Back to Journals » International Journal of Nanomedicine » Volume 13
Smart polymeric nanoparticles with pH-responsive and PEG-detachable properties for co-delivering paclitaxel and survivin siRNA to enhance antitumor outcomes
Authors Jin M ![]() , Jin G, Kang L, Chen L, Gao Z
, Jin G, Kang L, Chen L, Gao Z ![]() , Huang W
, Huang W
Received 3 January 2018
Accepted for publication 17 February 2018
Published 20 April 2018 Volume 2018:13 Pages 2405—2426
DOI https://doi.org/10.2147/IJN.S161426
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Linlin Sun
Mingji Jin,1 Guangming Jin,2 Lin Kang,1 Liqing Chen,1 Zhonggao Gao,1 Wei Huang1
1State Key Laboratory of Bioactive Substance and Function of Natural Medicines, Institute of Materia Medica, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing, China; 2Department of Diagnostic Radiology 2, Yanbian University Hospital, Yanji, Jilin, China
Background: The co-delivery of chemotherapeutic agents and small interfering RNA (siRNA) within one cargo can enhance the anticancer outcomes through its synergistic therapeutic effects.
Materials and methods: We prepared smart polymeric nanoparticles (NPs) with pH-responsive and poly(ethylene glycol) (PEG)-detachable properties to systemically co-deliver paclitaxel (PTX) and siRNA against survivin gene for lung cancer therapy. The cationic polyethyleneimine-block-polylactic acid (PEI-PLA) was first synthesized and characterized, with good biocompatibility. PTX was encapsulated into the hydrophobic core of the PEI-PLA polymers by dialysis, and then the survivin siRNA was loaded onto the PTX-loaded NPs (PEI-PLA/PTX) through electrostatic interaction between siRNA and PEI block. Finally, the negatively charged poly(ethylene glycol)-block-poly(l-aspartic acid sodium salt) (PEG-PAsp) was coated onto the surface of NPs by electrostatic interaction to form final smart polymeric NPs with mean particle size of 82.4 nm and zeta potential of 4.1 mV. After uptake of NPs by tumor cells, the PEG-PAsp segments became electrically neutral owing to the lower endosome pH and consequently detached from the smart NPs. This process allowed endosomal escape of the NPs through the proton-sponge effect of the exposed PEI moiety.
Results: The resulting NPs achieved drug loading of 6.04 wt% and exhibited good dispersibility within 24 h in 10% fetal bovine serum (FBS). At pH 5.5, the NPs presented better drug release and cellular uptake than at pH 7.4. The NPs with survivin siRNA effectively knocked down the expression of survivin mRNA and protein owing to enhanced cell uptake of NPs. Cell counting kit-8 (CCK-8) assay showed that the NPs presented low systemic toxicity and improved antiproliferation effect of PTX on A549 cells. Moreover, in vivo studies demonstrated that accumulated NPs in the tumor site were capable of inhibiting the tumor growth and extending the survival rate of the mice by silencing the survivin gene and delivering PTX into tumor cells simultaneously.
Conclusion: These results indicate that the prepared nano-vectors could be a promising co-delivery system for novel chemo/gene combination therapy.
Keywords: PEG detachable, co-delivery, survivin siRNA, paclitaxel, pH responsive
Introduction
Combination therapy with an anticancer drug and siRNA has been suggested to be an effective and synergistic strategy for cancer treatment with the advantages of enhanced therapeutic effects as well as improved quality of life for patients.1 To produce a maximal effect, the anticancer drug and siRNA should be delivered simultaneously into the same tumor cells after systemic administration.2 Moreover, co-delivery of the drug and siRNA into the same tumor cells achieves an additive therapeutic effect by using a lower dose of the drug and thereby reducing many serious side effects.2–4 However, there are some challenges regarding co-loading as well as the overall efficiency of using an anticancer drug and siRNA. For example, most anticancer drugs are hydrophobic, nonspecific, and toxic to healthy tissue,5–7 while siRNA on its own is rather ineffective because of its high molecular weight, hydrophilicity, and anionic characteristics, which impede penetration of the cell membrane.8,9 Furthermore, when siRNA is exposed to plasma or other media full of protein, it becomes unstable and is quickly degraded by nucleases, resulting in poor transfection efficiency.10 Therefore, development of an efficient drug delivery system that could effectively co-load as well as release the anticancer drug and siRNA has become the focus of research.
Amphiphilic copolymers have received great attention for the co-delivery of anticancer drug and siRNA in recent years because of their advantages, such as efficient drug encapsulation, enhanced drug accumulation in tumor tissue, maximized therapeutic efficacy, and minimized adverse effects.11–13 Owing to their tuneability, biocompatibility, and high transfection efficacy of siRNA, polymeric nanoparticles (NPs) are the preferred drug-loading (DL) formulations for co-delivery of anticancer drugs and siRNA.7 On one hand, these co-loaded polymeric NPs can enhance the chemotherapeutic drug effect by the addition of siRNA;14 on the other hand, the siRNA has the ability to specifically silence or inhibit target proteins that are crucial for modulating cellular pathways in cancer, thereby inducing a dual inhibitory effect on tumor cell proliferation.15
Survivin is an antiapoptotic protein that is overexpressed in many kinds of cancer tissue including non-small cell lung cancer (NSCLC).16,17 Previous studies have reported that inhibition of survivin significantly increased the cytotoxicity of paclitaxel (PTX).18,19 In this study, a novel pH-responsive polymeric drug delivery system composed of polyethyleneimine-block-polylactic acid (PEI-PLA) and poly(ethylene glycol)-block-poly(L-aspartic acid sodium salt) (PEG-PAsp) was designed to co-deliver PTX and survivin siRNA in NSCLC. Branched polyethyleneimine (PEI) has been considered as the gold-standard vector in cationic polymer-based gene delivery because of its high transfection efficiency and endosome-escape mechanism via the “proton sponge effect.”20,21 The hydrophobic PLA block in the diblock polymer was able to provide increased colloidal stability by localizing the inner layer of the NPs and enhancing cell interaction as well as tissue permeability of the delivery platform.22–24 Accordingly, in our study, the cationic amphiphilic PEI-PLA copolymer was synthesized and self-assembled into a hybrid micelle structure, in which the PTX was encapsulated into the hydrophobic core, and the siRNA was then condensed with the cationic PEI block in the hydrophilic shell, forming co-loaded cationic NPs (Figure 1A). In general, cationic NPs are considered to have a high cellular uptake rate, and the unique structure of cationic NPs makes it convenient to load the anticancer drug and siRNA simultaneously.9 However, positively charged NPs have disadvantages compared to neutral NPs. For example, they may interact with the anionic plasma membrane of cells and can be easily absorbed by systemic serum proteins.25 The attachment of PEG chains to the surface of NPs can improve the stability and decrease the toxicity of the nano-complex by shielding the positive charge.26 But it has been reported that the presence of PEG may have disadvantages in controlling intracellular trafficking of cellular uptake and endosomal escape owing to steric hindrance, which is called the “PEG dilemma.”27,28 Therefore, new approaches are needed to overcome this problem. PEG-PAsp is a kind of anionic polymer whose aspartic acid moiety is negatively charged at pH 7.4. But at pH below 6.0, the aspartic acid moiety will become neutral.22,25,29 Hence, to take advantage of this characteristic in our delivery system, we attached PEG-PAsp copolymers onto the surface of cationic PEI-PLA NPs that had been preloaded with PTX and siRNA, yielding a layer-by-layer delivery system. Such PEG modification had been reported as an effective method for improving the stability of the nano-complex delivery system.30 Once they enter tumor cells via the enhanced permeation and retention (EPR) effect, the PEG-PAsp blocks become neutral in endosomes (pH 5.0–6.0) and therefore detach from the complex NPs (Figure 1B). This approach not only induces the proton sponge effect of PEI (the PEI component promotes endosomal/lysosomal escape, which allows siRNA to migrate into the cytoplasm for gene silencing), but also avoids the PEG dilemma caused by PEG. As a result, tumor cells are controlled under the combined action of chemotherapeutic drugs and siRNA silencing.
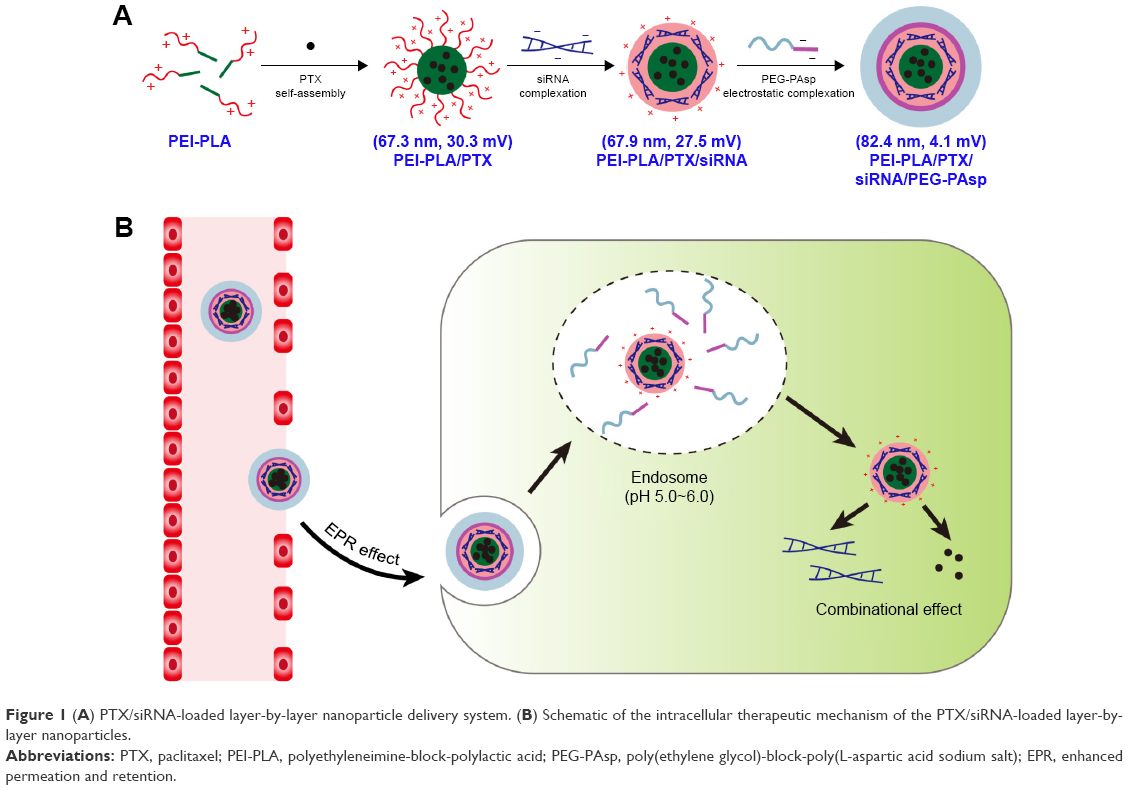 | Figure 1 (A) PTX/siRNA-loaded layer-by-layer nanoparticle delivery system. (B) Schematic of the intracellular therapeutic mechanism of the PTX/siRNA-loaded layer-by-layer nanoparticles. |
Materials and methods
Materials
Poly(lactic acid) with carboxyl groups on one end (PLA-COOH, MW=5–15 kDa) was purchased from Jinan Daigang Technology Co (Shandong, China). Branched polyethyleneimine (MW=1.8 kDa, bPEI1.8k) was purchased from Alfa Aesar (Ward Hill, MA, USA). Methoxyl-PEG-PAsp (MW=6.4 kDa) was purchased from Alamanda Polymers (Huntsville, AL, USA). 1-(3-Dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (EDC) and N-hydroxysuccinimide (NHS) were obtained from Sigma-Aldrich (Shanghai, China). RNase A was purchased from Solarbio (Beijing, China). DAPI and Hoechst 33258 were bought from the Beyotime Institute of Biotechnology (Jiangsu, China). The fluorescein isothiocyanate (FITC)-annexin V/propidium iodide (PI) apoptosis detection kit was purchased from KeyGEN Biosciences Company (Nanjing, China). Matrigel was purchased from Corning (NY, USA). Oregon Green® 488-conjugated paclitaxel (OG-PTX) was purchased from Life Technology (Thermo Scientific, Beijing, China). PTX was purchased from Meilun Bio Company (Dalian, China). Trypsin and RPMI 1640 media were obtained from Thermo Fisher Scientific Co. (Beijing, China). Cell counting kit-8 (CCK-8) was obtained from Dojindo Laboratories (Kumamoto, Japan). siRNA, targeting survivin (siSur): 5′-GCAUUCGUCCGGUUGCGCUTT-3′ (sense), and negative control siRNA (siNC): 5′-UUCUCCGAACGUGUCACGUTT-3′ (sense), Cy3 siRNA, and Cy5 siRNA were purchased from GenePharma Co. (Shanghai, China). All other reagents were of analytical grade.
The 4T1 cells, A549 and A549Luc cell lines were acquired from the Department of Pathology in the Institute of Medicinal Biotechnology at Peking Union Medical College and cultured in RPMI 1640 medium supplemented with 10% fetal bovine serum (FBS) at 37°C in a humidified atmosphere containing 5% CO2.
Male BALB/c nude mice (4–6 weeks old, 18–22 g) were acquired from Vital River Laboratory Animal Technology Co. (Beijing, China). All animal experiments were approved by the Laboratory Animal Ethics Committee in the Institute of Materia Medica in Peking Union Medical College. All the experimental procedures were performed in conformity with institutional guidelines and protocols for the care and use of laboratory animals.
Synthesis and characterization of PEI-PLA copolymer
The conjugation of PLA-COOH and bPEI1.8k was synthesized as previously reported.31,32 In brief, 1,300 mg of PLA-COOH (1 mmol) was dissolved in DMSO, then EDC (958.5 mg, 5 mmol) and NHS (575.4 mg, 5 mmol) were added and stirred at room temperature for 2 h. bPEI1.8k (360–1,800 mg, 0.2–1.0 mmol) was added into the DMSO solution and stirred for another 24 h at room temperature. The reacted mixture was dialyzed against 50% alcohol and distilled water at room temperature for 2 days to remove extra products. The PEI-PLA copolymer was obtained after freeze-drying of the solution. The structures and grafting degrees of PEI-PLA were characterized by 1H-nuclear magnetic resonance (NMR) spectroscopy (Varian Mercury-600 MHz spectrometer; Varian Medical Systems, Palo Alto, CA, USA) using D2O as a solvent, and further confirmed by Fourier transform infrared spectroscopy (Nicolet 5700; Thermo Fisher Scientific, Beijing, China).
