Back to Journals » International Journal of Nanomedicine » Volume 12
Skin-derived mesenchymal stem cells as quantum dot vehicles to tumors
Authors Dapkute D ![]() , Steponkiene S, Bulotiene D, Saulite L
, Steponkiene S, Bulotiene D, Saulite L ![]() , Riekstina U, Rotomskis R
, Riekstina U, Rotomskis R ![]()
Received 6 June 2017
Accepted for publication 14 September 2017
Published 6 November 2017 Volume 2017:12 Pages 8129—8142
DOI https://doi.org/10.2147/IJN.S143367
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Thomas Webster
Dominyka Dapkute,1,2 Simona Steponkiene,1 Danute Bulotiene,1 Liga Saulite,3 Una Riekstina,3 Ricardas Rotomskis1,4
1Biomedical Physics Laboratory, National Cancer Institute, Vilnius, Lithuania; 2Institute of Biosciences, Vilnius University, Vilnius, Lithuania; 3Faculty of Medicine, University of Latvia, Riga, Latvia; 4Biophotonics Group of Laser Research Center, Faculty of Physics, Vilnius University, Vilnius, Lithuania
Purpose: Cell-mediated delivery of nanoparticles is emerging as a new method of cancer diagnostics and treatment. Due to their inherent regenerative properties, adult mesenchymal stem cells (MSCs) are naturally attracted to wounds and sites of inflammation, as well as tumors. Such characteristics enable MSCs to be used in cellular hitchhiking of nanoparticles. In this study, MSCs extracted from the skin connective tissue were investigated as transporters of semiconductor nanocrystals quantum dots (QDs).
Materials and methods: Cytotoxicity of carboxylated CdSe/ZnS QDs was assessed by lactate dehydrogenase cell viability assay. Quantitative uptake of QDs was determined by flow cytometry; their intracellular localization was evaluated by confocal microscopy. In vitro tumor-tropic migration of skin-derived MSCs was verified by Transwell migration assay. For in vivo migration studies of QD-loaded MSCs, human breast tumor-bearing immunodeficient mice were used.
Results: QDs were found to be nontoxic to MSCs in concentrations no more than 16 nM. The uptake studies showed a rapid QD endocytosis followed by saturating effects after 6 h of incubation and intracellular localization in the perinuclear region. In vitro migration of MSCs toward MDA-MB-231 breast cancer cells and their conditioned medium was up to nine times greater than the migration toward noncancerous breast epithelial cells MCF-10A. In vivo, systemically administered QD-labeled MSCs were mainly located in the tumor and metastatic tissues, evading most healthy organs with the exception being blood clearance organs (spleen, kidneys, liver).
Conclusion: Skin-derived MSCs demonstrate applicability in cell-mediated delivery of nanoparticles. The findings presented in this study promise further development of a cell therapy and nanotechnology-based tool for early cancer diagnostics and therapy.
Keywords: mesenchymal stem cells, tumor tropism, quantum dots, nanoparticles, tumor-specific delivery, immunodeficient mice
Introduction
Despite advances made in cancer therapy, it is generally accepted that conventional treatment methods are not sufficient to permanently eliminate cancer and cancer stem-like cells.1 Nanotechnology emerged as one of the most promising solutions; however, nanoparticles themselves lack specificity to tumors, and thus, cell-based therapies were proposed. Out of all potential human cell sources, human mesenchymal stem cells (MSCs) were introduced as the most promising. MSCs do not evoke ethical controversy, have no tissue compatibility issues, and possess low tumorigenicity risk.2,3 MSCs are non-hematopoietic spindle-shaped fibroblast-like cells derived from the mesoderm.4 MSCs possess self-renewal and immunomodulatory properties, are capable of transdifferentiation, and participate in wound healing, bone regeneration, angiogenesis, and homeostasis.5–7 Due to these characteristics, MSCs hold great promise in the treatment of wounds, degenerative diseases, and other pathologies.8 Cancer cells secrete various chemokines similar to the ones produced in the lesion sites;9 therefore, tumors are sometimes called wounds that do not heal.10 MSCs have chemokine receptors11 and a tendency to migrate through the chemokine gradient toward the tumor.12,13 Owing to these tumor-tropic properties, MSCs could be used to transport therapeutic molecules directly to cancerous tissues. MSC-mediated transportation of different signaling molecules (interleukins, interferons, chemokines),14,15 genetically modified viruses,16 gene therapy components,17 chemotherapeutic drugs and prodrugs18,19 are already topics of research. There are also successful studies investigating the use of MSCs in transportation of nanoparticles – one of the main achievements was the delivery of optically active nanoparticles via MSCs across the blood–brain barrier and homing to glioblastomas.20,21 Knowledge about MSC tumor-tropic properties and their potential to perform tumor-directed delivery of nanoparticles opens the way to novel therapy.
Of the wide variety of nanoparticles investigated, quantum dots (QDs) have perhaps the most extensive applications. QDs exhibit unique photophysical properties. Some of the most desired characteristics of QDs are broad excitation and a narrow photoluminescence (PL) emission spectra.22 Together with the quantum confinement effect, which implements the size-dependent PL emission of QDs, such spectral characteristics enable the use of several QDs with different wavelengths in a single experiment and the construction of nanocrystals that fluoresce in a tissue optical window.23,24 QDs are also characterized by their brightness and high photostability.22 These optical properties permit QDs to be used in long-term tissue imaging and in vivo cell tracking. QDs have already been employed to track MSCs in regenerative or cancer therapies due to their superior in vitro cell tracking capability compared to commercial cell trackers.25 The large surface area of nanoparticles allows the attachment of multiple biologically active molecules, such as proteins, drugs, photosensitizers, and/or antibodies.24 Thus, QDs could combine a cell-imaging probe and a tumor-targeting agent in one platform.
Most researchers use bone marrow19,26–28 or adipose tissue-derived18,29,30 MSCs. In this study, we used human dermal MSCs. The successful application of dermal MSCs for the delivery of nanoparticles would be highly advantageous because of the high tissue accessibility and tractability.31 MSCs are abundant in skin tissues and can be obtained with minimally invasive procedures. In addition, after some surgeries, the skin tissue is considered a surgical waste and, therefore, could be used for a cost-effective appliance in cancer treatment. Post-surgery material gathered after liposuction,32 amputations,33 or surgical debridement34 has already been suggested as a source of MSCs.
We used Invitrogen Qdot® 625 ITK™ Carboxyl QDs to investigate the effect of nanoparticles on MSC migration in vitro and in vivo. Here, we identified the optimal nontoxic concentration of QDs, internalization dynamics of QDs into MSCs, nanoparticle intracellular localization, and most importantly, the QD-loaded MSC ability to migrate toward cancer cells in vitro and in vivo. Altogether, the results presented in this article show the promising potential of MSCs to be used as nanoparticle carriers to the tumors.
Materials and methods
Materials
Qdot 625 ITK nontargeted carboxyl-coated QDs used in the study were purchased from Thermo Fisher Scientific (Waltham, MA, USA). QDs consisted of CdSe/ZnS core/shell and were coated by carboxylic acid–functionalized polyethylene glycol (PEG) lipids, and had a strong emission in the red region with the emission peak at 625 nm. Atomic force microscopy and dynamic light scattering data showed mean nanoparticle size to be ~14.5 nm (Figure 1).
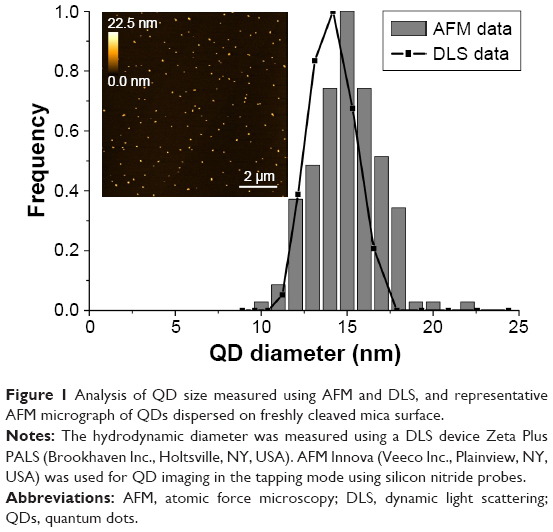 | Figure 1 Analysis of QD size measured using AFM and DLS, and representative AFM micrograph of QDs dispersed on freshly cleaved mica surface. |
Mouse anti-human monoclonal antibodies were the following: CD44 conjugated with Alexa Fluor® 488 (BioLegend, San Diego, CA, USA), CD90 conjugated with fluorescein isothiocyanate (FITC) (Dako Denmark A/S, Glostrup, Denmark), CD73 conjugated with phycoerythrin (PE; BD Biosciences, San Jose, CA, USA), CD105 conjugated with allophycocyanin (APC) (R&D Systems, Minneapolis, MN, USA), CD45 conjugated with FITC, CD34 conjugated with PE, CD14 conjugated with APC, CD184 (CXCR4) conjugated with PE (all four from BD Biosciences). The lactate dehydrogenase (LDH) cytotoxicity detection kit CytoTox 96® was purchased from Promega (Madison, WI, USA).
