Back to Journals » Drug Design, Development and Therapy » Volume 9
Small-molecule BH3 mimetic and pan-Bcl-2 inhibitor AT-101 enhances the antitumor efficacy of cisplatin through inhibition of APE1 repair and redox activity in non-small-cell lung cancer
Authors Ren T, Shan J, Li M, Qing Y, Qian C, Wang G, Li Q, Liu G, Li C, Peng Y, Luo H, Zhang S, Yang Y, Cheng Y, Wang D, Zhou S
Received 12 February 2015
Accepted for publication 9 March 2015
Published 8 June 2015 Volume 2015:9 Pages 2887—2910
DOI https://doi.org/10.2147/DDDT.S82724
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Wei Duan
Tao Ren,1–3,* Jinlu Shan,1,* Mengxia Li,1 Yi Qing,1 Chengyuan Qian,4 Guangjie Wang,5 Qing Li,1,3 Guoshou Lu,1 Chongyi Li,1 Yu Peng,1 Hao Luo,1 Shiheng Zhang,1 Yuxing Yang,1 Yi Cheng,1 Dong Wang,1 Shu-Feng Zhou3
1Cancer Center, Daping Hospital and Research Institute of Surgery, Third Military Medical University, Chongqing, 2Department of Oncology, The Affiliated Hospital, North Sichuan Medical College, Sichuan, People’s Republic of China; 3Department of Pharmaceutical Sciences, College of Pharmacy, University of South Florida, Tampa, FL, USA; 4Department of Oncology, The 97 Hospital of PLA, Jiangsu, 5Cancer Diagnosis and Treatment Center, Military District General Hospital of Chengdu Military Region, Sichuan, People’s Republic of China
*These authors contributed equally to this work
Abstract: AT-101 is a BH3 mimetic and pan-Bcl-2 inhibitor that has shown potent anticancer activity in non-small-cell lung cancer (NSCLC) in murine models, but failed to show clinical efficacy when used in combination with docetaxel in NSCLC patients. Our recent study has demonstrated that AT-101 enhanced the antitumor effect of cisplatin (CDDP) in a murine model of NSCLC via inhibition of the interleukin-6/signal transducer and activator of transcription 3 (STAT3) pathway. This study explored the underlying mechanisms for the enhanced anticancer activity of CDDP by AT-101. Our results show that, when compared with monotherapy, AT-101 significantly enhanced the inhibitory effects of CDDP on proliferation and migration of A549 cells and on tube formation and migration in human umbilical vein endothelial cells. AT-101 promoted the proapoptotic activity of CDDP in A549 cells. AT-101 also enhanced the inhibitory effect of CDDP on DNA repair and redox activities of apurinic/apyrimidinic endonuclease 1 (APE1) in A549 cells. In tumor tissues from nude mice treated with AT-101 plus CDDP or monotherapy, the combination therapy resulted in greater inhibition of angiogenesis and tumor cell proliferation than the monotherapy. These results suggest that AT-101 can enhance the antitumor activity of CDDP in NSCLC via inhibition of APE1 DNA repair and redox activities and by angiogenesis and induction of apoptosis, but other mechanisms cannot be excluded. We are now conducting a Phase II trial to examine the clinical efficacy and safety profile of combined use of AT-101 plus CDDP in advanced NSCLC patients.
Keywords: AT-101, cisplatin, non-small-cell lung cancer, apurinic/apyrimidinic endonuclease 1, angiogenesis, tumor
Introduction
Lung cancer is the most lethal malignancy worldwide, contributing 12.9% of the total new cancer cases (1.8 million) and 19.4% of cancer deaths (1.59 million of 9.2 million) globally in 2012.1 The disease remains the most common cancer in men worldwide (1.2 million, 16.7% of the total), with the highest estimated age-standardized incidence rates in Central and Eastern Europe (53.5 per 100,000) and Eastern Asia (50.4 per 100,000). In the USA, lung cancer is estimated to kill 158,040 patients, accounting for ~27% of all cancer deaths in 2015.2 During 2015, there will be an estimated 221,200 new cases of lung cancer, representing about 13% of all cancers diagnosed in the USA. In 2012, 86,740 male and 70,759 female Americans died due to lung cancer. In 2010, about 605,946 patients were diagnosed with lung cancer, including 416,333 men and 189,613 women, with a crude incidence rate of 46.08 per 100,000 accounting for 19.59% of overall new cancer cases in the People’s Republic of China.3 There were estimated 486,555 deaths in lung cancer (336,786 men and 149,769 women), with a crude mortality rate of 37 per 100,000, accounting for 23.33% of all cancer deaths in the People’s Republic of China in 2010.3 There has been a continuous rise in the incidence of lung cancer from 1973 to 2005 in the People’s Republic of China. Lung cancer incidence was higher in urban populations than in rural populations (52.52/100,000 versus 39.54/100,000) in 2010. Lung cancer is histologically classified into non-small-cell lung cancer (NSCLC), comprising 70%–85% of all cases, and small cell lung cancer (SCLC) constituting 15%–25% of cases.4–7 Only ~15% of lung cancer can be diagnosed in the early stage when it is operable, while platinum-based chemotherapy using cisplatin (CDDP) or carboplatin is recommended as the first-line regimen to treat advanced inoperable SCLC and NSCLC.5,8 Despite the development of comprehensive and individualized therapy, the overall 5-year survival rate, which has been improved to some extent, remains below 20% in Europe, is in the range of 15%–19% in North America, and is as low as 7%–9% in Mongolia and Thailand.9 The 5-year survival rate for lung cancer is 54% for cases detected when the disease is still localized, but drops to 4% when lung cancer becomes metastasized to other organs. The clinical outcome of treatment for advanced lung cancer remains disappointing due to intrinsic or acquired chemoresistance to platinum-based chemotherapy and severe dose-limiting organ toxicities.10 There is a clear need to identify and develop new therapeutic agents that can improve the outlook for NSCLC.
Apurinic/apyrimidinic (AP) sites are formed either spontaneously or due to DNA damage, and it is estimated that, under physiological conditions, 10,000 apurinic sites and 500 apyrimidinic may be generated in a cell daily.11 AP sites can also occur as intermediates in base excision repair (BER) initiated by a DNA glycosylase. If left unrepaired, AP sites can block DNA synthesis and lead to mutation during semiconservative replication.11 AP endonuclease 1 (APE1, also known as redox effector factor 1 [Ref-1]) is a multifunctional protein that not only repairs AP sites in DNA lesions via the BER pathway, but also plays a role in signal transduction by regulating DNA binding of a number of transcriptional factors, including activator protein-1, nuclear factor kappa B (NF-κB), early growth response 1, p53, paired box-containing factors 5 and 8 (PAX5 and PAX8), hepatic leukemia factor, nuclear factor erythroid-related factor 2, cAMP response element binding protein, nuclear respiration factor 1, activating transcription factor 1, nuclear transcription factor Y, v-myb avian myeloblastosis viral oncogene homolog, and polyomavirus enhancer binding protein 2.12 APE1/Ref-1 was also identified as a direct trans-acting factor for repressing human parathyroid hormone and renin genes by binding to the negative calcium-response element in their promoters. APE1/Ref-1 also interacted with several other trans-acting factors, including hypoxia-induced factor-1α, signal transducer and activator of transcription 3 (STAT3), Y box binding protein 1, histone deacetylase 1, and cAMP response element binding protein (CBP/p300), and formed distinct trans-acting complexes.12–14 Further, APE1 is an essential factor stabilizing telomeric DNA, and its deficiency is associated with telomere dysfunction and segregation defects in immortalized cells, maintaining telomeres by either alternative lengthening of the telomere pathway or telomerase expression, and in normal human fibroblasts.15 APE1 controls the intracellular redox state by acting as a redox coactivator of different transcription factors and inhibiting production of reactive oxygen species. The two biological activities of APE1 are located in two functionally distinct domains: the N-terminus containing the nuclear localization signal region, which is principally devoted to redox activity through Cys65, and the C-terminus, which possesses enzymatic activity on the abasic sites of DNA.12–14 The redox function of APE1 is found only in mammals and not in other vertebrates. APE1 is essential for the life process, as deletion of both alleles (Apex−/−) results in early-stage embryonic lethality in animals.16 Cell lines with complete deficiency of APE1 are also nonviable, further demonstrating its significance in maintaining essential cell activity.16 Altered APE1 expression and/or activity is observed in prostate, lung, cervical, ovarian, and colon cancers, as well as osteosarcoma and retinoblastoma.17–20 More importantly, overexpression of APE1 in malignancy is associated with a poor prognosis, chemoresistance or radioresistance, and increased angiogenesis.17,19,21 Nowadays, APE1 represents a promising target for cancer treatment. Targeting of this protein using antisense oligonucleotides, RNA interference, and natural and chemical agents has sensitized tumor cells to radiotherapy or chemotherapeutic drugs.17,22,23 Selective small-molecule inhibitors of APE1 have shown antitumor activity in murine models.24
