Back to Journals » International Journal of Nanomedicine » Volume 12
Sodium cholate-enhanced polymeric micelle system for tumor-targeting delivery of paclitaxel
Authors Zhang X, Wu Y, Zhang M, Mao J, Wu Y, Zhang Y, Yao J, Xu C, Guo W, Yu B
Received 28 August 2017
Accepted for publication 10 November 2017
Published 13 December 2017 Volume 2017:12 Pages 8779—8799
DOI https://doi.org/10.2147/IJN.S150196
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Linlin Sun
Xiaomin Zhang,1,2 Yibo Wu,1 Min Zhang,1 Jing Mao,3 Yun Wu,4 Yingxin Zhang,2 Ju Yao,2 Chang Xu,2 Wenli Guo,1 Bo Yu2
1Beijing Key Laboratory of Special Elastomeric Composite Materials, Beijing Institute of Petrochemical Technology, Beijing, 2Push-Kang Biotechnology, Hangzhou, 3Beijing Key Laboratory of Functional Materials for Building Structure and Environment Remediation, Beijing University of Civil Engineering and Architecture, Beijing, China; 4Department of Biomedical Engineering, University at Buffalo, State University of New York, Buffalo, NY, USA
Purpose: Polymeric micelles are attractive nanocarriers for tumor-targeted delivery of paclitaxel (PTX). High antitumor efficacy and low toxicity require that PTX mainly accumulated in tumors with little drug exposure to normal tissues. However, many PTX-loaded micelle formulations suffer from low stability, fast drug release, and lack of tumor-targeting capability in the circulation. To overcome these challenges, we developed a micellar formulation that consists of sodium cholate (NaC) and monomethoxy poly (ethylene glycol)-block-poly (D,L-lactide) (mPEG-PDLLA).
Methods: PTX-loaded NaC-mPEG-PDLLA micelles (PTX-CMs) and PTX-loaded mPEG-PDLLA micelles (PTX-Ms) were formulated, and their characteristics, particle size, surface morphology, release behavior in vitro, pharmacokinetics and in vivo biodistributions were researched. In vitro and in vivo tumor inhibition effects were systematically investigated. Furthermore, the hemolysis and acute toxicity of PTX-CMs were also evaluated.
Results: The size of PTX-CMs was 53.61±0.75 nm and the ζ-potential was –19.73±0.68 mV. PTX was released much slower from PTX-CMs than PTX-Ms in vitro. Compared with PTX-Ms, the cellular uptake of PTX-CMs was significantly reduced in macrophages and significantly increased in human cancer cells, and therefore, PTX-CMs showed strong growth inhibitory effects on human cancer cells. In vivo, the plasma AUC0–t of PTX-CMs was 1.8-fold higher than that of PTX-Ms, and 5.2-fold higher than that of Taxol. The biodistribution study indicated that more PTX-CMs were accumulated in tumor than PTX-Ms and Taxol. Furthermore, the significant antitumor efficacy of PTX-CMs was observed in mice bearing BEL-7402 hepatocellular carcinoma and A549 lung carcinoma. Results from drug safety assessment studies including acute toxicity and hemolysis test revealed that the PTX-CMs were safe for in vivo applications.
Conclusion: These results strongly revealed that NaC-mPEG-PDLLA micelles can tumor-target delivery of PTX and enhance drug penetration in tumor, suggesting that NaC-mPEG-PDLLA micelles are promising nanocarrier systems for anticancer drugs delivery.
Keywords: sodium cholate, polymeric micelles, enhanced, tumor-targeting delivery
Introduction
Taxane-based chemotherapy is one of the most widely used therapies for cancer treatment.1,2 Paclitaxel (PTX) is one of the best antineoplastic agents in the taxane family and has shown significant clinical activity against a wide variety of tumors, including ovarian cancer, lung cancer, and breast cancer.3 Most clinically available formulations are aspecific and have huge disadvantages, such as serious side effects, low stability, fast drug release, and lack of tumor-targeting capability.4
Due to these limitations, much effort has been devoted to developing effective drug carriers, such as polymer conjugates, liposomes, and micelles.5 Among these drug-delivery systems, polymeric micelles have gained considerable attention for their unique core–shell structure and high drug-loading capacity. They can be synthesized from polymers suitable for systemic administration, customized to encapsulate drugs at various doses, and optimized to achieve prolonged circulation kinetics.6,7 Several polymeric micelle drugs for cancer therapy have been evaluated in preclinical studies and clinical trials. Monomethoxy(polyethylene glycol)-block-poly(D,L-lactide) (mPEG-PDLLA) is one of the most widely used materials to construct micelles, and has been approved by the US Food and Drug Administration as a pharmaceutical excipient.8 Genexol PM, one type of mPEG-PDLLA-based PTX-loaded polymeric micelles, has been approved for the treatment of various cancers in several countries in Asia. This formulation has shown significantly decreased toxicity when compared to Taxol (a clinically available PTX formulation).6
Current PTX–polymeric micelle formulations have significantly improved PTX solubility and decreased its toxicity, but their therapeutic efficacy is comparable to Taxol.9 Instability and lack of tumor targeting in vivo are major factors that limit their antitumor efficacy.10 Effective ways to achieve controlled drug release from micelles include the conjugation of drugs to polymeric materials and the cross-linking of polymers by chemical reactions.11 Different from passive tumor targeting, which relies on the enhanced permeability and retention (EPR) effect, conjugating specific ligands to drug-loaded particles has become a commonly used method to achieve active tumor targeting. Various types of ligands, including small molecules, peptides, proteins, antibodies, engineered antibody fragments, and aptamers, have demonstrated their tumor-targeting capabilities.12,13 However, such approaches face significant technical challenges, cannot be easily scaled up for mass production, and thus are not suitable for industrial production.
Sodium cholate (NaC) is one kind of bile salt. NaC has a large, rigid, and hydrophobic steroid core, and has hydrophilic moieties of three hydroxyl groups and an ionic head of a carboxyl group, which make the amphiphilic properties of NaC quite different from ordinary aliphatic surfactant molecules.14 As a result of their amphiphilic properties, bile acids are widely applied in the development of micelle-based drug-delivery systems. The steroidal structures of bile acids can induce hydrophobic interaction with drugs and thus enhance drug-loading efficiency.15 Furthermore, intermolecular hydrogen bonding between hydroxyl and/or carboxyl groups may contribute a complementary mechanism to the micellization process.16 Bile acids can form mixed micelles when combined with polar lipids, conventional surfactants, or amphiphilic drugs, usually with lower critical micelle concentration (CMC) and better solubilization capacity than the individual components, due to synergistic interactions.17 In aqueous solutions, bile acid anions self-associate to form simple micelles.18 A wide variety of materials have been developed to form micelles with bile acids through noncovalent interactions, such as hydrogen-bond and electrostatic interaction.19 Studies have reported that the combined use of bile salts with compounds, such as phospholipids, fatty acids, and polyamines, may improve their effectiveness as absorption enhancers and allow a decrease in their concentrations, thus reducing the risks of membrane dissolution.20,21 Researchers have reported that the conjugation of cholic acid to anticancer drugs, such as carboplatin and cisplatin, resulted in significant antitumor efficiency in platinum-resistant tumor models.18,22 The organotropism of bile acids can also help overcome chemotherapy resistance of enterohepatic tumors and reduce noxious side effects to healthy tissue by selectively targeting drugs to tumor cells.23
In this study, we combined a bile acid (ie, NaC) with an amphiphilic polymer (ie, mPEG-PDLLA) to prepare PTX-loaded micelles for tumor-targeted delivery of PTX and enhanced drug penetration in tumors, resulting in a superior antitumor effect that cannot be achieved by clinically available PTX formulations, including Taxol, Abraxane, and liposomes. Bile acids were selected in our micelle formulation because they are biological surfactants naturally produced in humans and known to be beneficial for human health.24 Moreover, compared to other ordinary aliphatic surfactants, bile acids show absorption enhancement for drug delivery and a unique ability to facilitate drug permeation through biological membranes.25,26 We hypothesized that the addition of NaC to mPEG-PDLLA would improve the stability of PTX in micelles and enhance the accumulation of PTX in tumor tissue. The physicochemical properties of PTX-loaded NaC-mPEG-PDLLA micelles (PTX-CMs) were characterized. In vitro and in vivo tumor-inhibition effects were systematically investigated. Furthermore, hemolysis and acute toxicity were also evaluated in order to evaluate the safety of PTX-CMs.
Materials and methods
Materials
PTX was purchased from Jiangsu Yew Pharmaceutical (Wuxi, China). PTX Cremophor-based PTX injection was purchased from Yangtze River Pharmaceutical (Taizhou, China). MTT and 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindotricarbocyanine iodide (DiR) were purchased from Sigma-Aldrich Co. (St Louis, MO, China). Penicillin–streptomycin, RPMI 1640, FBS, 0.25% (w:v) trypsin, and 0.03% (w:v) EDTA solution were purchased from Thermo Fisher Scientific (Waltham, MA, USA). mPEG5,000-PDLLA16,000 was purchased from Advanced Polymer Materials (Montreal, QC, Canada). All reagents and buffer solutions were of analytical grade.
BEL7402 cells (human hepatocellular carcinoma cell line) and A549 cells (human lung adenocarcinoma cell line) were obtained from the Institute of Biochemistry and Cell Biology, Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences (Shanghai, China). Culture plates and dishes were purchased from Corning (Corning, NY, USA).
Male Sprague Dawley rats (250±20 g), male BALB/c nude mice (20±2 g), and specific pathogen-free male ICR mice were supplied by the Animal Experiment Center of Zhejiang Academy of Medical Sciences (Hangzhou, China) and kept under specific pathogen-free conditions. All animals received care in compliance with the guidelines outlined in the Guide for the Care and Use of Laboratory Animals. They were maintained in a room under aseptic conditions and 12-hour light/dark cycles with free access to autoclaved rodent diet and sterile water. All animals were housed at a temperature of 22°C±2°C. The entire animal protocol prior to conducting the experiments was reviewed and approved by the ethics committee of the Animal Experiment Center (approval ZJAMS20160616) and adhered to the aforementioned guidelines.
Preparation of PTX-loaded micelles
A modified titration method with an acetone–ethanol–water system was used to prepare the PTX-loaded micelles.27 PTX powder (2 mg) and 40 mg mPEG5,000-PDLLA15,000 were dissolved in 5 mL acetone. NaC (6 mg) was dissolved in 6 mL distilled water, then 5 mL ethanol was added to form the water phase (H2O:ethanol, v:v 6:5). Under magnetic stirring, 5 mL acetone solution containing PTX and mPEG5,000-PDLLA15,000 was dropped into 11 mL water phase containing NaC at a speed of 60 mL/h. Then, rotary vacuum evaporation was used to remove the organic solvent from the mixture at 37°C and the PTX-CMs were obtained. PTX-loaded mPEG-PDLLA micelles (PTX-Ms) were prepared in a similar manner without NaC addition.
