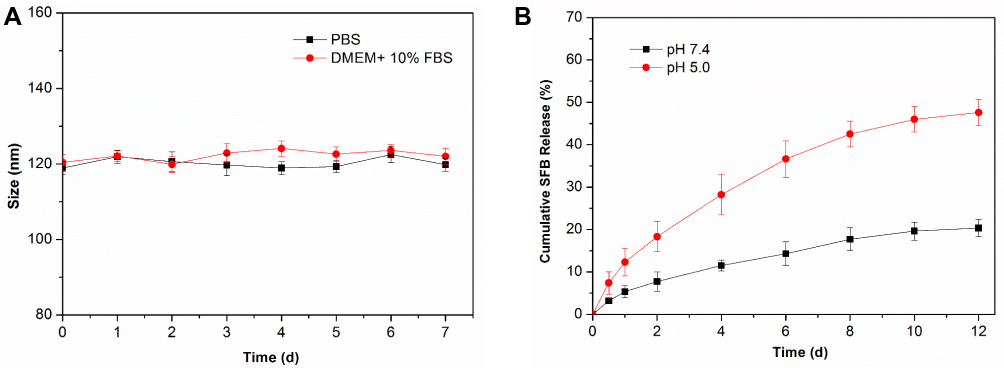 |
Figure 5 (A) Size changes of NP-TPGS-SFB incubated in PBS and DMEM + 10% FBS over 7 d. (B) Cumulative SFB release from NP-TPGS-SFB in different pH conditions over 12 days. |
Cellular Uptake of Coumarin 6-Loaded NPs
To investigate whether NP-TPGS-SFB can be uptaken by cancer cells, we used CLSM to observe the cellular uptake of coumarin 6-loaded NP-PEG and NP-TPGS in HepG2 cells after 3 hrs of culture. As shown in Figure 6, coumarin 6-loaded NP-TPGS cultured cells showed clear coumarin 6 fluorescence, indicating that NP-TPGS was efficiently ingested by HepG2 cells. In contrast, cells cultured with coumarin 6-loaded NP-PEG showed significantly lower fluorescence. These results demonstrated that TPGS-modified on the NP surface significantly increased the uptake of NPs by cancer cells.
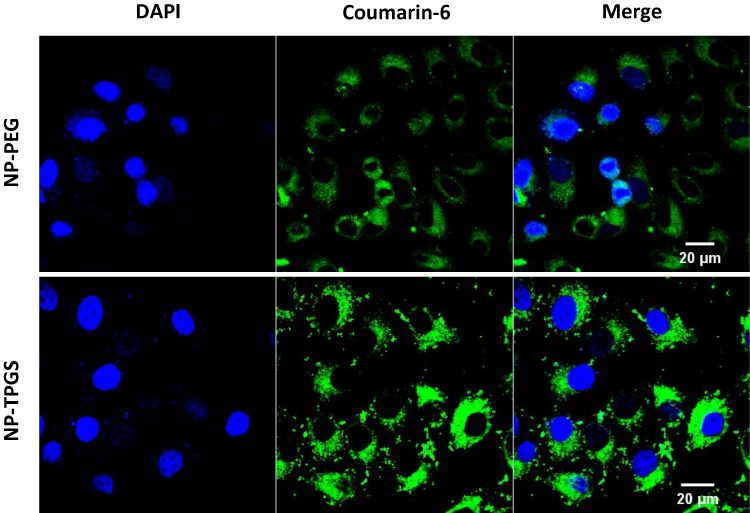 |
Figure 6 CLSM images of HepG2 cells after 3 h incubation with coumarin 6-loaded NPs at 37°C. |
Furthermore, we quantified the cellular uptake efficiency of coumarin 6-loaded NP-TPGS. As shown in Figure 7, after 3 hrs, the uptake level of NP-TPGS and NP-PEG was 75.5% and 42.7%, respectively, for a concentration of NPs of 100 μg/mL. This result shows that the uptake level of NP-TPGS is significantly higher than that of NP-PEG. However, as the NP concentration increased, the uptake efficiency of TPGS-modified NP decreased. This may be due to the cellular limit for the uptake of NPs. While the uptake of NPs in high concentrations may be almost the same as that in low concentration, the uptake efficiency in high NP concentrations is reduced. Nevertheless, these results confirm that conjugation of TPGS can remarkably increase the uptake of NP by cells.