Preparation of PTX-loaded cationic NPs
The PTX-loaded NPs (PEI-PLA/PTX) were prepared by a dialysis method.33 PTX (5 mg) and various grafting degrees of PEI-PLA (50 mg) were dissolved in DMSO (1 mL) and added dropwise to 25 mL of water under stirring. The mixture was stirred for another 30 min at room temperature and dialyzed against distilled water using a 7 kDa dialysis bag for 24 h. The unentrapped PTX was removed by filtration through a 0.45 μm filter (GE Healthcare, Beijing, China) and freeze-dried.34 The following equations were used to calculate the DL efficiency and encapsulation efficiency (EE). The concentration of PTX was detected by an Agilent 1200 LC (Agilent Tech, CA, USA) high-performance liquid chromatography (HPLC) system using a Diamonsil C18 column (5 μm, 4.6×250 mm). The mobile phase consisted of a mixture of methyl alcohol, water, and acetonitrile (23:41:36, v/v) delivered at a flow rate of 1.0 mL/min. The injection volume was 20 μL and the wavelength was set at 227 nm.
|
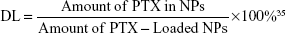
|
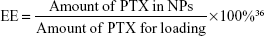
Preparation of PTX/siRNA-loaded cationic NPs
For the preparation of PEI-PLA/siRNA NPs, PEI-PLA was diluted with distilled water at different N/P ratios (N/P ratio: molar ratio of PEI-PLA nitrogen to siRNA phosphorus) and mixed with an equal volume of siRNA solution (concentration of 2 pmol/μL). The mixture was vortexed for 5 s and then kept at room temperature for 20 min to form the PEI-PLA/siRNA NPs. The PTX/siRNA-loaded NPs (PEI-PLA/PTX/siRNA) were prepared in the same way (N/P=30).
PEG-PAsp coating of PTX/siRNA-loaded NPs
PEG-PAsp was diluted with distilled water at various concentrations and added to the above prepared solutions including PTX-loaded, siRNA-loaded, and PTX/siRNA-loaded cationic NPs, separately. After 20 min, various complexes of PEI-PLA/PTX/PEG-PAsp, PEI-PLA/siRNA/PEG-PAsp, and PEI-PLA/PTX/siRNA/PEG-PAsp were formed at different C/N ratios (C/N ratio: molar ratio of carboxyl groups of PEG-PAsp to amino groups of PEI-PLA).
Characterization of the co-loaded NPs
The particle sizes and zeta potentials of the prepared cationic NPs and siRNA complexes with or without PEG-PAsp coating were measured at 25°C using a Malvern Zetasizer Nano ZS90 (Malvern Instruments, Malvern, UK). For the pH-sensitive analysis, the co-loaded NPs were kept in 10 nM HEPES buffer of different pH values. Furthermore, particle sizes, zeta potentials, and PTX contents of PEI-PLA/PTX/siRNA NPs were evaluated to screen the best grafting degree of PLA in the PEI-PLA copolymer.
The morphology of prepared NPs was characterized by transmission electron microscopy (TEM) (Hitachi H-7650; Hitachi, Tokyo, Japan) at a voltage of 80 kV. For TEM images, the suspension of PEI-PLA/PTX/siRNA/PEG-PAsp with a concentration of 0.1 mg/mL was applied dropwise onto a 400-mesh copper grid coated with carbon and negatively stained with 1% w/v phosphotungstic acid (PTA) solution.
The association of siRNA with different N/P ratios was evaluated by the gel retardation assay on 4% agarose gel, and electrophoresis was performed at 120 V for 20 min. Subsequently, the gel was stained with 0.5 mg/mL EtBr for 30 min and photographed under a ultraviolet imaging system (SIM135A; SIMON, Los Angeles, CA, USA).
To evaluate the serum stability of NPs, different formulations (N/P=30, C/N=1/5; PTX content: 6.04%) were prepared and incubated at 37°C in PBS supplemented with 10% FBS. The average particle size of the NPs was monitored by dynamic light scattering (DLS) over a period of 24 h. Each sample was performed in triplicate.
In vitro pH-sensitive drug release of PTX
The in vitro release of PTX in different formulations was evaluated using a dialysis diffusion technique. siRNA-loaded NPs without PEG-PAsp coating were formed at a fixed N/P ratio of 30, and those with PEG-PAsp coating at an N/P of 30 and a C/N of 1/5. Each sample of NPs (0.5 mL) containing 0.3 mg PTX was added into a dialysis bag (7 kDa) and tightly sealed. Then, the bags were immersed in 40 mL PBS (pH 7.4, containing 0.1% [v/v] Tween 80) or sodium acetate-buffered solution (pH 5.5, containing 0.1% [v/v] Tween 80), incubated in an orbital shaker at 37°C. At predetermined time points, 0.2 mL of each sample was withdrawn from the medium and the same volume of fresh medium was added. Each sample was centrifuged at 10,000 rpm and the supernatant was assayed by HPLC under the same conditions as described in “Preparation of PTX-loaded cationic NPs.”
Cellular uptake studies
To determine the uptake and localization of the NPs, Cy3 siRNA and Oregon Green-PTX were used in this study. A549 cells were seeded into a 12-well plate at a density of 5×104 cells per well and incubated at 37°C to allow for cell attachment. After 24 h, the medium was replaced with serum-free cell culture medium containing various formulations of NPs (N/P=30, C/N=1/5; Oregon Green-PTX content: 200 ng/mL) with 50 nM siRNA per well for 4 h. For the flow cytometry analysis, the cells were washed three times with PBS buffer and trypsinized, and the harvested cells were resuspended in fresh medium and washed with cold PBS. Finally, the cells were resuspended in 0.5 mL PBS buffer and analyzed with a FACSCalibur flow cytometer (Becton Dickinson, Franklin Lake, NJ, USA). For confocal laser scanning microscopy (CLSM), the cells were washed three times with cold PBS and fixed with 4% paraformaldehyde in PBS for 15 min. Then, the cell nuclei were stained with DAPI and imaged using a confocal fluorescence microscope (Carl Zeiss LSM 710; Carl Zeiss Microscopy, Jena, Germany).
In vitro gene silencing efficiency assay
To test the siRNA silencing efficacy on survivin expression, A549 cells were incubated in a six-well plate at a density of 1×105 cells per well for 24 h. Then, the cells were incubated in 2 mL of serum-free RPMI 1640 medium containing different formulations (N/P=30, C/N=1/5; PTX content: 6.04%) for 4 h. The final siRNA concentration was 100 nM in each well. After 4 h incubation, the transfection medium in each well was replaced by fresh medium containing 10% FBS and was incubated for another 20 h (for mRNA isolation) or 68 h (for protein extraction).37,38 To avoid cytotoxicity of PTX, the concentration of PTX in the NPs was as low as 2 nM,39 and all the results were presented as mean±SD of three measurements.
In reverse transcription polymerase chain reaction (RT-PCR) analysis, total RNA was extracted from the cells with TRIzol Reagent (Invitrogen, Carlsbad city, CA, USA). Total RNA was reverse transcribed to cDNA using the ReverAid First Strand cDNA Synthesis kit (Fermentas, Burlington, Ontario, Canada), and the mRNA levels of the target genes were quantified via real-time PCR using the SYBR Green qPCR kit (Takara Biotechnology Co., Dalian, China) along with selected DNA primer pairs. Primers used for the RT-PCR experiment were as follows: survivin-forward, 5′-AAACCAGACCCTCATGGCTAC-3′; survivin-reverse, 5′-TCCCAGACTCCACTCCAACTT-3′; GAPDH-forward, 5′-TGAAGGTCGGAGTCAACGG-3′; GAPDH-reverse, 5′-TCCTGGAAGATGGTGATGGG-3′.
For Western blot analysis, the transfected cells were washed twice with cold PBS, and then harvested using the lysis buffer (50 nM Tris-HCl pH=6.8, 2% sodium dodecyl sulfate [SDS], 6% glycerol, 1% β-mercapitalethanol, and 0.04% bromophenol blue). The cell lysates were incubated for 30 min at 4°C and then clarified by centrifugation for 15 min at 12,000× g. The protein concentration was determined using the BCA Protein Assay Kit (Lot: PA115-01; Tiangen Biotech (Beijing) Co., Beijing, China). Protein was separated by SDS polyacrylamide gel and transfected to a polyvinylidene difluoride membrane. Then, the membranes were incubated with β-actin (sc-47778; Santa Cruz Biotechnology, Shanghai, China) and survivin antibodies (1:1,000; Cell Signaling Technologies, Manhattan, NY, USA; 2808s) overnight at 4°C. After incubation with goat anti-rat immunoglobulin G (IgG) antibody for 1 h, bands were detected by the ECL system (ImageQuant LAS 4000 mini; Fuji, Tokyo, Japan).
In vitro cytotoxicity
CCK-8 assays on 4T1 cells and A549 cells were performed to evaluate the polymer toxicity. In brief, the cells were seeded in 96-well plates at a density of 3×103 cells per well and incubated for 24 h to allow for cell attachment. The cells were then treated with NPs as follows: (1) blank PEI-PLA NPs complexed with scrambled siRNA (siRNANC) of different N/P ratios and (2) blank PEI-PLA NPs complexed with siRNANC and then coated with different C/N ratios of PEG-PAsp. The polyplexes were added to the cells and incubated for 72 h. The final siRNA concentration was 20 nM in each well, and the OD was measured at 450 nm using the Synergy H1m Monochromator-Based Multi-Mode Microplate Reader (BioTek, Dallas, TX, USA). Untreated cells served as controls with 100% viability. The results are presented as the mean±SD of four measurements.
To study the cytotoxicity of PTX, PEI-PLA/PTX/siRNANC/PEG-PAsp, or PEI-PLA/PTX/siRNA/PEG-PAsp, A549 cells were seeded at 5×103 cells per well plate. After overnight incubation, the medium was replaced with fresh medium (pH 7.4/pH 5.5) containing different concentrations of PTX, PEI-PLA/PTX/siRNANC/PEG-PAsp, and PEI-PLA/PTX/siRNA/PEG-PAsp (N/P=30, C/N=1/5; PTX content: 6.04%). Cells treated with PBS, PEI-PLA/PEG-PAsp, PEI-PLA/siRNANC/PEG-PAsp, or PEI-PLA/siRNA/PEG-PAsp were used as controls (N/P=30, C/N=1/5). The final siRNA concentration was 20 nM in each well and the results are presented as the mean±SD of four measurements. To study the synergistic effect of PTX and survivin siRNA in the NPs, A549 cells were incubated with PEI-PLA/PTX/siRNANC/PEG-PAsp and PEI-PLA/siRNA/PEG-PAsp with different concentrations of PTX (0.004–0.128 μg/mL) or siRNA (0.041–1.320 μg/mL) for 48 h. Also, the co-loaded NPs of PEI-PLA/PTX/siRNA/PEG-PAsp at a fixed ratio (PTX/siRNA=1/10, w/w) were incubated with the cells at various concentrations based on PTX (0.004–0.128 μg/mL) for 48 h. The results are presented as the mean±SD of four measurements, and and the OD was measured at 450 nm using the Synergy H1m Monochromator-Based Multi-Mode Microplate Reader (BioTek, USA). Untreated cells served as controls with 100% viability.
Cell apoptosis and cell cycle analysis
A549 cells were seeded into a 12-well plate at a density of 5×104 cells per well, treated with PBS, siRNA, PTX, PEI-PLA/PEG-PAsp, PEI-PLA/siRNA/PEG-PAsp, PEI-PLA/PTX/siRNANC/PEG-PAsp, or PEI-PLA/PTX/siRNA/PEG-PAsp (N/P=30, C/N=1/5; PTX content: 6.04%; 50 nM of siRNA per well). The cell apoptosis study was performed 48 h after the administration of drugs, and the cell cycle analysis was performed 2 h after the administration of drugs. Cells without treatment were used as controls. For the Hoechst staining assay, cells were fixed with 4% paraformaldehyde for 15 min, then stained with Hoechst 33258 reagent at a concentration of 5 μg/mL for 10 min and washed with PBS. After drying, the nuclear morphology was observed with a confocal fluorescence microscope (Carl Zeiss LSM 710; Carl Zeiss Microscopy, Germany). For quantitative measurement of apoptosis, cells were harvested with 0.25% trypsin without EDTA, washed with PBS, resuspended in binding buffer, and stained with annexin V-FITC/PI for 15 min. To evaluate the cell cycle arrest induced by PTX-contained complex NPs, the treated cells were collected, washed, and fixed with 70% ethanol for 24 h at −20°C. Then, the cells were resuspended in PBS containing 100 μg/mL RNase A and incubated at 37°C for 30 min. PI was added (50 μg/mL) and the cells were incubated for another 1 h at room temperature. The cell apoptosis and cell cycle were analyzed by a FACSCalibur flow cytometer.
In vivo distribution
PEI-PLA/PEG-PAsp (N/P=30, C/N=1/5) mixed NPs were used in this experiment. The Luc-labeled lung cancer A549 (A549Luc) cells were suspended in BD Matrigel, and the male BALB/c nude mice (4–6 weeks old, 18–22 g) in each group were subcutaneously implanted with 2×106 cells in the right oxter to establish the subcutaneous tumor model. The tumor volume was calculated as ab2/2 (a: the length of the tumor; b: the width of the tumor). When the tumor volume reached around 200 mm3, mice were randomly divided into four groups (n=3 in each group): DIR, Cy5 siRNA (siRNACy5), PEI-PLA/DIR/siRNACy5, and PEI-PLA/DIR/siRNACy5/PEG-PAsp. Each group of mice was administered doses of DIR, Cy5 siRNA, DIR, and Cy5 siRNA co-loaded NPs (DIR=50 μg/kg, siRNACy5=2 mg/kg) via the tail vein. After 24 h, mice were killed by cervical dislocation, and the tumors and major organs (heart, liver, spleen, lung, and kidney) were isolated and imaged using Living Image Software (version 4.4; Caliper Life Sciences, Hopkinton, MA, USA).
In vivo anticancer efficacy
The A549Luc tumor-bearing BALB/c nude mice (4–6 weeks old, 18–22 g) model was established as described in the previous subsection. When the tumor volume reached around 200 mm3, mice were injected with one of the following eight preparations: saline, PEI-PLA/PEG-PAsp, Taxol®, PEI-PLA/siRNA/PEG-PAsp, PEI-PLA/PTX/siRNANC/PEG-PAsp, Taxol+PEI-PLA/siRNA/PEG-PAsp, PEI-PLA/PTX/siRNA, or PEI-PLA/PTX/siRNA/PEG-PAsp (N/P=30, C/N=1/5; PTX content: 6.04%). PTX was administered at a dose of 7.5 mg/kg, and survivin siRNA was administered at a dose of 3 mg/kg. All the formulations were given to mice via the tail vein every 3 days, four times, and the tumor volumes were measured every 2 days (n=5).
Another 48 tumor-bearing mice were grouped and administered with various drug formulations according to the same method (eight groups, six mice per group), and then carefully maintained for survival analysis.
Detection of survivin expression in tumor tissues
At the end of the in vivo anticancer experiments, animals were killed, and all the tumors were separated from the mice and photographed. The separated tumors were used for three purposes: survivin gene downregulation analysis, survivin protein content analysis, and immunohistochemical analysis. For the fluorescent quantitative PCR, total RNA was extracted from the tumor tissue with TRIzol Reagent (Invitrogen), treated, and evaluated as described in the “In vitro gene silencing efficiency assay” section. The content of survivin protein in tumor tissue was measured with an enzyme-linked immunosorbent assay (ELISA) kit for survivin (Cloud-Clone Corp., Katy, TX, USA). For the immunohistochemical analysis, the tumor sections were fixed with 4% neutral-buffered formaldehyde and treated with xylene, alcohol, and PBS. Then, the samples were incubated with a monoclonal rabbit polyclonal anti-survivin antibody (1:250; Abcam, Cambridge, UK) at 4°C overnight. The samples were then incubated with a secondary antibody at 1:1,000 (goat anti-rabbit IgG-HRP; Cell Signaling Technologies) for 45 min at room temperature. The sections were visualized and photographed under a light microscope.