Particle size and ζ-potential
The particle size and ζ-potential of PTX-loaded micelles were measured by dynamic light scattering (Zetasizer Nano ZS90; Malvern Instruments, Malvern, UK) at a detection angle of 90° at 25°C. Prior to measurement, PTX-loaded micelles were diluted three times in distilled water. Measurements were done in triplicate on independent batches. Morphological analysis of PTX-loaded micelles was performed by transmission electron microscopy (TEM; JEM-200C; JEOL, Tokyo, Japan). Briefly, 10 μL diluted sample (0.33 mg/mL) was placed on a carbon-coated grid and air-dried at room temperature. Fixed samples were examined using TEM after staining with 0.1% phosphotungstic acid (w:v).
Drug-loading capacity and encapsulation efficiency
Hydrophobic reverse-phase Thermo Fisher Scientific SOLA HRP solid-phase extraction cartridges were used for separation and determination of PTX-loaded micelles and free PTX, based upon the selective retention of micellar and free PTX on the cartridges described by He et al.28 Micellar PTX exhibited no retention, while free PTX was retained in the stationary phase and eluted only with acidified methanol. Briefly, after column conditioning with 0.5 mL methanol and equilibration with 0.5 mL water, 0.5 mL PTX-loaded micelles (30 μg/mL) in PBS (pH 7.4) was added, and then the column was washed with 0.5 mL PBS (pH 7.4) twice to collect PTX-loaded micelles. Next, the column was washed with 1 mL water:methanol (90:10 v:v) twice. Finally, free PTX was eluted using 0.5 mL methanol and 0.1% formic acid.
PTX concentration was measured by high-performance liquid chromatography (HPLC; Waters 2,695 separations module equipped with an autosampler and Waters 2,487 dual λ-absorbance detector). For PTX-loaded micelles, acetonitrile was added to disrupt micelle structure and release PTX prior to HPLC analysis. A reverse-phase column (Hypersil ODS2, 4.6×250 mm, 5 μm) was used and the detection wavelength was fixed at 227 nm. The mobile phase consisted of acetonitrile and water (50:50 v:v), and the flow rate was 1 mL/min at 30°C. Samples (20 μL) were injected into HPLC columns for analysis. For the calibration curve, six standard samples – 0.05, 0.5, 5, 10, 50, and 100 μg/mL – were used in duplicate. Drug-loading (DL) capacity and encapsulation efficiency (EE) of PTX-loaded micelles were calculated thus:
|
|
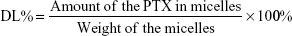
|
|
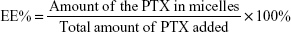
Critical micelle concentration
CMCs of PTX-loaded micelles were determined using a standard pyrene-fluorescence procedure.29 Briefly, 30 μL of pyrene solution in acetone (0.01 mg/mL) was added to standard vials and the solvent was allowed to evaporate. Calculated micellar dispersions were added to achieve varying concentrations of polymer (10−4–0.5 mg/mL) and a final concentration of pyrene at 15 μg/mL in each vial. Solutions were incubated at room temperature in the dark for 24 hours before measurement. Pyrene-fluorescence spectra were recorded using fluorescence spectrophotometry (F-4500; Hitachi, Tokyo, Japan). The excitation wavelength of the probe was 336 nm, and the slit openings were set at 5 nm. Emission spectra were recorded in the range of 350–600 nm. Then, the intensity ratios of the highest (373 nm, I1) and third-highest (384 nm, I3) energy bands in pyrene-emission spectra were analyzed for the calculation of CMC. Experiments were performed in triplicate. The ratio (I1:I3) was plotted against the logarithm of polymer concentration. CMC values corresponded to concentrations of the polymer where sharp increases in I1:I3 were observed. Under linear regression, equations describing the two linear parts of the plots were established. The CMC was then obtained from the intersection of these two lines.
In vitro release of PTX from micelles
To examine the release profile of PTX micelles, 1 mL PTX-loaded micelle solution (1 mg/mL PTX) was diluted with 9 mL PBS. Diluted PTX-loaded micelles (1 mL) were used to calculate the initial concentration. The rest (9 mL) was added to a dialysis tube (molecular weight cutoff 14 kDa) and dialyzed against 50 mL PBS (pH 7.4) containing 0.2% Tween 80 at 37°C. PBS-release medium (1 mL) was sampled at 0, 1, 2, 4, 8, 12, 24, 36, 48, and 62 hours, followed by immediate addition of an equal volume of fresh PBS. PTX concentration was measured by HPLC. Results are reported as cumulative weight percentage of PTX released as a function of time and calculated thus:
|
|
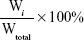
where Wi is the concentration of PTX in PBS release medium at time point i and Wtotal the total amount of PTX in equal volumes of micelle suspensions before performing release experiments. Results from three replicates are reported as mean ± SD.
Cell culture
A549 and BEL7402 cells were cultured in RPMI 1640 medium containing 10% (v:v) heat-inactivated FBS, 100 IU/mL penicillin G, 0.25 μg/mL amphotericin B, and 100 μg/mL streptomycin at 37°C with 5% CO2. Cells were subcultured every 2 days.
Extraction of bone-marrow macrophages
Extraction of bone-marrow macrophage (BMM) cells was performed according to a previously established protocol.30 Briefly, a Sprague Dawley rat was killed by CO2 asphyxiation followed by cervical dislocation. Femurs and tibiae were then surgically removed after stripping away skeletal muscles with forceps, and BM was flushed with PBS using a 10 mL syringe and 25-gauge needle. BM cells were dispersed into a single-cell suspension using a pipette. Cells were then washed with PBS and resuspended in DMEM complete. BM cells were counted and plated in P100 Petri dishes in DMEM complete and incubated at 37°C with 5% CO2.
In vitro cytotoxicity
In vitro cytotoxicity of PTX-loaded micelles was determined by standard MTT assay.31 Briefly, cells were seeded in a 96-well plate at a density of 5×103 cells per well and allowed to grow for 24 hours at 37°C with 5% CO2. Cells were then exposed to PTX-Ms, PTX-CMs, and Taxol at PTX concentrations of 0.05, 0.25, 2.5, 5, 10, 20, and 40 ng/mL. Culture medium was used as the blank control. After 48 hours’ incubation, 30 μL MTT solution (5 mg/mL) was added to the cells and incubated for another 4 hours. Then, the medium was removed and 200 μL dimethyl sulfoxide was added to each well to dissolve any purple formazan crystals formed. Plates were shaken vigorously before measurement. The optical density (OD) of each well was measured at 492 nm using a microplate reader (Multiskan FC; Thermo Fisher Scientific). Cell viability (CV) was determined:
|
|
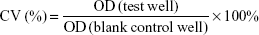
Results from five replicates are reported as mean ± SD. IC50 values were used to evaluate in vitro therapeutic effects of a pharmaceutical formulation quantitatively.32 IC50 values were calculated from concentration–effect curves, taking the OD of the blank control as 100%.
In vitro cellular uptake
Cellular uptake of PTX-loaded micelles was evaluated by incorporating the fluorescent dye fluorescein isothiocyanate (FITC) into the micelles to prepare FITC-Ms and FITC-CMs. A549 and BMM cells were seeded at 3×106 cells/well in six-well plates and incubated over night for adhesion. FITC-Ms and FITC-CMs (100 μL) containing 100 μg/mL FITC were added to each well. At 4 hours posttreatment, cells were washed with PBS to remove free FITC-Ms and FITC-CMs and then harvested for flow-cytometry analysis (BD Biosciences, San Jose, CA, USA). The fluorescence intensity of FITC in the cells was detected in the FITC channel. The uptake study was conducted in triplicate, and results are reported as mean ± SD.
Pharmacokinetic evaluation
Healthy Sprague Dawley rats (6 weeks, 200±20 g) were randomly divided into three groups (n=4) and treated with 8 mg/kg Taxol, PTX-Ms, and PTX-CMS through tail-vein injection. Blood samples (500 μL) were collected from eye sockets at 5, 15, and 30 minutes and 1, 2, 4, 8, 12, and 24 hours. Plasma was collected by centrifuging the blood samples at 4,000 rpm for 10 minutes; 200 μL plasma was used to determine PTX concentrations.
Extraction of PTX from plasma samples was performed prior to HPLC analysis. Briefly, 50 μL of the internal standard diazepam (10 μg/mL in acetonitrile) was added to 200 μL plasma and vortexed. Then, 350 μL acetonitrile was added to the mixture. After vigorous vortex mixing for 2 minutes, the mixture was centrifuged at 15,000 rpm for 10 minutes, and then 400 μL of the organic layer was filtered through a 0.22 μm filter and transferred to a glass tube. Samples (20 μL) were analyzed using HPLC. HPLC conditions were the same as in the vitro release study, except that the mobile phase was changed to a mixture of PBS (pH 3) and acetonitrile (50:50 v:v). Standard samples of 0.05–50 μg/mL (0.05, 0.1, 1, 5, 25, and 50 μg/mL) were prepared in duplicate and analyzed to generate the calibration curve. Main pharmacokinetic parameters–area under the plasma concentration-time curve from 0 to t h (AUC0–t), area under the plasma concentration-time curve from 0 to infinity (AUC0–∞), maximum plasma concentration (Cmax), mean residence time (MRT), apparent volume of distribution (Vz), total body clearance (Cl), and distribution half life (t1/2)–were calculated by non-compartmental analysis using the PKSolver program.33
In vivo biodistribution of PTX-loaded micelles
In vivo distributions of PTX-Ms and PTX-CMs in tumor-bearing nude mice were evaluated by encapsulating the fluorescent dye DiR in the micelles to prepare DiR-Ms and DiR-CMs. Male nude mice (6–8 weeks of age) were implanted with 107 BEL7402 human liver cancer cells in PBS by subcutaneous injection. When the tumor volume had reached about 400 mm3, the mice were randomly divided into three groups (n=9) and treated with 200 μL saline, DiR-Ms, and DiR-CMs via tail-vein injection at a DiR dose of 1 mg/kg. Three mice of each group were anesthetized at 2, 6, and 24 hours postinjection. Whole body, tumors, and major organs (heart, liver, spleen, lung, and kidney) were imaged with an in vivo imaging system (Clairvivo OPT; Shimadzu, Kyoto, Japan). Fluorescence intensity of DiR was used to calculate the biodistribution of PTX-loaded micelles.
Tissue distribution of PTX-loaded micelles
Tissue-distribution studies were performed to measure the concentrations of PTX quantitatively in different organs of tumor-bearing nude mice. BEL7402 cancer cells (107) were inoculated into male BALB/c nude mice (6–8 weeks, 20±2 g). When tumor volumes had reached ~200 mm3, the mice were randomly divided into three groups (n=5) and treated with Taxol, PTX-Ms, and PTX-CMs at 10 mg PTX/kg. Mice were killed at 2 hours posttreatment to harvest hearts, livers, spleens, lungs, kidneys, and tumors. Tissue samples were snap-frozen in liquid N2 and stored at −80°C until further analysis.