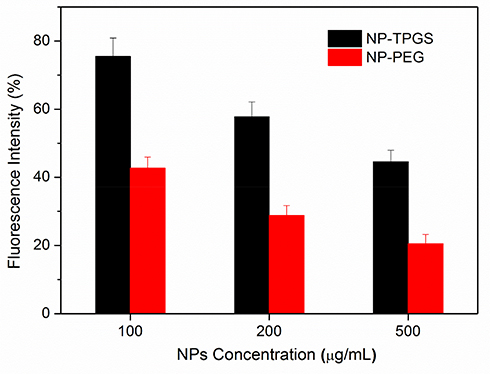 |
Figure 7 Cellular uptake efficiency of coumarin 6-loaded NPs by HepG2 cells under different NPs concentration over 3 h. |
In vitro Cytotoxicity of SFB-Loaded NPs
MTT assay was used to test the cytotoxicity of NP-TPGS-SFB in HepG2 cancer cells. The same concentrations of free SFB and NP-PEG-SFB and non-drug loaded NP-TPGS were used as controls. As shown in Figure 8A, after 24 hrs of cell culture, SFB induced significantly higher cell death than NP-PEG-SFB. However, NP-TPGS-SFB was significantly more lethal than SFB and NP-PEG-SFB. The IC50 values of SFB, NP-PEG-SFB and NP-TPGS-SFB were 6.8, 9.5 and 0.75 μg/mL, respectively (Table 2). After 48 hrs of culture, the cell-killing effect of NP-TPGS-SFB was significantly increased (Figure 8B), with a IC50 of 0.26 μg/mL, which was much lower than the IC50 of SFB and NP-PEG-SFB. This suggests that conjugation of TPGS can significantly enhance cytotoxicity, which is consistent with the results of cellular uptake. It also indicates that the NP can release enough SFB after entering the cell. In addition, the results of toxicity experiments showed that non-drug-loaded NPs had no effect on cell viability, indicating biosafety and potential for practical applications. Conversely, all drug formulations showed much weaker cytotoxicity in the LO2 normal liver cells than HepG2 cells, indicating their specific antitumor efficacy (Figure 8C). This might be attributed to the specificity of SFB to liver cancer cells as well as less cellular uptake of normal cells than cancer cells.42
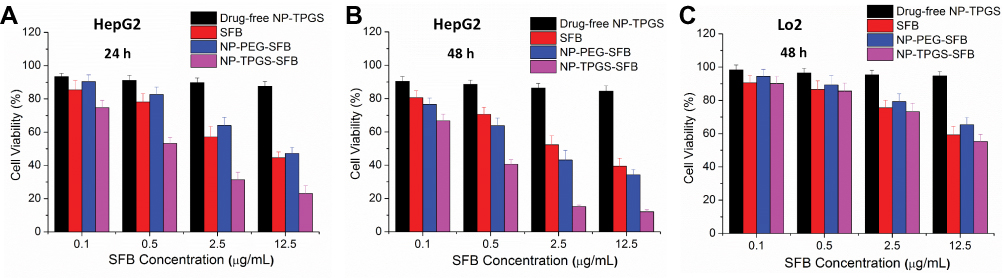 |
Figure 8 Viability of HepG2 and LO2 cells cultured with the SFB, NP-PEG-SFB and NP-TPGS-SFB at the same SFB dose and that of the drug-free NP-TPGS with the same polymer concentrations: (A) 24 h and (B) 48 h for HepG2 cells and (C) 48 h for LO2 cells. |
 |
Table 2 IC50 Values of SFB, NP-PEG-SFB and NP-TPGS-SFB on HepG2 Cells Following 24 and 48 h Incubation, Respectively |
Hemolysis Assay
The hemolysis assay was conducted to address the blood biocompatibility of NP-TPGS-SFB. As shown in Figure 9, the hemolytic results of SFB-loaded NPs including NP-TPGS-SFB and NP-TPGS-SFB were both low and negligible (<5%), within a SFB range of 5–50 μg/mL. This may be due to the electrorepulsion between neutral charge of the TPGS and PEG surface-modified NPs and the negatively charged blood cells. Generally, a hemolysis percentage of 5% is regarded as nontoxic and safe.43 These results indicated that SFB-NPs had good hemocompatibility for further biomedical applications.