Statistical analysis
We used SPSS 13.0 for the statistical analyses, and the results are presented as mean±SD. Statistical comparisons were performed to determine group differences through analysis of variance (ANOVA). The difference between two groups was evaluated by the Student’s t-test. Comparison among multiple groups was performed by one-way ANOVA with Bonferroni’s post hoc test. Asterisks indicate statistical significance as follows: **p<0.05, ***p<0.001.
Results
Synthesis and characterization of PEI-PLA copolymer
The PEI-PLA copolymer was synthesized by an amino reaction between the carboxyl group of PLA-COOH and amino groups of PEI (Figure 2A). In the 1H-NMR spectrum (Figure 2B), the peak of PEI appeared at around 2.6 ppm.20 In PEI-PLA, a new broad peak appeared at 2.3–3.4 ppm, which is attributed to the presence of protons of methylene (−CH2CH2−) in PEI. The signals at δ=1.20 ppm and δ=4.08 ppm correspond to the −CH3 and (−CH) proton in PLA block of PEI-PLA, respectively. The results indicate that the target molecule is successfully synthesized according to the reaction.
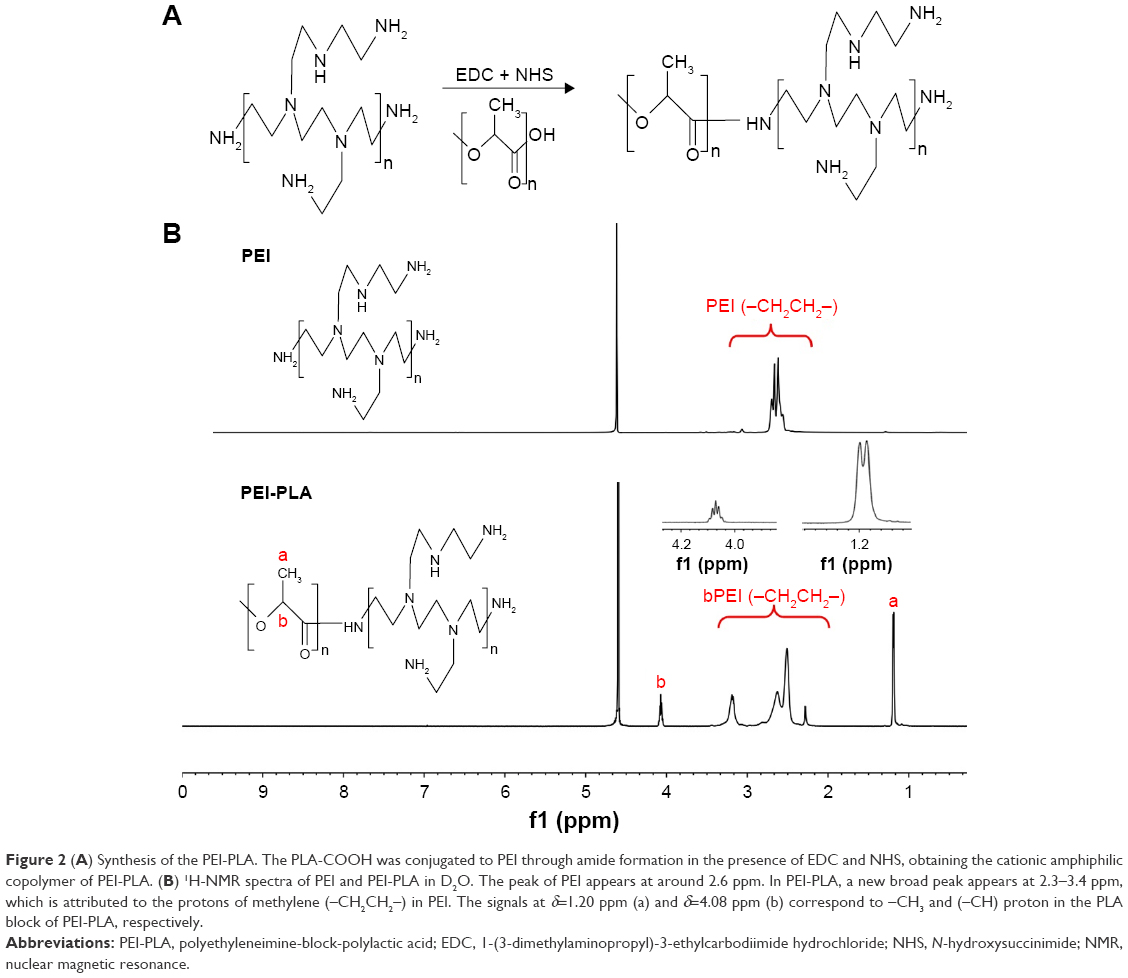 | Figure 2 (A) Synthesis of the PEI-PLA. The PLA-COOH was conjugated to PEI through amide formation in the presence of EDC and NHS, obtaining the cationic amphiphilic copolymer of PEI-PLA. (B) 1H-NMR spectra of PEI and PEI-PLA in D2O. The peak of PEI appears at around 2.6 ppm. In PEI-PLA, a new broad peak appears at 2.3–3.4 ppm, which is attributed to the protons of methylene (–CH2CH2–) in PEI. The signals at δ=1.20 ppm (a) and δ=4.08 ppm (b) correspond to –CH3 and (–CH) proton in the PLA block of PEI-PLA, respectively. |
Preparation and characterization of the NPs
As listed in Table 1, particle sizes and zeta potentials of PEI-PLA/PTX/siRNA NPs decreased with the increasing grafting degree of PLA, while the PTX content of the NPs increased with the increasing grafting degree of PLA. To attain the maximum drug loading, suitable particle size and zeta potential, it is important to determine the best grafting degree. Therefore, according to the results, we chose PEI-PLA with a grafting degree of 40% (67.3 nm, 30.3 mV, PTX content of 6.04%) as the final polymer.
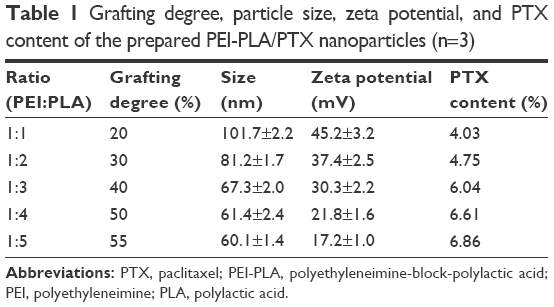 | Table 1 Grafting degree, particle size, zeta potential, and PTX content of the prepared PEI-PLA/PTX nanoparticles (n=3) |
As shown in Figure S1A, a decrease in N/P ratio was accompanied by enhanced particle size and reduced zeta potential. The particle size was greater than 100 nm when the N/P ratio was below 20. When the N/P ratio was 7.5 or higher, siRNA was completely condensed with PEI-PLA NPs (Figure S1C). The LC% and EE% of PTX in PEI-PLA/PTX NPs were 6.04% and 93.03%, respectively. As shown in Table 2, the zeta potential of PEI-PLA/PTX NPs decreased from 30.3 mV to 27.5 mV after being complexed with siRNA at an N/P ratio of 30. Further electrostatic coating of PEG-PAsp led to a decreased zeta potential of the PEI-PLA/PTX/siRNA/PEG-PAsp. But the electrostatic coating did not affect the gel retardation of the prepared NPs in the C/N range of 1/50 to 1/5 (Figure S1D). When the C/N ratio increased, there was a general increase in particle size but a decrease in zeta potential (Figure S1B). At the N/P ratio of 30 and C/N ratio of 1/5, the PEI-PLA/PTX/siRNA/PEG-PAsp NPs presented suitable size and zeta potential (82.4 nm, 4.10 mV) (Table 2, Figure 3A). The morphological images of PEI-PLA/PTX/siRNA/PEG-PAsp NPs from TEM (Figure 3B) further demonstrated that the NPs were spherical with smooth surfaces. The in vitro colloidal stability of the complex NPs was evaluated in the presence of PBS or PBS added with 10% FBS. The particle size of the PEI-PLA/PTX/siRNA in PBS without FBS changed from 94 nm to 243 nm, and that of PEI-PLA/PTX/siRNA in PBS with FBS changed from 96 nm to 242 nm (Figure S2). Differently from PEI-PLA/PTX/siRNA, the size of the PEI-PLA/PTX/siRNA/PEG-PAsp in this study showed no significant change when it was incubated in PBS, either with or without FBS.
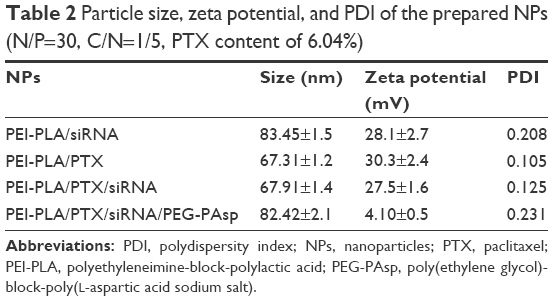 | Table 2 Particle size, zeta potential, and PDI of the prepared NPs (N/P=30, C/N=1/5, PTX content of 6.04%) |
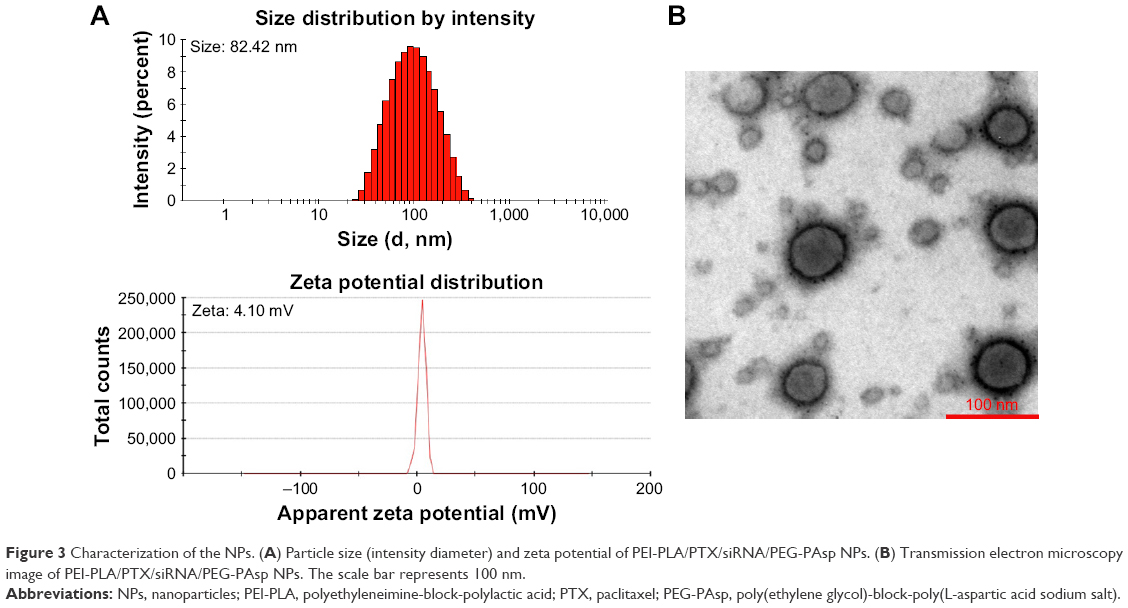 | Figure 3 Characterization of the NPs. (A) Particle size (intensity diameter) and zeta potential of PEI-PLA/PTX/siRNA/PEG-PAsp NPs. (B) Transmission electron microscopy image of PEI-PLA/PTX/siRNA/PEG-PAsp NPs. The scale bar represents 100 nm. |
pH responsiveness and in vitro release of PTX
As shown in Table 3, in the pH range 5.0–8.0, the lower the pH, the higher the zeta potential. This indicates that the PEG-PAsp copolymer was detached from the NPs and therefore exposed the cationic PEI moiety when the pH was below 6.0.
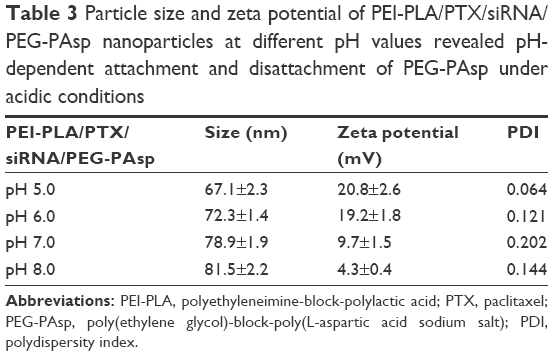 | Table 3 Particle size and zeta potential of PEI-PLA/PTX/siRNA/PEG-PAsp nanoparticles at different pH values revealed pH-dependent attachment and disattachment of PEG-PAsp under acidic conditions |
The pH-responsive release behavior of PTX from the NPs was investigated at pH 5.5 and 7.4, simulating the acidic endosome and the normal physiological environment, respectively. There was no significant difference in the sustained-release tendency of PTX from PEI-PLA/PTX/siRNA and PEI-PLA/PTX/siRNA/PEG-PAsp at either pH (Figure S3). But at pH 5.5, after 72 h, the release of PTX from the PEI-PLA/PTX/siRNA NPs and PEI-PLA/PTX/siRNA/PEG-PAsp NPs was about 86% and 75%, respectively, after 72 h of release at pH 5.5. In comparison, at pH 7.4, the cumulative release of PTX from the same NPs was 55% and 41%, respectively.
Cellular uptake studies
To exhibit a synergistic anticancer effect, the drug and siRNA encapsulated within the NPs must be released into the same cancer cells. To examine the intracellular efficiency, two-color flow cytometry was used to measure the cellular uptake of various NPs. The NPs in the experiment were prepared by replacing survivin siRNA with Cy3 siRNA, and PTX with Oregon Green-PTX. Cells without treatment were used as controls. The siRNA-loaded NPs of PEI-PLA/siRNA/PEG-PAsp, PEI-PLA/PTX/siRNA, and PEI-PLA/PTX/siRNA/PEG-PAsp delivered more siRNA into cells, with the percentages of siRNA-positive cells treated with these NPs being 98.69%, 99.16%, and 98.65%, respectively, compared to the free siRNA group which resulted in only 0.1% siRNA-positive cells (Figure 4A, b, c, f, g). The fact that the siRNA-loaded NPs showed much higher cellular uptake efficiency than naked siRNA indicates improved particle stability and enhanced cell membrane interaction caused by the PEI copolymer. Similarly, the PEI-PLA/PTX/PEG-PAsp, PEI-PLA/PTX/siRNA, and PEI-PLA/PTX/siRNA/PEG-PAsp NPs delivered more PTX into the cells, with the percentages of PTX-positive cells being 99.83%, 98.22%, and 94.24%, respectively, while only 16.51% PTX-positive cells were detected in the free PTX group (Figure 4A, d, e, f, g). These results further indicate that co-delivery of PTX and siRNA did not affect each other’s cellular uptake when compared with single-drug delivery.