Tissue concentrations of PTX were determined by HPLC as described previously.34 Briefly, tissue samples were homogenized with a PRO200 (PRO Scientific, Oxford, CT, USA) with physiological saline at a ratio of 1:2 g/mL. Tissue homogenate samples (200 μL) were mixed with 50 μL internal standard diazepam (10 μg/mL in acetonitrile) by vortex mixing for 30 seconds. Then, samples were extracted with 750 μL acetonitrile using vortex shaking for 3 minutes and centrifuged at 15,000 g for 10 minutes. The upper organic layer was transferred to autosampler vials and analyzed by HPLC. Standard samples were prepared in duplicate from 0.05 to 50 μg/mL (0.05, 0.1, 1, 5, 25, 50 μg/mL). Analysis conditions were same as the in pharmacokinetic evaluation.
In vivo antitumor activity
The in vivo antitumor efficacy of PTX-Ms and PTX-CMs was evaluated in A549 cell-bearing BALB/c nude mice and BEL7402 cell-bearing BALB/c nude mice. Cells (107) were injected subcutaneously into the right flanks of nude mice to establish A549 and BEL7402 xenografts. Two sets of experiments were performed: early-stage tumor model, in which the treatment started when tumor volume reached about 100 mm3; and late-stage tumor model, in which the treatment started when tumor volume reached >500 mm3.
In the early-stage tumor model, mice were randomly divided into four groups (n=8) and administered PBS (control group), Taxol, PTX-Ms, and PTX-CMs at PTX dose of 10 mg/kg through tail-vein injection. In the late-stage tumor model, PTX-CMs were given to mice at PTX dose of 30 mg/kg through intravenous injection. Mice were treated every 3 days for a total of four injections. Tumor volume and mouse body weight were monitored at predetermined time points. Tumor volume was calculated by the equation V = (L × W2)/2, where L represents the longest diameter and W the shortest diameter perpendicular to length. Mice were killed when their tumor volume exceeded a predetermined threshold of 1,500 mm3.
Hemocompatibility assays
For hemolysis studies, blood samples were collected from healthy Sprague Dawley rats (200–250 g, 4–6 weeks) through the retro-orbital plexus and placed into test tubes containing 2% potassium oxalate solution (1:9 volume ratios of potassium oxalate solution and blood). Collected blood samples were centrifuged at 1,500 rpm for 5 minutes to collect red blood cells (RBCs). Plasma was discarded. RBC pellets were washed three times with saline and diluted to concentrations of 2% (v:v). Increasing concentrations (5–200 μg/mL) of PTX-Ms, PTX-CMs, and Taxol were added to the RBC suspensions (1:1 v:v). Normal saline was added to the RBC suspensions as negative control, while distilled water was added as positive control. After incubation for 1.5 hours at 37°C with gentle shaking, the mixture was centrifuged at 1,500 rpm for 10 minutes and 150 μL supernatant was added to 96-well plates. The microplate reader (Multiskan FC) was used to measure OD values at 545 nm, and hemolysis rate was calculated:35
|
|
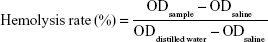
In vivo toxicity
In vivo toxicity of PTX-CMs was investigated in healthy male ICR mice (20±2 g, 6–8 weeks, three groups, n=10). ICR mice were administered PTX-CMs at PTX dose of 300 mg/kg through tail-vein injection. Saline was used as the control. All mice received only one treatment. Their body weights and behaviors were recorded and monitored for 2 weeks. Then, mice were killed at 2 weeks posttreatment. Blood was collected and centrifuged at 4,000 rpm for 10 minutes to obtain the serum. Levels of ALT, total protein, total bilirubin, γ-globulin, serum albumin, albumin/globulin, blood urea nitrogen, and uric acid were assayed as indicators of hepatic and renal function. RBC, white BCs, platelets, hemoglobin, and hematocrit were counted for detection of myelosuppression. Organs (liver, lung, kidney, and spleen) were fixed and sectioned for H&E staining to evaluate organ-specific toxicity.36
Results
Characterization of PTX-loaded micelles
Scheme 1 illustrates the preparation of PTX-CMs and the fate of PTX-CMs in vivo. The physicochemical characterization and DL efficiency of PTX-loaded micelles are summarized in Table 1. The mean particle size of PTX-Ms was 62.58±0.91 nm and PTX-CMs 53.61±0.75 nm (Figure 1A). Both PTX-Ms and PTX-CMs showed narrow size distribution. The polydispersity index of PTX-Ms was 0.12±0.01, and that of PTX-CMs 0.02±0.01. The morphology of PTX-Ms and PTX-CMs were visualized by TEM (Figure 1B and C). All results showed that the two types of PTX-loaded micelles were monodisperse and had spherical structures with the diameters of 50–100 nm. No significant difference in particle size or morphology of the PTX-loaded micelles was observed when NaC was introduced into the micelles. ζ-Potential played an important role in determining the stability of PTX-loaded micelles, and affected cellular uptake, drug release, and in vivo pharmacokinetic and biodistribution.37,38 The ζ-potential of PTX-CMs was −19.73±0.68 mV, which was much lower than that of the PTX-Ms (−3.13±0.06 mV), due to the negative charge of NaC. The calibration curve (y=29,698 x–5,054.8, R2=0.9998) of clozapine was used to calculate the concentration of PTX in the aqueous phase. The EE and DL of PTX-CMs were about 91.33%±0.72% and 3.82%±0.05%, respectively, which differed slightly from those of PTX-Ms (85.64%±2.07% and 4.11%±0.08%). When NaC was introduced into the micelles, the EE improved slightly and DL decreased slightly. However, no statistically significant difference was observed.
 | Scheme 1 Preparation of PTX-CMs and the fate of PTX-CMs in vivo. |
 | Table 1 Physicochemical characterization of PTX-loaded micelles (n=3) |
 | Figure 1 Particle-size distribution and morphology of PTX-Ms and PTX-CMs. |
Determination of critical micelle concentration
Amphiphilic block copolymers can form micelles in an aqueous phase. CMC, which is a measured physical parameter of micelles, refers to their thermodynamic stability.39 The CMC of the block copolymers was determined by a fluorescence technique with pyrene as the probe.40 Figure 1D shows the intensity ratios (I1:I3) of pyrene monomer emission spectra versus the concentrations of micelles and micelles containing NaC (CMs), respectively. From the intersection of two lines, CMC values of the micelles (0.014744 mg/mL) and CMs (0.011625 mg/mL) were obtained. Micelles and CMs showed comparable CMC values, suggesting that NaC did not affect the stability of micelles.
In vitro release of PTX
It is well known that the drug release from polymeric micelles is a complicated process and influenced by several factors.41 Figure 2A shows the in vitro release behavior of PTX from PTX-Ms, PTX-CMs, and Taxol in PBS solution (pH 7.4) at 37°C. Tween 80 (0.2%) was added to overcome the PTX dissolubility issue and provided pseudosink conditions, due to the extreme aqueous insolubility of PTX (<1 μg/mL). As shown in Figure 2A, PTX was released in a sustained manner from PTX-Ms and PTX-CMs in comparison with Taxol. Approximately 47.87% of PTX was rapidly released from Taxol in the first 4 hours and 78.37% within 12 hours. Approximately 22.23% of PTX was released from PTX-Ms within 4 hours and 51.14% within 12 hours. In comparison, about 20.85% of PTX was released from PTX-CMs in the first 4 hours, and the slow kinetics of release persisted for an extended period of 48 hours. Cumulative PTX release followed the order of PTX-CMs > PTX-Ms > Taxol. Compared with four clinically available PTX formulations (Taxol, liposomes, Abraxane, Genexol PM), PTX-CMs showed the slowest release properties (Figure S1).
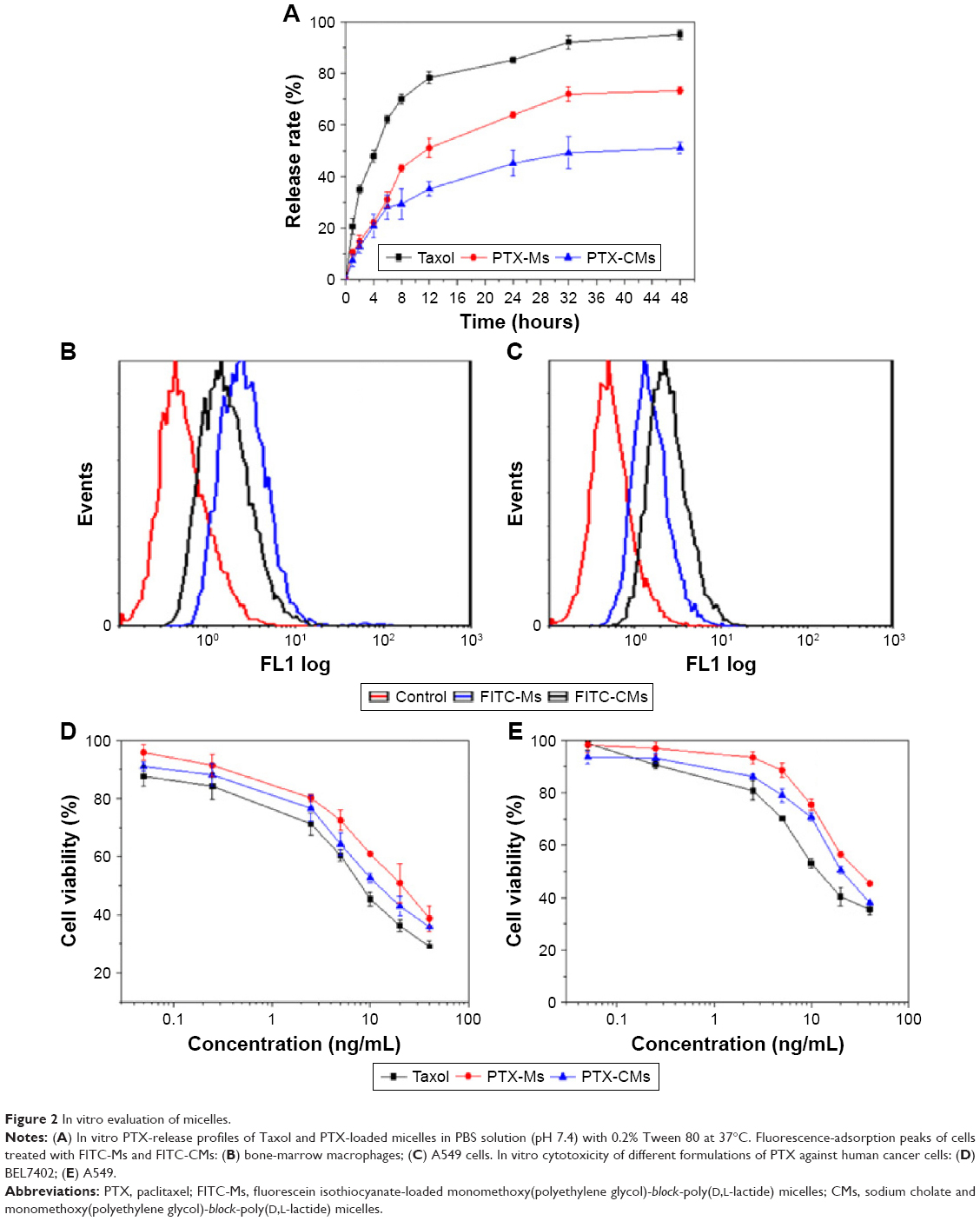 | Figure 2 In vitro evaluation of micelles. |
In vitro cellular uptake of PTX-loaded micelles
FITC was encapsulated in micelles to prepare FITC-Ms and FITC-CMs in the cell uptake study. BMMs and A549 cells were treated with FITC-Ms and FITC-CMs at FITC concentration of 100 μg/mL. At 4 hours posttreatment, cellular uptake of FITC-Ms and FITC-CMs was measured by flow cytometry. As shown in Figure 2B, the cellular uptake of FITC-CMs was 3.15-fold lower than FITC-Ms in BMM. However, the cellular uptake of FITC-CMs was 4.63-fold higher than FITC-Ms in A549 cells (Figure 2C). The successful uptake of FITC-CMs and FITC-Ms was confirmed by fluorescence microscopy imaging (Figure S2) and quantitative flow-cytometry analysis (Figure S3). These results demonstrated that introducing NaC into micelles enhanced cellular uptake in cancer cells, but reduced cellular uptake in macrophages.
