Back to Journals » International Journal of Nanomedicine » Volume 10 » Issue 1
Solid and liquid lipid-based binary solid lipid nanoparticles of diacerein: in vitro evaluation of sustained release, simultaneous loading of gold nanoparticles, and potential thermoresponsive behavior
Authors Rehman M ![]() , Madni A
, Madni A ![]() , Ihsan A, Khan W, Imran Khan M, Ahmad Mahmood M, Ashfaq M, Bajwa SZ, Shakir I
, Ihsan A, Khan W, Imran Khan M, Ahmad Mahmood M, Ashfaq M, Bajwa SZ, Shakir I
Received 20 August 2014
Accepted for publication 11 November 2014
Published 7 April 2015 Volume 2015:10(1) Pages 2805—2814
DOI https://doi.org/10.2147/IJN.S67147
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Thomas Webster
Mubashar Rehman,1 Asadullah Madni,1 Ayesha Ihsan,2 Waheed Samraiz Khan,2 Muhammad Imran Khan,1 Muhammad Ahmad Mahmood,1 Muhammad Ashfaq,1 Sadia Zafar Bajwa,2 Imran Shakir3
1Department of Pharmacy, Faculty of Pharmacy and Alternative Medicine, The Islamia University of Bahawalpur, Pakistan; 2Nanobiotechnology Group, Industrial Biotechnology Division, National Institute of Biotechnology and Genetic Engineering, Faisalabad, Pakistan; 3Sustainable Energy Technologies (SET) centre, College of Engineering, King Saud University, Riyadh, Saudi Arabia
Abstract: Binary fatty acid mixture-based solid lipid nanoparticles (SLNs) were prepared for delivery of diacerein, a novel disease-modifying osteoarthritis drug, with and without simultaneously loaded gold nanoparticles (GNPs). In order to optimize SLNs for temperature-responsive release, lipid mixtures were prepared using different ratios of solid (stearic acid or lauric acid) and liquid (oleic acid) fatty acids. SLNs were prepared by microemulsification (53 nm), hot melt encapsulation (10.4 nm), and a solvent emulsification-evaporation technique (7.8 nm). The physicochemical characteristics of SLNs were studied by Zetasizer, Fourier transform infrared, and X-ray diffraction analysis. High encapsulation of diacerein was achieved with diacerein-loaded and simultaneously GNP-diacerein-loaded SLNs. In vitro dissolution studies revealed a sustained release pattern for diacerein over 72 hours for diacerein-loaded SLNs and 12 hours for GNP-diacerein-loaded SLNs. An increase in diacerein payload increased the release time of diacerein while GNPs decreased it. In addition, rapid release of diacerein over 4 hours was observed at 40°C (melting point of optimized fatty acid mixture), demonstrating that these binary SLNs could be used for thermoresponsive drug delivery. Kinetic modeling indicated that drug release followed zero order and Higuchi diffusion models (R2>0.9), while the Korsmeyer-Peppas model predicted a diffusion release mechanism (n<0.5).
Keywords: diacerein, thermoresponsive, binary, gold nanoparticles, lipids, nanoparticles
Introduction
Osteoarthritis is a chronic disease of the joints characterized by progressive loss of articular cartilage, and is almost 15 times more prevalent than rheumatoid arthritis.1 Diacerein (DCN) is a novel disease-modifying drug with clinically proven chondroprotective effects in patients with osteoarthritis.2,3 Oral formulations of DCN have not been successful due to poor physicochemical properties (solubility <0.01 mg/mL) and an unfavorable pharmacokinetic profile, including low bioavailability (35%–56%) and a short half-life (4 hours).4,5 Its recommended oral dose is 40–50 mg twice daily. Patients generally become noncompliant when they are required to follow a twice-daily regimen of DCN for the recommended treatment period of 2–3 years.6 Moreover, clinical acceptance of DCN has been limited due to a high prevalence of severe gastrointestinal side effects associated with long-term oral administration. In most cases, these gastrointestinal side effects lead to treatment discontinuation and even the use of diacerein has been prohibited in some countries.7,8 In addition, current evidence of a hypoglycemic effect of DCN has raised concerns about the safety of the drug in long-term use.9
Solid lipid nanoparticles (SLNs) are widely used for efficient delivery of hydrophobic drugs.10 SLNs formulated from mixtures of lipids have better physicochemical properties and enhanced entrapment efficiency (EE). These properties appear to be dependent on heterogeneity of the lipid components.11 In addition, Muhlen et al reported that incorporation of a lipid with a low melting point in the lipid mixture prolonged drug release from SLNs.12
This study was designed to synthesize SLNs allowing sustained release of DCN using binary fatty acid mixtures (BFs) of solid and liquid lipids. Sustained release of DCN would provide better control of osteoarthritis and improve compliance due to the simplified regimen. Physical mixing of solid and liquid will decrease the melting point of BFs, providing an opportunity to formulate thermoresponsive SLNs. This study also evaluated the potential of DCN-loaded binary SLNs to simultaneously encapsulate gold nanoparticles (GNPs), which are used as anti-inflammatory agents in chrysotherapy.13
Materials and methods
Materials
DCN was provided by Consolidated Chemical Laboratories (Lahore, Pakistan) as a gift sample. Stearic acid, lauric acid, and oleic acid were sourced from Tokyo Chemical Industry (Tokyo, Japan). Gold chloride, Brij 98 (polyoxyethylene oleyl ether), Tween 80 (polysorbate 80), and soy lecithin were purchased from Acros Organics (Geel, Belgium). Chloroform (high-performance liquid chromatography grade) was purchased from Alfa Aesar (Ward Hill, MA, USA). All materials were of analytical grade.