Gossypol, a natural polyphenolic aldehyde isolated from the seeds of the cotton plant (Gossypium malvaceae), has shown antifertility, antioxidant, anticancer, antiviral, antiparasitic, and antimicrobial properties.25 Gossypol exists in two enantiomeric forms, (+) and (−), and naturally occurring gossypol exists as a racemic mixture of (+) and (−) enantiomers. The (−) enantiomer of gossypol (AT-101; Figure 1) has been found to have a more potent cytotoxic effect than the (+) enantiomer or racemic gossypol. Gossypol mimics BH3-only proteins, induces production of reactive oxygen species, and thus triggers apoptosis of cancer cells.26 The BH3-only proteins of the B-cell lymphoma 2 (Bcl-2) family (having only the Bcl-2 homology domain BH3) can trigger apoptosis by binding to prosurvival members of this family and neutralizing their functional activity.27,28 The ability of gossypol to bind to Bcl-2 and Bcl-extra large (Bcl-xl) was discovered using computer-assisted molecular modeling and fluorescence polarization assays.26 AT-101 possesses higher affinity for Bcl-2 and Bcl-xl than the (+) enantiomer or racemic gossypol. Fluorescence polarization binding studies and nuclear magnetic resonance structural analyses have revealed that racemic gossypol binds to the BH3 binding pocket of Bcl-xl with a Ki of ~0.3 μM and to the BH3 binding pocket of Bcl-2 with a Ki of ~10 μM.29 AT-101 inhibits Bcl-2 and disrupts heterodimerization of Bcl-2, myeloid cell leukemia 1 (Mcl-1), and Bcl-xl with proapoptotic family members.30 AT-101 was more efficient than etoposide at inducing caspase 3 activation and phosphatidylserine externalization in the setting of Bcl-2 or Bcl-xl overexpression.31 The cellular fate between apoptosis and survival depends on the balance between proapoptotic and antiapoptotic protein levels.28 Constitutively high levels of Bcl-2 or Bcl-xl have been associated with chemoresistance and more aggressive phenotypes in hematological malignancies and solid tumors.32 Overexpression of Bcl-2 and Bcl-xl has been found to stabilize the outer mitochondrial membrane and prevent release of cytochrome c following a variety of insults.33 As a pan-Bcl-2 inhibitor, gossypol has been investigated in patients with hematological malignancies and a number of solid tumors including NSCLC, SCLC, prostate cancer, and breast cancer, used alone or more often in combination with other cytotoxic agents.34–40 As a single agent in Phase I and Phase II trials, AT-101 was generally tolerated and showed cytoreductive activity in chronic lymphocytic leukemia, non-Hodgkin’s lymphoma, and castrate-resistant prostate cancer patients.35 However, in other Phase I/II studies in solid tumors, including advanced breast cancer, NSCLC, and SCLC, AT-101 either as a single agent or in combination therapy failed to show clinical efficacy, mainly due to dose-related organ toxicities, such as small intestinal obstruction, and for other unknown reasons.34,36–38 In a double-blind, placebo-controlled, randomized Phase II study of AT-101 plus docetaxel as second-line therapy for NSCLC, the median overall survival was 7.8 months for docetaxel plus AT-101 versus 5.9 months for docetaxel plus placebo (P=0.21).37 AT-101 plus docetaxel was well tolerated, with an adverse event profile indistinguishable from that of the base docetaxel regimen. In a Phase I study that included 20 cancer patients, oral dosing of AT-101 plus CDDP and etoposide resulted in a partial response in patients with extensive SCLC (n=7), high-grade neuroendocrine tumor, esophageal cancer, or NSCLC.40 AT-101 with CDDP and etoposide was well tolerated with growth factor support, and no pharmacokinetic interactions were observed. We hypothesized that AT-101-based combination regimens should be optimized with regard to dosage, dosing regimen, combined drugs, and dosing sequence to produce greater antitumor efficacy. Interestingly, we found that gossypol is a potent inhibitor of APE1/Ref-1, suppressing its DNA repair and redox activities via direct interactions.41 Gossypol also potentiates the antitumor effect of CDDP in nude mice bearing HeLa xenograft tumors.41 We observed that APE1/Ref-1 is involved in regulation of mitochondrial function by modulating the DNA-binding activity of nuclear respiration factor 1.42 Our recent study has shown that sequential AT-101 significantly enhances the sensitivity of A549 cells to CDDP via APE1-mediated modulation of the interleukin-6/STAT3 signaling pathway both in vitro and in vivo.43 AT-101 is a small-molecule inhibitor of antiapoptotic Bcl-2 family molecules,27 induces apoptosis in various types of cancer cells, and sensitizes cancer cells to chemotherapy and radiotherapy.25 Bcl-2 is a prosurvival molecule that functions as a proangiogenic signaling factor. Angiogenesis is a key process in the initiation, growth, development, and metastasis of cancer, and thus represents a useful target in anticancer treatment.44–46 Gossypol suppressed angiogenesis in human prostate cancer through modulation of NF-κB/activator protein-1 and vascular endothelial growth factor (VEGF) signaling pathways.47,48 AT-101 inhibited angiogenesis and tumor growth in human squamous cell carcinomas.49 Therefore, we hypothesized that combination of AT-101 and CDDP would inhibit angiogenesis and enhance cell apoptosis in NSCLC. In this study, we tested our hypothesis and found that AT-101 in combination with CDDP promoted apoptosis and antiangiogenesis in NSCLC via inhibition of APE1 repair and redox activity.
 | Figure 1 Chemical structures of AT-101 and CDDP. |
Materials and methods
Chemicals and reagents
AT-101 (purity: 99.07%) was sourced from Selleckchem Inc (Shanghai, People’s Republic of China). CDDP was obtained from Jiangsu Hanson Pharmaceutical Co Ltd (Jiangsu, People’s Republic of China). Dulbecco’s Modified Eagle’s Medium (DMEM), Roswell Park Memorial Institute 1640 medium, and Western blotting substrate were purchased from Thermo Scientific Inc (Beijing, People’s Republic of China). Dimethyl sulfoxide (DMSO), D-glucose, propidium iodide, fetal bovine serum, ethylenediaminetetraacetic acid (EDTA), dithiothreitol (DTT), 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), and phosphate-buffered saline were purchased from Sigma-Aldrich (St Louis, MO, USA). The polyvinylidene difluoride membrane was purchased from EMD Millipore Inc (Bedford, MA, USA). The Cell Counting Kit-8 was obtained from Beyotime Inc (Jiangsu, People’s Republic of China). Transwell® chambers were purchased from Costar Inc (Costar, NY, USA). Matrigel™, propidium iodide, and the Annexin V-fluorescein isothiocyanate apoptosis detection kit were sourced from BD Biosciences Inc (New York, NY, USA). The Pierce™ bicinchoninic acid protein assay kit was obtained from Thermo Scientific Inc (Waltham, MA, USA). Primary antibodies against human APE1, STAT3, β-actin, and Ki-67 were purchased from Santa Cruz Biotechnology Inc (Dallas, TX, USA). Primary antibodies against phosphorylated H2A histone family member X (γH2AX) and CD34 were sourced from EMD Millipore Inc and ZSGB-Bio Inc (Beijing, People’s Republic of China), respectively.
Cell culture
The A549 human NSCLC cell line was purchased from the Cell Bank of the Chinese Academy of Sciences (Shanghai, People’s Republic of China) and cultured in Roswell Park Memorial Institute 1640 medium. Human umbilical vein endothelial cells (HUVECs) were obtained from the American Type Culture Collection (Manassas, VA, USA) and cultured in DMEM. Both media were supplemented with 10% fetal bovine serum. The cells were cultured in DMEM supplemented with 10% (volume/volume [v/v]) heat-inactivated fetal bovine serum, 100 U/mL penicillin, and 100 μg/mL streptomycin in a humidified 5% CO2/95% air incubator at 37°C.
Cell viability assay
Cell viability was determined using the Cell Counting Kit-8. This assay used Dojindo’s tetrazolium salt, WST-8 [2-(2-methoxy-4-nitrophenyl)-3-(4-nitrophenyl)-5-(2,4-disulfophenyl)-2H-tetrazolium, monosodium salt], which produces a water-soluble formazan dye upon bioreduction in the presence of an electron carrier, 1-methoxy-5-methylphenazinium methyl sulfate. A549 cells were seeded into 96-well plates (7,000 cells per well) and incubated overnight, and then treated with AT-101 at 1.25, 2.5, 5, 10, and 20 μM, CDDP alone, or 5 μM AT-101 plus CDDP at 1.25, 2.5, 5, 10, and 20 μM for 48 hours. The drugs were dissolved in DMSO with a final DMSO concentration of 0.05% (v/v). The cells were washed and 110 μL was added to each well and incubated for a further 4 hours at 37°C in 5% CO2. The optical density was measured by reading the plates at an absorbance of 490 nm on a microplate reader.
Cell migration assay
Cell migration was determined using a Transwell (8 μm pore size) assay as described previously.19 In brief, A549 cells were suspended in serum-free medium containing 5 μM AT-101, 5 μM CDDP, or 5 μM AT-101 plus 5 μM CDDP dissolved in 0.05% DMSO. HUVECs were suspended in serum-free DMEM. The A549 or HUVEC cell suspensions were seeded to the upper chambers, and the lower chambers were filled with Roswell Park Memorial Institute 1640 medium containing 10% fetal bovine serum or tissue culture medium (TCM). After 18 hours of incubation at 37°C, the cells were fixed using 4% paraformaldehyde solution and stained using crystal violet. The number of migrated cells was counted using an inverted microscope (CKX41, Olympus, Tokyo, Japan). The assay was conducted three times.
Analysis of cell apoptosis using flow cytometry
Cell apoptosis was confirmed using flow cytometry. In brief, A549 cells were plated in 6-well plates, incubated overnight, and then treated with the vehicle control, 5 μM AT-101, 5 μM CDDP, or 5 μM AT-101 plus 5 μM CDDP dissolved in 0.05% DMSO. After 24 hours of treatment, the cells were harvested and washed twice with phosphate-buffered saline. The cell suspensions were double-stained with 50 μL of propidium iodide and 50 μL of Annexin V-fluorescein isothiocyanate for 15 minutes at room temperature in the dark. Apoptotic cells were determined by flow cytometry and 10,000 events were collected for analysis.
Matrigel tube formation assay in vitro
The Matrigel tube formation assay was conducted as described by us previously.19 In brief, HUVECs were cultured in TCM (800 μL) from the supernatant of A549 cells treated with the drugs at a density of 120,000 cells per well in a 24-well plate precoated with 350 μL thick Matrigel. The HUVECs were then maintained at 37°C in a 5% CO2 atmosphere for 24 hours. Capillary tube formation was calculated in five random areas using an CKX41 inverted microscope.
Tumor tissues from xenografted BALB/c nude mice
Tumor tissues were collected from xenografted BALB/c nude mice treated with the different drugs. The experimental protocol using the nude mice was approved by the ethics committee of the Third Military Medical University, Chongqing, People’s Republic of China. A549 cells were inoculated subcutaneously in the left anterior axilla of each nude mouse. When the tumor volume reached 200 mm3, the mice were treated with vehicle control (sesame oil, China Oil Food Co, Beijing, People’s Republic of China) by oral gavage for 10 consecutive days, CDDP at 4 mg/kg/day by intraperitoneal injection on days 3, 5, 7, and 9, AT-101 dissolved in sesame oil at 35 mg/kg/day by oral gavage for 10 consecutive days, or AT-101 at 35 mg/kg/day pretreated for 2 days plus CDDP at 4 mg/kg/day by intraperitoneal injection on days 3, 5, 7, and 9.