In vitro cytotoxicity of PTX-loaded micelles
BEL7402 and A549 cells were treated with PTX-Ms, PTX-CMs, and Taxol at PTX concentrations of 0.05–40 ng/mL. MTT assay was used to evaluate cytotoxicity at 48 hours posttreatment. Results showed that PTX-CMs exhibited higher toxicity in both BEL7402 cells (Figure 2D) and A549 cells (Figure 2E) compared to PTX-Ms. Taxol showed the highest cytotoxicity, which is mainly due to the presence of ethanol and Cremophor EL in the solvent.42 Table 2 shows the IC50 values of PTX-Ms, PTX-CMs, and clinically available Taxol in BEL7402 and A549 cells. The IC50 values of PTX-Ms and PTX-CMs in BEL7402 cells were 23.47±0.38 ng/mL and 14.81±0.27 ng/mL, respectively. The IC50 values of PTX-Ms and PTX-CMs in A549 cells were 28.72±0.33 ng/mL and 22.86±0.23 ng/mL, respectively. Significantly lower IC50 values of PTX-CMs than PTX-Ms may have been due to the higher cellular uptake of PTX-CMs than PTX-Ms.
 | Table 2 IC50 of various PTX-loaded formulations against BEL7402 cells and A549 cells after 48 hours’ incubation |
Pharmacokinetics
PTX standard curves of blood samples were found to be linear (y=0.2772x–0.0007, R2=1) over a concentration range of 0.05–50 μg/mL. The pharmacokinetics of PTX-Ms and PTX-CMs were investigated in healthy Sprague Dawley rats and compared with Taxol. After intravenous administration of Taxol, PTX-Ms, and PTX-CMs at PTX dose of 8 mg/kg, PTX concentration in plasma was measured at 5, 15, and 30 minutes and 1, 2, 4, 8, 12, and 24 hours posttreatment. As shown in Figure 3, PTX-CMs was more slowly eliminated from the circulation after intravenous administration compared with Taxol and PTX-Ms. At 24 hours postinjection, Taxol-treated rats showed the lowest PTX concentration in plasma. The order of PTX clearance from blood was: Taxol > PTX-Ms > PTX-CMs. Table 3 summarizes the pharmacokinetic parameters of PTX-Ms, PTX-CMs, and Taxol. There was no significant difference between Cmax values of PTX-Ms and PTX-CMs (16.06 μg/mL and 18.48 μg/mL, respectively), but their Cmax values were much higher than that of Taxol (10.21 μg/mL). Values for t½ and mean residence time in the PTX-CM-treated group were 17.19 and 19.96 hours, which were much higher than the PTX-M-treated group (8.97 and 9.72 hours, respectively) and the Taxol-treated group (2.36 and 3.25 hours, respectively). The AUC0–t value of PTX-CMs (46.46 mg/L·h) was also significantly higher than PTX-Ms (26.27 mg/L·h) and Taxol (8.92 mg/L·h). The results showed that t½, mean residence time, and AUC0–t of PTX-CMs were about twice as high as PTX-Ms and about sevenfold higher than Taxol, demonstrating the enhanced therapeutic efficacy of PTX-CMs.
 | Figure 3 Time courses of PTX levels in rat plasma after intravenous administration of PTX-Ms, PTX-CMs, and Taxol at PTX dose of 8 mg/kg (n=4). |
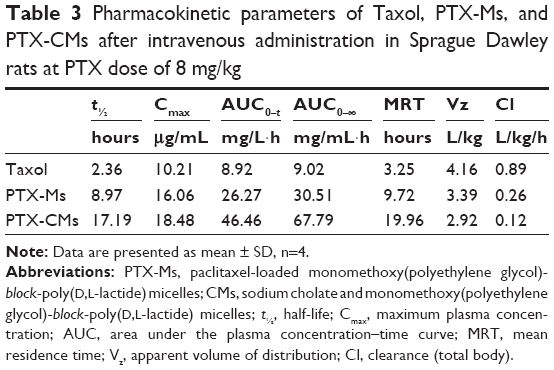 | Table 3 Pharmacokinetic parameters of Taxol, PTX-Ms, and PTX-CMs after intravenous administration in Sprague Dawley rats at PTX dose of 8 mg/kg |
In vivo biodistribution of PTX-loaded micelles
In vivo imaging is viewed as a promising technique in drug development in antitumor studies, and noninvasive near-infrared fluorescence imaging is widely used to investigate the in vivo fate of micelles in tumor-bearing live mice.43 Here, DiR-Ms and DiR-CMs were intravenously injected in BEL7402 xenograft mice at DiR 1 mg/kg. At 2, 6, and 24 hours postinjection, whole-body imaging was performed to examine biodistribution. The absence of animal fur, skin, and muscle would result in a more accurate detection of fluorescence signals.44 Therefore, tumors and major organs were harvested for ex vivo imaging after whole-body imaging. As shown in Figure 4, DiR-CMs showed higher accumulation in tumors and lower accumulation in livers than DiR-Ms.
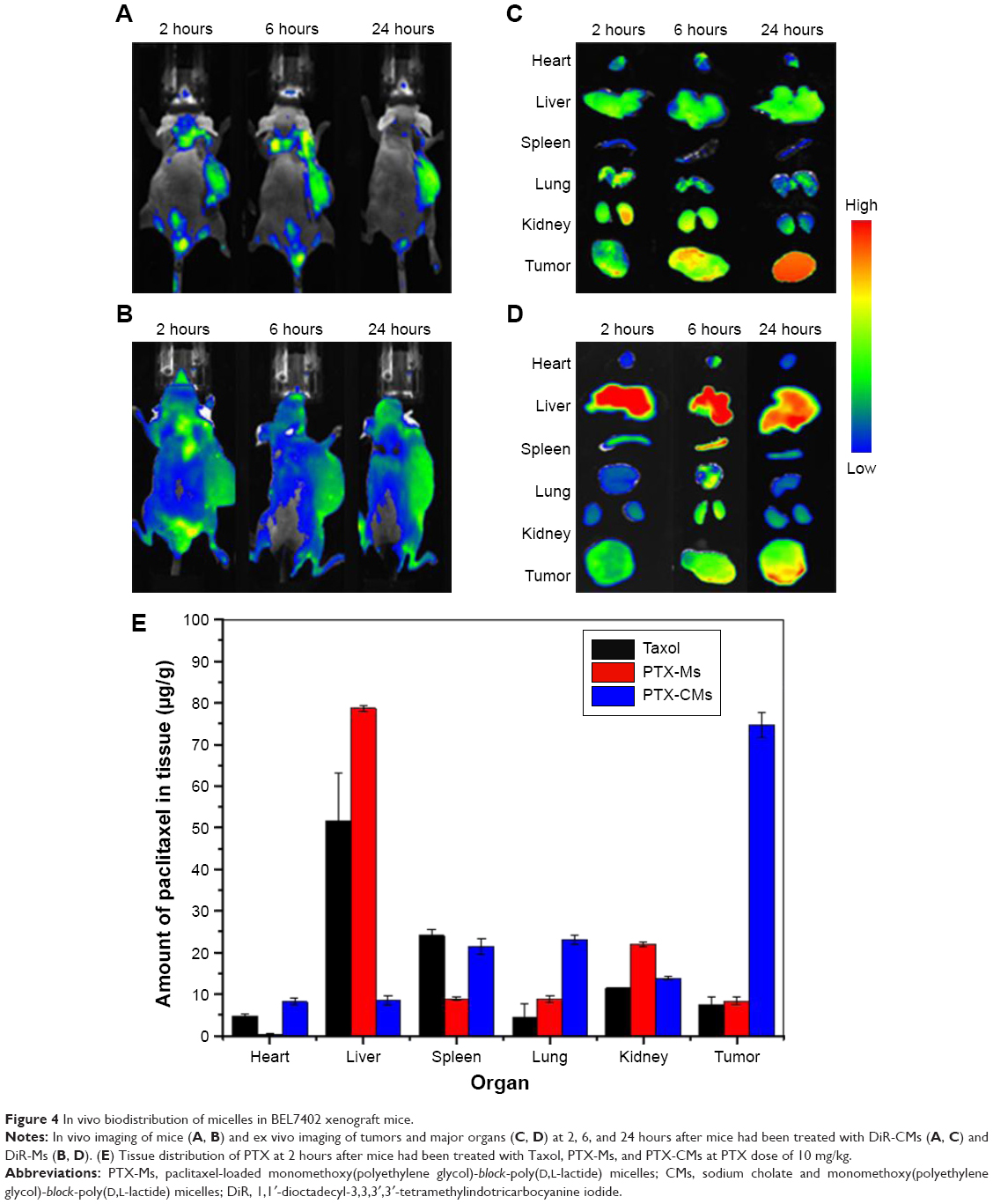 | Figure 4 In vivo biodistribution of micelles in BEL7402 xenograft mice. |
In order to analyze the distribution of PTX-loaded micelles in vivo, BEL7402-xenograft mice were treated with PTX-Ms, PTX-CMs, and Taxol through intravenous injection with PTX 10 mg/kg. At 2 hours postinjection, tumors and major organs were harvested to measure the concentration of PTX in each tissue type. As shown in Figure 4, the highest concentration of PTX was found in the livers of mice treated with Taxol and PTX-Ms, followed by kidney and spleen. However, for the PTX-CM-treated group, the highest concentration of PTX was found in tumors, followed by kidneys, lungs, and spleens. Quantitative results of PTX distribution in vivo were consistent with those obtained from whole-body and ex vivo imaging. These results demonstrated that CMs are a more efficient nanocarrier system than micelles or Taxol, and hold great promise in providing better therapeutic efficacy.