Preparation of binary lipid mixtures
BFs were prepared by homogeneously mixing melted fatty acids in different ratios from 8:1 to 2:1. The solid fatty acids were heated to at least 10°C above melting point and added to liquid fatty acids maintained at the same temperature. Melting to a higher temperature reduced the viscosity of the lipids and ensured homogenous mixing. The melting point of all the fatty acid ratios was measured by melting point apparatus (Mel-Temp, Bibby Scientific, Stone, UK). BFs were taken in capillary tubes with one closed end (Stuart, SMP10/1, Bibby Scientific). The temperature was slowly increased and melting was observed with an eye piece. The temperature at which melting started was noted and recorded until the lipid was completely melted.
Synthesis of gold nanoparticles
GNPs were synthesized by a method already developed for our group by Hussain et al.14 Briefly, 10 mL of a 20 mM gold chloride solution was put into a round bottom flask and placed in a heating oil bath maintained at 100°C. Next, 4 mL of 10 mM lecithin solution was added. The stirring rate was set to 300 rpm and water vapors were cooled by cold water reflux. After 5 minutes, the heater was switched off and the reaction system was allowed to cool to room temperature. The GNPs were recovered by centrifugation (Himac CS150GXL, Hitachi, Tokyo, Japan) at 10,000 rpm for 10 minutes and washed three times with deionized water following the same procedure. These GNPs had a lecithin bilayer and were termed B-GNPs. Lecithin monolayer-coated GNPs were obtained by dropwise addition of 0.1 M hydrochloric acid. In acidic pH, the outer layer of lecithin is removed and the particles become lecithin monolayer-coated GNPs (M-GNPs). The B-GNPs remain suspended in water, while the M-GNPs precipitate readily as soon as they are formed.
Development of SLNs
SLNs were prepared using a microemulsification technique (MET), Hot Melt Encapsulation (HME), and a solvent emulsification-evaporation technique (SEET) by the following procedures. The composition of each of the three model formulations is given in Table 1.
 | Table 1 Size and PDI for the three preparation methods |
Microemulsion technique
BFs were heated to 5°C above melting point. Next, 1 mL of melted BF was added to 20 mL of an aqueous solution of Brij-98 (4%) under magnetic stirring (1,500 rpm), and heat stirring was continued for 5 minutes to form a melted microemulsion. After 5 minutes, this hot melted microemulsion was added slowly to cold water (5°C) in a ratio of 1:50. Stirring was continued for 5 minutes and an SLN dispersion was obtained. For the preparation of drug loaded SLNs, DCN may be added to melted lipid in the first step.
Hot melt encapsulation method
The microemulsion was prepared as in the previous method; however, HME differs from MET in that the melted microemulsion was cooled to room temperature by switching off the heater, and magnetic stirring (15,000 rpm) was continued until room temperature was reached.
Solvent emulsification evaporation method
This method involved solubilizing 1 mL of BF and 150 mg of lecithin (as surfactant), with or without drug, into chloroform to make the organic phase (50 mL). The organic phase was added to 100 mL of an aqueous solution of 2% hydrophilic surfactant (Brij 98) under magnetic stirring (1,500 rpm) to form a milky phase or cold microemulsion. This was an oil-in-water (o/w) emulsion in which the aqueous phase contained droplets of chloroform containing lipid and drug. The emulsion was subjected to evaporation under vacuum by a rotary evaporator (Julabo USA Inc, Allentown, PA, USA) to evaporate chloroform until the total volume remaining was less than 100 mL. As the chloroform evaporated, the lipid-lecithin-drug started precipitating as SLNs.
Characterization of SLNs
Representative formulations were characterized for physicochemical parameters like particle size, zeta potential, and drug-polymer interaction.
Particle size and zeta potential
Dynamic light scattering was used for size and electrophoretic mobility studies for the zeta potential using a Zetasizer Nano (ZS-90, Malvern Instruments, Malvern, UK). First, 1 mL of the SLN preparation was put into Zetasizer cuvettes and diluted with 1 mL of deionized water. Readings were then taken for size, polydispersity index (PDI), and zeta potential.
Drug-polymer interaction
The compatibility of the formulation components was studied by Fourier transform infrared spectroscopy. Spectra of the individual lipid, drug, and representative formulation were taken. The characteristic peaks of the bond stretches of individual components were assessed in Fourier transform infrared spectra for each formulation.
X-ray diffraction analysis
The crystalline behavior of the SLNs was studied by X-ray diffraction analysis. The X-ray diffraction pattern was recorded using an X-ray diffractometer (Bruker Corporation, Billerica, MA, USA) with Cu Kα radiation (λ=0.1541nm) in the scan range of 2θ=15°–70°.