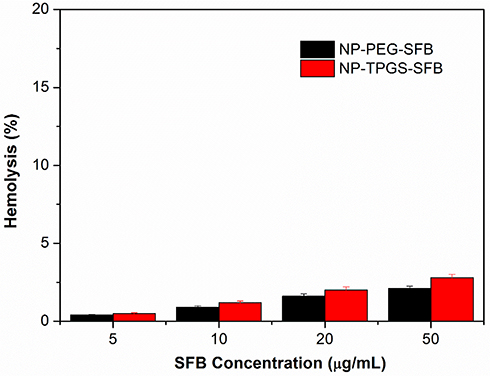 |
Figure 9 In vitro hemolysis assay of SFB-loaded NPs. |
In vivo Antitumor Efficacy
In light of the positive results obtained in the previous experiments, we assessed the anticancer efficacy of NP-TPGS-SFB in vivo. When the tumor volumes of HepG2 xenograft-bearing nude mice grew to 50 mm3, saline, SFB, NP-PEG-SFB and NP-TPGS-SFB were injected three times every 4 days. As shown in Figure 10, after treatment with saline, SFB and NP-PEG-SFB, the tumor volumes of mice increased to about 360, 228 and 181 mm3 for the 14 days after the initial injection. In contrast, treatment with NP-TPGS-SFB inhibits tumor growth to a maximum of 87 mm3. This indicates that NP-TPGS-SFB can inhibit tumor growth, and its effect significantly higher that of SFB and NP-PEG-SFB, which is consistent with the previous cytotoxicity results. Moreover, NP-TPGS-SFB treated mice showed no significant body weight changes compared to the other three control groups (Figure 8B), suggesting that the NPs were not significantly toxic.
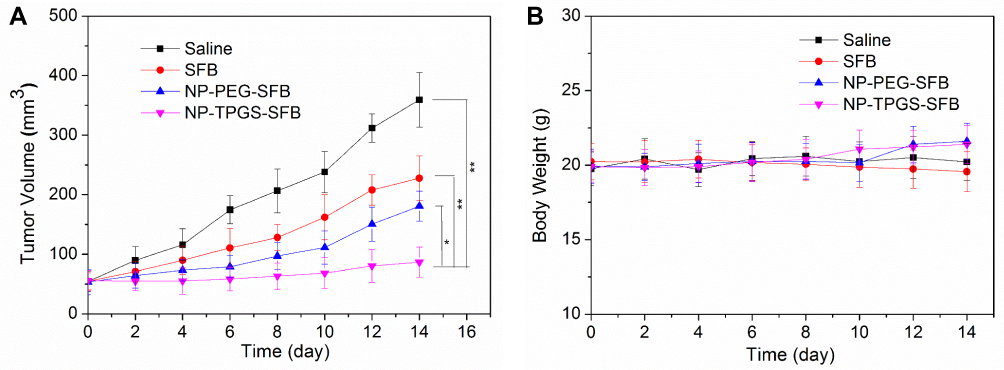 |
Figure 10 Antitumor efficacy of NP-TPGS-SFB upon HepG2 xenograft-bearing nude mice. (A) Tumor volume changes and (B) body weight changes of tumor-bearing mice after treatment with Saline, SFB, NP-PEG-SFB and NP-TPGS-SFB (n = 5). |
In order to study the tumor and tissue damage after treatment, tumor and major organs (heart, lung, liver, spleen and kidney) were collected after the mice were sacrificed. Tissue sections were stained with H&E and analyzed by optical microscopy. In H&E, normal tissues display a indigo blue-dyed nuclei by hematoxylin purple, and pink-dyed cytoplasm and extracellular matrix by eosin. The damaged cells had no obvious cellular morphology. As shown in Figure 11, all tumor tissues in SFB group were seriously damaged, while that in the two SFB-loaded NPs groups were even more damaged, with no obvious cell morphology observed. These results coincided with tumor inhibition results in vivo. In addition, the H&E results of major organs demonstrated that there were no obvious side effects of SFB-loaded NP administration on mice, implying the prospect of practical applications.