Evaluation of in vivo antitumor activity
The in vivo antitumor efficacy of PTX-CMs was systemically evaluated in BEL7402 early-stage (tumor volume ~100 mm3) and late-stage (tumor volume >500 mm3) and A549 late-stage (tumor volume >500 mm3) xenograft mouse models. In the BEL7402 early-stage model, mice were treated with PBS, PTX-Ms, PTX-CMs, blank CMs, and three clinically available PTX formulations: Taxol, Abraxane, and liposomes. Mice received injections every 3 days at PTX 10 mg/kg for a total of four injections. Tumor volume and body weight were monitored for 22 days. As shown in Figure 5A, PTX-CMs exhibited almost 100% tumor inhibition, which was far superior to PTX-Ms, Taxol, liposomes, and Abraxane. Among the three clinically available PTX formulations, Abraxane inhibited tumor growth by ~50%, while Taxol and liposomes showed limited tumor growth inhibition, which was comparable to PTX-Ms. Figure 5B shows body weight changes in tumor-bearing mice during this study. There was no significant loss in body weight, indicating that PTX-CMs were well tolerated by mice and did not have systemic toxicity.
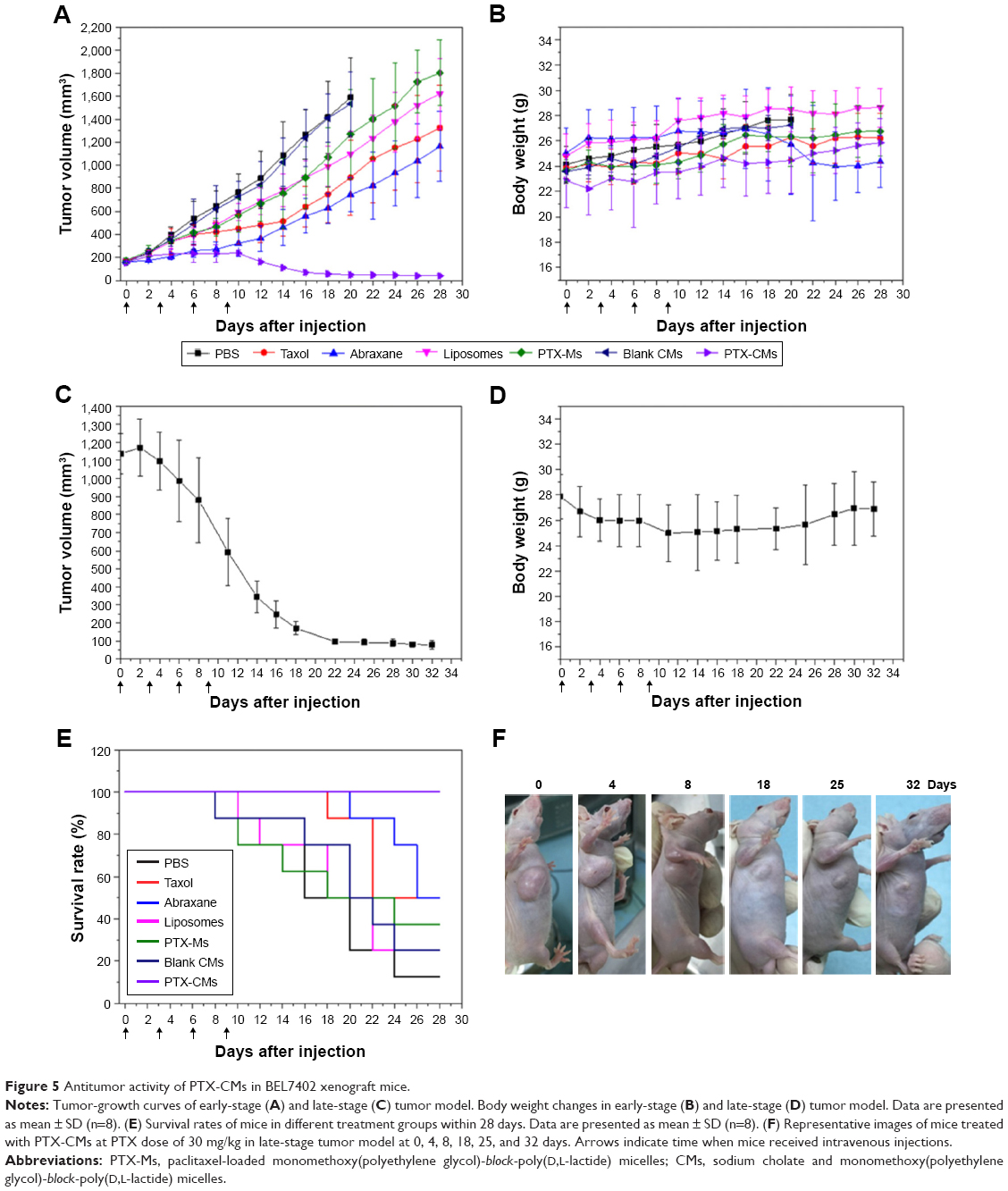 | Figure 5 Antitumor activity of PTX-CMs in BEL7402 xenograft mice. |
The antitumor efficacy of PTX-CMs also showed a dose–response effect. When early-stage BEL7402 tumors (~150 mm3) were treated four times with PTX-CMs at PTX 30 mg/kg, the tumors disappeared and never recurred thereafter (data not shown). Superior antitumor activity was further demonstrated in both BEL7402 and A549 late-stage models. In the BEL7402 late-stage model, tumors were allowed to grow to ~1,150 mm3. Then, mice were treated with PTX-CMs at PTX 30 mg/kg every 3 days for a total of four injections. As shown in Figure 5C and F, quick reduction in tumor volume was observed right after the second injection of PTX-CMs. At day 32, tumor volume was remarkably reduced to only ~50 mm3 from ~1,150 mm3 at the beginning. Also, no significant loss in body weight was observed (Figure 5D). In the A549 late-stage model, tumors were allowed to reach ~600 mm3. Similarly, mice were treated with PTX-CMs at PTX 30 mg/kg every 3 days for a total of four injections. Superior antitumor activity was also observed in the A549-xenograft model. At day 15, the tumor volume was reduced to ~100 mm3 (Figure S4). The remarkable antitumor efficacy of PTX-CMs in both early-stage and late-stage xenograft models demonstrated that CMs are a promising anticancer drug-delivery system. Pharmacokinetic and biodistribution results also fully supported this conclusion.
Hemocompatibility evaluation
Hemolytic activity, an in vitro toxicity screening test, has been suggested to serve as a simple and reliable measure for estimating membrane damage caused by formulations in vivo.45 Taxol is known to cause hemolysis due to the use of a high percentage of ethanol (50% v:v) and Cremophor EL for dissolving the PTX.46 NaC is a biosurfactant and can damage cell membrane when intravenously administrated. Membrane damage by PTX-CMs was investigated by measuring in vitro hemolytic activity. Taxol and PTX-Ms were selected as controls. Results from hemolysis assays (Figure 6) showed that Taxol caused severe hemolysis at PTX concentrations as low as 25 μg/mL. As Taxol concentration increased, more serious hemolysis was observed. Much less hemolysis was observed in PTX-CMs and PTX-Ms when PTX concentration was ≤125 μg/mL (equivalent to about 2.5 mg micelles). When the concentration of PTX was increased to 200 μg/mL (equivalent to about 4 mg micelles), the hemolysis ratio of PTX-CMs was 60.92%±3.37%, slightly higher than PTX-Ms (48.12%±1.54%), but lower than Taxol (67.77%±1.94%). Therefore, in terms of hemolysis, PTX-CMs were much safer than Taxol.
 | Figure 6 In vitro hemolysis of PTX-CMs. |
Evaluation of systemic toxicity
Myelosuppression is one of the major side effects of chemotherapy in cancer treatment.47 Routine blood tests were performed on healthy male ICR mice at day 14 after a single injection of PBS and PTX-CMs at PTX 300 mg/kg (n=10). During the 14 days, no significant loss in body weight in the treated groups was observed (Figure 7A). Compared to the PBS-treated group and reference values, there was no noticeable blood toxicity in the PTX-CM-treated group. WBC, RBC, hematocrit, and hemoglobin values of PTX-CM-treated mice were within the normal range, and were similar to those of the PBS-treated group (Table S1).
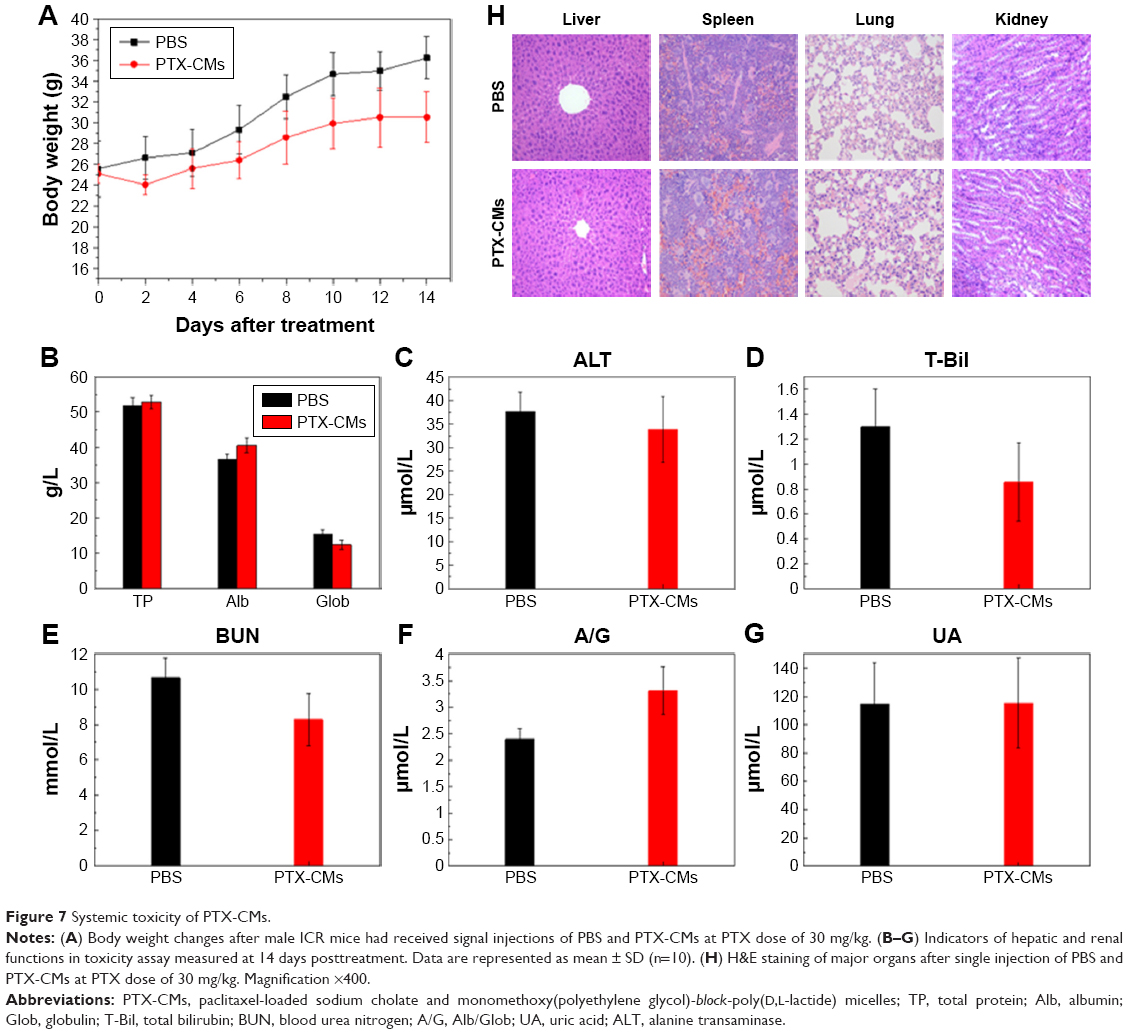 | Figure 7 Systemic toxicity of PTX-CMs. |
Other hematological parameters also showed no detectable toxicity. ALT, total protein, total bilirubin, γ-globulin, serum albumin, albumin/globulin, blood urea nitrogen, and uric acid analyses were all within normal range (Figure 7B–G). No noticeable histological changes were seen in H&E-stained tissue sections of the liver, spleen, lung, or kidney (Figure 7H). These studies demonstrated that PTX-CMs exhibited no significant toxicity to major organs. Therefore, PTX-CMs are promising therapeutics for cancer treatment.