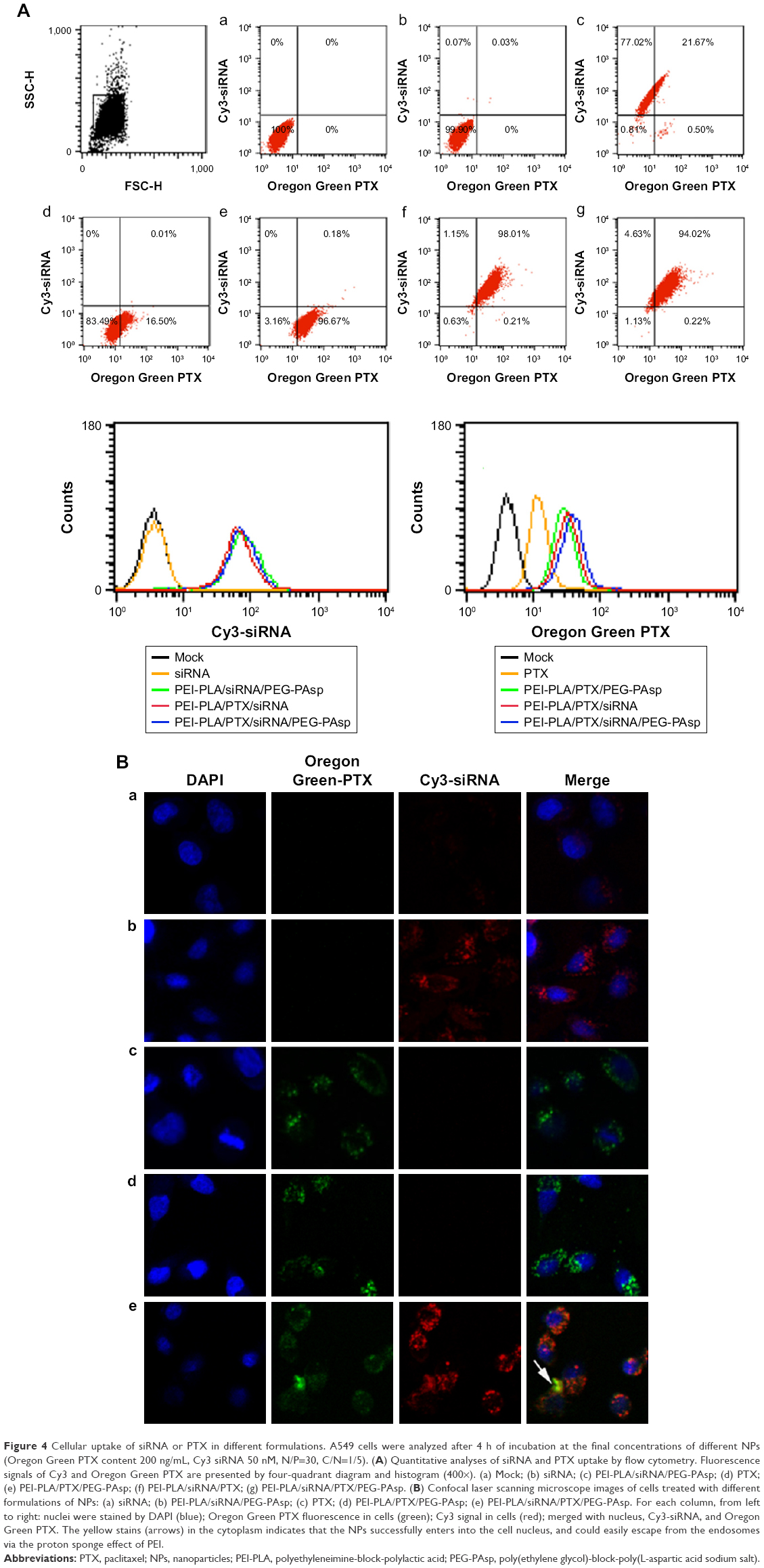 | Figure 4 Cellular uptake of siRNA or PTX in different formulations. A549 cells were analyzed after 4 h of incubation at the final concentrations of different NPs (Oregon Green PTX content 200 ng/mL, Cy3 siRNA 50 nM, N/P=30, C/N=1/5). (A) Quantitative analyses of siRNA and PTX uptake by flow cytometry. Fluorescence signals of Cy3 and Oregon Green PTX are presented by four-quadrant diagram and histogram (400×). (a) Mock; (b) siRNA; (c) PEI-PLA/siRNA/PEG-PAsp; (d) PTX; (e) PEI-PLA/PTX/PEG-PAsp; (f) PEI-PLA/siRNA/PTX; (g) PEI-PLA/siRNA/PTX/PEG-PAsp. (B) Confocal laser scanning microscope images of cells treated with different formulations of NPs: (a) siRNA; (b) PEI-PLA/siRNA/PEG-PAsp; (c) PTX; (d) PEI-PLA/PTX/PEG-PAsp; (e) PEI-PLA/siRNA/PTX/PEG-PAsp. For each column, from left to right: nuclei were stained by DAPI (blue); Oregon Green PTX fluorescence in cells (green); Cy3 signal in cells (red); merged with nucleus, Cy3-siRNA, and Oregon Green PTX. The yellow stains (arrows) in the cytoplasm indicates that the NPs successfully enters into the cell nucleus, and could easily escape from the endosomes via the proton sponge effect of PEI. |
Simultaneous delivery of PTX and siRNA was verified by confocal microscopy imaging after double-labeled (Cy3 siRNA with red, and Oregon Green-PTX with green) NPs in A549 cells. The cell nuclei were stained with DAPI (blue). As shown in Figure 4B, Cy3 siRNA in the cytoplasm of cells in both PEI-PLA/siRNA/PEG-PAsp and PEI-PLA/PTX/siRNA/PEG-PAsp groups was significantly increased compared with the siRNA group. In the PEI-PLA/PTX/siRNA/PEG-PAsp group, yellow stains in the cytoplasm (Figure 4B, e) suggest that the NPs entered cells most likely by endocytosis and subsequently escaped from the endosomes via the proton sponge effect of PEI in the acidic environment.
To investigate the detachment ability of PEG-PAsp in an acidic environment, we studied the cellular ability of PEI-PLA/PTX/siRNA/PEG-PAsp NPs in A549 tumor cells at different pHs using flow cytometry. A549 cells were treated with the NPs at pH 5.5 or 7.4 for 4 h in the serum-free medium. As shown in Figure S4, the cellular uptake of PTX and siRNA at pH 7.4 was 32.55% and 101.94%, respectively while the cellular uptake at pH 5.5 was 48.07% and 264.16%.
In vitro gene silencing efficiency and cytotoxicity study
Next, we investigated whether the NPs could effectively knockdown the expression of the therapeutic target gene survivin. We incubated A549 cells in different formulations for 24 h and then detected the survivin mRNA level using real-time PCR. The survivin protein in the cell lysates was also detected by Western blot analysis 48 h after transfection. The results showed that both mRNA (Figure 5A) and protein levels (Figure 5B and C) of survivin in A549 cells remarkably decreased (**p<0.05) when treated with PEI-PLA/siRNA/PEG-PAsp or PEI-PLA/PTX/siRNA/PEG-PAsp NPs. In contrast, treatment with siRNA, PTX, PEI-PLA/PEG-PAsp, or PEI-PLA/PTX/siRNANC/PEG-PAsp NPs failed to show any knockdown effect in A549 cells.
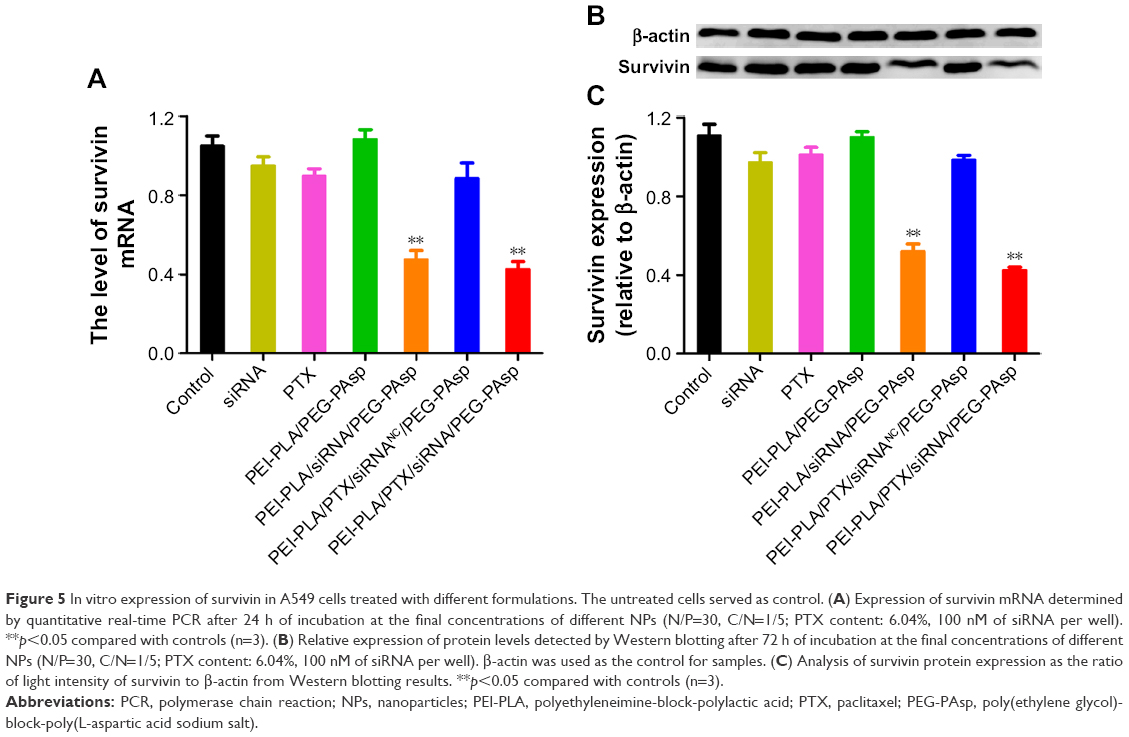 | Figure 5 In vitro expression of survivin in A549 cells treated with different formulations. The untreated cells served as control. (A) Expression of survivin mRNA determined by quantitative real-time PCR after 24 h of incubation at the final concentrations of different NPs (N/P=30, C/N=1/5; PTX content: 6.04%, 100 nM of siRNA per well). **p<0.05 compared with controls (n=3). (B) Relative expression of protein levels detected by Western blotting after 72 h of incubation at the final concentrations of different NPs (N/P=30, C/N=1/5; PTX content: 6.04%, 100 nM of siRNA per well). β-actin was used as the control for samples. (C) Analysis of survivin protein expression as the ratio of light intensity of survivin to β-actin from Western blotting results. **p<0.05 compared with controls (n=3). |
The cytotoxicity of blank NPs against 4T1 and A549 cells with different N/P and C/N ratios was evaluated to seek the safe concentration of nanocarriers. When the positively charged nonviral vectors are used to facilitate the cellular uptake of siRNA, toxicity from excessive positive charge becomes a critical issue. As different N/P ratios are associated with different charges, it is important to evaluate the cytotoxicity of NPs with different N/P ratios. The CCK-8 assay was used here to determine the cytotoxicity of siRNA-loaded NPs with or without PEG-PAsp coating. To make the data comparable, cytotoxic PTX was not encapsulated in the NPs and the concentration of scrambled siRNA was 20 nM. Figure S1 indicates that as the N/P ratio increased, the positive charge of NPs increased as well. Although the cytotoxicity was enhanced as the N/P ratio increased, cells transfected with PEI-PLA/siRNANC still retained a relatively high viability of about 70% (Figure S5A). The effect of PEG-PAsp coating on the cytotoxicity of siRNA-loaded PEI-PLA NPs (N/P=30) is shown in Figure S5B. With the increase in C/N ratio, the cytotoxicity of the system slightly decreased, and the cell viability reached 96% when the C/N ratio was 1/5.
After incubating the A549 cells overnight, the medium was replaced with fresh medium (pH 7.4 or pH 5.5) containing different formulations of NPs. The cells were incubated for another 48 h with various formulations at a fixed siRNA content (20 nM) and varying doses of PTX (0.004–0.128 μg/mL). As shown in Figure 6A, among the groups treated with PTX-free formulations, the cell inhibitory rate in the PEI-PLA/siRNA/PEG-PAsp treated group was remarkably higher than that of the PBS, PEI-PLA/PEG-PAsp or PEI-PLA/siRNANC/PEG-PAsp-treated group (**p<0.05). PTX and PTX-loaded NPs exhibited a dose-dependent cytotoxicity with increasing concentrations (Figure 6B). The cytotoxicity of PTX was significantly increased when the A549 cells were incubated with PTX-loaded NPs, compared with free PTX. This is probably due to the increased drug solubility and enhanced cellular uptake. At pH 7.4, the IC50 values of PEI-PLA/PTX/siRNA/PEG-PAsp (0.00368 μg/mL) and PEI-PLA/PTX/siRNANC/PEG-PAsp (0.01736 μg/mL) were much lower than that of free PTX (0.0522 μg/mL). Furthermore, the IC50 value of PEI-PLA/PTX/siRNA/PEG-PAsp at pH 5.5 was 0.00148 μg/mL, which was much lower than that at pH 7.4 (0.00368 μg/mL).