Calculation of encapsulation efficiency
Drug loading and EE were calculated for all formulations as shown in Table 2. The freshly prepared SLN formulation was centrifuged in a micro ultracentrifuge (Himac CS150GXL, Hitachi) at 30,000 rpm for 5 minutes to separate the SLNs. The amount of encapsulated drug was calculated indirectly from the amount of nonentrapped drug in the supernatant using the following formula:
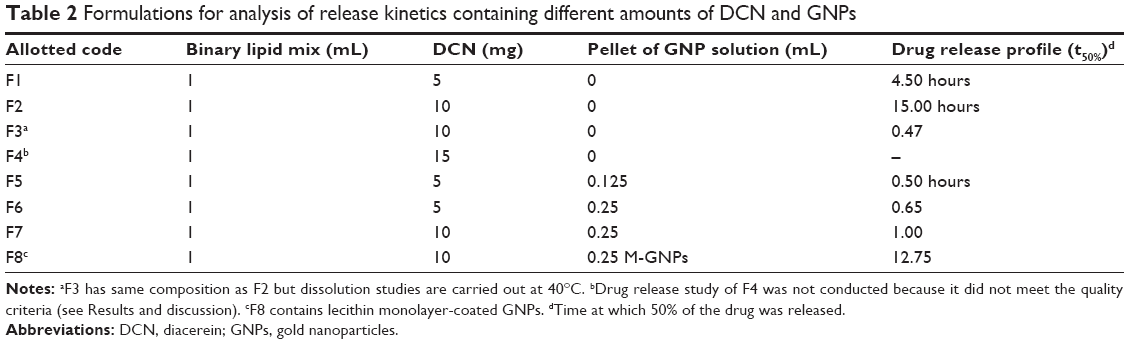 | Table 2 Formulations for analysis of release kinetics containing different amounts of DCN and GNPs |
Amount of loaded drug = Total amount of drug − Amount of nonentrapped drug |
The EE of the SLNs was calculated by the following formula:
|
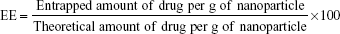
In vitro drug release studies
The in vitro release studies for all formulations (shown in Table 2) were carried out using the dialysis tube method. A cellulose ester dialysis tube with a molecular weight cutoff value of 10 kDa was filled with SLN formulation equivalent to 1 mg of drug and placed in 250 mL of phosphate-buffered saline dissolution medium. Dissolution conditions were set to a stirring rate of 50 rpm at 37°C±0.5°C. Samples (1 mL) were withdrawn at predetermined time points and an equivalent volume of fresh medium was added to the dissolution cells after each sample was taken. Dissolution of formulation F3 was carried out at 40°C. Absorbance of each sample was measured at 341 nm to calculate the drug concentration from the standard curve. The standard curve was prepared by measuring the absorbance of different concentrations of DCN in phosphate-buffered saline and constructing a graph of concentration versus absorbance.
Results and discussion
Binary SLNs were prepared and loaded with DCN, GNPs, and DCN-GNPs using binary fatty acid mixtures. In vitro characterization was done for physicochemical characteristics and the drug release profile.
Preparation of binary lipid mixture
BFs of solid and liquid fatty acids are known to have superior physicochemical properties and enhance drug EE. In addition, physical mixing of a liquid fatty acid is hypothesized to lower the melting point of the resulting BF. In the present work, the melting point of BFs was reduced in a linear fashion with addition of liquid oleic acid. A desirable melting point range of 38°C–39°C was achieved with a binary mixture of lauric acid and oleic acid at a ratio of 6:1 (39.25°C) and 5:1 (38.5°C). Addition of oleic acid to stearic acid in similar proportions produced BFs with a relatively higher melting point (62°C and 61°C) due to the higher melting point of stearic acid. Stearic acid and oleic acid at 2:1 showed a melting point of 41°C but, owing to a higher oleic acid content, may not be suitable for formulation of SLNs due to formation of oil clusters and reduced stability.15 On the basis of these results, BF3 (melting point 39.25°C) was selected for synthesis of all the SLN formulations (F1–F8), which are assumed to undergo solid-liquid transition above 39°C for thermoresponsive drug release.
Preparation of gold nanoparticles
GNPs were prepared by heating a solution of gold chloride and lecithin. This method yields a small (4.4±0.2 nm) and more homogenous (PDI <0.01) particle size. These GNPs remain suspended in aqueous solution due to the lecithin bilayer formed around gold nanoparticles. Colloidal gold has been reported to have anti-inflammatory properties. It has been reported that gold microbeads have antiarthritic effects in the knees;16 small GNPs may enhance this therapeutic effect due to a large surface area and reduced beads-associated joint damage observed in some patients.
M-GNPs were prepared by treating B-GNPs with gradually increasing acidic pH. Around an isoelectric point of pH 2.7, the lecithin outer layer loses its lamellar configuration due to loss of charge. This removes the outer unbound layer of lecithin from GNPs and the inner covalently bound monolayer remains on the GNPs. Removal of the outer lecithin layer was confirmed by a reduction in size from 4.4±0.2 nm to 3.6±0.2 nm.
Comparison of SLN preparation methods
Comparative results for the three preparation methods showed that MET (M1) produced SLNs with an average particle size of 53±2 nm and a PDI of 0.412. However, a large volume of cold water (50 times the volume of the primary product) was required to form SLNs, which diluted the final formulation and an additional recovery step was needed to separate SLNs. HME (M2) and SEET (M3) showed an average particle size of 10.4 nm and 7.8 nm. PDI was also relatively low, in the range of 0.214 and 0.246, respectively. This is evident from results that the two formulations prepared by HME and SEET were comparable with regard to production of relatively smaller SLNs. Penetration of nanoparticles into regenerating cartilage network is desirable for disease-modifying osteoarthritis drugs like DCN. The surfaces in a regenerating joint contain a fine mesh of cartilage, and particles as small as 38 nm are able to penetrate the growing cartilage meshwork.17 Keeping this in mind, HME and SEET appear to be suitable candidates for preparation of DCN-loaded SLNs. HME appears to be comparatively advantageous in that it does not involve use of potentially toxic organic solvents as in SEET,18 and no dilution step is required as in MET.19 HME uses very simple equipment, and water (as the solvent) was the only reagent used in addition to the formulation components.
Physicochemical characterization of SLNs
A total of eight SLN formulations with varying concentrations of DCN and GNPs were prepared by HME and characterized for size, chemical stability, and drug loading.