Primary cell cultures and cell lines
Primary human skin MSCs used for this study were supplied by a frozen primary cell bank established from cultivated dermis-derived adherent cells, isolated from post-surgery materials as described earlier.35 MSCs were used in accordance with authorized approval from the Institute of Experimental and Clinical Medicine Ethics committee, University of Latvia (issued on 04.06.2014). Skin MSCs were grown in DMEM with F12 Nutrient mixture (DMEM/F12, 3:1 v/v; Thermo Fisher Scientific) supplemented with 10% of fetal bovine serum (FBS; Sigma-Aldrich, St Louis, MO, USA) and 1% antibiotics (100 U/mL penicillin, 100 μg/mL streptomycin; Biochrom, Berlin, Germany). MSCs used in the experiments were between passages 4 and 8. To ensure the consistency of the results and prevent donor-dependent variations, MSCs from a single donor were used in all the experiments, if not stated otherwise.
Human breast cancer cell line MDA-MB-231 (ATCC HTB-26™) and human mammary epithelial cell line MCF-10A (ATCC CRL-10317™) were obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA). MDA-MB-231 cells were cultured in DMEM (Thermo Fisher Scientific) supplemented with 10% FBS and 1% antibiotics. MCF-10A cells were cultured in HuMEC Basal Serum-Free Medium supplemented with 1% HuMEC supplement, 0.4% bovine pituitary extract (all from Thermo Fisher Scientific), and 1% antibiotics.
Cells were cultured and passaged in 25–75 cm2 cell culture flasks with up to 90% confluence with complete cell culture medium. Cells were grown in a humidified chamber at 37°C with 5% CO2.
Immunostaining of MSCs
MSCs were characterized by the criteria set by the International Society of Cellular Therapy.36 Immunophenotype of MSCs was detected by staining 1×105 cells using antibodies against mesenchymal markers CD90, CD73, CD105, and hematopoiesis markers CD45, CD34, CD14, according to the manufacturer’s instructions. Additionally, cells were stained with CD184 to detect the amount of CXCR4 on the membrane of MSCs. Stained and washed cells were analyzed with a flow cytometer.
Optimization and quantification of QD internalization
Maximum nontoxic QD concentration was determined using standard LDH cytotoxicity assay. MSCs were seeded into 96-well plates at a density of 5×103 cells per well in 100 μL of complete medium. After 24 h, the growth medium was supplemented with increasing concentrations of QDs varying from 0.8 to 80 nM. Cells were incubated with QDs for 24 and 48 h. A few wells were chosen as a positive control. Positive control cells were lysed with lysis buffer. After incubation, CytoTox 96 reagent was added to the wells. After 30 min, the stop solution terminated the reaction and the absorbance at 490 nm was measured using Infinite 200 PRO (Tecan, Männedorf, Switzerland). Toxicity was determined by the following formula:
|
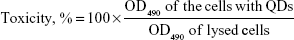
where OD is optical density.
Quantitative accumulation of QDs was performed seeding MSCs into six-well plates at a density of 1×105 cells per well in 2 mL of complete medium. After 24 h, QDs were added to the culture medium to a final concentration of 16 nM. The incubation was carried out from 1 min to 48 h. After the appropriate incubation time, the monolayer of the cells was washed three times with PBS (Biochrom), trypsinized, and centrifuged (200× g for 5 min). The cells were resuspended in 100 μL PBS and analyzed with a flow cytometer.
Intracellular localization
MSCs were seeded in eight-well chamber slides (Nunc Lab-Tek II; Thermo Fisher Scientific) at a density of 3×103 cells per well in 400 μL of complete medium. After 24 h, the QDs were diluted in the complete growth medium to a concentration of 16 nM and poured over the cells. The cells were incubated for various time points ranging from 15 min to 48 h. After incubation, the cells were washed a few times with Dulbecco’s PBS (Thermo Fisher Scientific) to prevent cell detachment. Cells were fixed with 4% paraformaldehyde (Sigma-Aldrich) for 15 min, permeabilized with 0.2% Triton X-100 (Sigma-Aldrich) for 4 min, and blocked with 1% bovine serum albumin (Sigma-Aldrich) for 20 min. Cells were incubated with 15 U/mL Alexa Fluor 488 Phalloidin (Thermo Fisher Scientific) for 30 min to label actin filaments. Nuclei were stained with 25 μg/mL Hoechst 33258 (Sigma-Aldrich) for 30 min. Slides were mounted with Qdot Mounting media (Thermo Fisher Scientific).
In vitro migration
The tropism of MSCs to tumor cells was determined using Transwell® Permeable Support inserts (Corning Inc., Corning, NY, USA). MDA-MB-231 and MCF-10A (1×105 cells/well) cells were seeded onto lower wells of 24-well plates in 600 μL of a serum-free medium. The remaining wells contained MDA-MB-231–conditioned medium (filtered [0.22 μm filter] serum-free medium in which MDA-MB-231 cancer cells had been cultured for 24 h), MSC growth medium supplemented with 20% FBS (positive control), or serum-free medium (negative control). After 24 h, QD-loaded and unlabeled MSCs were resuspended in 100 μL of serum-free medium and placed onto polycarbonate membrane inserts with 8 μm pores (3×104 cells/insert). MSC-containing inserts were positioned in the lower wells. MSCs were allowed to migrate through the pores for 24 h under standard cultivation conditions (37°C with 5% CO2). Nonmigratory cells were wiped away from the inside of the insert using a wet cotton bud. Migratory cells were fixed with 4% paraformaldehyde for 15 min and stained with 25 μg/mL Hoechst overnight. The migrated MSCs were examined under the confocal microscope. Results were evaluated by directly counting the number of migrated cells in at least five fields. The data were normalized according to the MSC migration toward positive control, which represented 100% migration. Results are presented as a mean ± SD. To determine whether in vitro cell migration depends on the donor, MSC migration toward MDA-MB-231 cells, FBS-supplemented and FBS-free medium was tested with, overall, three different donors.
Animals and tumor model
Experiments were performed on 6-week-old female CB17 SCID mice (Taconic Biosciences, Lille Skensved, Denmark). Mice were maintained at a constant temperature (22°C±1°C), relative humidity 55%±10%, and a photoperiod (12 h light/dark cycle). Animals were acclimatized for 7 days before each experiment. The animals were provided with autoclaved rodent chow (Diet 4RF25; Mucedola, Milan, Italy) and purified water ad libitum. Animal experiments were approved by the Animal Care and Use Committee of the State Food and Veterinary Service (approval No G2-29), and all procedures were in accordance with the guidelines for animal research set out in the European Union Directive 2010/63/EU and national regulations. Mice were inoculated with 2×106 MDA-MB-231 cells in a volume of 200 μL growth medium into adipose tissue around the nipple using 23G needles. The tumor volume was estimated by measuring three orthogonal diameters (L [length], W [width], H [height]) with calipers; the volume (V) was calculated as follows: V = (L × W × H) ×0.523. The migration studies were performed when the tumor size reached 300 mm3, generally 50–60 days after inoculation. At least five mice were used in the experimental groups.
In vivo migration
After the tumors were formed, 5×105 QD-loaded MSCs were injected subcutaneously (in 200 μL growth medium) ~5 mm below the tumor with 23G needles. Control mice were injected with a sterile saline solution. The mice were left for 24 h or 7 days in standard conditions. After the appropriate time, mice were sacrificed and tumor, peritoneum from MSC injection site, lungs, kidneys, brain, heart, spleen, liver, and metastases (if found) were taken. The organs were either cut into slices with cryomicrotome (10 μm slice thickness) or homogenized and analyzed with flow cytometer. Slices were analyzed with confocal microscope to get fluorescent images and later stained with hematoxylin and eosin to get respective histologic images. For the flow cytometric analysis, the organs were minced and digested with 0.25% trypsin/EDTA (Biochrom) for 30 min with continuous shaking at 37°C. After enzymatic dissociation, the suspension was rigorously pipetted with a cut pipette tip. The digested tissues were passed through 70 μm filters to obtain a single cell suspension, mixed with 10% FBS/PBS solution, and centrifuged at 400× g for 5 min. The cells were resuspended in PBS and counted. Then, 1×106 cells were stained with anti-CD44 antibody for 30 min, washed, and analyzed with a flow cytometer.
Flow cytometry
Flow cytometric analyses were performed using Accuri C6 (Accuri Cytometers, Inc., Ann Arbor, MI, USA) or Guava EasyCyte (Merck Millipore, Billerica, MA, USA) flow cytometers. A minimum of 10,000 viable cells were acquired. The data were analyzed with FlowJo (Tree Star, Inc., Ashland, OR, USA) and Accuri C6 software (Accuri Cytometers, Inc.).
Laser scanning confocal microscopy
Samples were examined under the Nikon Eclipse TE2000-U microscope (Nikon, Yokohama, Japan) with the confocal laser scanning system C1si (capable of 32-bit spectral imaging). Imaging was performed by scanning with the beam of diode laser (404 nm) for Hoechst, argon ion laser (488 nm) for Alexa Fluor 488, and helium–neon laser (543 nm) for QDs using oil immersion 60× NA 1.4 objective (Plan Apo VC; Nikon). Three different band pass filters were used – 450/35 for Hoechst, 515/30 for Alexa Fluor 488, and 605/75 for QDs. Image processing was performed using EZ-C1 Bronze version 3.80 (Nikon) and ImageJ 1.48 (National Institute of Health, Bethesda, MD, USA) software.