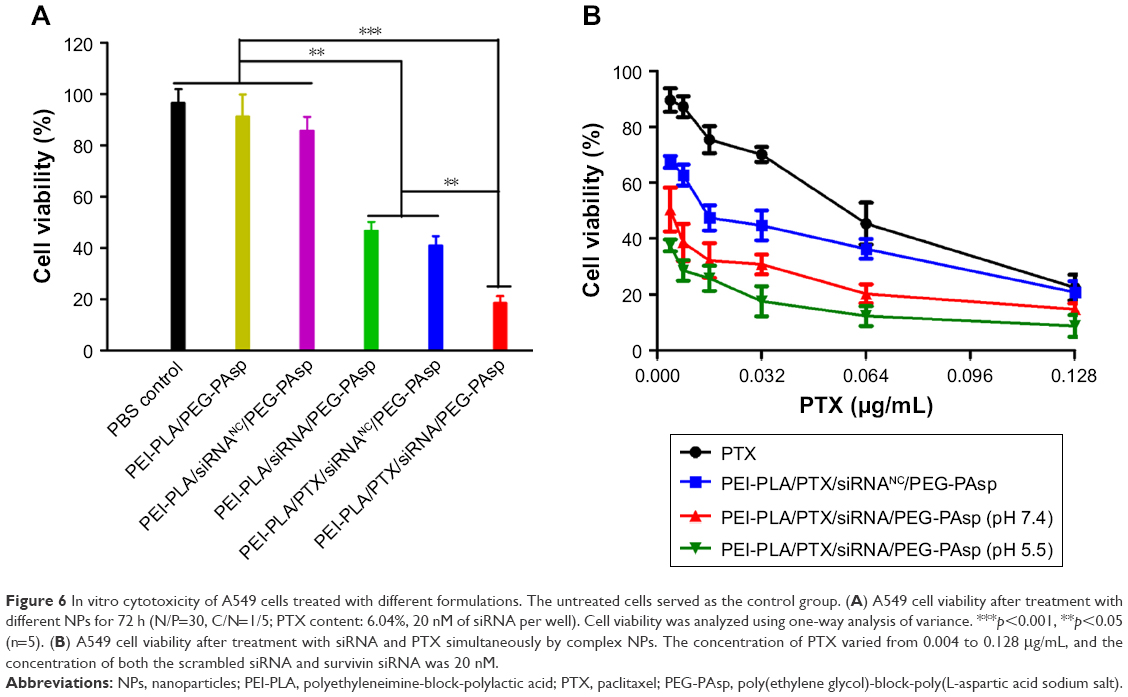 | Figure 6 In vitro cytotoxicity of A549 cells treated with different formulations. The untreated cells served as the control group. (A) A549 cell viability after treatment with different NPs for 72 h (N/P=30, C/N=1/5; PTX content: 6.04%, 20 nM of siRNA per well). Cell viability was analyzed using one-way analysis of variance. ***p<0.001, **p<0.05 (n=5). (B) A549 cell viability after treatment with siRNA and PTX simultaneously by complex NPs. The concentration of PTX varied from 0.004 to 0.128 μg/mL, and the concentration of both the scrambled siRNA and survivin siRNA was 20 nM. |
To confirm the synergistic effect of co-delivery of PTX and survivin siRNA, the combination index (CI) was calculated. As shown in Figure S6, PLA/PTX/siRNA/PEG-PAsp NPs exhibited CI values less than 1, derived from the effect of a range of PTX and siRNA concentrations in the A549 cell line, indicating a synergistic effect between PTX and survivin siRNA.
Cell apoptosis and cell cycle analyses
A549 cells were treated with different formulations for 48 h, and the untreated cells served as controls. As shown in Figure 7, cells treated with blank NPs (PEI-PLA/PEG-PAsp) showed 11.35% apoptosis, indicating that the blank polymeric delivery system produces a minor effect on normal cell progression compared with the drug-loaded groups. The cell apoptosis in siRNA- and PTX-treated groups was 13.07% and 14.53%, respectively, whereas PEI-PLA/siRNA/PEG-PAsp and PEI-PLA/PTX/siRNANC/PEG-PAsp-treated groups showed 51.73% and 69.67%, respectively. When we examined the nuclei of the cells after staining with Hoechst 33258, cells treated with naked siRNA or PTX displayed little or no apoptosis feature whereas the apoptotic effect significantly increased in both PEI-PLA/siRNA/PEG-PAsp and PEI-PLA/PTX/siRNANC/PEG-PAsp groups. Chromatin condensation, nuclear fragmentation, and chromosome abnormalities were found in the PEI-PLA/siRNA/PEG-PAsp and PEI-PLA/PTX/siRNANC/PEG-PAsp groups, indicating that the PEI-PLA/PEG-PAsp delivery system could effectively deliver and release siRNA and PTX to induce apoptosis, and sequentially inhibit cell proliferation. From the Hoechst staining test, we can see that much more nuclear fragmentation and chromosome abnormality appeared after treatment with PEI-PLA/PTX/siRNA/PEG-PAsp, and the apoptosis ratio was up to 80.81%. This is much higher than that of cells treated with either siRNA-loaded NPs (PEI-PLA/siRNA/PEG-PAsp, 51.73%) or PTX-loaded NPs (PEI-PLA/PTX/siRNANC/PEG-PAsp, 69.67%).
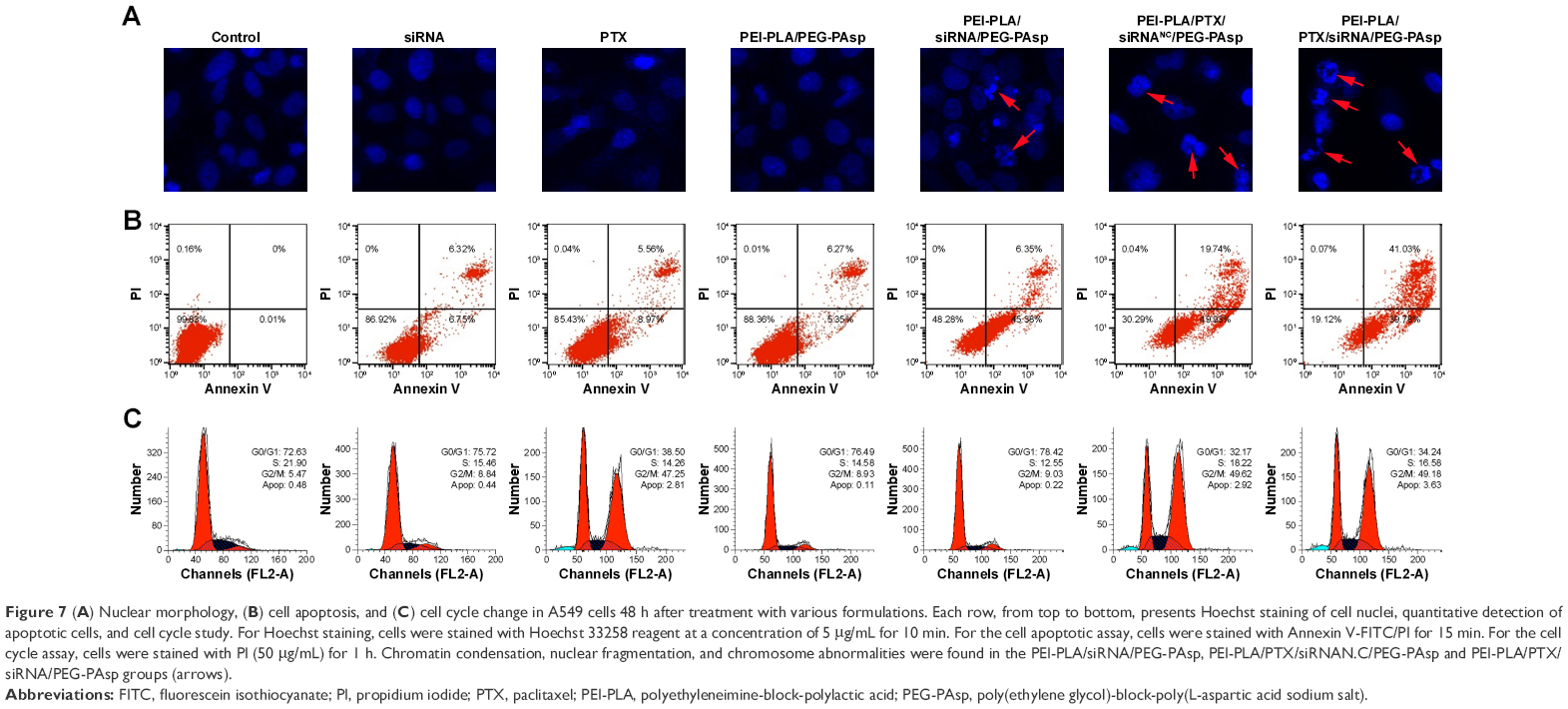 | Figure 7 (A) Nuclear morphology, (B) cell apoptosis, and (C) cell cycle change in A549 cells 48 h after treatment with various formulations. Each row, from top to bottom, presents Hoechst staining of cell nuclei, quantitative detection of apoptotic cells, and cell cycle study. For Hoechst staining, cells were stained with Hoechst 33258 reagent at a concentration of 5 μg/mL for 10 min. For the cell apoptotic assay, cells were stained with Annexin V-FITC/PI for 15 min. For the cell cycle assay, cells were stained with PI (50 μg/mL) for 1 h. Chromatin condensation, nuclear fragmentation, and chromosome abnormalities were found in the PEI-PLA/siRNA/PEG-PAsp, PEI-PLA/PTX/siRNAN.C/PEG-PAsp and PEI-PLA/PTX/siRNA/PEG-PAsp groups (arrows). |
It has been reported that PTX preferentially induces G2/M arrest of the cell cycle and thus causes apoptosis.40 Increased G2/M phase arrest indicates inhibited cell division and restrained cell growth. The results showed that changes in the cell cycle in A549 cells induced by blank NPs, siRNA, or PEI-PLA/siRNA were similar to that of the negative control (G2/M: 5.47%), which did not yield any change in cell cycle. But both PEI-PLA/PTX/siRNANC/PEG-PAsp (G2/M: 49.62%) and PEI-PLA/PTX/siRNA/PEG-PAsp (G2/M: 49.18%) NPs induced great G2/M arrest in A549 cells, and this is similar to that in the PTX group (G2/M: 47.25%). Data from the cell apoptosis and cell cycle analyses indicate that the enhanced accumulation of PTX and survivin siRNA by the PEI-PLA/PEG-PAsp drug delivery system facilitated drug entry into cells and induced improved apoptosis.
In vivo distribution
To monitor the biodistribution of the co-loaded NPs in the mice, fluorescence detection of the tumor and organs was performed with an in vivo imaging system. DIR accumulation level in tumor (Figures 8A and S7) is significantly higher in the PEI-PLA/DIR/siRNACy5/PEG-PAsp group than in both the DIR and PEI-PLA/DIR/siRNACy5 groups (**p<0.05). Moreover, the fluorescence intensity in all dissected organs in the PEI-PLA/DIR/siRNACy5/PEG-PAsp group was stronger than that of the other two groups. In the Cy5 channel (Figures 8B and S7), no fluorescent signal was detected in the naked siRNA group, indicating that the naked siRNA hardly achieves effective RNA interference without a proper delivery vector. The Cy5 intensity in the tumor of the mice injected with PEI-PLA/DIR/siRNACy5/PEG-PAsp is much stronger than that of the PEI-PLA/DIR/siRNACy5 group. The results suggest that the PEG-PAsp coating played a key role in this delivery system. This is in line with the previous finding that the PEG chain helps to prolong the circulating time of the NPs as well as enhance the drug accumulation in solid tumors.41–43
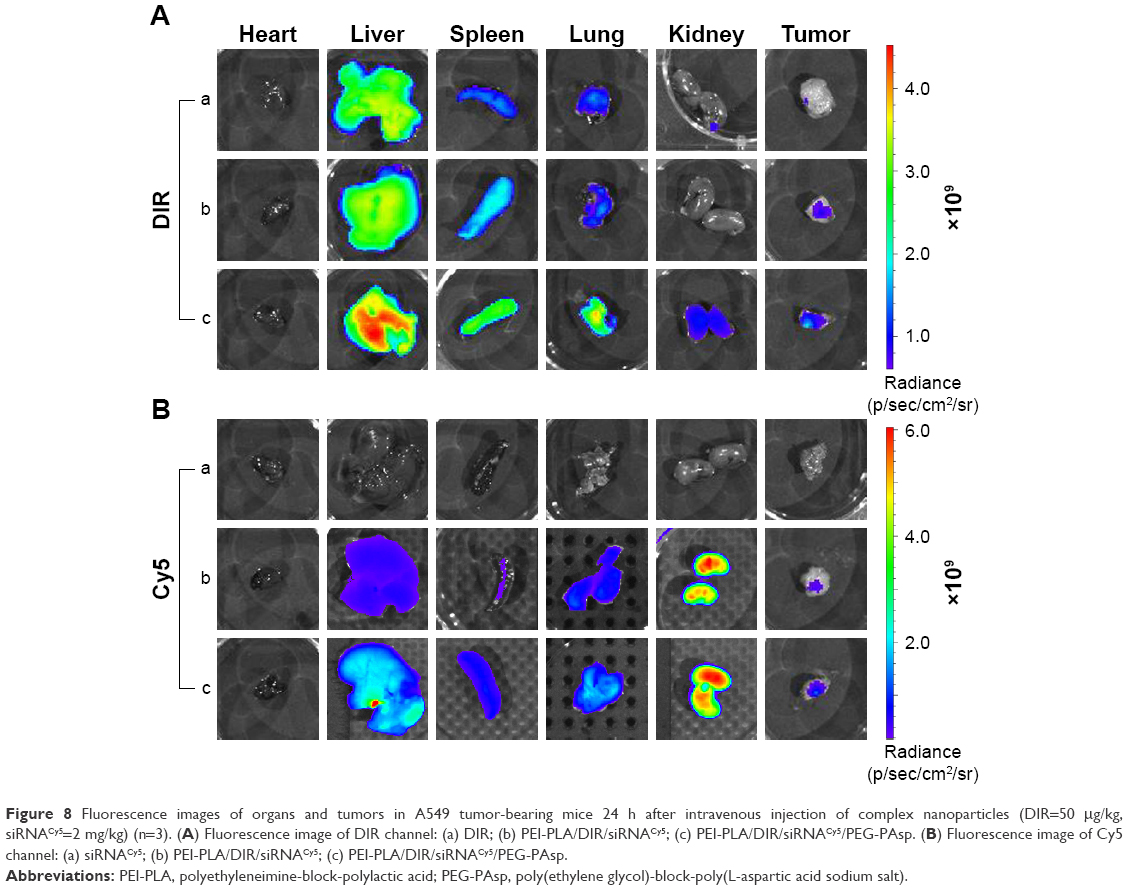 | Figure 8 Fluorescence images of organs and tumors in A549 tumor-bearing mice 24 h after intravenous injection of complex nanoparticles (DIR=50 μg/kg, siRNACy5=2 mg/kg) (n=3). (A) Fluorescence image of DIR channel: (a) DIR; (b) PEI-PLA/DIR/siRNACy5; (c) PEI-PLA/DIR/siRNACy5/PEG-PAsp. (B) Fluorescence image of Cy5 channel: (a) siRNACy5; (b) PEI-PLA/DIR/siRNACy5; (c) PEI-PLA/DIR/siRNACy5/PEG-PAsp. |
In vivo anticancer efficacy
To evaluate the antitumor efficacy of the co-loaded NPs, average tumor size was recorded during the experiment. As shown in Figure 9A and B, PEI-PLA/PTX/siRNA/PEG-PAsp resulted in the best tumor growth inhibition, whereas tumors in mice receiving PBS or blank NPs (PEI-PLA/PEG-PAsp) did not show any signs of tumor growth suppression after 25 days. Specifically, tumor volume decreased from 208±29 mm3 to 76±25 mm3 (***p<0.001) in the PEI-PLA/PTX/siRNA/PEG-PAsp group. However, tumor expanded from 206±34 mm3 to 880±169 mm3 in the saline group, and from 210±27 mm3 to 730±182 mm3 in the blank NPs group. This suggests that the co-loaded NPs have excellent antitumor efficacy, but the carrier itself does not.