Western blotting assay
The H2A histone family member X (H2AX) is a key factor in the repair process for damaged DNA. H2AX can be phosphorylated on Ser1, acetylated on Lys 5, and ubiquitinated on Lys119, but it differs from other H2A molecules because it is phosphorylated on the 139th serine residue in the presence of DNA damage.50,51 This phosphorylated H2AX protein is termed γH2AX which is an early indicator of formation of breaks in double-stranded DNA and the response to DNA damage.50,51 Detection and visualization of γH2AX allows assessment of DNA damage, related DNA damage proteins, and DNA repair. Expression levels of APE1 and γH2AX in A549 cells and tumor tissue were determined by Western blotting assay, as described previously.43,52 Briefly, an equal amount of protein was separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and then transferred onto a polyvinylidene difluoride membrane at 100 V for 2 hours at 4°C. The membrane was then blocked with 10% milk for one hour, and incubated with primary antibody for one hour, and then secondary antibody for a further hour. The following antibodies were used for immunoblotting: anti-APE1 (1:5,000) and anti-β-actin (1:5,000, Santa Cruz Biotechnology Inc), and anti-γH2AX (1:500, EMD Millipore Inc). The blots were exposed to X-ray films and visualization was performed using Quantity One imaging software (Bio-Rad Laboratories Inc, Hercules, CA, USA). The protein level was normalized to the matching densitometric value of β-actin.
Electrophoretic mobility shift assay of APE1/Ref-1 redox activity
An electrophoretic mobility shift assay (EMSA) was performed to assess the effect of combination of AT-101 and CDDP on APE1 redox activity-mediated STAT3-DNA binding in the nuclear extract (NE) from A549 cells as described previously.38,47 Briefly, biotin-labeled double-stranded oligonucleotide DNA was used as the probe that contains the STAT3 direct consensus sequence (5′-GATCCTTCTGGGAATTCCTAGATC-3′). To the reaction system, 12 μg NE from A549 cells was included and treated with vehicle control (DMSO), 50 μM AT-101, 20 μM CDDP, or 50 μM AT-101 plus 20 μM CDDP dissolved in 0.05% DMSO for 6 hours. In the redox activity assay, we used much higher concentrations of AT-101 and CDDP. We considered that this in vitro redox assay was cell-free, and the specific and non-specific binding of these two compounds to the enzyme APE1 and other proteins might be remarkable, so the free drugs available for the redox enzyme APE1 were much less than the concentrations used for other assays.
To determine the APE1/STAT3 interaction in A549 cells, we prepared a reduced APE1 solution (0.17 ng/μL) with 4 mM DTT at a ratio of 9:1 (purified APE1 protein to DTT) for 10 minutes. The APE1 was then added to NE for redox reactions, in which the final concentration of DTT was 0.04 mM. After incubation, the reaction was electrophoresed on 5% polyacrylamide gel at 100 V for one hour and then transferred to a Zeta-Probe GT nylon membrane (Bio-Rad Laboratories Inc). The probes were detected by horseradish peroxidase-conjugated streptavidin (1:300), and the bands were visualized by electrochemiluminescence reagents provided with the kit. The resultant bands were quantified using Quantity One imaging software (Bio-Rad Laboratories Inc).
AP endonuclease activity assay for APE1/Ref-1
To test the inhibition of AP endonuclease activity by AT-101, an oligonucleotide cleavage assay designed to monitor the cleavage of a substrate to product through electrophoretic separation was applied as described by us previously.41,42,53 An abasic site (dSpacer, ie, Int 1′,2′-dideoxyribose, used to introduce a stable abasic site within an oligonucleotide) containing 39-nucleotide oligo with IR700-labeled at the 5′ end (purchased from Integrated DNA Technologies Inc, Coralville, IA, USA) was annealed with the unlabeled complementary oligonucleotide through a standard annealing reaction. The activity assay system consisted of 1.0 pmol of IR700-labeled duplex oligonucleotide, 5× AP assay buffer (50 mM HEPES at pH 7.5, 100 mM KCl, 1 mM MgCl2, and 1 mM DTT), and 6 μg of APE1 (a kind gift from Dr David Wilson) in a 10 μL reaction system, and was incubated at 37°C for 10 minutes. The reaction was terminated by adding 2× stop buffer (90% formamide, 20 mM EDTA, and bromophenol blue/xylene cyanol) and denaturing at 95°C for 5 minutes. Equal volumes of the reaction products from the AP endonuclease activity assay were resolved on 15% polyacrylamide gel with 7 M urea in 5× Tris-borate EDTA buffer at 300 V for 70 minutes. Wet gels were photographed using Odyssey® Infrared imaging systems at IRDye-700 nm (Li-Cor Biotechnology Inc, Lincoln, NE, USA).
Tumor angiogenesis in vivo and immunohistochemistry analysis
Tumor tissues from A549-xenografted nude mice were fixed overnight in 4% paraformaldehyde, dehydrated, embedded in paraffin, and sectioned using a RM2235 rotary microtome (Leica Biosystems Inc, Wetzlar, Germany). The following was used for immunohistochemical assay: anti-Ki-67 (1:500), anti-APE1 (1:10,000), and anti-CD34 (1:200). Tissues were scored and microvessel density was counted as previously described.19 We counted the number of Ki-67-positive cells from 200 cancer cells and observed five areas for each section. For quantification of microvessel density, CD34-positive cells were counted under a light microscope at 40× magnification. For each section, three fields were captured and the results are expressed as the mean.
Statistical analysis
The data are presented as the mean ± standard deviation. Statistical analysis was carried out using one-way analysis of variance. P<0.05 was considered to be statistically significant.
Results
AT-101 potentiates cytotoxicity of CDDP in A549 cells
AT-101, as a BH3 mimic agent, is a promising anticancer drug that inhibits proliferation and growth of various cancer cells in vitro and in vivo.43,47,48 Our published data also show that sequential AT-101 plus CDDP can significantly inhibit proliferation and migration of A549 cells.43 Since the IC50 value for CDDP or AT-101 was 9.5 μM and 20 μM in A549 cells, respectively, we used lower concentrations to investigate the effect of the combination of AT-101 and CDDP on proliferation and migration of A549 cells. A549 cells were divided to be treated with 5 μM CDDP or 5 μM CDDP plus 5 μM AT-101. Cell viability was more significantly reduced by the combination treatment than by CDDP monotherapy (P<0.001, Figure 2). When CDDP was used alone, the IC50 was 8.7 μM, and the IC50 was brought down to 1.0 μM when the A549 cells were treated with AT-101 plus CDDP.
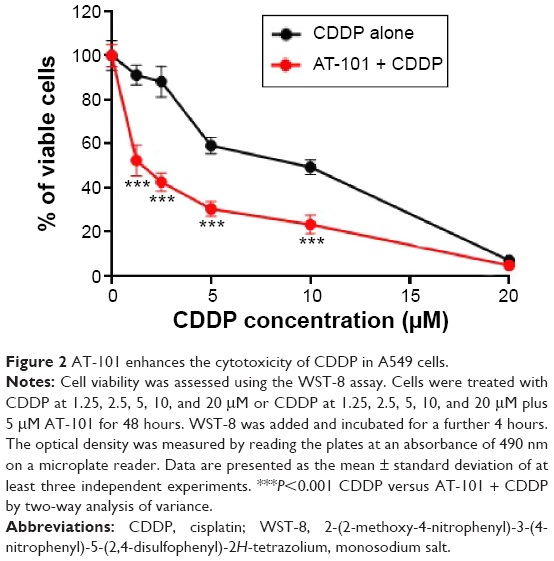 | Figure 2 AT-101 enhances the cytotoxicity of CDDP in A549 cells. |
AT-101 enhances the proapoptotic effect of CDDP in A549 cells
We used flow cytometry to evaluate the effect of AT-101 plus CDDP on apoptosis in NSCLC A549 cells. After 24 hours of treatment, the apoptotic rate of A549 cells treated with vehicle control, 5 μM CDDP, 5 μM AT-101, or 5 μM AT-101 plus 5 μM CDDP was 6.47%±0.43%, 14.78%±0.29%, 11.41%±0.49%, and 25.74%±0.52%, respectively. Our results show that the apoptosis of A549 cells was significantly increased when the cells were treated by AT-101 plus CDDP when compared with monotherapy using CDDP or AT-101 alone (P<0.001, Figure 3A and B).
 | Figure 3 AT-101 enhances the proapoptotic effect of CDDP in A549 cells. |
AT-101 enhances antimigration ability of CDDP in A549 cells
We conducted Transwell migration assays to determine the effect of AT-101 plus CDDP on the migration capacity of A549 cells. As shown in Figure 4A and B, the mean number of migratory A549 cells in the presence of TCM treated with vehicle control, 5 μM CDDP, 5 μM AT-101, 5 μM AT-101 plus 5 μM CDDP was 193.7±7.0, 150.3±2.9, 138.3±5.8, and 67.6±4.5, respectively. The migration activity of A549 cells was significantly reduced in the presence of TCM treated with the combination treatment than in AT-101 or CDDP alone (P<0.01).
 | Figure 4 AT-101 enhances the antimigration ability of CDDP in A549 cells exposed to A549 cell-conditioned medium. |
AT-101 enhances antimigration ability of CDDP in HUVECs exposed to A549 cell-conditioned medium
Next, we conducted Transwell migration assays to examine the effect of AT-101 plus CDDP on migration capacity in HUVECs. As shown in Figure 5A and B, after treatment for 18 hours, the number of migratory HUVECs in the presence of A549 cell-conditioned TCM treated with vehicle control, 5 μM CDDP, 5 μM AT-101, or 5 μM AT-101 plus 5 μM CDDP was 103.4±2.8, 99.5±2.1, 89.6±2.9, and 56.2±4.0, respectively. The data show that the migration activity of HUVECs was significantly decreased in the presence of A549 cell-conditioned TCM treated with the combination treatment than that in AT-101 or CDDP alone (P<0.01).