Discussion
The development of new drug-delivery systems that can effectively improve the stability of drugs, enhance tumor-targeting capability, and realize controlled release of drugs at tumor sites has attracted lots of research interest. Although techniques for controlled drug release, such as conjugating drugs to polymeric materials or cross-linking polymers by chemical reaction, have been developed, the tumor-targeted delivery of drugs still relies on the EPR effect or active tumor-targeting effect. Active tumor targeting has been realized through conjugation or decoration of specific ligands for drug-loaded particles.34–38 However, such approaches are limited by many technical challenges and not suitable for large-scale industrial production. The use of bile acid in polymer micelles as an enhancer to improve drug stability and targeted delivery has just begun.48 In this study, in order to develop a safe, stable, and tumor-targeted formulation that could improve the antitumor efficacy of PTX, we combined NaC with traditional mPEG-PDLLA micelles and prepared PTX-CMs.
PTX-CMs were prepared by a modified titration method. This method has advantages of simplicity and low energy consumption. Moreover, it usually leads to narrow distribution and good dispersibility of the resulting particles.49 NaC can be used as an emulsifier. It facilitates the incorporation of hydrophobic drugs into micelles when added in the preparation, and thus increases drug EE.50 PTX-CMs (53.61±0.75 nm) were a little smaller than PTX-Ms (62.58±0.91 nm). The surface charge of PTX-CMs (–19.73±0.68 mV) was more negative than that of PTX-Ms (–3.13±0.06 mV) (Figure 1, Table 1). This could be evidence that NaC participated in the formation of micelles. Nanocarriers in the size range of 20–200 nm can accumulate in tumors through the EPR effect, and smaller particles can extravasate more easily through poorly formed microvessels in angiogenic tumors.51 The negative surface charge of nanocarriers can reduce systemic toxicity, and is important for effective cancer therapy.52 As such, we hypothesized that PTX-CMs would be more effective than PTX-Ms in tumor growth inhibition. This hypothesis was tested systemically in in vitro and in vivo studies.
CMC is a very important property that needs to be considered not only in in vitro but also in in vivo studies. The dose for intravenous injection of polymeric micelles must be above the CMC to maintain micelle stability.53 Micelles with low CMC had high stability and were able to maintain integrity even upon dilution in the blood circulation.54 The CMC value of the CMs (0.011625 mg/mL) was lower than micelles (0.014744 mg/mL). Studies have shown that when combined with bile acids, amphiphilic drugs usually have low CMC and better solubilization capacity than the individual components, due to synergistic interactions.24 Therefore, the low CMC value of CMs was the result of synergistic interactions between NaC and mPEG-PDLLA, suggesting CMs may be more stable than micelles for anticancer drug delivery.
Drug release from micelles is generally dependent on degradation of the polymers and diffusion of the drug from the micelles, which leads to uncontrolled release of the encapsulated drug. Here, the release of PTX from PTX-CMs showed sustained release behavior compared with PTX-Ms and Taxol, liposomes and Abraxane (Figure 2A, Figure S1). Taxol released 50% of PTX within 4 hours, PTX-Ms 50% within 12 hours, and PTX-CMs 50% after 40 hours. The sustained PTX release from PTX-CMs was likely due to the hydrogen-bond and electrostatic interaction between mPEG-PDLLA and NaC.55 This interaction makes micellar cores compact and thus retards drug release.56 This behavior could protect drugs from degradation in the circulation system and improve therapeutic efficacy.
For PTX-based chemotherapy via intravenous infusion, developing a PTX-based formulation with high tumor-targeting capability and good therapeutic efficacy is one of the major challenges.27 To investigate tumor-targeting capability, we first measured the cellular uptake and sustained retention of FITC-loaded micelles in A549 lung cancer cells, which are closely related to therapeutic effects.57 Most nanomaterial formulations are rapidly sequestered by macrophages following intravenous administration.58 Therefore, we also investigated the uptake of FITC-Ms and FITC-CMs in macrophages. Compared with FITC-Ms, FITC-CMs showed significantly higher uptake in A549 cells and lower uptake in macrophages (Figure 2B and C, Figure S2). The different cellular uptake behavior between CMs and Ms was likely due to the incorporation of NaC in the micelles. NaC further reduced the ζ-potential of micelles. Studies have shown that negative ζ-potential is favorable for nanoparticle uptake in A549 cells.59 The other reason may be because NaC has biosurfactant properties, which can disturb the structure of cell membranes and facilitate the entry of micelles into cells.60 Because of high cellular uptake, PTX-CMs showed higher in vitro cytotoxicity than PTX-Ms in both BEL7402 and A549 cells (Figure 2D and E).
Long-circulating nanoparticles in the bloodstream provide the possibility of high accumulation of drugs in tumor.61 However, low stability of micelles in circulation remains the critical challenge.62 As shown in this study, PTX-CMs significantly prolonged systemic circulation time compared to PTX-Ms and Taxol (Figure 3, Table 3). Pharmacokinetic and in vitro release results demonstrated that NaC improved the stability of micelles and achieved sustained release of PTX in vitro and in vivo.
Solid tumors have high interstitial pressure, especially in tumor cores, and heterogeneous vascular supply,63 which limit the penetration of drugs into tumors after systemic administration. To demonstrate accumulation of CMs in tumors, in vivo and ex vivo imaging were used to visualize the biodistribution of CMs containing DiR fluorescent dye (Figure 4). We observed that DiR-Ms accumulated largely in the liver, and accumulation in tumors increased slightly with time. On the contrary, DiR-CMs accumulated mainly in tumors, and accumulation in tumors increased significantly with time. Also, the amount of DiR-CMs in liver was much lower than DiR-Ms. Since the liver is one of the major organs responsible for nonspecific clearance of nanoparticles,64 DiR-CMs were cleared out more slowly than DiR-Ms. Results from the pharmacokinetic study also supported this conclusion. Next, we quantitatively measured the distribution of PTX in tumors and major organs after the BEL7402 mice had been treated with PTX-CMs, PTX-Ms, and Taxol. Results from PTX tissue distribution agreed well with in vivo/ex vivo imaging results, ie, PTX-CM-treated mice showed much higher levels of PTX in tumors than the mice treated with PTX-Ms or Taxol (Figure 4E). With such enhanced tumor-targeting capability, the effective dose can be reduced and thus side effects in cancer therapy can be minimized. These results demonstrated that the addition of NaC in micelles can significantly improve tumor-targeting capability and reduce their distribution in major organs. In addition, the enhanced tumor accumulation of PTX-CMs might be due to the prolonged blood circulation time.
In vivo antitumor efficacy of PTX-CMs was investigated in BEL7402 and A549 mice. Remarkable antitumor activity of PTX-CMs was observed not only in the early-stage tumor model (tumor volume ~100 mm3) but also in the late-stage tumor model (tumor volume >500 mm3) (Figure 5, Figure S4). The mechanisms of improved anticancer efficiency of PTX-CMs can be summarized as follows. Firstly, NaC reduced the size of micelles. Solid tumors have leaky microvasculature. Smaller particles are favorable for passive targeting to the tumor through the EPR effect.65 Secondly, NaC improved the stability of micelles. PTX-CMs had lower CMC than PTX-Ms, which helped enhance the stability of micelles in the bloodstream. Bile acids have an enhanced affinity toward hydrophobic drugs.15 The interaction between NaC and PTX may protect PTX-CMs from degradation in blood circulation, and thus achieve better PTX delivery to tumors. Thirdly, NaC enhanced the circulation time of PTX-CMs in the bloodstream. Long circulation of PTX-CMs favored the accumulation of micelles in tumors via the EPR effect,66 and provided significantly higher PTX levels in tumors than PTX-Ms or Taxol. Finally, NaC may also affect membrane fluidity, increase transmembrane permeability, and thus enhance the cellular uptake of PTX-CMs.67,68 More importantly, micelles containing NaC showed unique tumor-targeting capability, which greatly improved the antitumor effect.17,23,24
The safety and toxicity of PTX-CMs were comprehensively investigated in this work. Because polysorbate 80 (Tween 80) used in PTX injection caused hemolysis69 and NaC is also a surfactant that has similar property as Tween 80, the hemolytic activity of PTX-CMs was studied. As shown in Figure 6, similarly to PTX-Ms, less hemolysis was observed in PTX-CMs compared to Taxol. Therefore, PTX-CMs were much safer than Taxol. Hematology and histological analysis were performed to investigate the toxicity of PTX-CMs further and explore potential side effects, such as BM depression, tissue damage, and inflammation. Hematological tests of whole blood collected from PTX-CM-treated mice was performed. All results were within normal range (Table S1). No significant difference was observed in liver or kidney functions between the PBS control group and the PTX-CM-treated group (Figure 7), demonstrating that no toxic syndrome, such as hemolytic anemia, acute infection, or BM dysfunction, was caused by PTX-CMs. Furthermore, H&E-stained sections of major organs were examined. As shown in Figure 7H, all major organs from PTX-CM-treated mice, including the heart, liver, spleen, lung, and kidney, were normal, with no obvious histopathological abnormalities, degenerations, or lesions, indicating PTX-CMs were nontoxic and well tolerated by mice.