Statistical analysis
Data are shown as the representative result or as the mean of at least three independent experiments ± SD. Statistical analyses were performed using the two-tailed Student’s t-test; differences were considered significant at p≤0.05, and p-values are shown in figures where needed.
Results
Characterization of MSCs
The cells isolated from donor skin tissue were plastic adherent and had fibroblast-like spindle-shaped morphology (Figure 2A). The ability of the cells to differentiate into adipocytes, osteocytes, and chondrocytes was evaluated earlier.37,38 The cultured cells were >95% positive for the expression of mesenchymal markers CD90, CD73, and CD105, and displayed no expression (≤0.5%) of hematopoietic markers CD45, CD34, and CD14 (Figure 2B). According to the criteria defined by the International Society for Cellular Therapy,36 cells used in this study were considered to be MSCs.
 | Figure 2 (A) Morphology of isolated dermal MSCs. Magnification 10×, scale bar 400 μm. (B) Immunophenotype of MSCs. |
Optimization and quantification of QD internalization
For MSCs to be used as nanoparticle vehicles, the optimal conditions for maximum QD loading must be established.
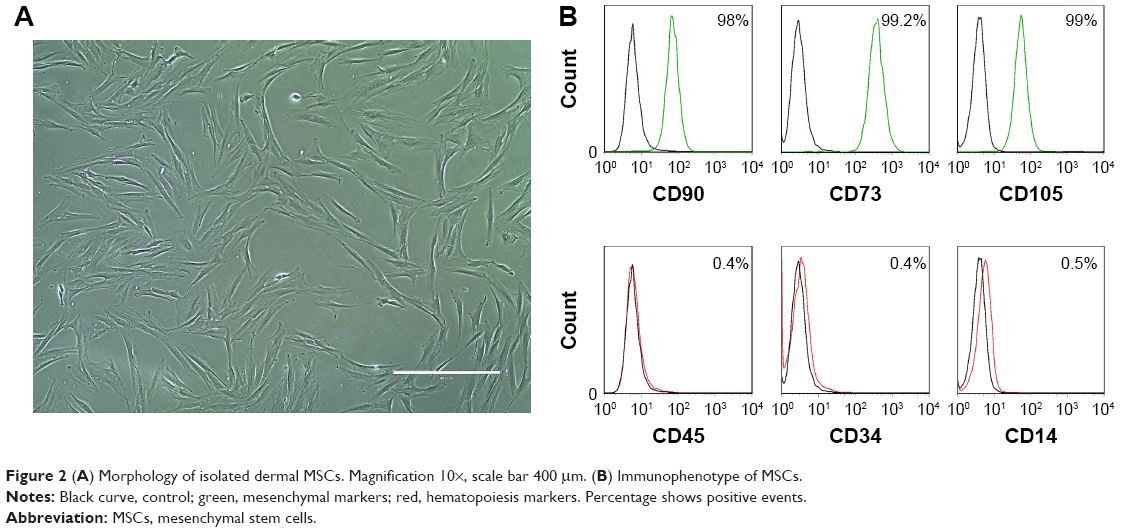
To evaluate the possible cytotoxic effect of nanoparticles and determine the maximum tolerated QD concentration, standard LDH cytotoxicity assay was used. Using various concentrations of nanoparticles and different incubation time points, we detected that 16 nM was the maximum QD concentration that did not cause any cytotoxicity to MSCs even after 48 h of treatment (Figure 3A). Higher concentrations of QDs caused only minor toxicity (1.7%–5%). Hence, 16 nM concentration was used in the following experiments. The negative value seen in Figure 3A is due to the increased metabolic activity of MSCs after incubation with nanoparticles, because the assay measures LDH activity in the cells. This shows the stimulating effect of nanoparticles on growth and proliferation of MSCs. Such an effect was also noticed by other authors.39–42
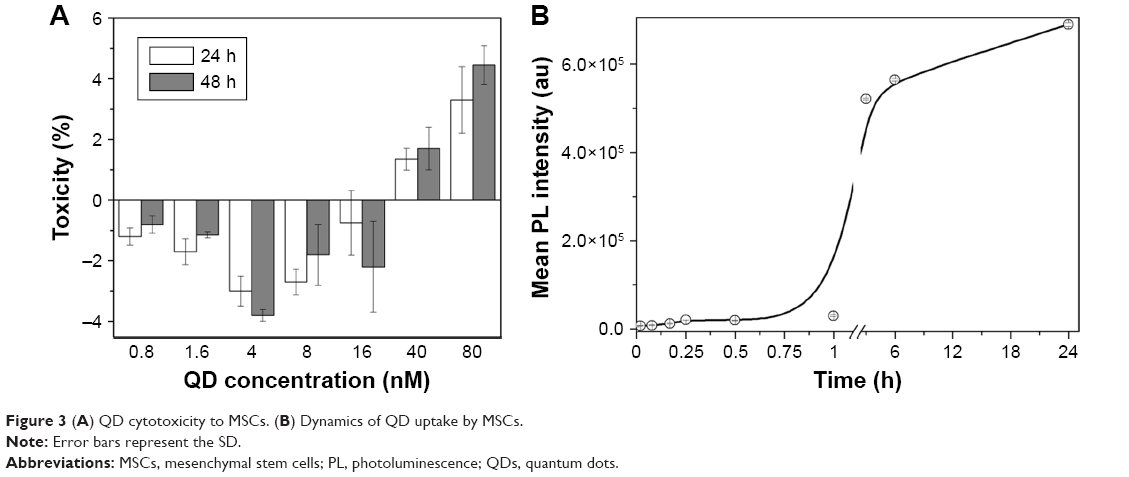 | Figure 3 (A) QD cytotoxicity to MSCs. (B) Dynamics of QD uptake by MSCs. |
To optimize the uptake of nanoparticles, the effect of the incubation time was assessed and a quantitative accumulation curve was acquired (Figure 3B). The curve corresponded to the standard S-shaped sigmoid QD accumulation curve (lag, growth, and saturation stages).43 For the first hour, the QD PL signal was low; after an hour, the PL signal sharply increased until 6 h at which time the transitional phase was reached. Since the results showed that incubation for 6 h is sufficient to obtain the early saturation phase of accumulation, 6 h incubation time point was chosen as the optimal QD incubation time (Figure 3B).
Intracellular localization
Using laser scanning confocal microscope, the uptake and intracellular localization of QDs was detected (Figure 4). The attachment of QDs to the cell membrane was rapid and occurred as early as 15 min incubation (Figure 4, insert a), and was followed by the endocytosis of nanoparticles. The uptake was first detected after 1 h and increased gradually with the incubation time. QDs were localized inside the vesicles, demonstrating that endocytosis was their entry pathway (Figure 4, insert b) – our previous study on QD microinjection, mimicking passive diffusion of nanoparticles, demonstrated that after microinjection or membrane damage, the QDs were uniformly dispersed in the cytoplasm of the cells and did not form any vesicular structures, proving endocytosis is responsible for vesicle formation.43 After 6 h, all MSCs had incorporated QDs and the endocytic vesicles with nanoparticles inside were spread all over the cytoplasm. After 24 h, the vesicles with QDs merged into multivesicular body-like structures that localized mostly in the perinuclear region (Figure 4, insert d). Along with the intracellular localization, the size of the vesicles containing QDs also changed over time (Figure 4, inserts). QDs were not detected in the nuclei.
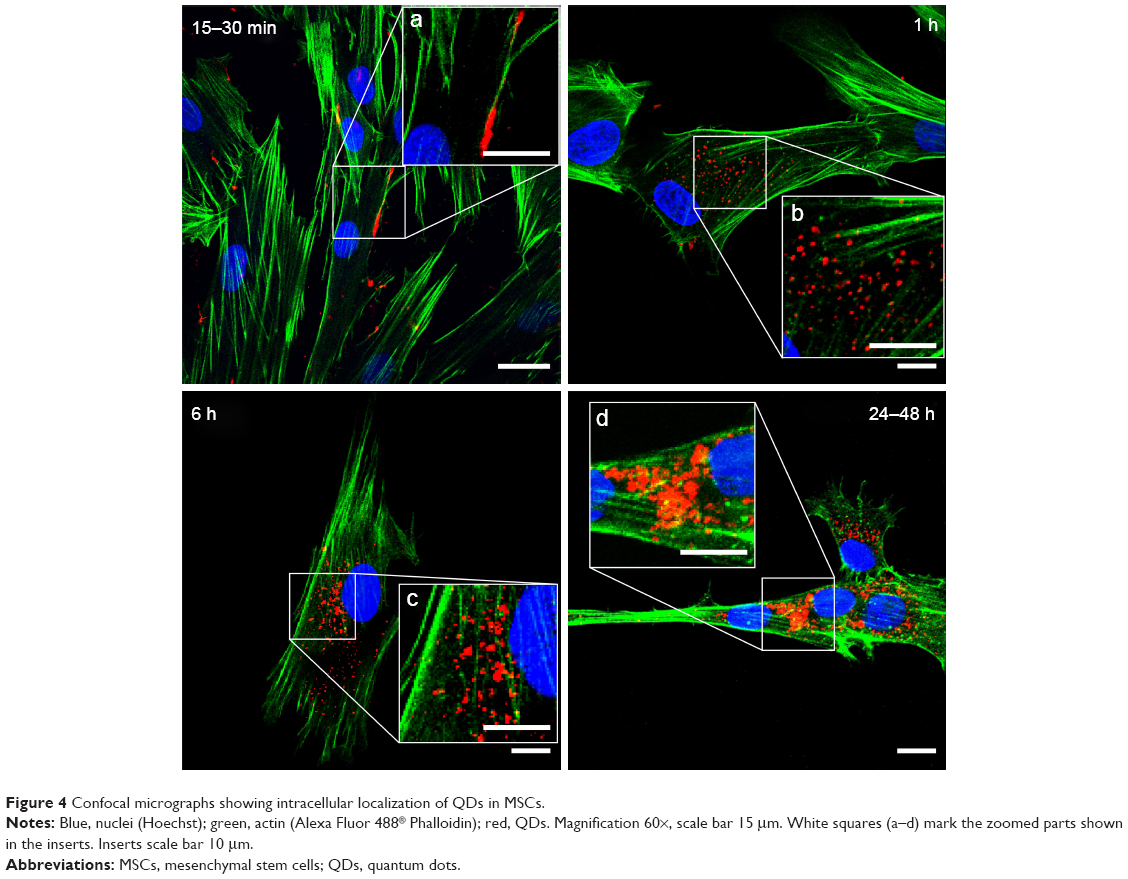 | Figure 4 Confocal micrographs showing intracellular localization of QDs in MSCs. |
In vitro migration
Several studies44–46 demonstrate the tendency of MSCs to migrate to the sites of the tumor. Therefore, to investigate whether skin MSCs possess the same properties, we simulated the migration of MSCs toward cancer cells (MDA-MB-231) and noncancerous breast epithelial cells (MCF-10A) using Transwell migration assay. During the assay, all the cells were incubated in serum-free medium to prevent nonspecific chemotactic movement toward the serum.