 | Figure 5 AT-101 enhances the antimigration ability of CDDP in HUVECs exposed to A549 cell-conditioned medium. |
AT-101 enhances inhibitory effect of CDDP on capacity of A549 cells to induce HUVEC tube formation
Tube formation of human endothelial cells is an extremely important step in tumor-associated angiogenesis. Therefore, we examined the effect of a combination of AT-101 and CDDP on tumor angiogenesis in HUVECs treated with A549 cell-conditioned TCM. The Matrigel tube formation assay was performed in HUVECs exposed to TCM. Twenty-four hours later, the mean number of tubular structure of HUVECs in the presence of TCM treated with vehicle control, 5 μM CDDP, 5 μM AT-101, 5 μM AT-101 plus 5 μM CDDP was 23.7±1.7, 20.0±1.4, 19.3±1.2, and 1.0±0.8, respectively. Monotherapy with AT-101 or CDDP only slightly reduced capillary tube formation, but the combination therapy significantly inhibited the angiogenic capacity of HUVECs in the presence of TCM from A549 cells treated with AT-101 plus CDDP (P<0.001, Figure 6A and B).
 | Figure 6 AT-101 enhances the inhibitory activity of CDDP on the capillary morphogenesis of HUVECs. |
AT-101 enhances the inhibitory effect of CDDP on DNA repair and redox activity of APE1 in A549 cells
APE1 plays an important role in angiogenesis.12,19 On the other hand, overexpression of APE1 has been found in various types of cancer cells, which correlates with tumor resistance and a poor prognosis.17,49 Our previous data have shown that AT-101 is a potent APE1 inhibitor.41 However, the effect of AT-101 plus CDDP on expression of APE1 remains unclear. Using the Western blotting assay, we did not observe any significant effect of AT-101 plus CDDP or monotherapy on the expression of APE1 in A549 cells (Figure 7).
 | Figure 7 AT-101 plus CDDP does not affect expression levels of APE1/Ref-1 in A549 cells determined by Western blotting assay. |
APE1 has two key functions, ie, DNA repair and redox activity.12 The redox-regulating activity of APE1 affects the responses to stress, DNA repair, cell survival, inflammation, and angiogenesis.12 APE1 directly regulates STAT3 transcriptional activity.54 Our previous study revealed that sequential treatment of AT-101 sensitizes A549 cells to CDDP via regulation of STAT3-DNA binding when APE1 was at reduced status induced by dithiothreitol.43 Therefore, we performed EMSA to investigate whether combination of AT-101 and CDDP influenced the redox function of APE1. Our data show that STAT3-DNA binding activity in the NE extracted from A549 cells treated with 50 μM AT-101 or 50 μM AT-101 plus 20 μM CDDP was decreased by 60% and 90%, respectively, as compared with the vehicle control (P<0.01 or P<0.001, Figure 8A and C). AT-101 alone inhibited DNA binding of STAT3 in A549 cells, but inhibition of STAT3 transcriptional activity was more marked when treated with AT-101 plus CDDP, as compared with CDDP or AT-101 alone.
 | Figure 8 AT-101 enhances the attenuating effect of CDDP on APE1/Ref-1 redox activity and thus promotes STAT3-DNA binding as determined by electrophoretic mobility shift assay. |
Furthermore, the EMSA was used to explore whether APE1 redox activity directly regulated STAT3-DNA binding. To test the APE1/STAT3 interaction, we prepared reduced APE1 as previously described.43 As shown in Figure 8B and D, addition of reduced APE1 to the NE of A549 cells stimulated and activated the transcriptional activity of STAT3. On the other hand, when NE supplemented with reduced APE1 status was treated with 50 μM AT-101 or 50 μM AT-101 plus 20 μM CDDP, the transcriptional activity of STAT3 was decreased by 50% and 80%, respectively, when compared with unreduced APE1 status (P<0.01 and P<0.001). Addition of 0.04 mM DTT increased the transcriptional activity of STAT3 by 80%. The EMSA results showed that the combination of AT-101 and CDDP was more effective in attenuating the redox ability of APE1 and stimulated STAT3-DNA binding, compared with the monotherapy.
APE1 is an essential BER enzyme that is responsible for repair of DNA damage resulting from oxidative stress, chemotherapy, and radiotherapy.14 Since we had observed that combination of AT-101 and CDDP promoted apoptosis of A549 cells, we hypothesized that the repair function of APE1 might be reduced or abrogated in A549 cells treated with AT-101 plus CDDP. First, we determined the level of γH2AX in A549 cells treated with AT-101 plus CDDP using Western blotting assay. The data showed that both monotherapy and combination therapy significantly increased γH2AX expression compared with the vehicle control (P<0.01, Figure 9A), suggesting that increased DNA damage contributed to apoptosis of A549 cells exposed to AT-101, CDDP, or AT-101 plus CDDP.
 | Figure 9 AT-101 enhances the inhibitory effect of CDDP on APE1/Ref-1 DNA repair activity in A549 cells as indicated by the increased γH2AX level determined by Western blotting assay. |
Second, we performed an AP endonuclease activity assay to testify the abrogation of repair function in A549 cells treated with AT-101 plus CDDP. As shown in Figure 10A and B, AP endonuclease activity was significantly inhibited by monotherapy and by combination therapy, and AP endonuclease activity was more significantly decreased in A549 cells receiving the combination therapy than the monotherapy (P<0.01). APE1 cleaves the phosphodiester backbone on the 5′ side of the AP site via a hydrolytic mechanism.11,13 CDDP acting as an inhibitor of APE1 may strengthen its DNA-damaging effect. It is unknown how CDDP attenuates the AP endonuclease activity of APE1. The APE1 redox function is required in promotion of angiogenesis.19,55,56
 | Figure 10 AT-101 enhances the inhibitory effect of CDDP on APE1 repair activity in A549 cells determined by the oligonucleotide cleavage assay. |
AT-101 plus CDDP does not affect tumor expression of APE1 in xenografted mice
We assessed APE1 expression in A549 xenograft tumor tissues using immunohistochemistry. The result showed that APE1 expression levels remained unchanged in xenograft tumor tissues in all groups (Figure 11A and B).
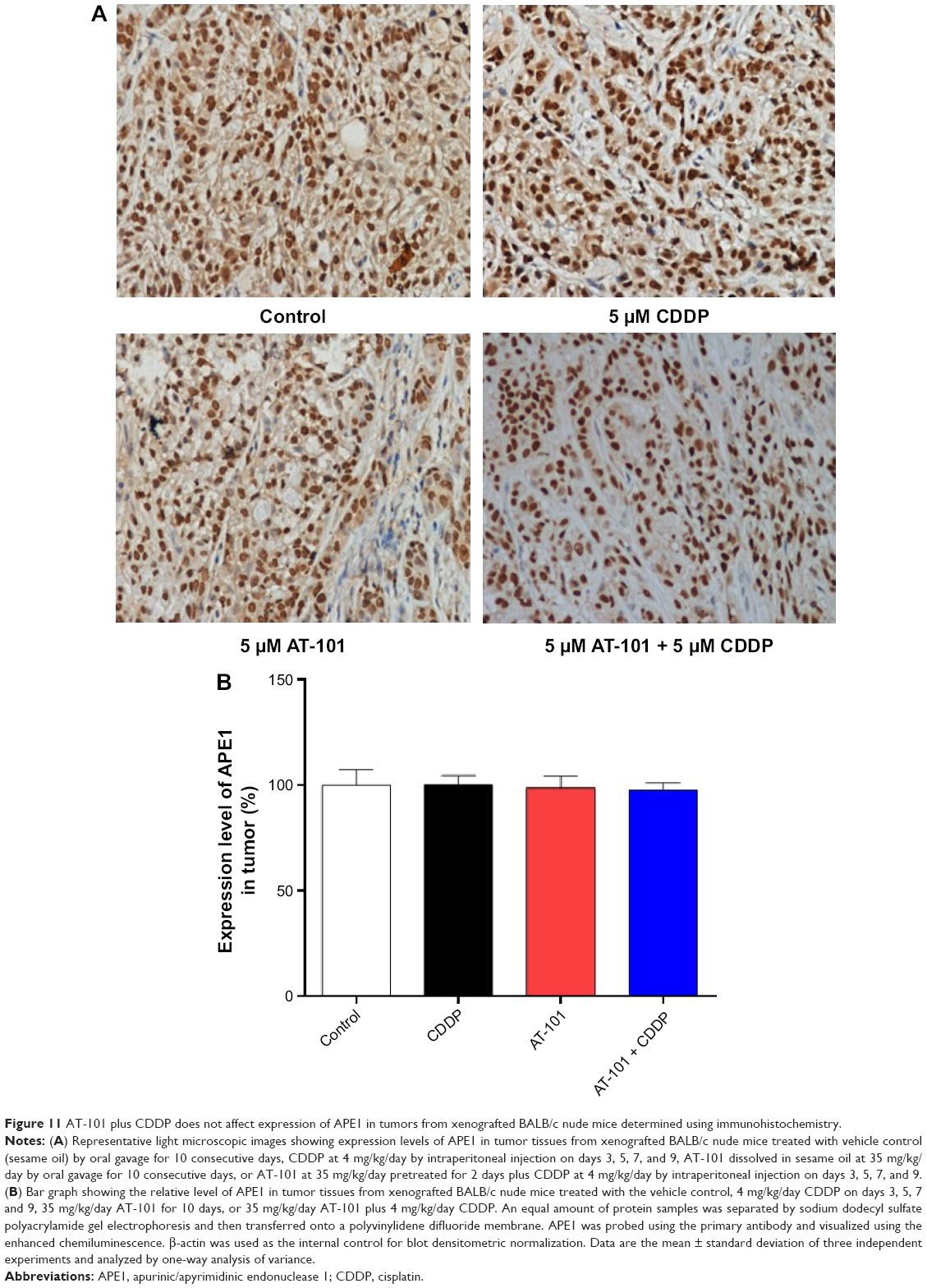 | Figure 11 AT-101 plus CDDP does not affect expression of APE1 in tumors from xenografted BALB/c nude mice determined using immunohistochemistry. |
AT-101 enhances the antiangiogenic effect of CDDP in xenografted mice
We have already observed enhanced anticancer efficacy of combination therapy using AT-101 and CDDP in a xenograft mouse model,41,43 but the impact on angiogenesis is unknown. Thus, we used the xenograft tumor tissues to further analyze the effect of AT-101 plus CDDP on angiogenesis using immunohistochemistry. Representative images are shown in Figure 12A and B. The number of CD34-positive microvessels in the groups treated with vehicle control, either CDDP or AT-101 alone, and AT-101 plus CDDP was 12.0±1.6, 10.7±1.2, 7.7±0.9, and 1.3±0.5, respectively. As expected, the density of CD34-positive microvessels in tumor sections was much lower in tumors treated with the combination of AT-101 and CDDP than in the other groups (P<0.01).