Size of DCN-loaded and GNP-loaded SLNs
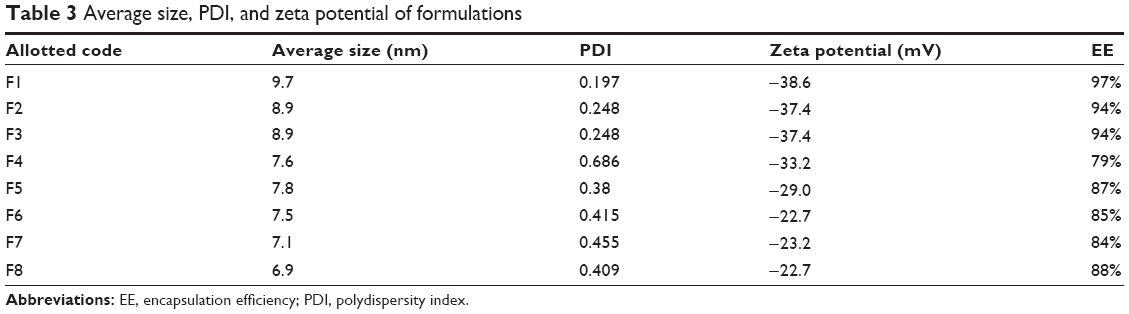
The size of blank as well as DCN-loaded and GNP-loaded SLNs was measured by dynamic light scattering. A reduction in size of the SLNs was observed with an increase in payload. The size of the SLNs decreased from 9.7 nm to 8.9 nm and 7.6 nm as the DCN payload was increased from 5 mg to 10 mg and 15 mg, respectively. A similar reduction in size was observed from 7.8 nm to 7.5 nm as the GNP load was increased from 0.125 mL to 0.250 mL. This reduction in SLN size with increase in drug payload can be described on the basis of the free lipid content. As the amount of payload increases, the proportion of free lipids and hence the size of the SLNs was decreased.20 Reduction in size was also observed as the amount of DCN was increased in GNP-loaded SLNs. Similarly, the size of the M-GNP-loaded SLNs was slightly smaller than that of the B-GNP-loaded SLNs due to the smaller size of M-GNPs.
An inverse relationship between size and PDI was observed, ie, the PDI increased with reduction in the size of all formulations (Table 3). The PDI of F4 containing 15 mg of DCN was 0.686, which is beyond acceptable limits. This may be attributed to the incorporation of DCN more than its solubility limit in the melted lipid mixture. Such formulations with excessive drug lead to heterogeneous dispersion and the PDI would be greater than 0.5.21
 | Table 3 Average size, PDI, and zeta potential of formulations |
Zeta potential of DCN-loaded and GNP-loaded SLNs
The surface charge on particles helps them to repel each other and stabilize colloidal dispersions. SLNs must have a zeta potential, a measure of surface charge, sufficient to prevent their aggregation during storage. The zeta potential of all formulations containing DCN was in the acceptable range of −40 mV to −30 mV, which is prerequisite for long-term stability of a SLN dispersion.22 However, the zeta potential decreased from −38.6 mV to −37.4 mV and −33.2 mV as the DCN payload increased from 5 mg to 10 mg and 15 mg, respectively. In GNP-loaded SLNs, the zeta potential decreased from −29.0 mV to −22.7 mV as the GNP payload increased from 0.125 mL to 0.25 mL. The zeta potential values of F5 and F6 were lower than the corresponding values for DCN-loaded SLNs. This reduction in negative zeta potential is due to the positive charge of gold which tends to neutralize the negative charge of SLNs. The zeta potential of GNP-loaded formulations (F5–F7) was observed to be in the range of −20 mV to −30 mV, indicating that these formulations will form mild to moderately stable colloidal dispersions.
Chemical stability of DCN and polymers in SLNs
Fourier transform infrared analysis is a reliable way of assessing the chemical compatibility of components in a formulation. In the event of a chemical reaction, certain absorption peaks of the components disappear and absorption peaks of new bonds appear.23 The absorption peaks of DCN were C=O and C-O stretch at 1,764 cm−1 and 1,024 cm−1, as shown in Figure 1A. FTIR Spectra of lauric and oleic acid are shown in Figure 1B and 1C, respectively. These characteristic peaks of DCN were present in the spectra of DCN-loaded SLNs with slight shifts at 1,766 cm−1 and 1,026 cm−1 (Figure 1D). These results demonstrate that DCN was present physically in SLNs without any chemical interaction.
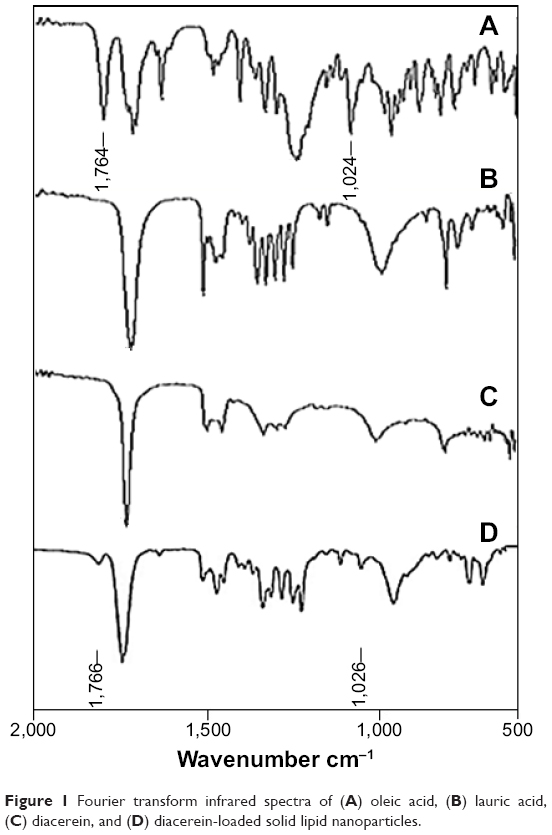 | Figure 1 Fourier transform infrared spectra of (A) oleic acid, (B) lauric acid, (C) diacerein, and (D) diacerein-loaded solid lipid nanoparticles. |
X-ray analysis
X-ray diffraction analysis is an efficient tool for assessing crystallinity in SLNs. In this study, it was postulated that addition of liquid oleic acid would suppress the crystallinity of BFs. The results revealed two sharp peaks for lauric acid in the 2θ range of 20°–30°. In SLNs made from BF3, the intensity of the first peak was suppressed from 23,594 absorbance units to 11,344 absorbance units, indicating reduced crystal formation in binary SLNs (Figure 2). Crystal formation in nanoparticles is undesirable because drug entrapped in the crystals is not released or is released very slowly. Reduced intensity of peaks in X-ray diffraction indicates that fewer crystals are formed and more drug is present in amorphous form to be released in vitro and in vivo.24 The width of all peaks remained the same, indicating no significant change in crystal size.