The excessive migration toward positive control (growth medium supplemented with 20% serum) showed that even after the prolonged ex vivo cultivation, MSCs retained their migratory capabilities (Figure 5A, B). The migration toward MDA-MB-231 cancer cells was thirty times greater than the random undirected cell migration toward serum-free medium (negative control), as shown in Figure 5C. In order to verify that MSCs do not migrate toward all types of cells including healthy tissue, cell migration toward nonmalignant breast tissue epithelial cells MCF-10A was tested. The results demonstrate that the migration toward healthy breast tissue cells was the same as an undirected MSC migration (<2%). The migration toward MDA-MB-231–conditioned medium (~33%) proved that cancer cells secrete soluble molecules stimulating MSC migration (Figure 5C). Furthermore, the impact of nanoparticles on the migration of MSCs was evaluated when comparing the migration of QD-loaded and unlabeled MSCs. QDs did not reduce the MSCs’ ability to migrate, as there were no statistically significant differences between nanoparticle-labeled and unlabeled MSCs (Figure 5A–C).
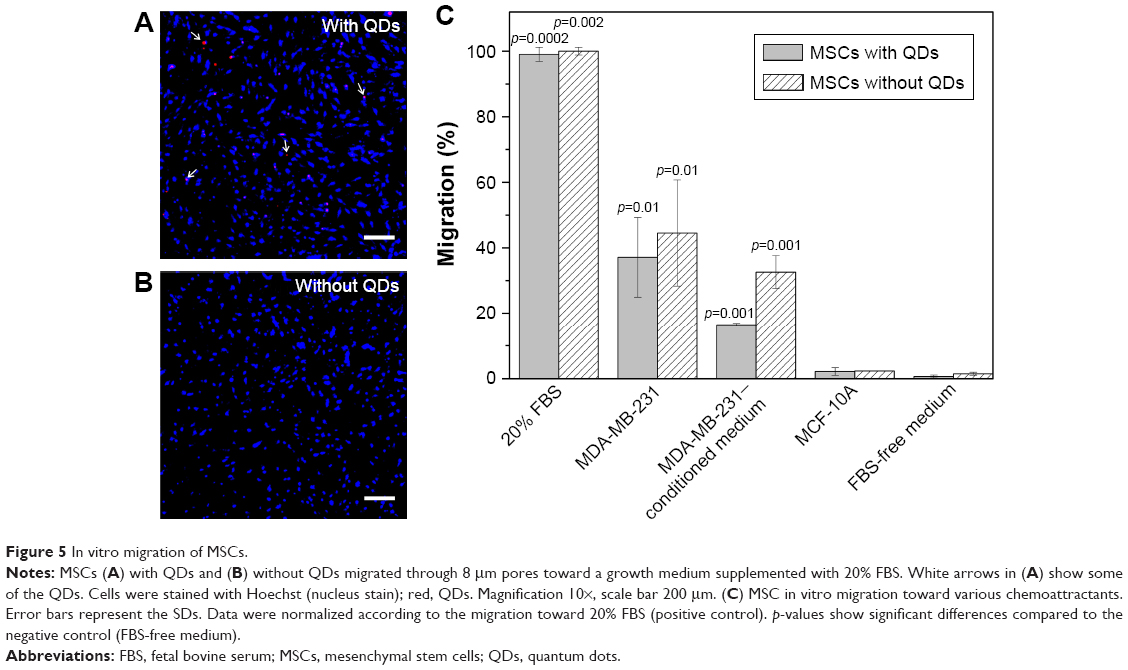 | Figure 5 In vitro migration of MSCs. |
To detect whether in vitro migration of MSCs depends on the donor, we tested the migration of MSCs extracted from three different donor skin tissues (Figure 6). The results showed no differences in migration toward positive and negative controls between different donor cells. Yet, some differences arose in migration toward cancer cells – one out of three donor MSCs displayed higher in vitro migration both with and without QDs.
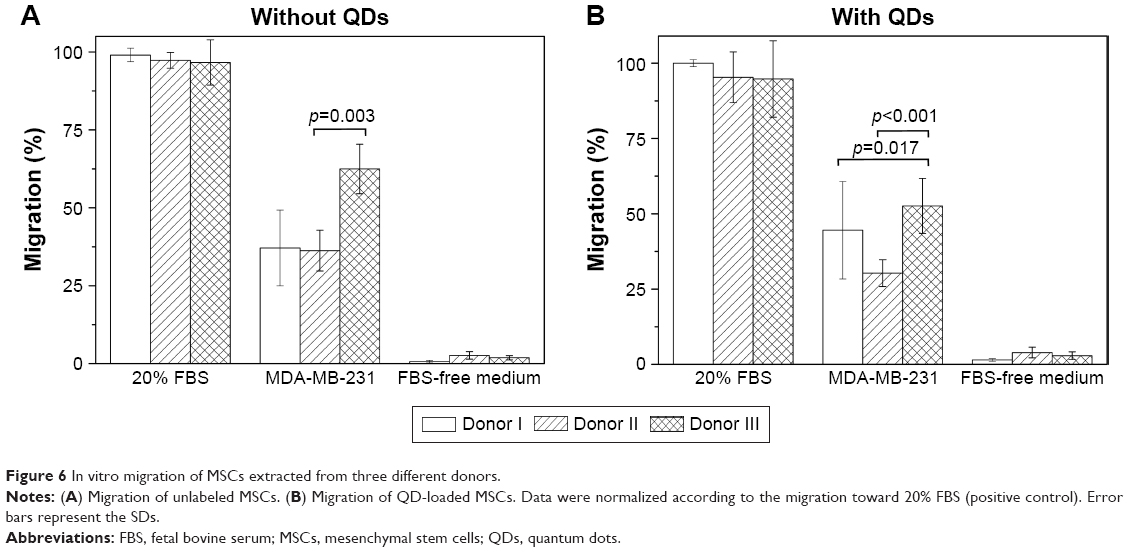 | Figure 6 In vitro migration of MSCs extracted from three different donors. |
In vivo migration
In vitro migration experiment showed that MSCs have a predisposition to migrate specifically to cancer cells while avoiding healthy cells (Figure 5); thus, the in vivo experiment with human breast tumor-bearing immunodeficient mice was performed. Confocal micrographs of tumor and metastasis cryosections showed red QD PL (Figure 7A and D). Interestingly, QD-labeled MSCs were distributed throughout the entire tumor (Figure 7A–C), while in the metastatic tissue, most QDs were detected in cells of different morphology (Figure 7D–F).