 | Figure 12 AT-101 enhances the antiangiogenic activity of CDDP in tumors from xenografted BALB/c nude mice as determined by counting of CD34-positive microvessels in tumor tissues. |
AT-101 enhances the inhibitory effect of CDDP on tumor cell proliferation in xenografted mice
Ki-67 is a cancer antigen that is found in growing and dividing cells but is absent in the resting phase of cell growth. We stained Ki-67 protein to investigate tumor cell proliferation levels. Figure 13A and B shows that the mean number of Ki-67 positive cells per 200 cells in the vehicle control, either CDDP or AT-101 alone, and AT-101 plus CDDP groups was 96.6±18.4, 65.8±5.8, 60.6±5.2, and 12.2±4.1, respectively. In accordance with the results for angiogenesis, tumor cell proliferation in the combination treatment group was significantly inhibited when compared with the other groups (P<0.001, Figure 13A and B).
 | Figure 13 AT-101 enhances the inhibitory effect of CDDP on tumor cell proliferation as determined by counting the number of Ki-67-positive cells in tumors from xenografted BALB/c nude mice. |
Discussion
Sensitizing tumor cells to DNA damaging agents by targeting DNA repair pathways is a possible strategy to kill cancer cells, since efficient DNA repair is an important mechanism by which tumor cells exert chemoresistance.57,58 In the present study, we assessed the effects of combined therapy using AT-101 and CDDP on apoptosis and angiogenesis in NSCLC and elucidated the underlying molecular mechanisms for the enhancement of the antitumor activity of CDDP by AT-101, a BH3 mimetic and pan-inhibitor of Bcl-2. We demonstrated that AT-101 combined with CDDP significantly inhibited A549 cell proliferation and migration and promoted apoptosis. We also found for the first time that a combination of AT-101 and CDDP markedly decreased the capacity of A549 cells to induce HUVEC tube formation. CDDP, as a first-line therapy, is one of the most frequently used chemotherapeutic drugs for treatment of advanced NSCLC.8,59 As a DNA intercalator, CDDP can crosslink with the purine bases in DNA, interfering with DNA repair mechanisms, causing DNA damage, and subsequently inducing apoptosis in cancer cells.10 The main DNA pathways involved in the repair of platinum-induced DNA damage are nucleotide excision repair and homologous recombination.57 However, the efficacy of CDDP is markedly limited due to chemoresistance and severe organ toxicities following repeated CDDP treatment. The chemoresistance of NSCLC to CDDP may involve altered drug transport, enhanced drug detoxification, enhanced cell survival, ineffective induction of cell death, altered DNA repair, and altered apoptotic and autophagic cell death.59–61 There is an urgent need to identify and develop efficient reversing agents and modulators to overcome resistance to platinum-based chemotherapy. AT-101, a BH3 mimic agent and Bcl-2 pan-inhibitor derived from the extract of cotton seeds, has shown promising anticancer activity in murine studies but failed to have clinical efficacy in cancer patients in Phase I and II trials when used alone or in combination with other cytotoxic drugs.34–40 To date, there is only one Phase I report on the use of AT-101 with CDDP plus etoposide in 20 patients with advanced solid tumors.40
AT-101 may act by directly binding to Bcl-2 or Bcl-xl and inhibiting their antiapoptotic activity by displacing bound proapoptotic factors such as Bak, Bax, or BH3-domain only proteins, but it may also interact directly with proapoptotic Bcl-2 family members such as Bak to promote apoptotic activity.31,41 Bak resides in the mitochondrial outer membrane and has been shown to contain a homologous BH3 binding pocket critical for interactions with the Bak-activating, BH3-domain only protein Bid.28 Once activated, Bak oligomerizes in the mitochondrial outer membrane and induces both defects in membrane integrity and release of apoptogenic factors, including cytochrome c. Our present study has clearly demonstrated that AT-101 enhanced the proapoptotic effect of CDDP in A549 cells (Figure 3). Dysregulation of apoptotic mechanisms results in a myriad pathologic conditions, including autoimmune disorders, neurologic diseases, and cancer.62,63 Defective apoptosis is often observed in both solid and hematologic malignancies. The antiapoptotic members of the Bcl-2 family (Bcl-2, Bcl-xl, Bcl-w, Mcl-1, and A-1) inhibit apoptosis by binding to multidomain proapoptotic members (Bax and Bak), which are embedded in the mitochondrial outer membrane.32 Many cancers overexpress antiapoptotic members of the Bcl-2 family and are resistant to death stimuli. Overexpression of either Bcl-2 and Bcl-xl serves to suppress the mitochondria-mediated pathway of apoptosis, and contributes to development of chemoresistance and radioresistance.33,63,64 As a BH3-mimetic, AT-101 acts like BH3-only proteins and interacts with the BH3 binding groove of antiapoptotic Bcl-2 proteins, thereby preventing their interaction with the proapoptotic Bcl-2 family proteins, Bax and Bak. Gossypol has been found to inhibit the antiapoptotic function of Bcl-2, Bcl-xl, and Mcl-1 and thus trigger apoptosis of cancer cells.41,65 Interestingly, gossypol was previously examined as a male antifertility agent in clinical studies.66 The antifertility effect of gossypol is possibly due to inhibition of the cellular energy metabolism of spermatogonia67 rather than its activity as a BH3 mimetic. The mechanisms for the antiproliferative and proapoptotic activity of gossypol and AT-101 in cancer cells are complicated. They may include inhibition of DNA synthesis, disrupted cell cycle control, altered intracellular calcium hemostasis, inhibition of protein kinase C, interaction with steroid receptor coactivators, and modulation of several components of mitochondrial apoptotic signaling.25,26,41,47,48 Gossypol can activate the intrinsic apoptotic pathway by increasing the generation of reactive oxygen species,68 activating p53,69 and suppressing NF-κB activity.70,71 Gossypol can upregulate the expression of proapoptotic BH3-only proteins and induce a conformational change in Bcl-2, which renders it proapoptotic.72,73 As a potent apoptosis inducer, AT-101 may be used as a chemosensitizer to overcome drug resistance of cancer cells. AT-101 has been shown to effectively induce cell death in chemoresistant leukemia cell lines overexpressing Bcl-2 and Bcl-xl.31 AT-101 induced marked apoptosis with high efficiency in CDDP-resistant head and neck cancer cell lines that express high levels of Bcl-xl.74 There is a synergistic drug interaction between gossypol and gemcitabine in gemcitabine-resistant cancer cells with high Bcl-2 expression, and reversal of gemcitabine resistance by gossypol was associated with upregulation of the proapoptotic proteins phorbol-12-myristate-13-acetate-induced protein 1 (Noxa) and Mcl-1 and downregulation of the antiapoptotic proteins Bcl-2 and Bcl-xl.72 Activation of Noxa could lead to BH3 motif-dependent localization to the mitochondria and interaction with antiapoptotic Bcl-2 family members, resulting in activation of apoptosis. Macoska et al75 reported a synergistic interaction in bladder cancer cell lines treated with AT-101 in combination with gemcitabine or carboplatin, resulting in an increased apoptosis via decreased expression of prosurvival Bcl-xl and Mcl-1 and increased expression of proapoptotic genes. Microarray gene expression analysis also revealed that expression of proapoptotic genes such as Bax, Bak, apoptosis-inducing factor, Noxa, carbamoyl-phosphate synthetase 2, aspartate transcarbamylase, and dihydro-orotase, poly ADP ribose polymerase, and cytochrome c were increased when cancer cells were treated with gossypol.69 Gossypol and AT-101 can also induce autophagy of multiple types of cancer cells which interplays with the apoptotic pathway.76–81 Gossypol can disrupt the Bcl-2/beclin 1 complex and induce autophagy through a beclin 1-independent mechanism.81 Given these multiple actions, the primary mechanism by which gossypol or AT-101 induces cell death in any particular tumor type is likely to vary depending on levels of antiapoptotic Bcl-2 proteins and other tumor cell-specific factors.