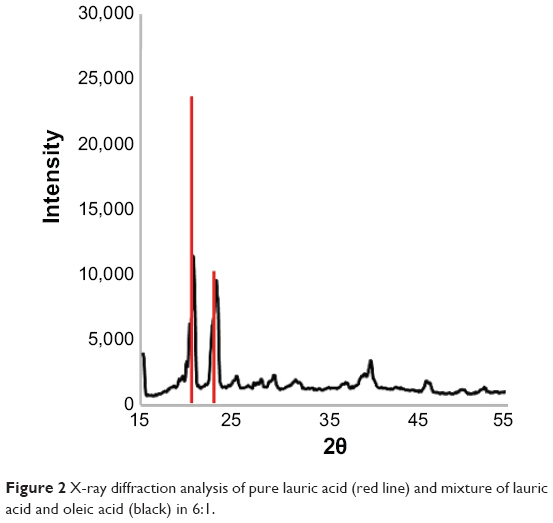 | Figure 2 X-ray diffraction analysis of pure lauric acid (red line) and mixture of lauric acid and oleic acid (black) in 6:1. |
Encapsulation efficiency
SLNs show high encapsulation efficiency of lipophilic drugs. In the present study, DCN was successfully loaded into SLNs and high percentage encapsulation was achieved for all formulations (Table 3). However, the EE was reduced from 97% to 94% and 79% as the DCN payload increased from 5 mg (F1) to 10 mg (F2) and 15 mg (F4). The sudden fall in EE in F4 might be due to incorporation of drug above its saturation level because the nonentrapped fraction of the drug was visible at the bottom of the beaker when cooled to room temperature. Muller et al found that all hydrophobic drugs can achieve supersaturation in lipids when the lipids are in a melted state; upon cooling, the saturation level of the drug decreases and an excessive amount of drug tends to partition into the outer shell or into the external solvent.25 The influence of simultaneous loading of GNPs and DCN on EE was evaluated for formulations F5–F8. As the GNP payload increased from 0.125 mL to 0.25 mL, the EE decreased from 87% to 85%. However, it should be noted that the EE of DCN was relatively higher in the presence of M-GNPs as compared with B-GNPs due to hydrophobic nature of the M-GNPs. This may also be due to the smaller size of M-GNPs, which allows more space in the lipid for incorporation of the drug.
In vitro release profile of DCN from SLNs
The USP dissolution test is widely used for in vitro evaluation of drug release behavior in different drug delivery systems. In the case of nanoparticles, the dialysis tube method is used so that nanoparticles can be enclosed in a cellulose tube. This study aimed to achieve sustained release of DCN from SLNs. Dissolution studies for all formulations were carried out in phosphate-buffered saline (pH 7.4) at 37°C and cumulative drug release was plotted against time. The results showed that the F1 formulation containing 5 mg of DCN released 95% of the drug in almost 39 hours. The dissolution time was found to be directly dependent on the drug payload, as sustained release of DCN was achieved for up to 72 hours when the payload was increased to 10 mg (Figure 3).
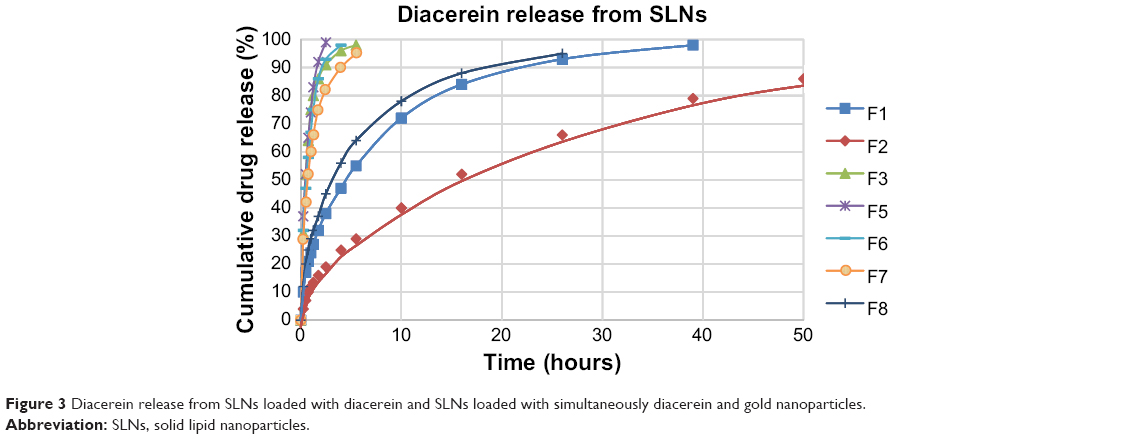 | Figure 3 Diacerein release from SLNs loaded with diacerein and SLNs loaded with simultaneously diacerein and gold nanoparticles. |
All formulations showed a high initial release of DCN. This burst release is due to movement of the drug in the shell during cooling. This proportion of drug is immediately released due to a short path length of diffusion.26 Simultaneous loading of GNPs (0.125 mL) enhanced drug release and reduced release time from 39 hours to 4 hours (F1 and F5). Drug release was increased further when gold load was increased to double (F5 and F6). On the other hand, release time was improved from 4 hours to 10 hours when DCN load was increased from 5 mg to 10 mg in the presence of a constant GNP load (F6 and F7). In simultaneously loaded SLNs, the whole drug is distributed in the periphery of the SLNs because the central portion is occupied by GNPs. This reduces the diffusion path length of DCN, so the release pattern of simultaneously loaded SLNs resembles the initial burst release observed with DCN-loaded SLNs. Formulations F7 and F8 compared the drug release pattern of B-GNP-loaded and M-GNP-loaded SLNs, respectively. Formulation F7 released more than 90% of the drug in 10 hours while F8 released 90% of the drug in 24 hours. This observation can also be explained on the basis of the smaller size of the M-GNPs when compared with B-GNPs. In the case of M-GNPs, a relatively larger lipid proportion was available to incorporate the drug which provided a larger diffusion path than B-GNPs and more sustained drug release. It was also noted that whole drug was not released from the SLNs. Drug release was less than 93% from DCN-loaded SLNs and less than 96% in simultaneously loaded SLNs. Some previous studies have reported that crystal formation suppresses drug release in SLNs because drug entrapped in crystals in not released.15 In our study, most of the drug was released because addition of liquid oleic acid suppressed crystal formation in SLNs and drug was present in an amorphous form in binary SLNs.