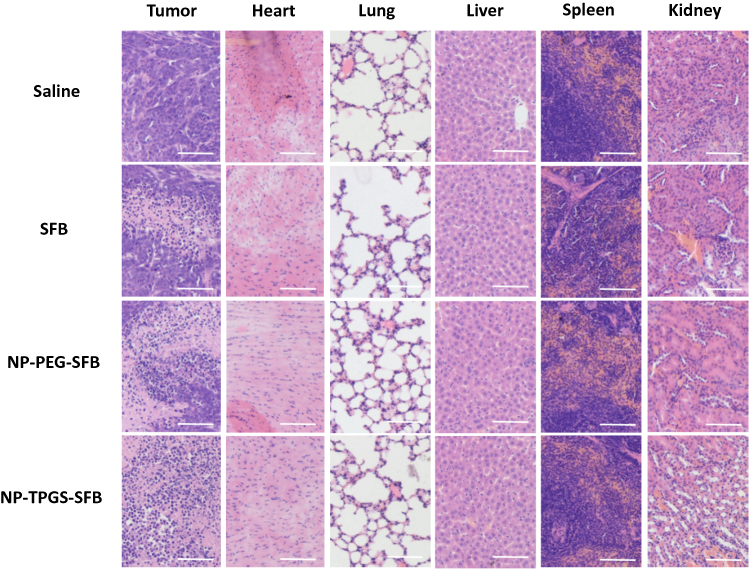 |
Figure 11 Representative H&E stained images of tumor, heart, lung, liver, spleen, and kidney of mice after treated with Saline, SFB, NP-PEG-SFB and NP-TPGS-SFB (scale bar=100 μm). |
Conclusions
A novel SFB-loaded polymeric nanoparticle NP-TPGS-SFB was successfully developed for enhanced therapy of liver cancer. We demonstrated NP-TPGS-SFB holds robust stability and achieves greater efficacy by releasing the anticancer drug SFB in a pH-dependent manner. The TPGS-conjugated NP also exhibited much higher cellular uptake than PEG-conjugated NP, thus causing much greater cytotoxicity than NP-PEG-SFB and free SFB. In vivo results confirmed that NP-TPGS-SFB significantly inhibits tumor growth without obvious side effects. Therefore, this polymeric nanoparticle shows promising potential as a novel platform for enhanced liver therapy.
Acknowledgments
We are grateful for the financial support from Guangdong Medical Science and Technology Research Fund Project (Nos. B2019132), Science, Technology & Innovation Commission of Shenzhen Municipality (Nos. JCYJ20180305180540801), a key scientific research project for young people in Shenzhen Municipal People’s Hospital (Nos. SYKYPY201931).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Torre LA, Bray F, Siegel RL, et al. Global cancer statistics, 2012. CA: Cancer J Clin. 2015;65:87–108. doi:10.3322/caac.21262
2. Semela D, Dufour JF. Angiogenesis and hepatocellular carcinoma. J Hepatol. 2004;41:864–880. doi:10.1016/j.jhep.2004.09.006
3. Cheng A, Kang Y, Chen Z, et al. Efficacy and safety of sorafenib in patients in the Asia-Pacific region with advanced hepatocellular carcinoma: a phase III randomised, double-blind, placebo-controlled trial. Lancet Oncol. 2009;10:25–34. doi:10.1016/S1470-2045(08)70285-7
4. Wang Y, Gao J, Zhang D, et al. New insights into the antifibrotic effects of sorafenib on hepatic stellate cells and liver fibrosis. J Hepatol. 2010;53:132–144. doi:10.1016/j.jhep.2010.02.027
5. Llovet JM, Ricci S, Mazzaferro V, et al. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med. 2008;359:378–390. doi:10.1056/NEJMoa0708857
6. Blanchet B, Billemont B, Cramard J, et al. Validation of an HPLC-UV method for sorafenib determination in human plasma and application to cancer patients in routine clinical practice. J Pharmaceut Biomed. 2009;49:1109–1114. doi:10.1016/j.jpba.2009.02.008
7. Carmeliet P, Jain RK. Molecular mechanisms and clinical applications of angiogenesis. Nature. 2011;473:298–307. doi:10.1038/nature10144
8. Semenza GL. Hypoxia-inducible factors in physiology and medicine. Cell. 2012;148:399–408. doi:10.1016/j.cell.2012.01.021
9. Jain RK. Normalizing tumor microenvironment to treat cancer: bench to bedside to biomarkers. J Clin Oncol. 2013;31:2205–2210. doi:10.1200/JCO.2012.46.3653
10. Ferrari M. Cancer nanotechnology: opportunities and challenges. Nat Rev Cancer. 2005;5:161–171. doi:10.1038/nrc1566
11. Chauhan VP, Jain RK. Strategies for advancing cancer nanomedicine. Nat Mater. 2013;12:958–962. doi:10.1038/nmat3792
12. Shi J, Kantoff PW, Wooster R, et al. Cancer nanomedicine: progress, challenges and opportunities. Nat Rev Cancer. 2016;17:20–37. doi:10.1038/nrc.2016.108
13. Zeng X, Liu G, Tao W, et al. A drug-self-gated mesoporous antitumor nanoplatform based on pH-sensitive dynamic covalent bond. Adv Funct Mater. 2017;27:1605985. doi:10.1002/adfm.201605985