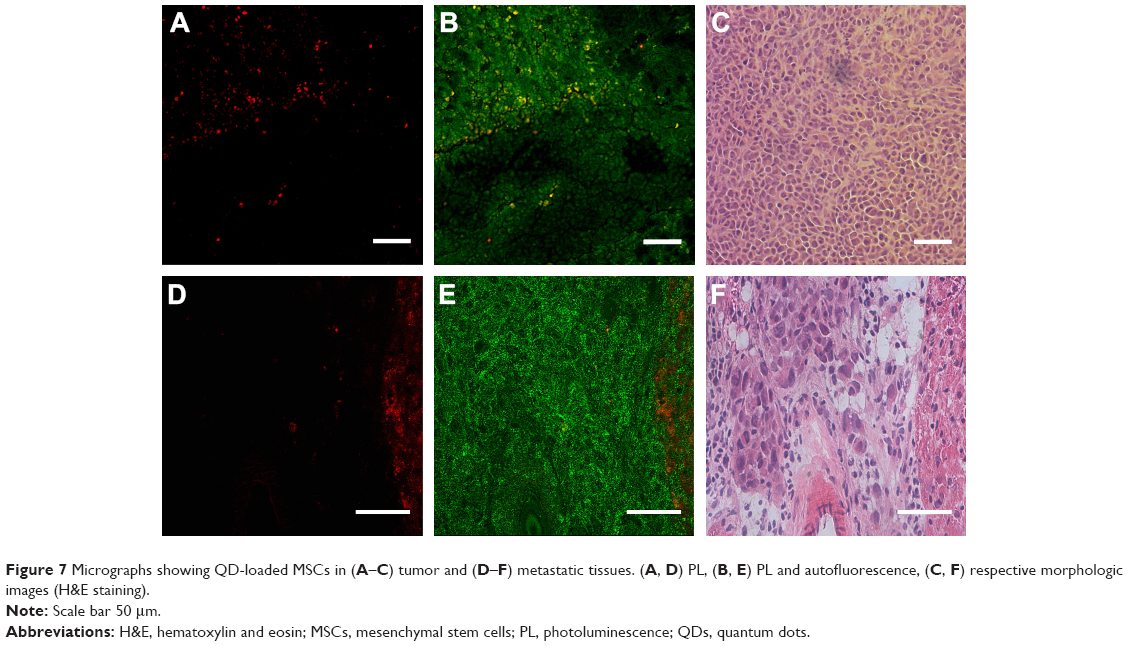 | Figure 7 Micrographs showing QD-loaded MSCs in (A–C) tumor and (D–F) metastatic tissues. (A, D) PL, (B, E) PL and autofluorescence, (C, F) respective morphologic images (H&E staining). |
In parallel, tissue homogenates were analyzed with a flow cytometer. To accurately evaluate QD-positive cells, homogenized samples were stained with anti-human CD44 antibody that binds only to cancer cells and MSCs. It was discovered that up to 19% of tumor cells were QD-positive cells (Figure 8A). During the tumor formation period, several mice developed spontaneous metastases due to rapid tumor growth. It was a good chance to investigate the accumulation of MSCs at the metastasis site. Up to almost 7% of the cells constituting the metastases were identified as QD positive (Figure 8B). A comprehensive flow cytometric analysis was carried out with healthy mice organs as well. Set side by side, it became obvious that the largest number of QD-positive cells was detected in the tumor. However, after a week, the amount of tumor-residing QD-labeled cells decreased approximately two times (Figure 8C). Twenty-four hours after the injection of MSCs, a statistically significant amount of QD-loaded MSCs was detected in the spleen and kidneys. After 7 days, the amount of QD-positive cells decreased and of all healthy mice organs, they were detected mainly in the liver. The highest accumulation of MSCs in mice organs was in the body parts responsible for filtration and clearance of blood and lymph (liver, spleen, kidneys), which indicates that after 7 days MSCs redistributed into the liver or were cleared from the mice (Figure 8C). The amount of MSCs found in the MSC injection site remained similar throughout the experiment.
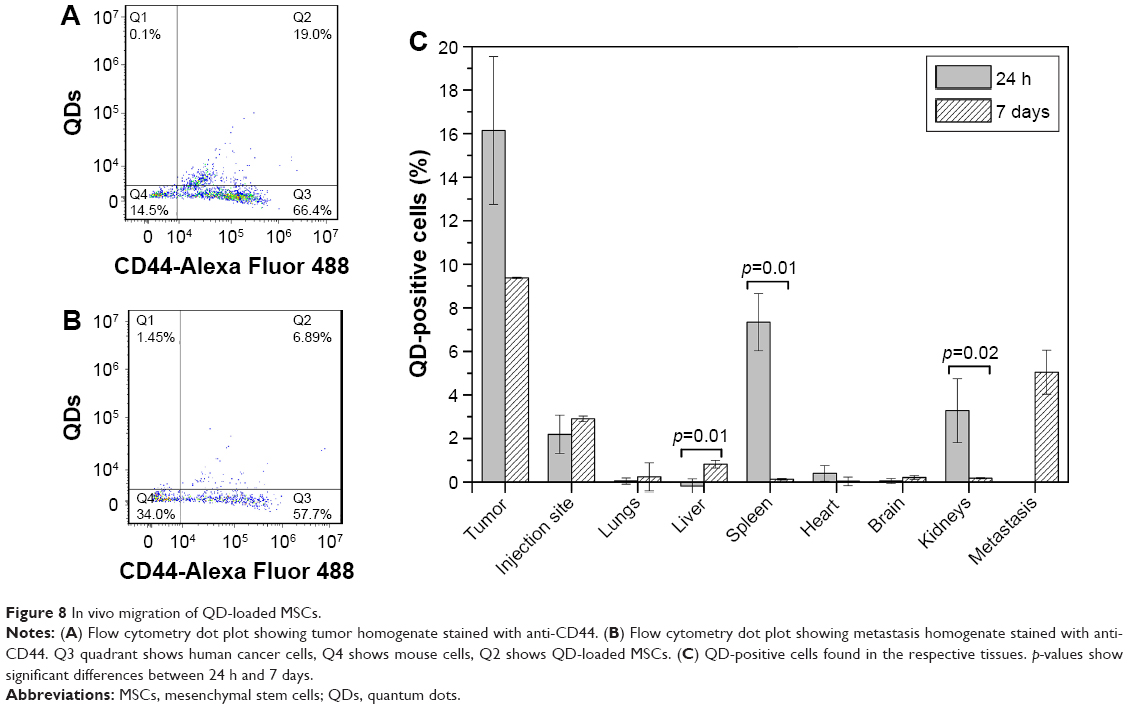 | Figure 8 In vivo migration of QD-loaded MSCs. |
Discussion
Nanoparticles might become a preferential choice in the future instead of conventional cancer treatment. Due to exceptional physical, chemical, and optical characteristics, nanoparticles are ideal candidates for early cancer diagnostics and great benefits are anticipated for cancer therapy. Although it is claimed that nanoparticles should accumulate specifically inside the tumor due to the enhanced permeability and retention effect,47 passive accumulation lacks specificity to tumors.48 As a result, functionalization of nanoparticles using various targeting molecules – ligands, peptides, antibodies, and so on – became a natural choice. There were attempts to use antibody-conjugated QDs for cancer cell imaging and tracking.49,50 We previously showed that the antibody–QD complex not only recognizes its specific antigen, but also is engulfed inside the cells, while antibodies conjugated with commonly used organic fluorophore FITC remained on the cell membrane.50 Although these results show promising potential of antibody–nanoparticle complexes to be used in both diagnostics and therapy, the success of such therapy is limited due to lack of tumor-specific antigens.51 Ergo, nanoparticle transportation using cells that are naturally attracted to tumors emerged as a potential treatment method. MSCs have natural stealth properties and are able to cross biologic barriers.52,53 Thus, MSCs hold a tremendous potential for being employed in anticancer therapy as drug delivery vehicles. In addition, nanoparticle-labeled MSC homing to cancerous tissue would add great benefit to visualization of tumors and/or metastases and their response to treatment.
In this study, we analyzed the toxicity, accumulation patterns, and both in vitro and in vivo migration of QD-loaded dermal MSCs. We have shown that commercially available QDs are nontoxic to MSCs (Figure 3A), are efficiently and rapidly taken up by the cells (Figure 3B), and are localized in endocytic vesicles in the perinuclear region (Figure 4). Despite studies showing nanoparticle anchoring to the outer cell membrane,54,55 QD internalization by MSCs is more common.56–58 Nanoparticle uptake and localization inside the cytoplasm is more advantageous as it leaves the cell membrane unaltered since the nanoparticles do not block cellular receptors. Internalization can also protect QDs from interacting with healthy tissue in vivo and from the unpredictable detachment during cargo transportation.59 QDs did not penetrate into the nucleus of MSCs (Figure 4), hence minimizing the potential genotoxicity caused by the binding of nanoparticles to DNA.60
Most importantly, our results showed that MSCs, extracted from the skin connective tissue, were able to migrate toward cancer cells in vitro and in vivo (Figures 5–8). The migration capability toward cancerous tissue is a prerequisite for MSCs to be used in targeting tumor cells. We successfully simulated directional migration of MSCs toward human breast cancer cells and their conditioned medium in vitro. Our results complement the evidence that MSCs tend to migrate toward cancer cells,13,18 cancer cell-conditioned medium,61 and growth medium, supplemented with recombinant proteins found in cancer cell-conditioned medium,62 verifying that cancer cells secrete molecules promoting MSC migration. The key molecules inducing MSC migration toward tumors supposedly are vascular endothelial growth factor,63 fibroblast growth factor 2,64 stromal cell derived factor 1 (SDF-1),65 chemokine (C-C motif) ligand 2, also known as monocyte chemotactic protein-1,66 and macrophage migration inhibitory factor.13 MSC migration is suppressed by antibodies against chemokine (C-X-C motif) receptor 4 (CXCR4)67 or by receptor antagonists,13 proving SDF-1/CXCR4 to be the main axis mediating MSC migration to cancerous tissue.51 Our research shows that in comparison to the chemotaxis toward highly potent chemoattractant (growth medium supplemented with 20% serum), the migration induced by cancer cells was mediocre (Figures 5C and 6). However, this may be related to the fact that the conditions under which MSCs are cultured in vitro are different from the ones found in their natural in vivo niches.68 Therefore, longer ex vivo cultivation leads to the reduced expression of receptors responsible for cell migration toward malignant cells (eg, CXCR4).69 The receptor for SDF-1 – CXCR4 – is found on both the cancer cell membrane as well as MSCs; thus, it participates in tumor metastasis if overexpressed in cancer cells70 or partakes in MSC migration toward the tumor if found on MSCs.71 To detect the level of CXCR4 in skin MSCs, we performed additional flow cytometric analysis on MSCs using anti-CXCR4 monoclonal antibody. The study revealed that our cultured MSCs have only 4%±2.1% CXCR4-positive cells (Figure S1). Shen et al also found 4.1% CXCR4 expression in umbilical cord MSCs,72 while Shi et al found an almost negative CXCR4 cell surface expression in fetal bone marrow MSCs; whereas, upon cytokine stimulation, the amount of CXCR4 increased to 55.7%.73 The poor expression of CXCR4 might be the reason for weak MSC migration to cancer cells both in vitro and in vivo and should be increased before in vivo manipulation. Hypoxia,74 cellular modifications,75 and preincubation with cytokines, for example, interleukin-6 or hepatocyte growth factor,76 are several strategies enhancing the migration of MSCs. Additionally, MSC migration potential might be donor dependent. Figure 6 shows that MSC migration depends slightly on the donor – one donor’s MSCs possessed higher than usual migration toward cancer cells and such an effect must be taken into account before choosing a donor as it would lead to better outcome of the potential therapy.