Thermoresponsive drug release from binary SLNs
Development of thermoresponsive drug delivery systems has attracted much research interest in the medical field. This strategy enables specific release of a drug at target sites that exhibited relatively high temperature than normal body, eg, cancer. Many novel materials have been chemically engineered to be capable of thermoresponsive drug release by undergoing conformational or chemical changes. In this study, binary SLNs of solid and liquid lipids were evaluated for thermoresponsive drug release. It was hypothesized that physical mixtures of lauric acid and oleic acid can be optimized to melt, ie, undergo solid-liquid phase transition, above 39°C. The thermoresponsive potential of binary SLNs was evaluated for formulations F2 and F3. Both formulations had the same composition (Table 2) and differed only in that dissolution of F2 was carried out at 37°C and dissolution of F3 was carried out at 40°C, 1°C above the phase transition temperature of SLNs. In case of F3, 50% of the drug was released from melted SLNs in 0.5 hours and whole drug (T95%) was released in almost 4 hours. In contrast, F2 released whole drug in more than 72 hours. These findings suggest thermoresponsive drug release from binary SLNs. Sustained release profile of DCN at physiological body temperature may be transformed to an immediate release pattern in response to elevated body temperature faced during various pathological conditions. This pattern of thermoresponsive release above phase transition temperature is due to the fact that solid nanoparticles show prolong drug release as compared with a liquid nanosuspension of the same oil.27 To best of our knowledge, this study reports for the first time that SLNs based on binary lipid mixtures can be optimized for thermoresponsive drug release. Figure 4 graphically represents three different patterns of drug release from our binary SLN formulations. In osteoarthritis, thermoresponsive SLNs will offer enhanced drug release in response to local hyperthermia at swollen joints or generalized hyperthermia in febrile cases.28,29 In addition, hyperthermia at the joint may also be achieved by application of heat-producing products conventionally employed in osteoarthritis.30
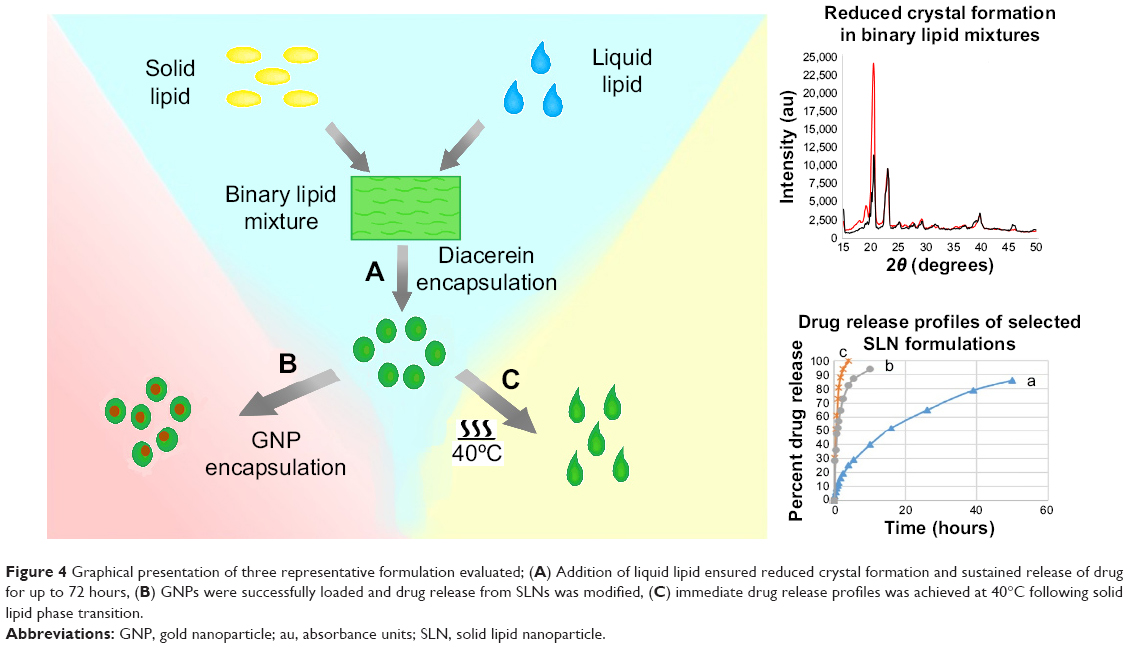 | Figure 4 Graphical presentation of three representative formulation evaluated; (A) Addition of liquid lipid ensured reduced crystal formation and sustained release of drug for up to 72 hours, (B) GNPs were successfully loaded and drug release from SLNs was modified, (c) immediate drug release profiles was achieved at 40°C following solid lipid phase transition. |
Kinetic modeling of drug release
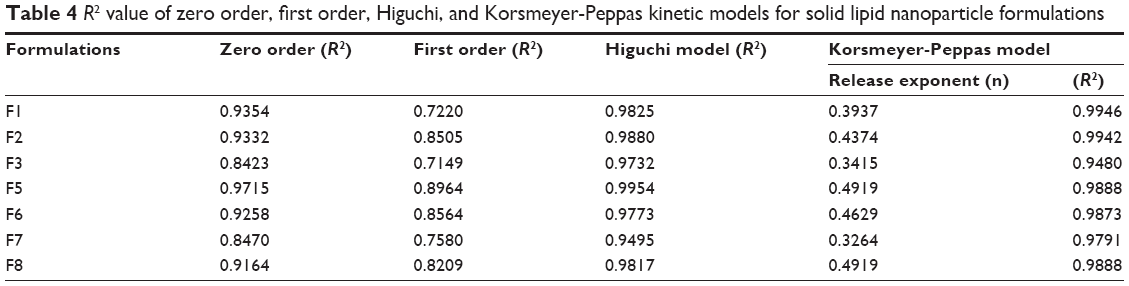