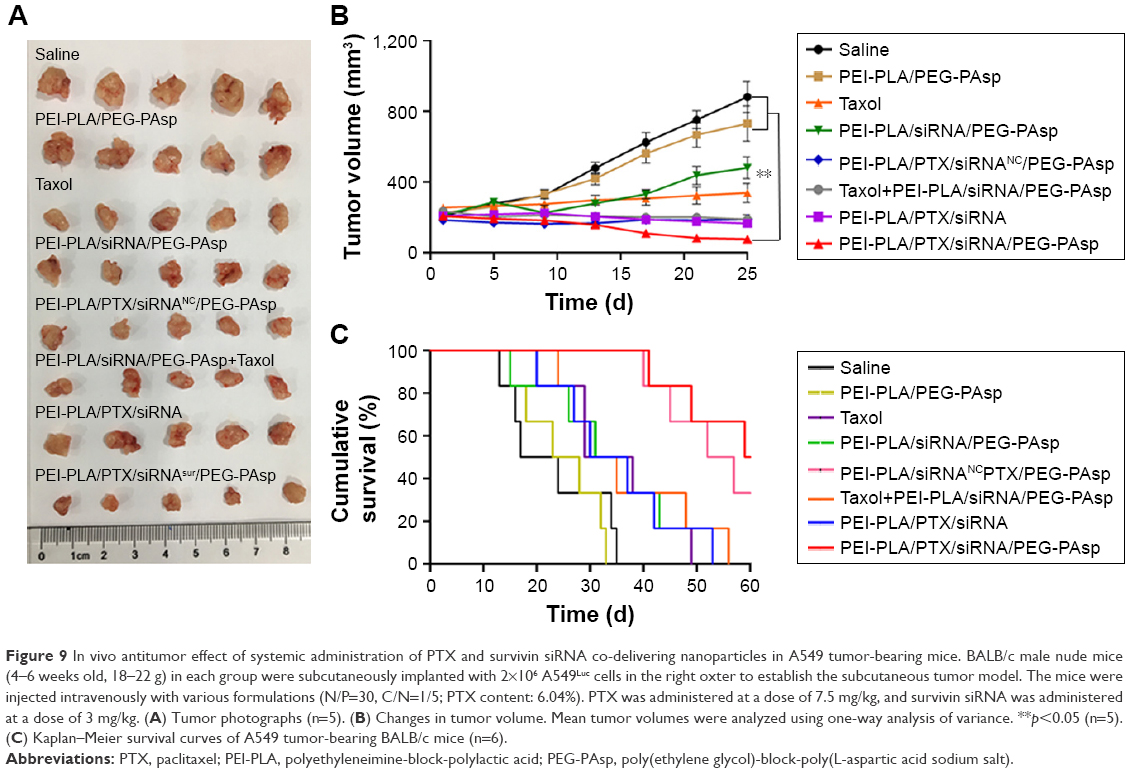 | Figure 9 In vivo antitumor effect of systemic administration of PTX and survivin siRNA co-delivering nanoparticles in A549 tumor-bearing mice. BALB/c male nude mice (4–6 weeks old, 18–22 g) in each group were subcutaneously implanted with 2×106 A549Luc cells in the right oxter to establish the subcutaneous tumor model. The mice were injected intravenously with various formulations (N/P=30, C/N=1/5; PTX content: 6.04%). PTX was administered at a dose of 7.5 mg/kg, and survivin siRNA was administered at a dose of 3 mg/kg. (A) Tumor photographs (n=5). (B) Changes in tumor volume. Mean tumor volumes were analyzed using one-way analysis of variance. **p<0.05 (n=5). (C) Kaplan–Meier survival curves of A549 tumor-bearing BALB/c mice (n=6). |
Based on the effective inhibition efficacy on tumor growth, we further evaluated the influence of PEI-PLA/PTX/siRNA/PEG-PAsp NPs on the survival rates of A549 tumor-bearing BALB/c mice. As shown in Figure 9C, mice in the saline and PEI-PLA/PEG-PAsp groups all died within 35 days of administration. The survival rate of PEI-PLA/PTX/siRNANC/PEG-PAsp group remained at 33.33% up to 60 days, while that of the PEI-PLA/PTX/siRNA/PEG-PAsp group was 50%.
As shown in Figure 10A, relative survivin mRNA level in the PEI-PLA/PTX/siRNA/PEG-PAsp group (0.38±0.14) was significantly decreased compared to the control group (1.15±0.18), PEI-PLA/PEG-PAsp group (1.02±0.13), and Taxol group (0.97±0.14) (***p<0.001). There was also a great difference in mRNA level between the PEI-PLA/PTX/siRNA/PEG-PAsp, PEI-PLA/PTX/siRNANC/PEG-PAsp, Taxol+PEI-PLA/siRNA/PEG-PAsp, and PEI-PLA/PTX/siRNA groups. Similarly, the relative survivin protein level (Figure 10B) in the PEI-PLA/PTX/siRNA/PEG-PAsp group (2.51±0.71 ng/mg) was significantly lower than in the other groups (***p<0.001). Consistent with the results, the immunohistochemistry study showed that the PTX and survivin siRNA-combined treatment using PEI-PLA/PTX/siRNA/PEG-PAsp resulted in the lowest level of survivin expression (Figure 10C). This indicates that the NPs first accumulated in tumor sites and delivered anti-survivin siRNA and PTX in sufficiently high amounts to mediate a potent and specific survivin down-regulation, as well as to improve the anticancer ability of PTX.
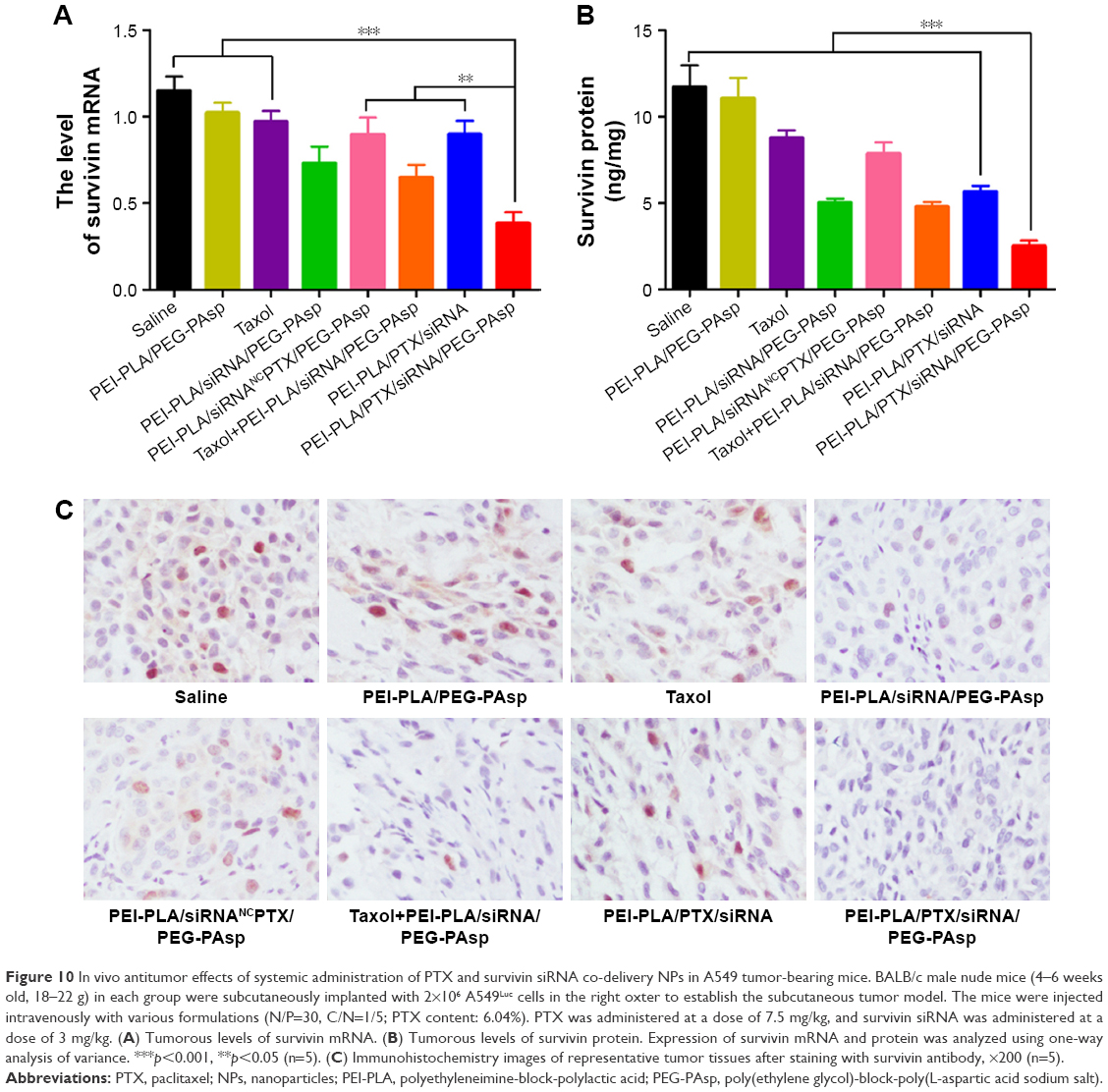 | Figure 10 In vivo antitumor effects of systemic administration of PTX and survivin siRNA co-delivery NPs in A549 tumor-bearing mice. BALB/c male nude mice (4–6 weeks old, 18–22 g) in each group were subcutaneously implanted with 2×106 A549Luc cells in the right oxter to establish the subcutaneous tumor model. The mice were injected intravenously with various formulations (N/P=30, C/N=1/5; PTX content: 6.04%). PTX was administered at a dose of 7.5 mg/kg, and survivin siRNA was administered at a dose of 3 mg/kg. (A) Tumorous levels of survivin mRNA. (B) Tumorous levels of survivin protein. Expression of survivin mRNA and protein was analyzed using one-way analysis of variance. ***p<0.001, **p<0.05 (n=5). (C) Immunohistochemistry images of representative tumor tissues after staining with survivin antibody, ×200 (n=5). |
Discussion
The prepared PEI-PLA/PTX/siRNA/PEG-PAsp NPs presented the most suitable size and zeta potential (82.4 nm, 4.10 mV) for passive targeting of the EPR effect. Size and surface charge are two critical parameters that dictate the disposition of NPs in the body.44 Although strong positively charged NPs have better cellular uptake efficacy, they may easily interact with negatively charged proteins during circulation. Thus, it was believed that NPs with a weak positive charge and suitable size range (<100 nm) could optimally take advantage of the EPR effect and avoid this issue.26,45,46 In the stability study, we showed that the PEI-PLA/PTX/siRNA/PEG-PAsp NPs presented no change when incubated in PBS with or without FBS as opposed to PEI-PLA/PTX/siRNA, suggesting that the coupling amount of PEG-PAsp as well as the weak positive charge of the NPs increased the stability in plasma.
To verify the sheddable ability of PEG-PAsp at different pH values, we examined the particle size and zeta potential of PEI-PLA/PTX/siRNA/PEG-PAsp NPs in various pH environments. We showed that the NPs released PTX more quickly in an acidic environment, which is probably due to faster degradation of the PLA core and proton sponge effect of PEI. Such behavior presumably enhanced intracellular drug release once the complex NPs entered tumor cells via endocytosis and became trapped within the acidic endosomal compartments.26 At pH 7.4, the cellular uptake of PTX and siRNA was 32.55% and 101.94%, respectively, while at pH 5.5, the cellular uptake rates were 48.07% and 264.16%, respectively. We believe the reason why drug accumulation increased in A549 cells at pH 5.5 is that the PEG-PAsp coating was able to readily detach from the outer layer of the NPs, and thus expose the cationic co-loaded NPs. The positively charged NPs bound to anionic phospholipids in the membrane via electrostatic interaction and subsequently entered the cell carrying drug and siRNA.26,47 Therefore, along with PEI-PLA, the PEG-PAsp copolymer provided an excellent drug-delivery platform for combining PTX and siRNA. In the drug release study, we showed that PTX was released more rapidly in NPs with or without PEG-PAsp coating at pH 5.5 compared with the same at pH 7.4. In the CCK-8 assay, the PEI-PLA/PTX/siRNA/PEG-PAsp exhibited a better cytotoxic effect against the A549 cells at pH 5.5 (IC50 value of 1.48 mg/mL) than at pH 7.4 (IC50 value of 3.68 mg/mL) at corresponding doses. These results indicate that low pH could help to increase the cellular uptake of complex NPs, trigger the release of the drugs, and finally target and kill the cancer cells.
We further sought to determine whether simultaneous delivery of siRNA and PTX by the complex NPs can synergistically lower the survivin expression as well as inhibit the cell proliferation. The results showed that PEI-PLA/PTX/siRNA/PEG-PAsp NPs could preferentially downregulate the survivin expression and further remarkably inhibit the cell proliferation. As a member of the inhibitor of apoptosis family, survivin inhibits apoptosis and promotes cell proliferation.48 Also, the PEI-PLA/PTX/siRNANC/PEG-PAsp NPs showed better inhibitory effect than that of PTX-free formulations (**p<0.05), indicating that PTX-loaded NPs can enhance the cytotoxicity of PTX at a certain dose. It is worth nothing that the PEI-PLA/PTX/siRNA/PEG-PAsp NPs presented the lowest cell viability of less than 15%, which indicates that simultaneous delivery of siRNA and PTX achieved a synergistic inhibitory effect on A549 cancer cells.
Previous work has suggested survivin as a target for cancer treatment given that inhibition of survivin promotes apoptosis and suppresses cancer cell proliferation.49–51 Mitotic arrest of PTX-treated cells related to apoptosis has also been reported, and this has been considered as the cause of PTX-induced cytotoxicity.52,53 PTX preferentially induces G2/M arrest of the cell cycle and thus causes apoptosis.40 In the cell cycle assay, similarly to the negative control, there was no change in the cell cycle of A549 cells in blank NPs, siRNA, or PEI-PLA/siRNA. But both PEI-PLA/PTX/siRNANC/PEG-PAsp and PEI-PLA/PTX/siRNA/PEG-PAsp NPs induced great G2/M arrest in A549 cells, which was comparable to that of the PTX group. These results indicate that the change in cell cycle is mostly related to PTX release from the NPs, rather than any other components of the complex NPs.
The in vivo studies demonstrate that the complex NPs effectively accumulated in tumor and were capable of inhibiting the tumor growth and extending the survival rate of the mice. The PTX in NPs is toxic to cancer cells, and thus affects the cell proliferation, while the function of survivin siRNA in NPs is to silence the gene expression. Thus, the anticancer effect of co-loaded NPs is more obvious than single-loaded NPs (PEI-PLA/siRNA/PEG-PAsp or PEI-PLA/PTX/siRNANC/PEG-PAsp) owing to the combinational antitumor behavior. The fact that tumor volume in the PEI-PLA/PTX/siRNA group showed less decrease than in the PEI-PLA/PTX/siRNA/PEG-PAsp group indicates that although the drug content was the same in both groups, the lack of PEG-PAsp coating greatly affected the antitumor effect when the formulations were intravenously injected. PEG-PAsp is able to provide a “protective” surface to improve the drug stability and help to enhance the accumulation of the NPs at the tumor site. The survivin expression at both the mRNA and protein levels in the tumors was evaluated to determine whether reduced tumor growth was associated with survivin gene silencing. Further molecular biological evidence for the synergistic effect of PTX and survivin siRNA in the complex NPs was provided from the immunohistochemistry assay. At all levels, we successfully showed that survivin siRNA was downregulated in the tumor. In a previous study, it was reported that downregulating survivin was associated with enhanced chemotherapeutic efficacy in the treatment of NSCLC.54 Thus, together with our results, we can conclude that in accordance with the tumor reduction, the intratumorol survivin expression in PEI-PLA/PTX/siRNA/PEG-PAsp provided superiority in terms of anticancer efficacy.