Another important factor for low MSC engraftment is the retaining properties of QDs. QD excretion from MSCs is a topic of controversy. Although it has been noted that QDs might leak from intact stem cells,56,58,77 it was also reported that QD-labeled cells retain bright QD PL for a few weeks and do not transfer nanoparticles to adjacent cells and host tissues.25,78,79 Such disagreements might be explained by different QD surface charges, as positively charged nanoparticles were used in most studies that showed QD leakage from cells,56,77 while negatively charged nanoparticles were retained inside the cells better.78,79 Our previous in vitro studies showed that QD PL decreases by 30% after the first 24 h most probably due to some degradation in lysosomes and does not change after additional 24 hours.38
Our in vitro migration study showed that incubation with QDs did not diminish cell migration, showing the biocompatibility of the nanoparticles used (Figures 5 and 6). These results are in agreement with previous studies using MSCs and nanoparticles.80,81
In vivo migration results with SCID mice revealed that most MSCs were located in tumor tissue in comparison to healthy organs and MSC injection site. The highest number of MSCs was detected in the tumor 24 h postinjection, suggesting that short periods are sufficient and most suitable for MSCs to reach the tumor. We observed minor engraftment of subcutaneously administered MSCs in organs comprising the mononuclear phagocyte system – liver, spleen, kidneys (Figure 8C). Previous studies also have shown localization of exogenously administered MSCs in the aforementioned tissues.82 No distribution of MSCs was detected in other healthy organs, including heart, brain, lungs. Although MSC entrapment in the lungs is quite common due to the large size of the cells (20–24 μm) and the small lung capillaries of diameter 10–15 μm,83 we detected only a slight engraftment of MSCs in the lungs. Our findings concerning poor distribution of exogenously administered MSCs in healthy tissues are in agreement with earlier reports.84–86 This property adds to the benefits of MSC application in tumor-tropic therapy, while avoiding multiorgan toxicity.
The role of MSCs in cancer development is a topic of controversy. Although we did find encouraging data that QD-loaded MSCs migrate toward breast tumor in vivo, it must be taken into account that MSCs could act as pro-tumorigenic due to paracrine stimulation that promotes metastases, angiogenesis, immune system suppression, expansion of cancer stem cells,87–89 as well as tumor suppressor agents through the inhibition of migration, suppression of cell cycle, and apoptosis induction.90–92 Thus, to prevent therapy-induced tumor progression, QDs ought to act as theranostic agents, combining diagnostics due to PL and therapeutics due to the attachment of anticancer agents, for example, photosensitizers. Such complexes have been already designed and tested as effective tools for pancreatic cancer.93
Although we chose breast cancer as a tumor model, recent studies show the applicability of MSC-based therapy for the treatment of tumors of various locations, for example, ovarian,94 liver,46 lung,95 and so on. A considerable amount of research has been done on glioma models, as MSCs are able to cross the blood–brain barrier and localize in intracranial neoplasms.20,53,62
Adipose tissue- or bone marrow-derived MSCs are most commonly used in tumor-tropic cell therapy researches. To our knowledge, we are the first ones to use skin connective tissue MSCs for tumor-tropic nanoparticle delivery. Using skin MSCs for the delivery of anticancer agents would be advantageous, since the skin tissue composes the widest area and has the easiest accessibility in the human body.
Conclusion
In this study, we tested the ability of skin-derived MSCs to be used as cellular nanoparticle vehicles and evaluated their tumor-tropic migration toward breast cancer cells in vitro and in vivo. We demonstrated that skin connective tissue MSCs are able to selectively deliver QDs to human tumor xenograft models. To our knowledge, this is the first time that skin MSCs have been used in tumor-tropic in vitro and in vivo studies. We suggest that QD-loaded skin MSCs could be a useful tool to target cancer cells. However, further studies are necessary.
Acknowledgments
This study was supported by the Lithuanian-Latvian-Taiwanese joint project Grant No TAP LLT 03/2014 and State Research program ‘Improvement of early cancer diagnostics’ approved by the Ministry of Education and Science of Lithuania (No V-273). We are very grateful to Vilius Poderys for measuring QD size distribution using atomic force microscopy and dynamic light scattering.
Disclosure
The authors report no conflicts of interest in this work.
References
Pattabiraman DR, Weinberg RA. Tackling the cancer stem cells-what challenges do they pose? Nat Rev Drug Discov. 2014;13(7):497–512. | ||
Wei X, Yang X, Han ZP, Qu FF, Shao L, Shi YF. Mesenchymal stem cells: a new trend for cell therapy. Acta Pharmacol Sin. 2013;34(6):747–754. | ||
Barkholt L, Flory E, Jekerle V, et al. Risk of tumorigenicity in mesenchymal stromal cell-based therapies-bridging scientific observations and regulatory viewpoints. Cytotherapy. 2013;15(7):753–759. | ||
Ren G, Chen X, Dong F, et al. Concise review: mesenchymal stem cells and translational medicine: emerging issues. Stem Cell Transl Med. 2012;1(1):51–58. | ||
Williams AR, Hare JM. Mesenchymal stem cells biology, pathophysiology, translational findings, and therapeutic implications for cardiac disease. Circ Res. 2011;109(8):923–940. | ||
Rojas M, Gupta N, Majka SM. Mesenchymal Stem Cells: Complex Players in Lung Repair and Injury. In: Hayat MA, ed. Stem Cells and Cancer Stem Cells, Volume 7: Therapeutic Applications in Disease and Injury. Dordrecht: Springer Netherlands; 2012:145–154. | ||
Wang S, Hu F, Li J, et al. Design of electrospun nanofibrous mats for osteogenic differentiation of mesenchymal stem cells. Nanomedicine. Epub 2017 May 26. | ||
Uccelli A, Moretta L, Pistoia V. Mesenchymal stem cells in health and disease. Nat Rev Immunol. 2008;8(9):726–736. | ||
Arwert EN, Hoste E, Watt FM. Epithelial stem cells, wound healing and cancer. Nat Rev Cancer. 2012;12(3):170–180. | ||
Dvorak HF. Tumors: wounds that do not heal-redux. Cancer Immunol Res. 2015;3(1):1–11. | ||
Jung Y, Kim JK, Shiozawa Y, et al. Recruitment of mesenchymal stem cells into prostate tumours promotes metastasis. Nat Commun. 2013;4:1795. | ||
Hocking AM. The role of chemokines in mesenchymal stem cell homing to wounds. Adv Wound Care (New Rochelle). 2015;4(11):623–630. | ||
Lourenco S, Teixeira VH, Kalber T, Jose RJ, Floto RA, Janes SM. Macrophage migration inhibitory factor-CXCR4 is the dominant chemotactic axis in human mesenchymal stem cell recruitment to tumors. J Immunol. 2015;194(7):3463–3474. | ||
Xin H, Kanehira M, Mizuguchi H, et al. Targeted delivery of CX3CL1 to multiple lung tumors by mesenchymal stem cells. Stem cells. 2007;25(7):1618–1626. | ||
Kosztowski T, Zaidi HA, Quinones-Hinojosa A. Applications of neural and mesenchymal stem cells in the treatment of gliomas. Expert Rev Anticancer Ther. 2009;9(5):597–612. | ||
Suzuki T, Kawamura K, Li Q, et al. Mesenchymal stem cells are efficiently transduced with adenoviruses bearing type 35-derived fibers and the transduced cells with the IL-28A gene produces cytotoxicity to lung carcinoma cells co-cultured. BMC Cancer. 2014;14:713. | ||