Different pharmacokinetic models were used to predict drug release behavior from nanoparticles. In this study, kinetic modeling was performed for all formulations to predict the rate and mechanism of drug release (Table 4). To eliminate the effect of initial burst release, kinetic models were applied to drug release data after 25% of the drug was released. All formulation containing DCN and GNPs showed higher R2 values for a zero order kinetics model (0.8423–0.9716) as compared with a first-order kinetics model (0.7149–0.8964). All formulations followed the Higuchi model of drug release, with R2 values for all formulations being in the range of 0.9495–0.9954.
 | Table 4 R2 value of zero order, first order, Higuchi, and Korsmeyer-Peppas kinetic models for solid lipid nanoparticle formulations |
The above results demonstrate that drug release from binary SLNs tends to follow zero order kinetics, ie, drug release is independent of the amount of drug remaining in SLNs to be released. All formulation follow Higuchi model which means drug release is diffusion-controlled; however, R2 values did not fit the model perfectly. The Korsmeyer-Peppas model was followed by all formulations as R2 values were between 0.9480 and 0.9960. Values for the release exponent (n) were found to be below 0.5, which also predicts diffusion-controlled release from SLNs.31 Therefore, it is concluded from results of kinetic modeling that drug release from binary SLNs was diffusion controlled and followed zero order release rate.
Conclusion
Binary SLNs made from binary mixtures of solid and liquid lipids had superior physicochemical properties and enabled sustained release of DCN, a new disease-modifying drug, for up to 3 days. GNPs were simultaneously loaded into DCN-loaded binary SLNs and the sustained release pattern for DCN was found to be directly related to the DCN payload and inversely related to the gold payload. The strategy of mixing solid and liquid lipids also provides an opportunity to optimize binary SLNs for thermoresponsive drug release. Drug release from binary SLNs was diffusion-controlled and followed zero order kinetics.
Acknowledgments
The author would like to extend their sincere appreciation to the Deanship of Scientific Research at King Saud University for its funding to support this research through the Research Group Project no. RGP-VPP-312. The authors also acknowledge the Faculty of Pharmacy and Alternative Medicine, The Islamia University of Bahawalpur and Nanobiotechnology Group of National Institute of Biotechnology and Genetic Engineering (NIBGE), Faisalabad, PAKISTAN for technical support for this research work. They also acknowledge Asma Rehman (NIBGE) and Ibrahim Javed (Bahauddin Zakariya University, Multan) for useful contribution.
Disclosure
The authors report no conflicts of interest in this work.