Conclusion
In this study, we successfully prepared and characterized polymeric NPs for co-delivery of PTX and survivin siRNA. The efficient co-delivery system enables 1) high drug loading, 2) extended drug release that allows prolonged exposure of the tumor to the treatment, 3) passive targeting and tumor cell uptake owing to proper particle size and zeta potential, 4) low systemic toxicity and improved antiproliferation effect of PTX on A549 cells, and 5) enhanced antitumor efficacy by co-delivery of siRNA and PTX. The PEI-PLA/PEG-PAsp-based co-delivery system could be a promising vector to pave the way for a novel chemo/gene therapeutic treatment modality.
Acknowledgments
This work was financially supported by CAMS Innovation Fund for Medical Sciences (CAMS-2017-I2M-1-011), the Scientific Research Foundation of Graduate School of Peking Union Medical College (No. 1007-08), PUMC Youth Fund and the Fundamental Research Funds for the Central Universities (2017350003).
Disclosure
The authors declare that no competing interests exist. The authors alone are responsible for the content and writing of the paper.
References
Guan X, Li Y, Jiao Z, et al. Codelivery of antitumor drug and gene by a pH-sensitive charge-conversion system. ACS Appl Mater Interfaces. 2015;7(5):3207–3215. | ||
Sun TM, Du JZ, Yao YD, et al. Simultaneous delivery of siRNA and paclitaxel via a “two-in-one” micelleplex promotes synergistic tumor suppression. ACS Nano. 2011;5(2):1483–1494. | ||
Wiradharma N, Tong YW, Yang YY. Self-assembled oligopeptide nanostructures for co-delivery of drug and gene with synergistic therapeutic effect. Biomaterials. 2009;30(17):3100–3109. | ||
Han K, Chen S, Chen WH, et al. Synergistic gene and drug tumor therapy using a chimeric peptide. Biomaterials. 2013;34(19):4680–4689. | ||
Ferguson SM, Norton CS, Watson SJ, Akil H, Robinson TE. Amphetamine-evoked c-fos mRNA expression in the caudate-putamen: the effects of DA and NMDA receptor antagonists vary as a function of neuronal phenotype and environmental context. J Neurochem. 2003;86(1):33–44. | ||
Primeau AJ, Rendon A, Hedley D, Lilge L, Tannock IF. The distribution of the anticancer drug doxorubicin in relation to blood vessels in solid tumors. Clin Cancer Res. 2005;11(24 Pt 1):8782–8788. | ||
Creixell M, Peppas NA. Co-delivery of siRNA and therapeutic agents using nanocarriers to overcome cancer resistance. Nano Today. 2012;7(4):367–379. | ||
Tsouris V, Joo MK, Kim SH, Kwon IC, Won YY. Nano carriers that enable co-delivery of chemotherapy and RNAi agents for treatment of drug-resistant cancers. Biotechnol Adv. 2014;32(5):1037–1050. | ||
Chitkara D, Singh S, Mittal A. Nanocarrier-based co-delivery of small molecules and siRNA/miRNA for treatment of cancer. Ther Deliv. 2016;7(4):245–255. | ||
Lee SY, Yang CY, Peng CL, et al. A theranostic micelleplex co-delivering SN-38 and VEGF siRNA for colorectal cancer therapy. Biomaterials. 2016;86:92–105. | ||
Xiao B, Ma L, Merlin D. Nanoparticle-mediated co-delivery of chemotherapeutic agent and siRNA for combination cancer therapy. Expert Opin Drug Deliv. 2017;14(1):65–73. | ||
Peer D, Karp JM, Hong S, Farokhzad OC, Margalit R, Langer R. Nanocarriers as an emerging platform for cancer therapy. Nat Nanotechnol. 2007;2(12):751–760. | ||
Hu Q, Sun W, Wang C, Gu Z. Recent advances of cocktail chemotherapy by combination drug delivery systems. Adv Drug Deliv Rev. 2016;98:19–34. | ||
Eldar-Boock A, Polyak D, Scomparin A, Satchi-Fainaro R. Nano-sized polymers and liposomes designed to deliver combination therapy for cancer. Curr Opin Biotechnol. 2013;24(4):682–689. | ||
Salzano G, Costa DF, Torchilin VP. siRNA delivery by stimuli-sensitive nanocarriers. Curr Pharm Des. 2015;21(31):4566–4573. | ||
Zaffaroni N, Daidone MG. Survivin expression and resistance to anticancer treatments: perspectives for new therapeutic interventions. Drug Resist Updat. 2002;5(2):65–72. | ||
Coumar MS, Tsai FY, Kanwar JR, Sarvagalla S, Cheung CH. Treat cancers by targeting survivin: just a dream or future reality? Cancer Treat Rev. 2013;39(7):802–811. | ||
Wang S, Huang X, Lee CK, Liu B. Elevated expression of erbB3 confers paclitaxel resistance in erbB2-overexpressing breast cancer cells via upregulation of Survivin. Oncogene. 2010;29(29):4225–4236. | ||
Wang S, Huang J, Lyu H, et al. Therapeutic targeting of erbB3 with MM-121/SAR256212 enhances antitumor activity of paclitaxel against erbB2-overexpressing breast cancer. Breast Cancer Res. 2013;15(5):R101. | ||
Ding X, Wang W, Wang Y, et al. Versatile reticular polyethylenimine derivative-mediated targeted drug and gene codelivery for tumor therapy. Mol Pharm. 2014;11(10):3307–3321. | ||
Liu J, Jiang X, Xu L, Wang X, Hennink WE, Zhuo R. Novel reduction-responsive cross-linked polyethylenimine derivatives by click chemistry for nonviral gene delivery. Bioconjug Chem. 2010;21(10):1827–1835. | ||
Lim C, Youn YS, Lee KS, et al. Development of a robust pH-sensitive polyelectrolyte ionomer complex for anticancer nanocarriers. Int J Nanomedicine. 2016;11:703–713. | ||
Sun TM, Du JZ, Yan LF, Mao HQ, Wang J. Self-assembled biodegradable micellar nanoparticles of amphiphilic and cationic block copolymer for siRNA delivery. Biomaterials. 2008;29(32):4348–4355. | ||
Oh KT, Bronich TK, Bromberg L, Hatton TA, Kabanov AV. Block ionomer complexes as prospective nanocontainers for drug delivery. J Control Release. 2006;115(1):9–17. | ||
Tran TH, Ramasamy T, Choi JY, et al. Tumor-targeting, pH-sensitive nanoparticles for docetaxel delivery to drug-resistant cancer cells. Int J Nanomedicine. 2015;10:5249–5262. | ||
Cao N, Cheng D, Zou S, Ai H, Gao J, Shuai X. The synergistic effect of hierarchical assemblies of siRNA and chemotherapeutic drugs co-delivered into hepatic cancer cells. Biomaterials. 2011;32(8):2222–2232. | ||
Guo S, Huang L. Nanoparticles escaping RES and endosome: challenges for siRNA delivery for cancer therapy. J Nanomater. 2011;2011:742895. | ||
Hatakeyama H, Akita H, Harashima H. A multifunctional envelope type nano device (MEND) for gene delivery to tumours based on the EPR effect: a strategy for overcoming the PEG dilemma. Adv Drug Deliv Rev. 2011;63(3):152–160. | ||
Ramasamy T, Haidar ZS, Tran TH, et al. Layer-by-layer assembly of liposomal nanoparticles with PEGylated polyelectrolytes enhances systemic delivery of multiple anticancer drugs. Acta Biomater. 2014;10(12):5116–5127. | ||
Shuai XT, Merdan T, Unger F, Wittmar M, Kissel T. Novel biodegradable ternary copolymers hy-PEI-g-PCL-b-PEG: synthesis, characterization, and potential as efficient nonviral gene delivery vectors. Macromolecules. 2003;36(15):5751–5759. | ||