Our previous study showed that AT-101 significantly enhanced the antitumor activity of CDDP in nude mice bearing A549 xenografts through APE1-mediated modulation of the interleukin-6/STAT3 signaling pathway.43 To further explore the underlying mechanisms for the enhancement of the antitumor effect of CDDP by AT-101, we examined the effects of AT-101 plus CDDP on angiogenesis and tumor cell proliferation in xenograft tumor tissues. In vitro, we found that greater inhibition of cell migration and A549 cell-inducing HUVEC angiogenesis was achieved when cells were treated with the combination of AT-101 and CDDP when compared with treatment using AT-101 or CDDP alone. To further validate the data, we investigated the expression of CD-34 and Ki-67 in xenograft tumor tissues from nude mice treated with AT-101, CDDP, or AT-101 plus CDDP. As expected, the density of CD34-positive microvessels in the tumor sections was much lower in tumors treated with the combination of AT-101 and CDDP than in the other treatment groups. In accordance with the results for angiogenesis, expression of Ki-67 protein, being a marker of tumor cell proliferation, was markedly decreased in the combination therapy group when compared with the monotherapy groups. To our knowledge, tube formation is an important step in tumor-associated angiogenesis in human endothelial cells. Recently accumulated evidence has demonstrated that gossypol monotherapy or combined treatment can not only inhibit proliferation of tumor cells, but may also be associated with inhibition of tumor-associated angiogenesis through modulating NF-κB/activator protein-1-dependent/independent signaling or other mechanisms.48,49,82
Angiogenesis is important for tumor growth, invasion, and metastases.83,84 It is a multistep process that includes degradation of the basement membrane by proteases, endothelial cell migration and proliferation, formation of a new basement membrane by recruitment of supporting pericytes and assembly of the new vessels. Angiogenesis is regulated by a tight balance between antiangiogenic agents, such as angiostatin, and proangiogenic agents, like VEGF. Apoptosis is tightly linked to angiogenesis in the development of cancer and chemotherapy for the disease.85 During cytotoxic chemotherapy, apoptosis of endothelial cells in the vascular bed of tumors precedes apoptosis of tumor cells, even when the tumor has become chemoresistant. Administration of an angiogenesis inhibitor which is not directly cytotoxic to tumor cells can increase apoptosis of tumor cells and inhibit tumor growth by inhibiting proliferation and migration of endothelial cells and/or by inducing endothelial apoptosis.46,84,86–88 Further, oncogene expression and loss of tumor suppressor gene activity can at once protect tumor cells against apoptosis and increase their angiogenic output. Inhibition of apoptosis and angiogenesis can be overcome by angiogenesis inhibitors or low-dose metronomic chemotherapy, which continuously exposes endothelial cells in the tumor bed to the drug.89
Currently, the most widely preferred approach for arresting tumor angiogenesis is blockade of the VEGF pathway. VEGF acts through two high affinity receptor tyrosine kinases, VEGF receptor 1 and VEGF receptor 2; both are expressed on normal vascular endothelial cells and are upregulated during angiogenesis. Most of the VEGF isoforms also bind to a third, non-tyrosine kinase receptor on vascular endothelium, neuropilin-1. Mice with knockout of VEGF receptors 1 and 2 are embryonic lethal, indicating that both receptors are essential for angiogenesis. VEGF receptor 2 is essential for the earliest steps of angiogenesis, like formation of blood islands and large vessels, whereas VEGF receptor 1 plays a role in the later course. VEGF signaling in tumor angiogenesis is mainly mediated by VEGF receptor 2. Upon ligand binding, VEGF receptors undergo homodimerization and oligomerization that activate their intrinsic tyrosine kinase activity, leading to phosphorylation of major signaling molecules and kinases, such as stress-activated protein kinase/p38, Src family kinase, focal adhesion kinase, phosphatidylinositol 3-kinases/protein kinase B kinase, extracellular signal-related kinase, and STAT3 in endothelial cells.90,91 Major autophosphorylation sites on the VEGF receptor 2 include Tyr1175 and Tyr1214; other putatively important phosphorylated sites include Tyr951 and Tyr996 in the kinase insert domain and Tyr1054 and Tyr1059 in the tyrosine kinase catalytic domain. These tyrosine residues, when phosphorylated, are involved as docking sites to recruit molecules containing SH2, SH3, or phosphotyrosine-binding domains and to convey signals to downstream pathways. Paracrine VEGF signaling is essential for the angiogenic cascade, proliferation, survival, permeability responses, and endothelial differentiation, while autocrine VEGF signaling only conveys survival signals.90 Drugs targeting the VEGF pathway, such as bevacizumab, sunitinib and aflibercept, have shown activity in certain settings.46,86,87 However, inhibition of VEGF signaling is not effective in all cancers, prompting the need to further understand how the vasculature can be effectively targeted in tumors. The clinical use of current angiogenesis inhibitors is further limited by several factors, such as adverse reactions, acquired drug resistance, and lack of valid biomarkers. AT-101 induces cancer cell apoptosis and inhibits angiogenesis in tumors, and thus represents a new promising anticancer agent. Pang et al47 have found that AT-101 inhibited the expression of VEGF, Bcl-2, and Bcl-xl in human PC-3 prostate cancer and DU 145 cells and primary cultured HUVECs. The growth of human prostate tumor PC-3 xenografts in mice was significantly suppressed by AT-101 at 15 mg/kg/day for 50 days, which was accompanied by significantly decreased VEGF expression and microvessel density. AT-101 also inhibited VEGF-induced chemotactic motility and capillary tube formation in HUVECs and human microvascular endothelial cells and suppressed microvessel sprouting from rat aortic rings ex vivo.47 AT-101 at 5–10 μM also blocked activation of VEGF receptor 2 with an IC50 of 2.38 μM in HUVECs and inhibited phosphorylation of key intracellular proangiogenic kinases, including Src family kinase, focal adhesion kinase, extracellular signal-related kinase, and protein kinase B induced by VEGF at 50 ng/mL.47 These findings indicate that AT-101 elicits its antiangiogenic activity through inhibition of the VEGF/VEGF receptor 2 signaling cascade in endothelial cells.
AT-101 may inhibit the function of VEGF via Bcl-2-mediated pathways. VEGF from paracrine/autocrine of tumor cells and endothelial cells induces expression of Bcl-2 in tumor-associated microvascular endothelial cells.92 Upregulated Bcl-2 expression in microvascular endothelial cells is sufficient to enhance intratumoral angiogenesis and to accelerate tumor growth.93 Interestingly, Bcl-2 in turn functions as a proangiogenic molecule through its ability to activate the NF-κB signaling pathway and to induce expression of the proangiogenic chemokine (C-X-C motif) ligand 8 (CXCL8) and CXCL1 from endothelial cells to affect nearby tumor cells.94 Therefore, the VEGF/Bcl-2/CXCL8 pathway may be a new target for development of antiangiogenic agents.
APE1 has two major functions, including DNA repair and redox regulation of gene transcription, the reduction–oxidation activity of which plays a role in tumor cell survival mechanisms, including growth, proliferation, invasion, metastasis, and angiogenesis.12 APE1 redox function regulates a number of transcription factors, such as hypoxia-induced factor-1α, activator protein-1, NF-κB, early growth response 1, nuclear respiration factor 1, activating transcription factor 1, nuclear transcription factor Y, v-myb avian myeloblastosis viral oncogene homolog, cAMP response element binding protein, polyomavirus enhancer binding protein 2, PAXs 5 and 8, hepatic leukemia factor, p53, and STAT3. Many of these genes are known to contribute to regulation of angiogenesis. We found that combined treatment with APE1 knockdown by RNA interference and recombinant human endostatin promoted apoptosis and had potent antiangiogenic effects in osteosarcoma through APE1-mediated regulation of VEGF expression.95 Our present data indicate that APE1 expression remained unchanged when A549 cells were treated with AT-101 or CDDP alone, or a combination of AT-101 and CDDP as compared with the control. However, the EMSA results showed that combination of AT-101 and CDDP was more effective in attenuating APE1 redox ability as indicated by enhanced STAT3-DNA binding compared with monotherapy. These data suggest that combined therapy of AT-101 plus CDDP promotes apoptosis and inhibits angiogenesis in NSCLC through inhibition of APE1 redox activity, but not APE1 expression.
Nowadays, the popular viewpoint is that selective inhibition of the redox activity of APE1 can induce greater tumor growth inhibition and alterations in the tumor microenvironment, as well as promote tumor cell killing. On the other hand, APE1 is an important BER protein that is responsible for repair of DNA damage induced by chemotherapeutic drugs, oxidative stress, and radiation. In this role, APE1 identifies the abasic site, and then cooperates with other enzymes to cut off the damaged base, generate the correct base, and connect the repair. All these roles show the underlying importance of APE1 as a promising molecule for targeted therapy. Some APE1 inhibitors have shown antitumor activity.14,23 Our previous data indicated that gossypol effectively inhibits APE1 DNA repair and redox activity.41 Therefore, we further investigated whether AT-101 combined with CDDP also affects APE1 DNA repair function. Our results showed that a combination of AT-101 and CDDP significantly decreased AP endonuclease activity, thus affecting the APE1 DNA repair function and contributing to increased DNA damage. Taken together, AT-101 plus CDDP enhances apoptosis and antiangiogenesis in A549 cells in vitro and in vivo through inhibition of APE1 DNA repair and redox activity.
Our study has confirmed that AT-101 enhances the antitumor efficacy of CDDP via inhibition of APE1 DNA repair and redox activity in NSCLC (Figure 14). However, involvement of other pharmacodynamic mechanisms should be taken into account. Pharmacokinetic interactions of AT-101 and CDDP are possible and should be checked. The effect of AT-101 on CDDP-induced neurotoxicity and nephrotoxicity should be explored. We are conducting clinical trials to see whether AT-101 potentiates the antitumor effect of CDDP in patients with advanced NSCLC.
 | Figure 14 A scheme of possible mechanisms by which AT-101 enhances the anticancer activity of CDDP in NSCLC. AT-101 is a BH3 mimetic and pan inhibitor of Bcl-2 and CDDP acts as a DNA intercalator. CDDP can crosslink with the purine bases on the DNA, interfering with DNA repair, causing DNA damage, and eventually inducing apoptosis of cancer cells. AT-101 can enhance the antitumor effects of CDDP in NSCLC via inhibition of APE1/Ref-1 DNA repair and redox activities. As a primary DNA repair protein, APE1/Ref-1 recognizes and repairs mutagenic noncoding AP sites in DNA molecules resulting from spontaneous, chemical, or DNA glycosylase-mediated hydrolysis of the N-glycosyl bond via the BER pathway. APE1/Ref-1 mainly possesses 5′-endonuclease activity, but it also shows minor 3′-phosphodiesterase, 3′-phosphatase, and 3′→5′ exonuclease activities. Accumulation of unrepaired AP sites will promote cell death by serving as blocks to DNA replication, stalling RNA transcription, or promoting double-stranded DNA breaks. As a redox protein, APE1/Ref-1 can activate multiple transcriptional factors including AP-1, p53, NF-κB, NRF1, ATF1, NF-Y, Myb, CREB, PEBP2, STAT3, NRF2, HIF-1α, PAXs 5 and 8, HLF, and Egr-1. Activation of APE1/Ref-1 involves the reduction of Cys65 to a sulfhydryl state. Therefore, APE1/Ref-1 is involved in the regulation of cellular growth, proliferation and differentiation, cell cycle control, apoptosis, autophagy, and angiogenesis. Elevated levels of APE1/Ref-1 have been linked to drug resistance, poor prognosis, and poor survival in cancer patients. Inhibition of APE1/Ref-1 may represent a useful approach to sensitize cancer cells to DNA intercalators such as CDDP and carboplatin. AT-101 also inhibits tumor angiogenesis via downregulation of the VEGF/VEGF receptor 2 pathway. In addition, AT-101 can induce or inhibit autophagy of cancer cells. |
In conclusion, our data demonstrate, for the first time, that a combination of AT-101 and CDDP significantly inhibits A549 cell proliferation and migration, promotes tumor cell apoptosis, and suppresses A549 cell-stimulating angiogenesis of HUVECs (Figure 14). These effects are realized through significant inhibition of APE1 DNA repair and redox activity in A549 cells in vitro and in a xenograft mouse model treated with combined therapy of AT-101 and CDDP. Therefore, after preclinical studies with AT-101, AT-101 can be considered to be a potent selective inhibitor of APE1 dual functions for the treatment of patients with NSCLC overexpressing APE1.