References
Myasoedova E, Cynthia SC, Hilal M, Terry MT, Sherine EG. Is the incidence of rheumatoid arthritis rising? Results from Olmsted County, Minnesota, 1955–2007. Arthritis Rheum. 2010;62:1576–1582. | ||
Sanchez C, Mathy-Hartert M, Deberg MA, Ficheux H, Reginster JY, Henrotin YE. Effects of rhein on human articular chondrocytes in alginate beads. Biochem Pharmacol. 2003;65:377–388. | ||
Alvarez-Soria MA, Herrero-Beaumont G, Sanchez-Pernaute O, Bellido M, Largo R. DCN has a weak effect on the catabolic pathway of human osteoarthritis synovial fibroblast – comparison to its effects on osteoarthritic chondrocytes. Rheumatology. 2008;47:627–633. | ||
Maski N, Kumaran A, Girhepunje K, Ghode P, Randive S, Pal R. Studies on the preparation, characterization and solubility of β-cyclodextrin-DCN inclusion complexes. Int J Pharm Pharm Sci. 2009;1:121–135. | ||
Nicolas P, Tod M, Padoin C, Petitjean O. Clinical pharmacokinetics of diacerein. Clin Pharmacokinet. 1998;35:347–359. | ||
Dougados M, Nguyen M, Berdah L, Maziéres B, Vignon E, Lequesne M; ECHODIAH Investigators Study Group. Evaluation of the structure-modifying effects of DCN in hip osteoarthritis: ECHODIAH, a three-year, placebo-controlled trial. Evaluation of the chondromodulating effect of diacerein in OA of the hip. Arthritis Rheum. 2001;44:2539–2547. | ||
Brahmachari B, Chatterjee S, Ghosh A. Efficacy and safety of DCN in early knee osteoarthritis: a randomized placebo-controlled trial. Clin Rheumatol. 2009;8:1193–1198. | ||
European Medicine Agency. PRAC recommends suspension of diacerein-containing medicines. London, UK: European Medicine Agency Online Resources, Inc; 2013. Available from: http://www.ema.europa.eu/ema/index.jsp?curl=pages/news_and_events/news/2013/11/news_detail_001947.jsp&mid=WC0b01ac058004d5c1. Accessed November 10, 2013. | ||
Ramos-Zavala MGGonzález-Ortiz M, Martínez-Abundis E, Robles-Cervantes JA, González-López R, Santiago-Hernández NJ. Effect of diacerein on insulin secretion and metabolic control in drug-naive patients with type 2 diabetes: a randomized clinical trial. Diabetes Care. 2011;34:1591–1594. | ||
Mehnert W, Mäder K. Solid lipid nanoparticles. Production, characterization and applications. Adv Drug Deliv Rev. 2001;47:165–196. | ||
Khalil RM, El-Bary AA, Kassem MA, Ghorab MM, Ahmed MB. Solid lipid nanoparticles for topical delivery of meloxicam: development and in vitro characterization. Presented at the Annual International Interdisciplinary Conference, April 24–26, 2013, Azores Islands, Portugal. | ||
Mühlen A, Schwarz C, Mehnert W. Solid lipid nanoparticles (SLN) for controlled drug delivery – drug release and release mechanism. Eur J Pharm Biopharm. 1998;45:149–155. | ||
Joh DY, Kao GD, Murty S, et al. Theranostic gold nanoparticles modified for durable systemic circulation effectively and safely enhance the radiation therapy of human sarcoma cells and tumors. Transl Oncol. 2013;6:722–731. | ||
Hussain I, Hussain SZ, Habib-ur-Rehman, et al. In situ growth of gold nanoparticles on latent fingerprints – from forensic applications to inkjet printed nanoparticle patterns. Nanoscale. 2010;2:2575–2578. | ||
Jenning V, Mäder K, Gohla SH. Solid lipid nanoparticles (SLN™) based on binary mixtures of liquid and solid lipids: a 1H-NMR study. Int J Pharm. 2005;205:15–21. | ||
Research Unit of General Practice, Copenhagen. Implantation of gold beads to relieve discomfort from knee osteoarthritis. Bethesda, MD, USA: National Institutes of Health; 2007. Available from: http://clinicaltrials.gov/show/NCT00487370. Accessed November 23, 2014. | ||
Rothenfluh DA, Bermudez H, O’Neil CP, Hubbell JA. Biofunctional polymer nanoparticles for intraarticular targeting and retention in cartilage. Nat Mater. 2008;7:248–254. | ||
Yeo Y, Baek N, Park K. Microencapsulation methods for delivery of protein drugs. Biotechnol Bioprocess Eng. 2001;6:213–230. | ||
Mao SR, Wang YZ, Ji HY, Bi DZ. [Preparation of solid lipid nanoparticles by microemulsion technique]. Yao Xue Xue Bao. 2003;38:624–626. Chinese. | ||
Kumar PP, Gayatri P, Sunil R, Jaganmohan S, Rao YM. Atorvastatin loaded solid lipid nanoparticles: formulation, optimization, and in vitro characterization. IOSR J Pharm. 2013;2:23–32. | ||
Chavda H, Patel J, Chavada G, Dave S, Patel A, Patel C. Self-nanoemulsifying powder of isotretinoin: preparation and characterization. J Powder Technol. 2013;2013:108569. | ||
Clogston JD, Patri AK. Zeta potential measurement. Methods Mol Biol. 2011;697:63–70. | ||
Jain A, Kumar SS, Yuvraj S, Sanjay S. Development of lipid nanoparticles of DCN, an antiosteoarthritic drug for enhancement in bioavailability and reduction in its side effects. J Biomed Nanotechnol. 2013;9:891–900. | ||
Akbari B, Tavandashti MP, Zandrahimi M. Particle size characterization of nanoparticles – a practical approach. Iran J Mater Sci Eng. 2011;8:48–56. | ||
Muller RH, Mader K, Gohla S. Solid lipid nanoparticle (SLN) for controlled drug delivery – a review of the state of the art. Eur J Pharm Biopharm. 2000;50:161–177. | ||
Lobovkina T, Jacobson GB, Gonzalez EG, et al. In vivo sustained release of siRNA from solid lipid nanoparticles. ACS Nano. 2011;5:9977–9983. | ||
Friedrich I, Reichl S, Müller-Goymann CC. Drug release and permeation studies of nanosuspensions based on solidified reverse micellar solutions (SRMS). Int J Pharm. 2005;305:167–175. | ||
Collins JA, Cosh JA. Temperature and biochemical studies of joint inflammation. Ann Rheum Dis. 1970;29:386–389. | ||
McAlindon T, Formica M, Schmid CH, Fletcher J. Changes in barometric pressure and ambient temperature influence osteoarthritis pain. Am J Med. 2007;120:429–434. | ||
Robinson V, Brosseau L, Casimiro L, et al. Thermotherapy for treating rheumatoid arthritis. Cochrane Database Syst Rev. 2002;2:CD002826. | ||
Barzegar-Jalali M, Adibkia K, Valizadeh H, et al. Kinetic analysis of drug release from nanoparticles. J Pharm Pharm Sci. 2008;11:167–177. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.