Kang L, Fan B, Sun P, et al. An effective tumor-targeting strategy utilizing hypoxia-sensitive siRNA delivery system for improved anti-tumor outcome. Acta Biomater. 2016;44:341–354. | ||
Thambi T, Deepagan VG, Yoon HY, et al. Hypoxia-responsive polymeric nanoparticles for tumor-targeted drug delivery. Biomaterials. 2014;35(5):1735–1743. | ||
Tang S, Yin Q, Su J, et al. Inhibition of metastasis and growth of breast cancer by pH-sensitive poly (β-amino ester) nanoparticles co-delivering two siRNA and paclitaxel. Biomaterials. 2015;48:1–15. | ||
Navarro G, Sawant RR, Biswas S, Essex S, Tros de Ilarduya C, Torchilin VP. P-glycoprotein silencing with siRNA delivered by DOPE-modified PEI overcomes doxorubicin resistance in breast cancer cells. Nanomedicine (Lond). 2012;7(1):65–78. | ||
Michaud LB, Valero V, Hortobagyi G. Risks and benefits of taxanes in breast and ovarian cancer. Drug Saf. 2000;23(5):401–428. | ||
Lee J, Lee SC, Acharya G, Chang CJ, Park K. Hydrotropic solubilization of paclitaxel: analysis of chemical structures for hydrotropic property. Pharm Res. 2003;20(7):1022–1030. | ||
Yin T, Wang L, Yin L, Zhou J, Huo M. Co-delivery of hydrophobic paclitaxel and hydrophilic AURKA specific siRNA by redox-sensitive micelles for effective treatment of breast cancer. Biomaterials. 2015;61:10–25. | ||
Zheng C, Zheng M, Gong P, et al. Polypeptide cationic micelles mediated co-delivery of docetaxel and siRNA for synergistic tumor therapy. Biomaterials. 2013;34(13):3431–3438. | ||
Feng Q, Yu MZ, Wang JC, et al. Synergistic inhibition of breast cancer by co-delivery of VEGF siRNA and paclitaxel via vapreotide-modified core-shell nanoparticles. Biomaterials. 2014;35(18):5028–5038. | ||
Trielli MO, Andreassen PR, Lacroix FB, Margolis RL. Differential Taxol-dependent arrest of transformed and nontransformed cells in the G1 phase of the cell cycle, and specific-related mortality of transformed cells. J Cell Biol. 1996;135(3):689–700. | ||
Klibanov AL, Maruyama K, Torchilin VP, Huang L. Amphipathic polyethyleneglycols effectively prolong the circulation time of liposomes. FEBS Lett. 1990;268(1):235–237. | ||
Alexis F, Pridgen E, Molnar LK, Farokhzad OC. Factors affecting the clearance and biodistribution of polymeric nanoparticles. Mol Pharm. 2008;5(4):505–515. | ||
Maeda H, Wu J, Sawa T, Matsumura Y, Hori K. Tumor vascular permeability and the EPR effect in macromolecular therapeutics: a review. J Control Release. 2000;65(1–2):271–284. | ||
Jhaveri A, Torchilin V. Intracellular delivery of nanocarriers and targeting to subcellular organelles. Expert Opin Drug Deliv. 2016;13(1):49–70. | ||
Ishida O, Maruyama K, Sasaki K, Iwatsuru M. Size-dependent extravasation and interstitial localization of polyethyleneglycol liposomes in solid tumor-bearing mice. Int J Pharm. 1999;190(1):49–56. | ||
Petros RA, DeSimone JM. Strategies in the design of nanoparticles for therapeutic applications. Nat Rev Drug Discov. 2010;9(8):615–627. | ||
Zeng Q, Jiang H, Wang T, Zhang Z, Gong T, Sun X. Cationic micelle delivery of Trp2 peptide for efficient lymphatic draining and enhanced cytotoxic T-lymphocyte responses. J Control Release. 2015;200:1–12. | ||
Ryan BM, O’Donovan N, Duffy MJ. Survivin: a new target for anti-cancer therapy. Cancer Treat Rev. 2009;35(7):553–562. | ||
Duffy MJ, O’Donovan N, Brennan DJ, Gallagher WM, Ryan BM. Survivin: a promising tumor biomarker. Cancer Lett. 2007;249(1):49–60. | ||
Altieri DC. Validating survivin as a cancer therapeutic target. Nat Rev Cancer. 2003;3(1):46–54. | ||
Ambrosini G, Adida C, Altieri DC. A novel anti-apoptosis gene, survivin, expressed in cancer and lymphoma. Nat Med. 1997;3(8):917–921. | ||
Wang TH, Wang HS, Soong YK. Paclitaxel-induced cell death: where the cell cycle and apoptosis come together. Cancer. 2000;88(11):2619–2628. | ||
Jordan MA, Toso RJ, Thrower D, Wilson L. Mechanism of mitotic block and inhibition of cell proliferation by taxol at low concentrations. Proc Natl Acad Sci U S A. 1993;90(20):9552–9556. | ||
Wang S, Zhu L, Zuo W, et al. MicroRNA-mediated epigenetic targeting of survivin significantly enhances the antitumor activity of paclitaxel against non-small cell lung cancer. Oncotarget. 2016;7(25):37693–37713. |
Supplementary materials
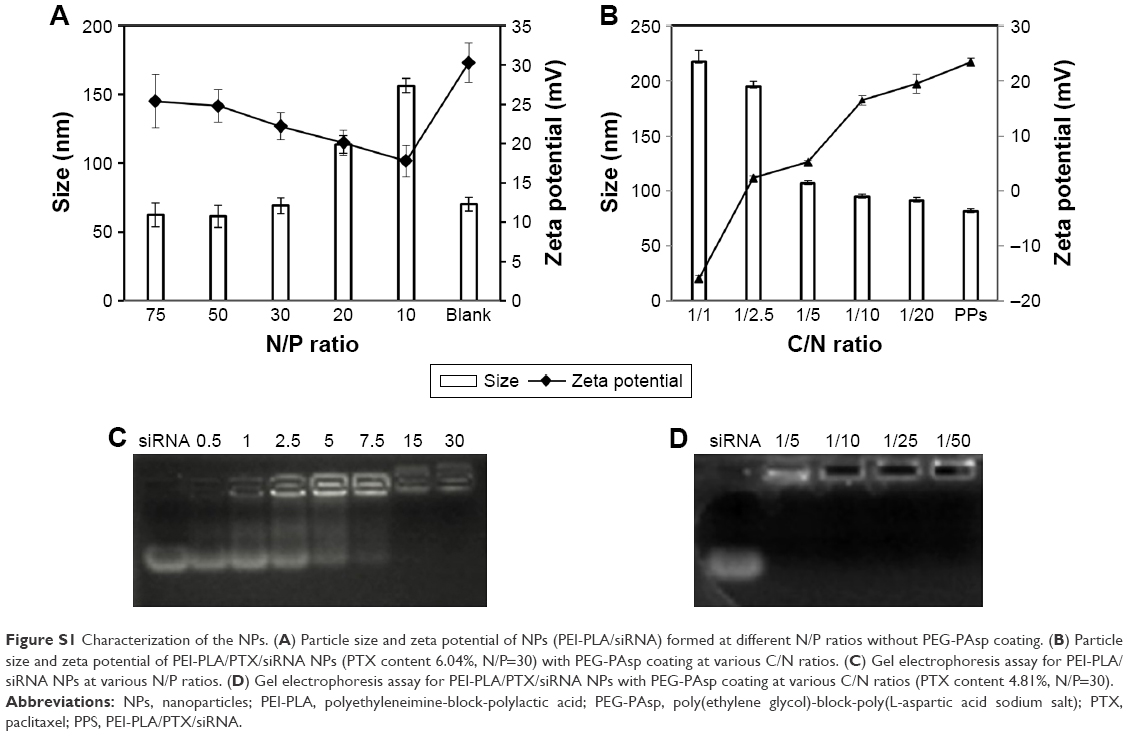 | Figure S1 Characterization of the NPs. (A) Particle size and zeta potential of NPs (PEI-PLA/siRNA) formed at different N/P ratios without PEG-PAsp coating. (B) Particle size and zeta potential of PEI-PLA/PTX/siRNA NPs (PTX content 6.04%, N/P=30) with PEG-PAsp coating at various C/N ratios. (C) Gel electrophoresis assay for PEI-PLA/siRNA NPs at various N/P ratios. (D) Gel electrophoresis assay for PEI-PLA/PTX/siRNA NPs with PEG-PAsp coating at various C/N ratios (PTX content 4.81%, N/P=30). |
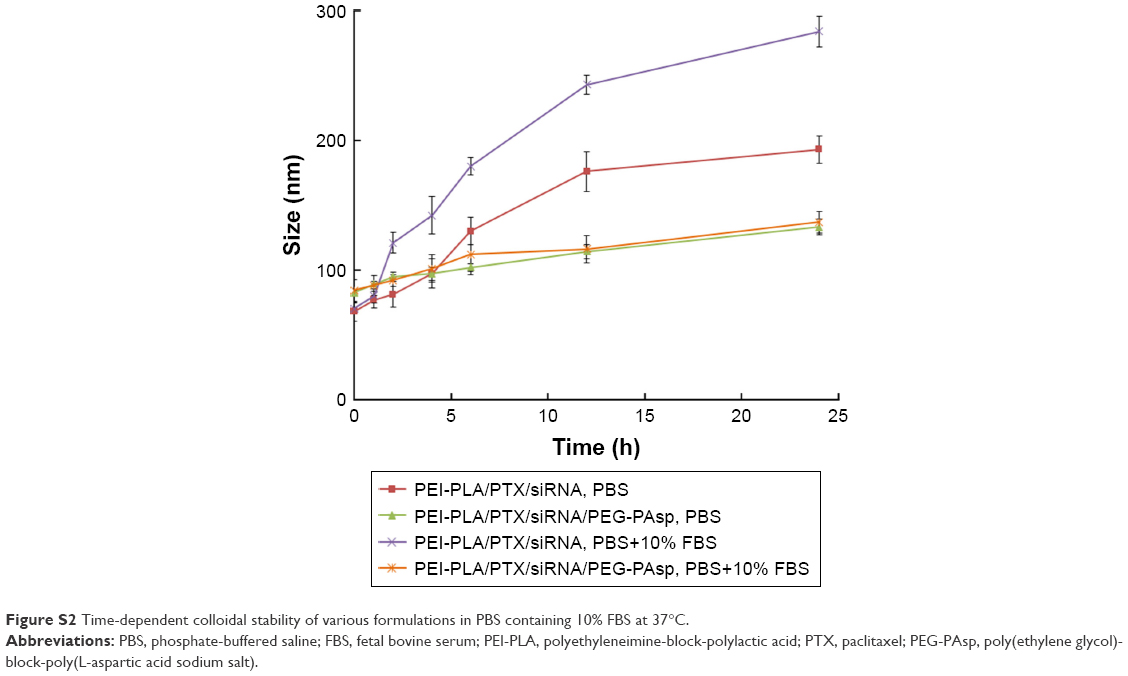 | Figure S2 Time-dependent colloidal stability of various formulations in PBS containing 10% FBS at 37°C. |
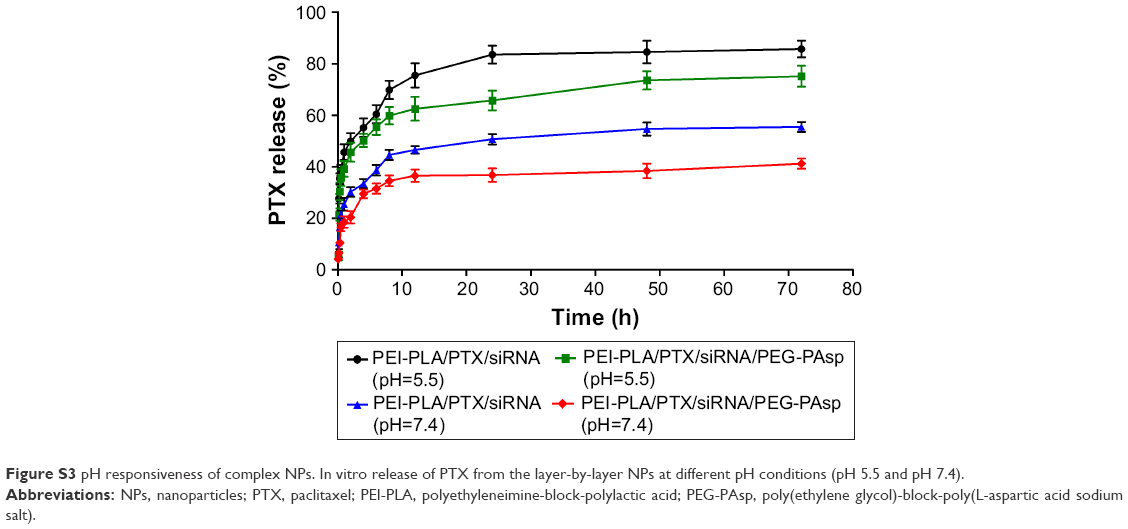 | Figure S3 pH responsiveness of complex NPs. In vitro release of PTX from the layer-by-layer NPs at different pH conditions (pH 5.5 and pH 7.4). |
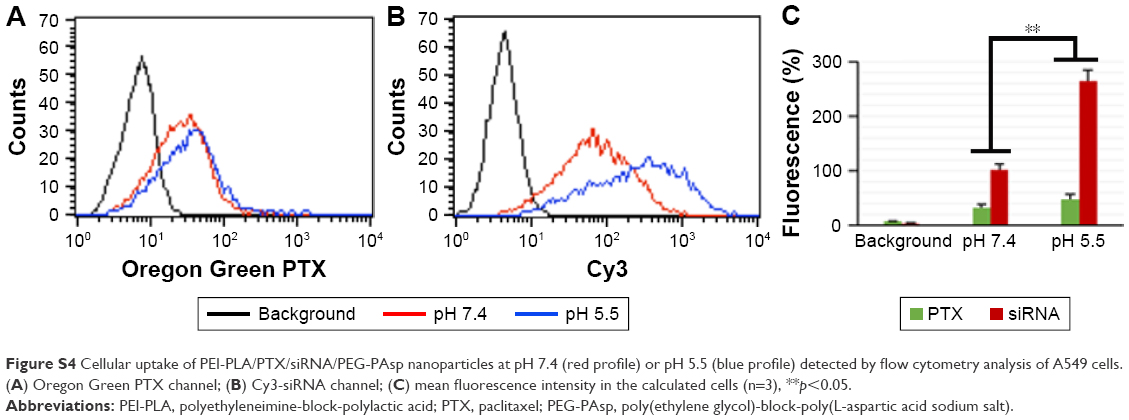 | Figure S4 Cellular uptake of PEI-PLA/PTX/siRNA/PEG-PAsp nanoparticles at pH 7.4 (red profile) or pH 5.5 (blue profile) detected by flow cytometry analysis of A549 cells. (A) Oregon Green PTX channel; (B) Cy3-siRNA channel; (C) mean fluorescence intensity in the calculated cells (n=3), **p<0.05. |
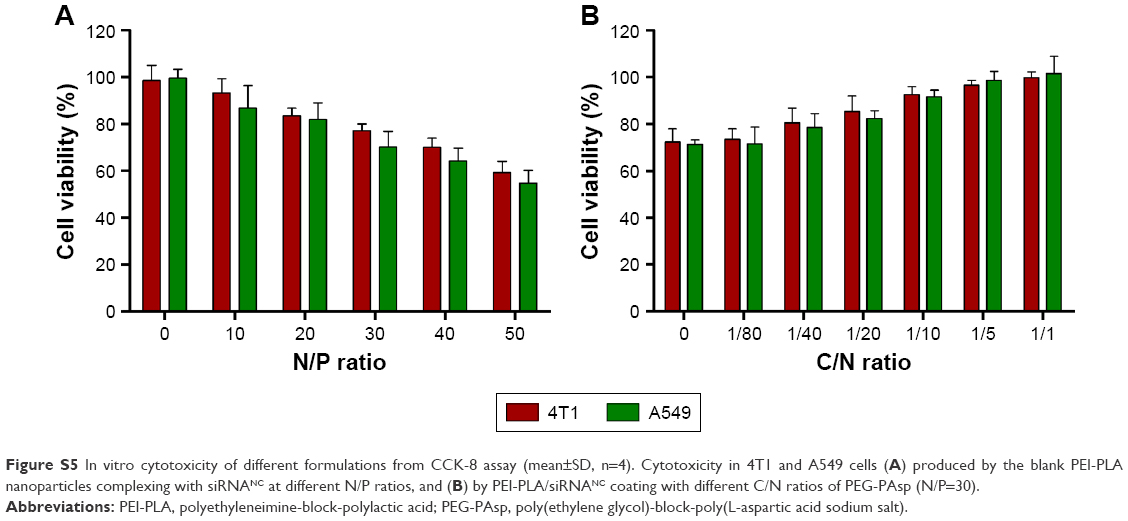 | Figure S5 In vitro cytotoxicity of different formulations from CCK-8 assay (mean±SD, n=4). Cytotoxicity in 4T1 and A549 cells (A) produced by the blank PEI-PLA nanoparticles complexing with siRNANC at different N/P ratios, and (B) by PEI-PLA/siRNANC coating with different C/N ratios of PEG-PAsp (N/P=30). |
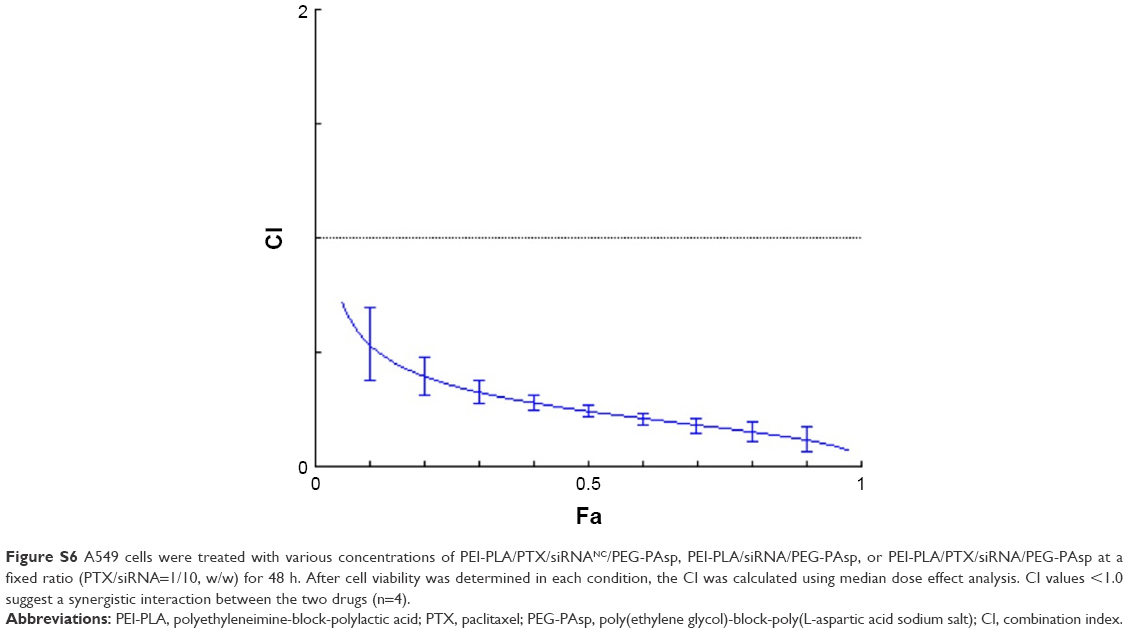 | Figure S6 A549 cells were treated with various concentrations of PEI-PLA/PTX/siRNANC/PEG-PAsp, PEI-PLA/siRNA/PEG-PAsp, or PEI-PLA/PTX/siRNA/PEG-PAsp at a fixed ratio (PTX/siRNA=1/10, w/w) for 48 h. After cell viability was determined in each condition, the CI was calculated using median dose effect analysis. CI values <1.0 suggest a synergistic interaction between the two drugs (n=4). |
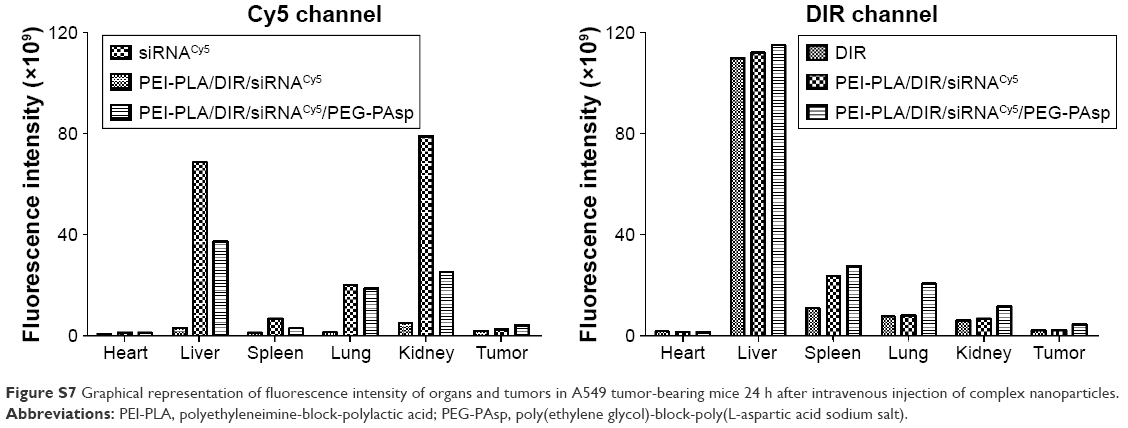 | Figure S7 Graphical representation of fluorescence intensity of organs and tumors in A549 tumor-bearing mice 24 h after intravenous injection of complex nanoparticles. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.