14. Liu G, Tsai H, Zeng X, et al. Phosphorylcholine-based stealthy nanocapsules enabling tumor microenvironment-responsive doxorubicin release for tumor suppression. Theranostics. 2017;7:1192–1203. doi:10.7150/thno.17881
15. Cheng W, Nie J, Gao N, et al. A multifunctional nanoplatform against multidrug resistant cancer: merging the best of targeted chemo/gene/photothermal therapy. Adv Funct Mater. 2017;27:1704135. doi:10.1002/adfm.201704181
16. Liu G, Tsai H, Zeng X, et al. Black phosphorus nanosheets-based stable drug delivery system via drug-self-stabilization for combined photothermal and chemo cancer therapy. Chem Eng J. 2019;375:121917. doi:10.1016/j.cej.2019.121917
17. Luo M, Cheng W, Zeng X, et al. Folic acid-functionalized black phosphorus quantum dots for targeted chemo-photothermal combination cancer therapy. Pharmaceutics. 2019;11:242. doi:10.3390/pharmaceutics11050242
18. Kydd J, Jadia R, Velpurisiva P, et al. Targeting strategies for the combination treatment of cancer using drug delivery systems. Pharmaceutics. 2017;9:46. doi:10.3390/pharmaceutics9040046
19. Lee KS, Chung HC, Im SA, et al. Multicenter phase II trial of genexol-PM, a cremophor-free, polymeric micelle formulation of paclitaxel, in patients with metastatic breast cancer. Breast Cancer Res Tr. 2008;108:241–250. doi:10.1007/s10549-007-9591-y
20. Liu J, Zeng F, Allen C. In vivo fate of unimers and micelles of a poly(ethylene glycol)-block-poly(caprolactone) copolymer in mice following intravenous administration. Eur J Pharm Biopharm. 2007;65:309–319. doi:10.1016/j.ejpb.2006.11.010
21. Cabral H, Kataoka K. Progress of drug-loaded polymeric micelles into clinical studies. J Control Release. 2014;190:465–476. doi:10.1016/j.jconrel.2014.06.042
22. Eetezadi S, Ekdawi SN, Allen C. The challenges facing block copolymer micelles for cancer therapy: in vivo barriers and clinical translation. Adv Drug Deliv Rev. 2015;91:7–22. doi:10.1016/j.addr.2014.10.001
23. Letchford K, Burt HM, Micelles C. Copolymer micelles and nanospheres with different in vitro stability demonstrate similar paclitaxel pharmacokinetics. Molecular Pharmaceutics. 2012;9:248–260. doi:10.1021/mp2002939
24. Prabaharan M, Grailer JJ, Pilla S, et al. Amphiphilic multi-arm-block copolymer conjugated with doxorubicin via pH-sensitive hydrazone bond for tumor-targeted drug delivery. Biomaterials. 2009;30:5757–5766. doi:10.1016/j.biomaterials.2009.07.020
25. Li X, Qian Y, Liu T, et al. Amphiphilic multiarm star block copolymer-based multifunctional unimolecular micelles for cancer targeted drug delivery and MR imaging. Biomaterials. 2011;32:6595–6605. doi:10.1016/j.biomaterials.2011.05.049
26. Wurm F, Frey H. Linear-dendritic block copolymers: the state of the art and exciting perspectives. Prog Polym Sci. 2011;36:1–52. doi:10.1016/j.progpolymsci.2010.07.009
27. Zeng X, Tao W, Mei L, et al. Cholic acid-functionalized nanoparticles of star-shaped PLGA-vitamin E TPGS copolymer for docetaxel delivery to cervical cancer. Biomaterials. 2013;34:6058–6067. doi:10.1016/j.biomaterials.2013.04.052
28. Guo J, Hong H, Chen G, et al. Image-guided and tumor-targeted drug delivery with radiolabeled unimolecular micelles. Biomaterials. 2013;34:8323–8332. doi:10.1016/j.biomaterials.2013.07.085