Zhang TY, Huang B, Yuan ZY, Hu YL, Tabata Y, Gao JQ. Gene recombinant bone marrow mesenchymal stem cells as a tumor-targeted suicide gene delivery vehicle in pulmonary metastasis therapy using non-viral transfection. Nanomedicine. 2014;10(1):257–267. | ||
Kucerova L, Altanerova V, Matuskova M, Tyciakova S, Altaner C. Adipose tissue-derived human mesenchymal stem cells mediated prodrug cancer gene therapy. Cancer Res. 2007;67(13):6304–6313. | ||
Zhang X, Yao S, Liu C, Jiang Y. Tumor tropic delivery of doxorubicin-polymer conjugates using mesenchymal stem cells for glioma therapy. Biomaterials. 2015;39:269–281. | ||
Roger M, Clavreul A, Venier-Julienne MC, et al. Mesenchymal stem cells as cellular vehicles for delivery of nanoparticles to brain tumors. Biomaterials. 2010;31(32):8393–8401. | ||
Huang X, Zhang F, Wang H, et al. Mesenchymal stem cell-based cell engineering with multifunctional mesoporous silica nanoparticles for tumor delivery. Biomaterials. 2013;34(7):1772–1780. | ||
Zrazhevskiy P, Gao X. Quantum dot imaging platform for single-cell molecular profiling. Nat Commun. 2013;4:1619. | ||
Maestro LM, Ramirez-Hernandez JE, Bogdan N, et al. Deep tissue bio-imaging using two-photon excited CdTe fluorescent quantum dots working within the biological window. Nanoscale. 2012;4(1):298–302. | ||
Wegner KD, Hildebrandt N. Quantum dots: bright and versatile in vitro and in vivo fluorescence imaging biosensors. Chem Soc Rev. 2015;44(14):4792–4834. | ||
Rak-Raszewska A, Marcello M, Kenny S, Edgar D, Sée V, Murray P. Quantum dots do not affect the behaviour of mouse embryonic stem cells and kidney stem cells and are suitable for short-term tracking. PLoS One. 2012;7(3):e32650. | ||
Ho IA, Toh HC, Ng WH, et al. Human bone marrow-derived mesenchymal stem cells suppress human glioma growth through inhibition of angiogenesis. Stem Cells. 2013;31(1):146–155. | ||
Kidd S, Spaeth E, Dembinski JL, et al. Direct evidence of mesenchymal stem cell tropism for tumor and wounding microenvironments using in vivo bioluminescent imaging. Stem Cells. 2009;27(10):2614–2623. | ||
Ghaedi M, Soleimani M, Taghvaie NM, et al. Mesenchymal stem cells as vehicles for targeted delivery of anti-angiogenic protein to solid tumors. J Gene Med. 2011;13(3):171–180. | ||
Ding D, Mao D, Li K, et al. Precise and long-term tracking of adipose-derived stem cells and their regenerative capacity via superb bright and stable organic nanodots. ACS Nano. 2014;8(12):12620–12631. | ||
Pendleton C, Li Q, Chesler DA, Yuan K, Guerrero-Cazares H, Quinones-Hinojosa A. Mesenchymal stem cells derived from adipose tissue vs bone marrow: in vitro comparison of their tropism towards gliomas. PLoS One. 2013;8(3):e58198. | ||
Potdar PD, Prasannan P. Differentiation of human dermal mesenchymal stem cells into cardiomyocytes by treatment with 5-azacytidine: concept for regenerative therapy in myocardial infarction. ISRN Stem Cells. 2013;2013:9. | ||
Mahmoudifar N, Doran PM. Mesenchymal Stem Cells Derived from Human Adipose Tissue. In: Doran MP, ed. Cartilage Tissue Engineering: Methods and Protocols. New York, NY: Springer New York; 2015:53–64. | ||
Brückner S, Tautenhahn HM, Winkler S, et al. Isolation and hepatocyte differentiation of mesenchymal stem cells from porcine bone marrow “surgical waste” as a novel MSC source. Transplant Proc. 2013;45(5):2056–2058. | ||
Jackson WM, Aragon AB, Djouad F, et al. Mesenchymal progenitor cells derived from traumatized human muscle. J Tissue Eng Regen Med. 2009;3(2):129–138. | ||
Riekstina U, Cakstina I, Parfejevs V, et al. Embryonic stem cell marker expression pattern in human mesenchymal stem cells derived from bone marrow, adipose tissue, heart and dermis. Stem Cell Rev. 2009;5(4):378–386. | ||
Dominici M, Le Blanc K, Mueller I, et al. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy. 2006;8(4): 315–317. | ||
Riekstina U, Parfejevs V, Muceniece R, Cakstina I, Boroduskis M, Ancans J. FGF-2 Effect on the Self-Renewal of Human Dermal Stem Cell Population in vitro. Medicine. 2009;7502009:117–128. | ||
Saulite L, Dapkute D, Pleiko K, et al. Nano-engineered skin mesenchymal stem cells: potential vehicles for tumour-targeted quantum-dot delivery. Beilstein J Nanotechnol. 2017;8:1218–1230. | ||
Chellappa M, Anjaneyulu U, Manivasagam G, Vijayalakshmi U. Preparation and evaluation of the cytotoxic nature of TiO(2) nanoparticles by direct contact method. Int J Nanomedicine. 2015;10(Suppl 1):31–41. | ||
Chung TH, Wu SH, Yao M, et al. The effect of surface charge on the uptake and biological function of mesoporous silica nanoparticles in 3T3-L1 cells and human mesenchymal stem cells. Biomaterials. 2007;28(19):2959–2966. | ||
Huang DM, Hung Y, Ko BS, et al. Highly efficient cellular labeling of mesoporous nanoparticles in human mesenchymal stem cells: implication for stem cell tracking. FASEB J. 2005;19(14):2014–2016. | ||
Lu CW, Hung Y, Hsiao JK, et al. Bifunctional magnetic silica nanoparticles for highly efficient human stem cell labeling. Nano Lett. 2007;7(1):149–154. | ||
Damalakiene L, Karabanovas V, Bagdonas S, Valius M, Rotomskis R. Intracellular distribution of nontargeted quantum dots after natural uptake and microinjection. Int J Nanomedicine. 2013;8:555–568. | ||
Song C, Li G. CXCR4 and matrix metalloproteinase-2 are involved in mesenchymal stromal cell homing and engraftment to tumors. Cytotherapy. 2011;13(5):549–561. | ||
Shah K. Mesenchymal stem cells engineered for cancer therapy. Adv Drug Deliv Rev. 2012;64(8):739–748. | ||
Gao Y, Zhou Z, Lu S, et al. Chemokine CCL15 mediates migration of human bone marrow-derived mesenchymal stem cells toward hepatocellular carcinoma. Stem Cells. 2016;34(4):1112–1122. | ||
Nakamura H, Jun F, Maeda H. Development of next-generation macromolecular drugs based on the EPR effect: challenges and pitfalls. Expert Opin Drug Deliv. 2015;12(1):53–64. | ||
Durymanov MO, Rosenkranz AA, Sobolev AS. Current approaches for improving intratumoral accumulation and distribution of nanomedicines. Theranostics. 2015;5(9):1007–1020. | ||
Pang L, Cui H, Liu Y, Zhong W. Anti-VEGF antibody conjugated CdHgTe quantum dots as a fluorescent probe for imaging in living mouse. J Lumin. 2016;173:274–278. | ||
Steponkiene S, Dapkute D, Riekstina U, Rotomskis R. Accumulation and distribution of non-targeted and Anti-CD44-conjugated quantum dots in distinct phenotypes of breast cancer. J Nanomed Nanotechnol. 2015;6(6):1000341. | ||
Dragu DL, Necula LG, Bleotu C, Diaconu CC, Chivu-Economescu M. Therapies targeting cancer stem cells: current trends and future challenges. World J Stem Cells. 2015;7(9):1185–1201. | ||
Schmidt A, Ladage D, Steingen C, et al. Mesenchymal stem cells transmigrate over the endothelial barrier. Eur J Cell Biol. 2006;85(11):1179–1188. | ||
Liu L, Eckert MA, Riazifar H, Kang DK, Agalliu D, Zhao W. From blood to the brain: can systemically transplanted mesenchymal stem cells cross the blood-brain barrier? Stem Cells Int. 2013;2013:435093. | ||
Li L, Guan Y, Liu H, et al. Silica nanorattle-doxorubicin-anchored mesenchymal stem cells for tumor-tropic therapy. ACS Nano. 2011;5(9): 7462–7470. | ||
Cheng H, Kastrup CJ, Ramanathan R, et al. Nanoparticulate cellular patches for cell-mediated tumoritropic delivery. ACS Nano. 2010;4(2): 625–631. | ||
Ranjbarvaziri S, Kiani S, Akhlaghi A, Vosough A, Baharvand H, Aghdami N. Quantum dot labeling using positive charged peptides in human hematopoetic and mesenchymal stem cells. Biomaterials. 2011;32(22):5195–5205. | ||