Disclosure
The authors report no conflicts of interest in this work.
References
Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65(2):87–108. | ||
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin. 2015;65(1):5–29. | ||
Chen WQ, Zheng RS, Zhang SW, Zeng HM, Zou XN. The incidences and mortalities of major cancers in China, 2010. Chin J Cancer. 2014; 33(8):402–405. | ||
Pasche B, Grant SC. Non-small cell lung cancer and precision medicine: a model for the incorporation of genomic features into clinical trial design. JAMA. 2014;311(19):1975–1976. | ||
Herbst RS, Heymach JV, Lippman SM. Lung cancer. N Engl J Med. 2008;359(13):1367–1380. | ||
Goldstraw P, Ball D, Jett JR, et al. Non-small-cell lung cancer. Lancet. 2011;378(9804):1727–1740. | ||
van Meerbeeck JP, Fennell DA, De Ruysscher DK. Small-cell lung cancer. Lancet. 2011;378(9804):1741–1755. | ||
Reck M, Heigener DF, Mok T, Soria JC, Rabe KF. Management of non-small-cell lung cancer: recent developments. Lancet. 2013;382(9893):709–719. | ||
Allemani C, Weir HK, Carreira H, et al. Global surveillance of cancer survival 1995–2009: analysis of individual data for 25,676,887 patients from 279 population-based registries in 67 countries (CONCORD-2). Lancet. 2015;385(9972):977–1010. | ||
Dasari S, Tchounwou PB. Cisplatin in cancer therapy: molecular mechanisms of action. Eur J Pharmacol. 2014;740:364–378. | ||
Dianov GL, Sleeth KM, Dianova II, Allinson SL. Repair of abasic sites in DNA. Mutat Res. 2003;531(1–2):157–163. | ||
Li M, Wilson DM 3rd. Human apurinic/apyrimidinic endonuclease 1. Antioxid Redox Signal. 2014;20(4):678–707. | ||
Abbotts R, Madhusudan S. Human AP endonuclease 1 (APE1): from mechanistic insights to druggable target in cancer. Cancer Treat Rev. 2010;36(5):425–435. | ||
Kelley MR, Georgiadis MM, Fishel ML. APE1/Ref-1 role in redox signaling: translational applications of targeting the redox function of the DNA repair/redox protein APE1/Ref-1. Curr Mol Pharmacol. 2012;5(1):36–53. | ||
Madlener S, Strobel T, Vose S, et al. Essential role for mammalian apurinic/apyrimidinic (AP) endonuclease Ape1/Ref-1 in telomere maintenance. Proc Natl Acad Sci U S A. 2013;110(44):17844–17849. | ||
Xanthoudakis S, Smeyne RJ, Wallace JD, Curran T. The redox/DNA repair protein, Ref-1, is essential for early embryonic development in mice. Proc Natl Acad Sci U S A. 1996;93(17):8919–8923. | ||
Wang D, Xiang DB, Yang XQ, et al. APE1 overexpression is associated with cisplatin resistance in non-small cell lung cancer and targeted inhibition of APE1 enhances the activity of cisplatin in A549 cells. Lung Cancer. 2009;66(3):298–304. | ||
Al-Attar A, Gossage L, Fareed KR, et al. Human apurinic/apyrimidinic endonuclease (APE1) is a prognostic factor in ovarian, gastro-oesophageal and pancreatico-biliary cancers. Br J Cancer. 2010;102(4):704–709. | ||
Ren T, Qing Y, Dai N, et al. Apurinic/apyrimidinic endonuclease 1 induced upregulation of fibroblast growth factor 2 and its receptor 3 induces angiogenesis in human osteosarcoma cells. Cancer Sci. 2014;105(2):186–194. | ||
Lou D, Zhu L, Ding H, Dai HY, Zou GM. Aberrant expression of redox protein Ape1 in colon cancer stem cells. Oncol Lett. 2014;7(4):1078–1082. | ||
Woo J, Park H, Sung SH, Moon BI, Suh H, Lim W. Prognostic value of human apurinic/apyrimidinic endonuclease 1 (APE1) expression in breast cancer. PLoS One. 2014;9(6):e99528. | ||
Cun Y, Dai N, Xiong C, et al. Silencing of APE1 enhances sensitivity of human hepatocellular carcinoma cells to radiotherapy in vitro and in a xenograft model. PLoS One. 2013;8(2):e55313. | ||
Thakur S, Sarkar B, Cholia RP, Gautam N, Dhiman M, Mantha AK. APE1/Ref-1 as an emerging therapeutic target for various human diseases: phytochemical modulation of its functions. Exp Mol Med. 2014;46:e106. | ||
Al-Safi RI, Odde S, Shabaik Y, Neamati N. Small-molecule inhibitors of APE1 DNA repair function: an overview. Curr Mol Pharmacol. 2012;5(1):14–35. | ||
Keshmiri-Neghab H, Goliaei B. Therapeutic potential of gossypol: an overview. Pharm Biol. 2014;52(1):124–128. | ||
Kitada S, Leone M, Sareth S, Zhai D, Reed JC, Pellecchia M. Discovery, characterization, and structure-activity relationships studies of proapoptotic polyphenols targeting B-cell lymphocyte/leukemia-2 proteins. J Med Chem. 2003;46(20):4259–4264. | ||
Billard C. BH3 mimetics: status of the field and new developments. Mol Cancer Ther. 2013;12(9):1691–1700. | ||
Shore GC, Nguyen M. Bcl-2 proteins and apoptosis: choose your partner. Cell. 2008;135(6):1004–1006. | ||
Zhang M, Liu H, Guo R, et al. Molecular mechanism of gossypol-induced cell growth inhibition and cell death of HT-29 human colon carcinoma cells. Biochem Pharmacol. 2003;66(1):93–103. | ||
Mohammad RM, Wang S, Aboukameel A, et al. Preclinical studies of a nonpeptidic small-molecule inhibitor of Bcl-2 and Bcl-XL [(-)-gossypol] against diffuse large cell lymphoma. Mol Cancer Ther. 2005;4(1):13–21. | ||
Oliver CL, Miranda MB, Shangary S, Land S, Wang S, Johnson DE. (-)-Gossypol acts directly on the mitochondria to overcome Bcl-2- and Bcl-XL-mediated apoptosis resistance. Mol Cancer Ther. 2005;4(1):23–31. | ||
Lomonosova E, Chinnadurai G. BH3-only proteins in apoptosis and beyond: an overview. Oncogene. 2008;27(Suppl 1):S2–S19. | ||
Johnstone RW, Ruefli AA, Lowe SW. Apoptosis: a link between cancer genetics and chemotherapy. Cell. 2002;108(2):153–164. | ||
Van Poznak C, Seidman AD, Reidenberg MM, et al. Oral gossypol in the treatment of patients with refractory metastatic breast cancer: a phase I/II clinical trial. Breast Cancer Res Treat. 2001;66(3):239–248. | ||
Liu G, Kelly WK, Wilding G, Leopold L, Brill K, Somer B. An open-label, multicenter, phase I/II study of single-agent AT-101 in men with castrate-resistant prostate cancer. Clin Cancer Res. 2009;15(9):3172–3176. | ||
Heist RS, Fain J, Chinnasami B, et al. Phase I/II study of AT-101 with topotecan in relapsed and refractory small cell lung cancer. J Thorac Oncol. 2010;5(10):1637–1643. | ||
Ready N, Karaseva NA, Orlov SV, et al. Double-blind, placebo-controlled, randomized phase 2 study of the proapoptotic agent AT-101 plus docetaxel, in second-line non-small cell lung cancer. J Thorac Oncol.2011;6(4):781–785. | ||
Baggstrom MQ, Qi Y, Koczywas M, et al. A phase II study of AT-101 (gossypol) in chemotherapy-sensitive recurrent extensive-stage small cell lung cancer. J Thorac Oncol. 2011;6(10):1757–1760. | ||
Sonpavde G, Matveev V, Burke JM, et al. Randomized phase II trial of docetaxel plus prednisone in combination with placebo or AT-101, an oral small molecule Bcl-2 family antagonist, as first-line therapy for metastatic castration-resistant prostate cancer. Ann Oncol. 2012;23(7):1803–1808. | ||
Schelman WR, Mohammed TA, Traynor AM, et al. A phase I study of AT-101 with cisplatin and etoposide in patients with advanced solid tumors with an expanded cohort in extensive-stage small cell lung cancer. Invest New Drugs. 2014;32(2):295–302. | ||
Qian C, Li M, Sui J, et al. Identification of a novel potential antitumor activity of gossypol as an APE1/Ref-1 inhibitor. Drug Des Devel Ther. 2014;8:485–496. | ||
Li M, Vascotto C, Xu S, et al. Human AP endonuclease/redox factor APE1/ref-1 modulates mitochondrial function after oxidative stress by regulating the transcriptional activity of NRF1. Free Radic Biol Med. 2012;53(2):237–248. | ||
Ren T, Shan J, Qing Y, et al. Sequential treatment with AT-101 enhances cisplatin chemosensitivity in human non-small cell lung cancer cells through inhibition of apurinic/apyrimidinic endonuclease 1-activated IL-6/STAT3 signaling pathway. Drug Des Devel Ther. 2014;8: 2517–2529. | ||
Potente M, Gerhardt H, Carmeliet P. Basic and therapeutic aspects of angiogenesis. Cell. 2011;146(6):873–887. | ||
Wang Z, Dabrosin C, Yin X, et al. Broad targeting of angiogenesis for cancer prevention and therapy. Semin Cancer Biol. January 16, 2015. [Epub ahead of print]. | ||
Moserle L, Jimenez-Valerio G, Casanovas O. Antiangiogenic therapies: going beyond their limits. Cancer Discov. 2014;4(1):31–41. | ||
Pang X, Wu Y, Wu Y, et al. (-)-Gossypol suppresses the growth of human prostate cancer xenografts via modulating VEGF signaling-mediated angiogenesis. Mol Cancer Ther. 2011;10(5):795–805. | ||