29. Liu G, Gao H, Zuo Y, et al. DACHPt-loaded unimolecular micelles based on hydrophilic dendritic block copolymers for enhanced therapy of lung cancer. ACS Appl Mater Interfaces. 2017;9:112–119. doi:10.1021/acsami.6b11917
30. Tsai H, Jiang L, Zeng X, et al. DACHPt-loaded nanoparticles self-assembled from biodegradable dendritic copolymer polyglutamic acid-b-D-alpha-tocopheryl polyethylene glycol 1000 succinate for multidrug resistant lung cancer therapy. Front Pharmacol. 2018;9:119. doi:10.3389/fphar.2018.00119
31. Marti Coma-Cros E, Biosca A, Marques J, et al. Polyamidoamine nanoparticles for the oral administration of antimalarial drugs. Pharmaceutics. 2018;10:225. doi:10.3390/pharmaceutics10040225
32. Shi Y, van Steenbergen MJ, Teunissen EA, et al. Π–Π stacking increases the stability and loading capacity of thermosensitive polymeric micelles for chemotherapeutic drugs. Biomacromolecules. 2013;14:1826–1837. doi:10.1021/bm400234c
33. Shi Y, van der Meel R, Theek B, et al. Complete regression of xenograft tumors upon targeted delivery of paclitaxel via Π–Π stacking stabilized polymeric micelles. ACS Nano. 2015;9:3740–3752. doi:10.1021/acsnano.5b00929
34. Tang X, Zhou S, Tao X, et al. Targeted delivery of docetaxel via Pi-Pi stacking stabilized dendritic polymeric micelles for enhanced therapy of liver cancer. Mat Sci Eng C-Mater. 2017;75:1042–1048. doi:10.1016/j.msec.2017.02.098
35. Zhang Z, Tan S, Feng S. Vitamin E TPGS as a molecular biomaterial for drug delivery. Biomaterials. 2012;33:4889–4906. doi:10.1016/j.biomaterials.2012.03.046
36. Mu L, Feng SS. A novel controlled release formulation for the anticancer drug paclitaxel (Taxol®): PLGA nanoparticles containing vitamin E TPGS. J Control Release. 2003;86:33–48. doi:10.1016/S0168-3659(02)00320-6
37. Kim RM, Jang D, Kim YC, et al. Flurbiprofen-loaded solid SNEDDS preconcentrate for the enhanced solubility, in-vitro dissolution and bioavailability in rats. Pharmaceutics. 2018;10:247. doi:10.3390/pharmaceutics10040247
38. Guo Y, Luo J, Tan S, et al. The applications of Vitamin E TPGS in drug delivery. Eur J Pharm Sci. 2013;49:175–186. doi:10.1016/j.ejps.2013.02.006
39. Liu G, Tsai H, Zeng X, et al. Phosphorylcholine-based stealthy nanocapsules decorating TPGS for combatting multi-drug-resistant cancer. ACS Biomater Sci Eng. 2018;4:1679–1686. doi:10.1021/acsbiomaterials.8b00152
40. Lin T, Gao D, Liu Y, et al. Development and characterization of sorafenib-loaded PLGA nanoparticles for the systemic treatment of liver fibrosis. J Control Release. 2016;221:62–70. doi:10.1016/j.jconrel.2015.11.003
41. Upadhyay KK, Bhatt AN, Mishra AK, et al. The intracellular drug delivery and anti tumor activity of doxorubicin loaded poly(gamma-benzyl L-glutamate)-b-hyaluronan polymersomes. Biomaterials. 2010;31:2882–2892. doi:10.1016/j.biomaterials.2009.12.043
42. Shoshan MS, Vonderach T, Hattendorf B, et al. Peptide-coated platinum nanoparticles with selective toxicity against liver cancer cells. Angew Chem Int Ed. 2019;58:4901–4905. doi:10.1002/anie.201813149
43. Yang S, Zhang B, Gong X, et al. In vivo biodistribution, biocompatibility, and efficacy of sorafenib-loaded lipid-based nanosuspensions evaluated experimentally in cancer. Int J Nanomed. 2016;11:2329. doi:10.2147/IJN.S104119
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.