The development of effective delivery systems for antitumor therapeutics remains one of the great challenges today. We developed NaC-based mPEG-PDLLA polymeric micelles to deliver PTX for cancer treatment. The PTX-CMs showed high tumor-targeting capability, sustained release of PTX in the blood circulation, improved cytotoxicity in tumor cells, and enhanced penetration in tumor tissue. As a result, PTX-CMs showed significant better tumor-inhibition effects than current PTX formulations (Taxol, liposomes, and Abraxane). Also, no significant toxicity was observed in vivo. Therefore, CMs represent a promising delivery system for anticancer drugs. However, the interaction between CMs and serum proteins and how the resulting protein corona affects therapeutic efficacy are not clear. Further investigations are required to address these questions. In addition, many other bile acids, such as cholic acid, deoxycholic acid, lithocholic acid, ursodeoxycholic acid, chenodeoxycholic acid,70 may be incorporated in micelles, and their roles in antitumor activity need to be explored.
Conclusion
In this study, we introduced NaC into the mPEG-PDLLA polymeric micelles and developed a promising nanocarrier system for PTX delivery in cancer treatment. The PTX-CMs were successfully formulated and their physicochemical properties characterized. PTX-CMs were monodisperse nanoparticles with size of 53.61 nm and ζ-potential of −19.73 mV. In vitro release profiles showed sustained release of PTX. In vitro cellular uptake studies showed better tumor-targeting capability of PTX-CMs than PTX-Ms or Taxol, which reduced the clearance of PTX-CMs by macrophages. In vivo biodistribution studies showed much higher accumulation of PTX-CMs in tumors than PTX-Ms or Taxol. In BEL7402 and A549 xenograft mouse models, PTX-CMs showed much higher (>90%) tumor growth inhibition than PTX-Ms or clinically available PTX formulations (Taxol, liposomes, and Abraxane). Systemic toxicity studies demonstrated that PTX-CMs had good biocompatibility and showed no significant toxicity. All results demonstrated that CMs are a promising nanocarrier system for the delivery of anticancer drugs.
Acknowledgments
This work was supported by the National Science Foundation of China (51573020), Beijing Natural Science Foundation (2172022), Undergraduate Training Program (2016-238), and URT Program (2016J00036). We also acknowledge the technical support from Hangxiang Wang for TEM imaging; Qi Lou and Honggang Guo for helping build tumor models and perform pharmacokinetic experiments; Chenhuan Yu for in vivo imaging; and Chengjun Jiang and Caiyun Deng for CMC analysis.
Disclosure
The authors report no conflicts of interest in this work.
References
Cui M, Naczynski DJ, Zevon M, et al. Multifunctional albumin nanoparticles as combination drug carriers for intra-tumoral chemotherapy. Adv Healthc Mater. 2013;2(9):1236–1245. | ||
Lv L, Guo Y, Shen Y, et al. Intracellularly degradable, self-assembled amphiphilic block copolycurcumin nanoparticles for efficient in vivo cancer chemotherapy. Adv Healthc Mater. 2015;4(10):1496–1501. | ||
Xiao K, Li Y, Lee JS, et al. “OA02” peptide facilitates the precise targeting of paclitaxel-loaded micellar nanoparticles to ovarian cancer in vivo. Cancer Res. 2012;72(8):2100–2110. | ||
Hucke A, Ciarimboli G. The role of transporters in the toxicity of chemotherapeutic drugs: focus on transporters for organic cations. J Clin Pharmacol. 2016;56(Suppl 7):S157–S172. | ||
Mikhail AS, Allen C. Poly(ethylene glycol)-b-poly(ε-caprolactone) micelles containing chemically conjugated and physically entrapped docetaxel: synthesis, characterization, and the influence of the drug on micelle morphology. Biomacromolecules. 2010;11(5):1273–1280. | ||
Shi Y, Meel RV, Theek B, et al. Complete regression of xenograft tumors upon targeted delivery of paclitaxel via π-π stacking stabilized polymeric micelles. ACS Nano. 2015;9(4):3740–3752. | ||
Kataoka K, Harada A, Nagasaki Y. Block copolymer micelles for drug delivery: design, characterization and biological significance. Adv Drug Deliv Rev. 2001;47(1):113–131. | ||
Kutikov AB, Jie S. Biodegradable PEG-based amphiphilic block copolymers for tissue engineering applications. ACS Biomater Sci Eng. 2015;1(7):463–480. | ||
Letchford K, Liggins R, Wasan KM, Burt H. In vitro human plasma distribution of nanoparticulate paclitaxel is dependent on the physicochemical properties of poly(ethylene glycol)-block-poly(caprolactone) nanoparticles. Eur J Pharm Biopharm. 2009;71(2):196–206. | ||
Miller T, Breyer S, van Colen G, et al. Premature drug release of polymeric micelles and its effects on tumor targeting. Int J Pharm. 2013;445(1–2):117–124. | ||
Yu TT, Gao J, Kwon GS. Oligo(lactic acid)n-paclitaxel prodrugs for poly(ethylene glycol)-block-poly(lactic acid) micelles: loading, release, and backbiting conversion for anticancer activity. J Am Chem Soc. 2016;138(28):8674–8677. | ||
Yu MK, Park J, Jon S. Targeting strategies for multifunctional nanoparticles in cancer imaging and therapy. Theranostics. 2012;2(1):3–44. | ||
Masood F. Polymeric nanoparticles for targeted drug delivery system for cancer therapy. Mater Sci Eng C Mater Biol Appl. 2016;60:569–578. | ||
Subuddhi U, Mishra AK. Micellization of bile salts in aqueous medium: a fluorescence study. Colloids Surf B Biointerfaces. 2007;57(1):102–107. | ||
Nurunnabi M, Zehedina K, Revuri V, et al. Design and strategies for bile acid mediated therapy and imaging. RSC Adv. 2016;6(78):73986–74002. | ||
Madenci D, Egelhaaf SU. Self-assembly in aqueous bile salt solutions. Curr Opin Colloid Interface Sci. 2010;15(1–2):109–115. | ||
Moghimipour E, Ameri A, Handali S. Absorption-enhancing effects of bile salts. Molecules. 2015;20(8):14451–14473. | ||
Hofmann AF. Bile acids: trying to understand their chemistry and biology with the hope of helping patients. Hepatology. 2009;49(49):1403–1418. | ||
Luo YL, Yuan JF, Shi JH, Gao QY. Synthesis and characterization of polyion complex micelles and their controlled release of folic acid. J Colloid Interface Sci. 2010;350(1):140–147. | ||
Maestrelli F, Cirri M, Mennini N, Zerrouk N, Mura P. Improvement of oxaprozin solubility and permeability by the combined use of cyclodextrin, chitosan, and bile components. Eur J Pharm Biopharm. 2011;78(3):385–393. | ||
Serafim C, Ferreira I, Rijo P, et al. Lipoamino acid-based micelles as promising delivery vehicles for monomeric amphotericin B. Int J Pharm. 2016;497(1–2):23–35. | ||
Alnouti Y. Bile acid sulfation: a pathway of bile acid elimination and detoxification. Toxicol Sci. 2009;108(2):225–246. | ||
Faustino C, Serafim C, Rijo P, Reis CP. Bile acids and bile acid derivatives: use in drug delivery systems and as therapeutic agents. Expert Opin Drug Deliv. 2016;13(8):1133–1148. | ||
Stojančević M, Pavlović N, Goločorbin-Kon S, Mikov M. Application of bile acids in drug formulation and delivery. Front Life Sci. 2014;7(3–4):112–122. | ||
Chen G, Fawcett JP, Mikov M, Tucker IG. Monoketocholate can decrease transcellular permeation of methotrexate across Caco-2 cell monolayers and reduce its intestinal absorption in rat. J Pharm Pharmacol. 2009;61(7):953–959. | ||
Chen G, Yang L, Zhang H, Tucker IG, Fawcett JP. Effect of ketocholate derivatives on methotrexate uptake in Caco-2 cell monolayers. Int J Pharm. 2012;433(1–2):89–93. | ||
Wang G, Chen Y, Wang P, et al. Preferential tumor accumulation and desirable interstitial penetration of poly(lactic-co-glycolic acid) nanoparticles with dual coating of chitosan oligosaccharide and polyethylene glycol-poly(d,l-lactic acid). Acta Biomater. 2016;29:248–260. | ||
He Z, Wan X, Schulz A, et al. A high capacity polymeric micelle of paclitaxel: implication of high dose drug therapy to safety and in vivo anti-cancer activity. Biomaterials. 2016;101:296–309. | ||
Zhang CY, Yang YQ, Huang TX, et al. Self-assembled pH-responsive MPEG-b-(PLA-co-PAE) block copolymer micelles for anticancer drug delivery. Biomaterials. 2012;33(26):6273–6283. | ||
Weischenfeldt J, Porse B. Bone marrow-derived macrophages (BMM): isolation and applications. CSH Protoc. 2008;3(12):pdb.prot5080. | ||
Wang C, Wang Y, Wang Y, Fan M, Luo F, Qian Z. Characterization, pharmacokinetics and disposition of novel nanoscale preparations of paclitaxel. Int J Pharm. 2011;414(1–2):251–259. | ||