Jiang J, Slivova V, Jedinak A, Sliva D. Gossypol inhibits growth, invasiveness, and angiogenesis in human prostate cancer cells by modulating NF-kB/AP-1 dependent- and independent-signaling. Clin Exp Metastasis. 2012;29(2):165–178. | ||
Imai A, Zeitlin BD, Visioli F, et al. Metronomic dosing of BH3 mimetic small molecule yields robust antiangiogenic and antitumor effects. Cancer Res. 2012;72(3):716–725. | ||
Valdiglesias V, Giunta S, Fenech M, Neri M, Bonassi S. gH2AX as a marker of DNA double strand breaks and genomic instability in human population studies. Mutat Res. 2013;753(1):24–40. | ||
Matthaios D, Hountis P, Karakitsos P, Bouros D, Kakolyris S. H2AX a promising biomarker for lung cancer: a review. Cancer Invest. 2013; 31(9):582–599. | ||
Fishel ML, Wu X, Devlin CM, et al. Apurinic/apyrimidinic endonuclease/redox factor-1 (APE1/Ref-1) redox function negatively regulates NRF2. J Biol Chem. 2015;290(5):3057–3068. | ||
Li MX, Wang D, Zhong ZY, et al. Targeting truncated APE1 in mitochondria enhances cell survival after oxidative stress. Free Radic Biol Med. 2008;45(5):592–601. | ||
Cardoso AA, Jiang Y, Luo M, et al. APE1/Ref-1 regulates STAT3 transcriptional activity and APE1/Ref-1-STAT3 dual-targeting effectively inhibits pancreatic cancer cell survival. PLoS One. 2012;7(10):e47462. | ||
Li Y, Liu X, Zhou T, et al. Inhibition of APE1/Ref-1 redox activity rescues human retinal pigment epithelial cells from oxidative stress and reduces choroidal neovascularization. Redox Biol. 2014;2:485–494. | ||
Li Y, Liu X, Zhou T, et al. Suppression of choroidal neovascularization through inhibition of APE1/Ref-1 redox activity. Invest Ophthalmol Vis Sci. 2014;55(7):4461–4469. | ||
Bonanno L, Favaretto A, Rosell R. Platinum drugs and DNA repair mechanisms in lung cancer. Anticancer Res. 2014;34(1):493–501. | ||
Housman G, Byler S, Heerboth S, et al. Drug resistance in cancer: an overview. Cancers (Basel). 2014;6(3):1769–1792. | ||
Kelland L. The resurgence of platinum-based cancer chemotherapy. Nat Rev Cancer. 2007;7(8):573–584. | ||
Galluzzi L, Vitale I, Michels J, et al. Systems biology of cisplatin resistance: past, present and future. Cell Death Dis. 2014;5:e1257. | ||
Holohan C, Van Schaeybroeck S, Longley DB, Johnston PG. Cancer drug resistance: an evolving paradigm. Nat Rev Cancer. 2013;13(10):714–726. | ||
Fuchs Y, Steller H. Programmed cell death in animal development and disease. Cell. 2011;147(4):742–758. | ||
Brown JM, Attardi LD. The role of apoptosis in cancer development and treatment response. Nat Rev Cancer. 2005;5(3):231–237. | ||
Nunnari J, Suomalainen A. Mitochondria: in sickness and in health. Cell. 2012;148(6):1145–1159. | ||
Kang MH, Reynolds CP. Bcl-2 inhibitors: targeting mitochondrial apoptotic pathways in cancer therapy. Clin Cancer Res. 2009;15(4):1126–1132. | ||
Waites GM, Wang C, Griffin PD. Gossypol: reasons for its failure to be accepted as a safe, reversible male antifertility drug. Int J Androl. 1998;21(1):8–12. | ||
White IG, Vishwanath R, Swan MA, Brown-Woodman PD. Studies of the mechanism of action of gossypol as a male antifertility agent. Contraception. 1988;37(3):269–277. | ||
Ko CH, Shen SC, Yang LY, Lin CW, Chen YC. Gossypol reduction of tumor growth through ROS-dependent mitochondria pathway in human colorectal carcinoma cells. Int J Cancer. 2007;121(8):1670–1679. | ||
Volate SR, Kawasaki BT, Hurt EM, et al. Gossypol induces apoptosis by activating p53 in prostate cancer cells and prostate tumor-initiating cells. Mol Cancer Ther. 2010;9(2):461–470. | ||
Moon DO, Kim MO, Lee JD, Kim GY. Gossypol suppresses NF-kB activity and NF-kB-related gene expression in human leukemia U937 cells. Cancer Lett. 2008;264(2):192–200. | ||
Song B, Huang G, Tong C, et al. Gossypol suppresses mouse T lymphocytes via inhibition of NFkB, NFAT and AP-1 pathways. Immunopharmacol Immunotoxicol. 2013;35(5):615–621. | ||
Wong FY, Liem N, Xie C, et al. Combination therapy with gossypol reveals synergism against gemcitabine resistance in cancer cells with high BCL-2 expression. PLoS One. 2012;7(12):e50786. | ||
Lei X, Chen Y, Du G, et al. Gossypol induces Bax/Bak-independent activation of apoptosis and cytochrome c release via a conformational change in Bcl-2. FASEB J. 2006;20(12):2147–2149. | ||
Bauer JA, Trask DK, Kumar B, et al. Reversal of cisplatin resistance with a BH3 mimetic, (-)-gossypol, in head and neck cancer cells: role of wild-type p53 and Bcl-xL. Mol Cancer Ther. 2005;4(7):1096–1104. | ||
Macoska JA, Adsule S, Tantivejkul K, Wang S, Pienta KJ, Lee CT. -(-)Gossypol promotes the apoptosis of bladder cancer cells in vitro. Pharmacol Res. 2008;58(5–6):323–331. | ||
Jang GH, Lee M. BH3-mimetic gossypol-induced autophagic cell death in mutant BRAF melanoma cells with high expression of p21Cip1. Life Sci. 2014;102(1):41–48. | ||
Kaza N, Kohli L, Graham CD, Klocke BJ, Carroll SL, Roth KA. BNIP3 regulates AT101 [(-)-gossypol] induced death in malignant peripheral nerve sheath tumor cells. PLoS One. 2014;9(5):e96733. | ||
Mei H, Lin Z, Wang Y, Wu G, Song Y. Autophagy inhibition enhances pan-Bcl-2 inhibitor AT-101-induced apoptosis in non-small cell lung cancer. Neoplasma. 2014;61(2):186–192. | ||
Ni Z, Dai X, Wang B, et al. Natural Bcl-2 inhibitor (-)-gossypol induces protective autophagy via reactive oxygen species-high mobility group box 1 pathway in Burkitt lymphoma. Leuk Lymphoma. 2013;54(10):2263–2268. | ||
Voss V, Senft C, Lang V, et al. The pan-Bcl-2 inhibitor (-)-gossypol triggers autophagic cell death in malignant glioma. Mol Cancer Res. 2010;8(7):1002–1016. | ||
Lian J, Karnak D, Xu L. The Bcl-2-Beclin 1 interaction in (-)-gossypol-induced autophagy versus apoptosis in prostate cancer cells. Autophagy. 2010;6(8):1201–1203. | ||
Jarzabek MA, Amberger-Murphy V, Callanan JJ, et al. Interrogation of gossypol therapy in glioblastoma implementing cell line and patient-derived tumour models. Br J Cancer. 2014;111(12):2275–2286. | ||
Mittal K, Ebos J, Rini B. Angiogenesis and the tumor microenvironment: vascular endothelial growth factor and beyond. Semin Oncol. 2014;41(2):235–251. | ||
Gacche RN, Meshram RJ. Angiogenic factors as potential drug target: efficacy and limitations of anti-angiogenic therapy. Biochim Biophys Acta. 2014;1846(1):161–179. | ||
Folkman J. Angiogenesis and apoptosis. Semin Cancer Biol. 2003; 13(2):159–167. | ||
Limaverde-Sousa G, Sternberg C, Ferreira CG. Antiangiogenesis beyond VEGF inhibition: a journey from antiangiogenic single-target to broad-spectrum agents. Cancer Treat Rev. 2014;40(4):548–557. | ||
Vasudev NS, Reynolds AR. Anti-angiogenic therapy for cancer: current progress, unresolved questions and future directions. Angiogenesis. 2014;17(3):471–494. | ||
Jain RK. Antiangiogenesis strategies revisited: from starving tumors to alleviating hypoxia. Cancer Cell. 2014;26(5):605–622. | ||
Andre N, Carre M, Pasquier E. Metronomics: towards personalized chemotherapy? Nat Rev Clin Oncol. 2014;11(7):413–431. | ||
Lee S, Chen TT, Barber CL, et al. Autocrine VEGF signaling is required for vascular homeostasis. Cell. 2007;130(4):691–703. | ||
Lichtenberger BM, Tan PK, Niederleithner H, Ferrara N, Petzelbauer P, Sibilia M. Autocrine VEGF signaling synergizes with EGFR in tumor cells to promote epithelial cancer development. Cell. 2010; 140(2):268–279. | ||
Nor JE, Christensen J, Mooney DJ, Polverini PJ. Vascular endothelial growth factor (VEGF)-mediated angiogenesis is associated with enhanced endothelial cell survival and induction of Bcl-2 expression. Am J Pathol. 1999;154(2):375–384. | ||
Nor JE, Christensen J, Liu J, et al. Upregulation of Bcl-2 in microvascular endothelial cells enhances intratumoral angiogenesis and accelerates tumor growth. Cancer Res. 2001;61(5):2183–2188. | ||
Karl E, Warner K, Zeitlin B, et al. Bcl-2 acts in a proangiogenic signaling pathway through nuclear factor-kB and CXC chemokines. Cancer Res. 2005;65(12):5063–5069. | ||
Wang D, Zhong ZY, Li MX, Xiang DB, Li ZP. Vector-based Ape1 small interfering RNA enhances the sensitivity of human osteosarcoma cells to endostatin in vivo. Cancer Sci. 2007;98(12):1993–2001. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.