Muller-Borer BJ, Collins MC, Gunst PR, Cascio WE, Kypson AP. Quantum dot labeling of mesenchymal stem cells. J Nanobiotechnology. 2007;5:9. | ||
Pietilä M, Lehenkari P, Kuvaja P, et al. Mortalin antibody-conjugated quantum dot transfer from human mesenchymal stromal cells to breast cancer cells requires cell–cell interaction. Exp Cell Res. 2013;319(18):2770–2780. | ||
Anselmo AC, Mitragotri S. Cell-mediated delivery of nanoparticles: taking advantage of circulatory cells to target nanoparticles. J Control Release. 2014;190:531–541. | ||
Fröhlich E. The role of surface charge in cellular uptake and cytotoxicity of medical nanoparticles. Int J Nanomedicine. 2012;7:5577–5591. | ||
Barcellos-de-Souza P, Comito G, Pons-Segura C, et al. Mesenchymal stem cells are recruited and activated into carcinoma-associated fibroblasts by prostate ancer microenvironment-derived TGF-β1. Stem Cells. 2016;34(10):2536–2547. | ||
Nakamizo A, Marini F, Amano T, et al. Human bone marrow-derived mesenchymal stem cells in the treatment of gliomas. Cancer Res. 2005;65(8):3307–3318. | ||
Niu CS, Dong YF, Gao G. Vascular endothelial growth factor/platelet-derived growth factor receptor pathway is involved in bone marrow mesenchymal stem cell differentiation and directional migration toward gliomas. Neural Regen Res. 2010;5(13):993–998. | ||
Ritter E, Perry A, Yu J, Wang T, Tang L, Bieberich E. Breast cancer cell-derived fibroblast growth factor 2 and vascular endothelial growth factor are chemoattractants for bone marrow stromal stem cells. Ann Surg. 2008;247(2):310–314. | ||
Sun Z, Wang S, Zhao RC. The roles of mesenchymal stem cells in tumor inflammatory microenvironment. J Hematol Oncol. 2014;7(1):14. | ||
Dwyer RM, Potter-Beirne SM, Harrington KA, et al. Monocyte chemotactic protein-1 secreted by primary breast tumors stimulates migration of mesenchymal stem cells. Clin Cancer Res. 2007;13(17):5020–5027. | ||
Ehtesham M, Yuan X, Kabos P, et al. Glioma tropic neural stem cells consist of astrocytic precursors and their migratory capacity is mediated by CXCR4. Neoplasia. 2004;6(3):287–293. | ||
Karp JM, Leng Teo GS. Mesenchymal stem cell homing: the devil is in the details. Cell Stem Cell. 2009;4(3):206–216. | ||
Marquez-Curtis LA, Janowska-Wieczorek A. Enhancing the migration ability of mesenchymal stromal cells by targeting the SDF-1/CXCR4 Axis. Biomed Res Int. 2013;2013:561098. | ||
Dillenburg-Pilla P, Patel V, Mikelis CM, et al. SDF-1/CXCL12 induces directional cell migration and spontaneous metastasis via a CXCR4/G alpha i/mTORC1 axis. FASEB J. 2015;29(3):1056–1068. | ||
Park SA, Ryu CH, Kim SM, et al. CXCR4-transfected human umbilical cord blood-derived mesenchymal stem cells exhibit enhanced migratory capacity toward gliomas. Int J Oncol. 2011;38(1):97–103. | ||
Shen C, Lie P, Miao T, et al. Conditioned medium from umbilical cord mesenchymal stem cells induces migration and angiogenesis. Mol Med Rep. 2015;12(1):20–30. | ||
Shi M, Li J, Liao L, et al. Regulation of CXCR4 expression in human mesenchymal stem cells by cytokine treatment: role in homing efficiency in NOD/SCID mice. Haematologica. 2007;92(7):897–904. | ||
Feng Y, Zhu M, Dangelmajer S, et al. Hypoxia-cultured human adipose-derived mesenchymal stem cells are non-oncogenic and have enhanced viability, motility, and tropism to brain cancer. Cell Death Dis. 2015;6:e1797. | ||
Kean TJ, Lin P, Caplan AI, Dennis JE. MSCs: delivery routes and engraftment, cell-targeting strategies, and immune modulation. Stem Cells Int. 2013;2013:732742. | ||
Sohni A, Verfaillie CM. Mesenchymal stem cells migration homing and tracking. Stem Cells Int. 2013;2013:130763. | ||
Pi QM, Zhang WJ, Zhou GD, Liu W, Cao Y. Degradation or excretion of quantum dots in mouse embryonic stem cells. BMC Biotechnol. 2010;10:36. | ||
Rosen AB, Kelly DJ, Schuldt AJ, et al. Finding fluorescent needles in the cardiac haystack: tracking human mesenchymal stem cells labeled with quantum dots for quantitative in vivo three-dimensional fluorescence analysis. Stem Cells. 2007;25(8):2128–2138. | ||
Slotkin JR, Chakrabarti L, Dai HN, et al. In vivo quantum dot labeling of mammalian stem and progenitor cells. Dev Dyn. 2007;236(12):3393–3401. | ||
Paris JL, de la Torre P, Manzano M, Victoria Cabanas M, Flores AI, Vallet-Regi M. Decidua-derived mesenchymal stem cells as carriers of mesoporous silica nanoparticles. In vitro and in vivo evaluation on mammary tumors. Acta Biomater. 2016;33:275–282. | ||
Sadhukha T, O’Brien TD, Prabha S. Nano-engineered mesenchymal stem cells as targeted therapeutic carriers. J Control Release. 2014;196:243–251. | ||
Kraitchman DL, Tatsumi M, Gilson WD, et al. Dynamic imaging of allogeneic mesenchymal stem cells trafficking to myocardial infarction. Circulation. 2005;112(10):1451–1461. | ||
Gao J, Dennis JE, Muzic RF, Lundberg M, Caplan AI. The dynamic in vivo distribution of bone marrow-derived mesenchymal stem cells after infusion. Cells Tissues Organs. 2001;169(1):12–20. | ||
Ren C, Kumar S, Chanda D, et al. Cancer gene therapy using mesenchymal stem cells expressing interferon-β in a mouse prostate cancer lung metastasis model. Gene Ther. 2008;15(21):1446–1453. | ||
Mouiseddine M, Francois S, Semont A, et al. Human mesenchymal stem cells home specifically to radiation-injured tissues in a non-obese diabetes/severe combined immunodeficiency mouse model. Br J Radiol. 2007;80(Spec No 1):S49–S55. | ||
Yun JW, Ahn JH, Kwon E, et al. Human umbilical cord-derived mesenchymal stem cells in acute liver injury: Hepatoprotective efficacy, subchronic toxicity, tumorigenicity, and biodistribution. Regul Toxicol Pharmacol. 2016;81:437–447. | ||
Karnoub AE, Dash AB, Vo AP, et al. Mesenchymal stem cells within tumour stroma promote breast cancer metastasis. Nature. 2007;449(7162):557–563. | ||
Huang WH, Chang MC, Tsai KS, Hung MC, Chen HL, Hung SC. Mesenchymal stem cells promote growth and angiogenesis of tumors in mice. Oncogene. 2013;32(37):4343–4354. | ||
Luo J, Ok Lee S, Liang L, et al. Infiltrating bone marrow mesenchymal stem cells increase prostate cancer stem cell population and metastatic ability via secreting cytokines to suppress androgen receptor signaling. Oncogene. 2014;33(21):2768–2778. | ||
Clarke MR, Imhoff FM, Baird SK. Mesenchymal stem cells inhibit breast cancer cell migration and invasion through secretion of tissue inhibitor of metalloproteinase-1 and -2. Mol Carcinog. 2015;54(10):1214–1219. | ||
Sun B, Roh KH, Park JR, et al. Therapeutic potential of mesenchymal stromal cells in a mouse breast cancer metastasis model. Cytotherapy. 2009;11(3):289–298. | ||
Zhu Y, Sun Z, Han Q, et al. Human mesenchymal stem cells inhibit cancer cell proliferation by secreting DKK-1. Leukemia. 2009;23(5):925–933. | ||
Steponkiene S, Valanciunaite J, Skripka A, Rotomskis R. Cellular uptake and photosensitizing properties of quantum dot-chlorin e6 complex: in vitro study. J Biomed Nanotechnol. 2014;10(4):679–686. | ||
Dembinski JL, Wilson SM, Spaeth EL, et al. Tumor stroma engraftment of gene-modified mesenchymal stem cells as anti-tumor therapy against ovarian cancer. Cytotherapy. 2013;15(1):20–32. | ||
Kolluri KK, Laurent GJ, Janes SM. Mesenchymal stem cells as vectors for lung cancer therapy. Respiration. 2013;85(6):443–451. |
Supplementary material
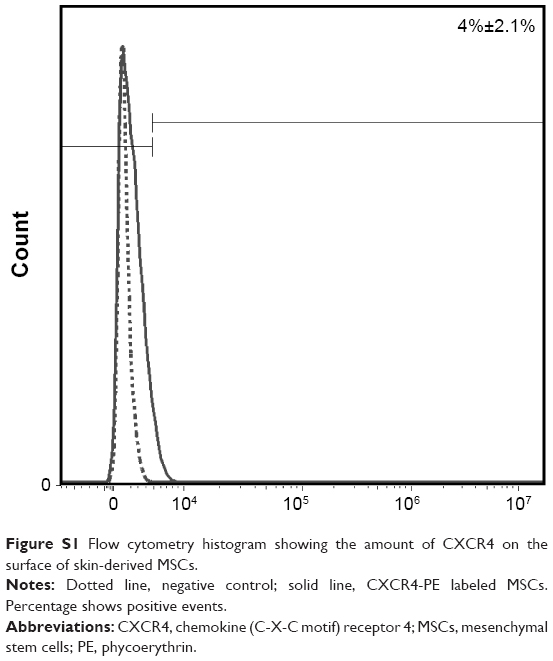 | Figure S1 Flow cytometry histogram showing the amount of CXCR4 on the surface of skin-derived MSCs. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.