Xie JW, Shen ZM, King CL, Danthi N. Tumor angiogenic endothelial cell targeting by a novel integrin-targeted nanoparticle. Int J Nanomedicine. 2007;2(3):479–485. | ||
Zhang Y, Huo M, Zhou J, Xie S. PKSolver: an add-in program for pharmacokinetic and pharmacodynamic data analysis in Microsoft Excel. Comput Methods Programs Biomed. 2010;99(3):306–314. | ||
Rezazadeh M, Emami J, Mostafavi A, et al. A rapid and sensitive HPLC method for quantitation of paclitaxel in biological samples using liquid-liquid extraction and UV detection: application to pharmacokinetics and tissues distribution study of paclitaxel loaded targeted polymeric micelles in tumor bearing mice. J Pharm Pharm Sci. 2015;18(5):647–660. | ||
Raza K, Thotakura N, Kumar P, et al. C60-fullerenes for delivery of docetaxel to breast cancer cells: a promising approach for enhanced efficacy and better pharmacokinetic profile. Int J Pharm. 2015;495(1):551–559. | ||
Miao L, Guo S, Zhang J, Kim WY, Huang L. Nanoparticles with precise ratiometric co-loading and co-delivery of gemcitabine monophosphate and cisplatin for treatment of bladder cancer. Adv Funct Mater. 2015;24(42):6601–6611. | ||
Sanna V, Roggio AM, Siliani S, et al. Development of novel cationic chitosan- and anionic alginate-coated poly(d,l-lactide-co-glycolide) nanoparticles for controlled release and light protection of resveratrol. Int J Nanomedicine. 2012;7:5501–5516. | ||
Xu G, Yu X, Zhang J, et al. Robust aptamer-polydopamine-functionalized M-PLGA-TPGS nanoparticles for targeted delivery of docetaxel and enhanced cervical cancer therapy. Int J Nanomedicine. 2016;11:2953–2965. | ||
Li GH, Cho CG. Synthesis and characterization of poly(vinyl alcohol-b-styrene) via atom transfer radical polymerization and saponification. Macromol Res. 2002;10(6):339–344. | ||
Kozlov MY, Melik-Nubarov NS, Batrakova EV, Kabanov AV. Relationship between Pluronic block copolymer structure, critical micellization concentration and partitioning coefficients of low molecular mass solutes. Macromolecules. 2000;33(9):3305–3313. | ||
Gu C, Le V, Lang M, Liu J. Preparation of polysaccharide derivates chitosan-graft-poly(ε-caprolactone) amphiphilic copolymer micelles for 5-fluorouracil drug delivery. Colloids Surf B Biointerfaces. 2014;116:745–750. | ||
Zhang JA, Gopal A, Lan M, et al. Development and characterization of a novel Cremophor EL free liposome-based paclitaxel (LEP-ETU) formulation. Eur J Pharm Biopharm. 2005;59(1):177–187. | ||
Han LM, Guo J, Zhang LJ, Wang QS, Fang XL. Pharmacokinetics and biodistribution of polymeric micelles of paclitaxel with Pluronic P123. Acta Pharmacol Sin. 2006;27(6):747–753. | ||
Lin Z, Gao W, Hu H, et al. Novel thermo-sensitive hydrogel system with paclitaxel nanocrystals: high drug-loading, sustained drug release and extended local retention guaranteeing better efficacy and lower toxicity. J Control Release. 2014;174:161–170. | ||
Utreja P, Jain S, Tiwary AK. Localized delivery of paclitaxel using elastic liposomes: formulation development and evaluation. Drug Deliv. 2011;18(5):367–376. | ||
Park JH, Chi SC, Woo JS. Preparation and evaluation of paclitaxel solid dispersion by supercritical antisolvent process. J Pharm Investig. 2008;38(4):241–247. | ||
Iwamoto T. Clinical application of drug delivery systems in cancer chemotherapy: review of the efficacy and side effects of approved drugs. Biol Pharm Bull. 2013;36(5):715–718. | ||
Xing T, Lai B, Ye X, Yan L. Disulfide core cross-linked PEGylated polypeptide nanogel prepared by a one-step ring opening copolymerization of N-carboxyanhydrides for drug delivery. Macromol Biosci. 2011;11(7):962–969. | ||
Ukawala M, Rajyaguru T, Chaudhari K, et al. Investigation on design of stable etoposide-loaded PEG-PCL micelles: effect of molecular weight of PEG-PCL diblock copolymer on the in vitro and in vivo performance of micelles. Drug Deliv. 2012;19(3):155–167. | ||
Chen W, Guo M, Wang S. Anti prostate cancer using PEGylated bombesin containing, cabazitaxel loading nano-sized drug delivery system. Drug Dev Ind Pharm. 2016;42(12):1968–1976. | ||
Torchilin V. Tumor delivery of macromolecular drugs based on the EPR effect. Adv Drug Deliv Rev. 2011;63(3):131–135. | ||
Ibrahim MM, Tawfique SA, Mahdy MM. Liposomal diltiazem HCl as ocular drug delivery system for glaucoma. Drug Dev Ind Pharm. 2014;40(6):765–773. | ||
Liu J, Zeng F, Allen C. In vivo fate of unimers and micelles of a poly(ethylene glycol)-block-poly(caprolactone) copolymer in mice following intravenous administration. Eur J Pharm Biopharm. 2007;65(3):309–319. | ||
Saxena V, Hussain MD. Poloxamer 407/TPGS mixed micelles for delivery of gambogic acid to breast and multidrug-resistant cancer. Int J Nanomedicine. 2012;7:713–721. | ||
Kuang H, Wu S, Xie Z, Meng F, Jing X, Huang Y. Biodegradable amphiphilic copolymer containing nucleobase: synthesis, self-assembly in aqueous solutions, and potential use in controlled drug delivery. Biomacromolecules. 2012;13(9):3004–3012. | ||
Ge YX, Zhao YL, Li LB. Preparation of sodium cholate-based micelles through non-covalent bonding interaction and application as oral delivery systems for paclitaxel. Drug Deliv. 2016;23(7):2555–2565. | ||
Xu A, Yao M, Xu G, et al. A physical model for the size-dependent cellular uptake of nanoparticles modified with cationic surfactants. Int J Nanomedicine. 2012;7:3547–3554. | ||
Fischer HC, Liu L, Pang KS, Chan WC. Pharmacokinetics of nanoscale quantum dots: in vivo distribution, sequestration, and clearance in the rat. Adv Funct Mater. 2006;16(10):1299–1305. | ||
Patil S, Sandberg A, Heckert E, Self W, Seal S. Protein adsorption and cellular uptake of cerium oxide nanoparticles as a function of zeta potential. Biomaterials. 2007;28(31):4600–4607. | ||
Taranto MP, Murga ML, Lorca G, Valdez GF. Bile salts and cholesterol induce changes in the lipid cell membrane of Lactobacillus reuteri. J Appl Microbiol. 2003;95(1):86–91. | ||
Wang AZ, Langer R, Farokhzad OC. Nanoparticle delivery of cancer drugs. Annu Rev Med. 2011;63:185–198. | ||
Rijcken CJ, Snel CJ, Schiffelers RM, Nostrum CF, Hennink WE. Hydrolysable core-crosslinked thermosensitive polymeric micelles: synthesis, characterisation and in vivo studies. Biomaterials. 2008;28(36):5581–5593. | ||
Ravar F, Saadat E, Gholami M, et al. Hyaluronic acid-coated liposomes for targeted delivery of paclitaxel, in-vitro characterization and in-vivo evaluation. J Control Release. 2016;229:10–22. | ||
Lu J, Huang Y, Zhao W, et al. PEG-derivatized embelin as a nanomicellar carrier for delivery of paclitaxel to breast and prostate cancers. Biomaterials. 2013;34(5):1591–1600. | ||
Xia B, Liu X, Zhou Q, et al. Disposition of orally administered a promising chemotherapeutic agent flavopiridol in the intestine. Drug Dev Ind Pharm. 2013;39(6):845–853. | ||
Deng C, Jiang Y, Cheng R, Meng F, Zhong Z. Biodegradable polymeric micelles for targeted and controlled anticancer drug delivery: promises, progress and prospects. Nano Today. 2012;7(7):467–480. | ||
Yang L, Fawcett JP, Østergaard J, Zhang H, Tucker IG. Mechanistic studies of the effect of bile salts on rhodamine 123 uptake into RBE4 cells. Mol Pharm. 2012;9(1):29–36. | ||
Coyne CB, Ribeiro CM, Boucher RC, Johnson LG. Acute mechanism of medium chain fatty acid-induced enhancement of airway epithelial permeability. J Pharmacol Exp Ther. 2003;305(2):440–450. | ||
Desai N, Trieu V, Yang A, et al. Enhanced efficacy and safety of nanoparticle albumin-bound nab-docetaxel versus taxotere. Cancer Res. 2006;66(8 Suppl):1277–1278. | ||
Gong Y, Zhang X, Zhang Y, et al. Bile acids, carriers of hepatoma-targeted drugs? Pharmazie. 2016;71(3):139–145. |
Supplementary materials
 | Figure S1 Cumulative PTX-release profile. |
 | Figure S2 Fluorescence microscopy images of bone-marrow macrophage cells and A549 cells at 4 hours after incubation with FITC-Ms and FITC-CMs. |
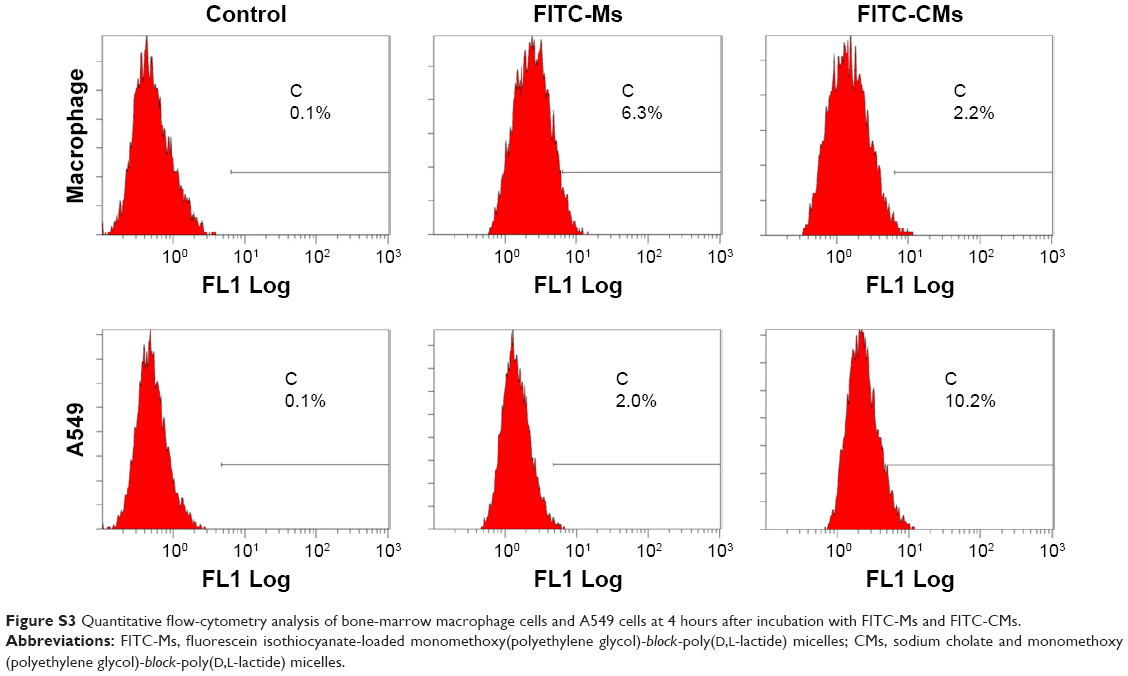 | Figure S3 Quantitative flow-cytometry analysis of bone-marrow macrophage cells and A549 cells at 4 hours after incubation with FITC-Ms and FITC-CMs. |
 | Figure S4 Antitumor effect of PTX-CMs in A549 late-stage tumor model. |
 | Table S1 Hematological test of whole blood collected from healthy ICR mice treated with single injections of PBS and PTX-CMs at PTX dose of 300 mg/kg (n=10) |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.