Back to Journals » International Journal of Nanomedicine » Volume 14
Single-dose Ag85B-ESAT-6 loaded–poly(lactic-co-glycolic acid) nanoparticles confer protective immunity against tuberculosis
Authors Malik A ![]() , Gupta M, Mani R
, Gupta M, Mani R ![]() , Bhatnagar R
, Bhatnagar R
Received 26 April 2018
Accepted for publication 17 July 2018
Published 1 May 2019 Volume 2019:14 Pages 3129—3143
DOI https://doi.org/10.2147/IJN.S172391
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Thomas Webster
Anshu Malik,* Manish Gupta,* Rajesh Mani, Rakesh Bhatnagar
Molecular Biology and Genetic Engineering Laboratory, School of Biotechnology, Jawaharlal Nehru University, New Delhi 110067, India
*These authors contributed equally to this work
Background: Bacillus Calmette–Guérin, the attenuated strain of Mycobacterium bovis, remains the only available vaccine against tuberculosis (TB). However, its ineffectiveness in adults against pulmonary TB and varied protective efficacy (0–80%) speak to an urgent need for the development of an improved and efficient TB vaccine. In this milieu, poly(lactic-co-glycolic acid) (PLGA), is a preferential candidate, due to such properties as biocompatibility, targeted delivery, sustained antigen release, and atoxic by-products.
Methods: In this study, we formulated PLGA nanoparticles (NPs) encapsulating the bivalent H1 antigen, a fusion of Mycobacterium tuberculosis (Mtb) Ag85B and ESAT6 proteins, and investigated its role in immunomodulation and protection against Mtb challenge. Using the classical water–oil–water solvent-evaporation method, H1-NPs were prepared, with encapsulation efficiency of 86.1%±3.2%. These spherical NPs were ~244.4±32.6 nm in diameter, with a negatively charged surface (ζ-potential -4±0.6 mV).
Results: Under physiological conditions, NPs degraded slowly and the encapsulated H1 antigen was released over a period of weeks. As a proof-of-concept vaccine candidate, H1 NPs were efficiently internalized by the THP-1 human macrophages. Six weeks after a single-dose vaccination, H1 NP–immunized C57BL/6J mice showed significant increase in the production of total serum IgG (P<0.0001) and its isotypes compared to H1 alone, IgG2a being the predominant one, followed by IgG1. Further, the cytokine-release profile of antigen-stimulated splenocyte-culture supernatant indicated a strong TH1-biased immunoresponse in H1 NP–vaccinated mice, with ~6.03- and ~2.8-fold increase in IFNγ and TNFα cytokine levels, and ~twofold and 1.6 fold increase in IL4 and IL10 cytokines, respectively, compared to H1 alone–immunized mice. In protection studies, H1 NP–vaccinated mice displayed significant reductions in lung and spleen bacillary load (P<0.05) at 5-week post–Mtb H37Rv challenge and prolonged survival, with a mean survival time of 177 days, compared to H1 alone–vaccinated mice (mean survival time 80 days).
Conclusion: Altogether, our findings highlight the significance of the H1-PLGA nanoformulation in terms of providing long-term protection in mice with a single dose.
Keywords: PLGA, nanoparticles, Ag85B, ESAT6, Mycobacterium tuberculosis, vaccine
Introduction
The increasing morbidity and mortality caused by tuberculosis (TB) continues to be a global health problem. According to the 2017 World Health Organization report,1 TB has surpassed HIV as the most lethal infectious disease in the world. In 2016, 10.4 million new cases of active TB were reported, eventually resulting in 1.8 million deaths worldwide.1 Hitherto, bacillus Calmette–Guérin (BCG), the attenuated strain of Mycobacterium bovis, remains the only currently available vaccine against TB.1 To date, with more than 4 billion BCG doses administered worldwide, this vaccine has efficiently provided protection in infants against TB infection.2 However, the global decline in the rate of TB occurrence was only 1.5% from 2014 to 2015, as BCG is ineffective in adults against pulmonary TB.1 Moreover, globally the protective efficacy of BCG varies from 0 to 85% in different models, prompting an urgent need for the development of an improved and efficient TB vaccine.3,4
Currently, 12 different vaccine candidates based upon recombinant BCG, attenuated Mycobacterium tuberculosis (Mtb), or M. vaccae strains and inactivated whole-cell or adjuvant subunit vaccines are in human clinical trials.5 The demand for an efficient immunocorrelate to provide protection against TB in both human and animal models gained momentum following the disappointing outcomes of the MVA85A vaccine’s phase IIB clinical trials in humans.6 Among these approaches, subunit vaccination using Mtb proteins as immunogens provides many advantages, such as increased safety and stability, less toxicity, and fewer booster immunizations.7 However, mycobacteria have evolved various defensive strategies to interfere with antigen presentation and associated protective immunoresponses.8 Therefore, for appropriate processing of antigens, efficient delivery mechanisms to antigen-presenting cells (APCs) like macrophages and dendritic cells is of utmost importance.
It is only from the last decade that protein- or peptide-loaded nanoparticles (NPs) or microparticles (MPs) have been employed as efficient and stable vaccine-delivery vehicles.9 These antigen-conjugated or -encapsulated polymeric NPs or MPs are far more effective than their antigen-alone counterparts, as they serve as depots for slow and sustained release of the antigen, leading to prolonged immunoexposure with reduced systemic side effects.10 In this context, the US Food and Drug Administration (FDA)-approved polymer poly(lactic-co-glycolic acid) (PLGA), which belongs to the aliphatic polyester family, has long been a popular choice for therapeutics and vaccine-delivery applications.9 Once injected into the bloodstream, the release of any encapsulated antigen relies on the PLGA-polymer degradation time, which varies from a few months to a few years, depending upon its molecular weight and copolymer ratio.11 The major advantages of using PLGA as an ideal delivery carrier are properties like biocompatibility, nonimmunogenicity, antigen stabilization, controlled and sustained antigen release, atoxic by-products (lactic acid and glycolic acid) and targeted delivery.
Besides cell targeting and antigen uptake, the nature of immunoresponse induced by a polymeric NP vaccine is governed by various physical parameters, such as size, shape, and surface properties.12–14 Dendritic cells preferentially engulf NPs of 20–200 nm in size through pinocytosis, whereas 0.2–5 μm particles are phagocytosed primarily by APCs (like macrophages), eventually to CD4+ or CD8+ T-cell proliferation.15 Interestingly, PLGA NPs (size 200–600 nm) are known to trigger higher levels of TH1-specific IFNγ cytokines, whereas 2–8 μm PLGA MPs display a TH2-biased immunoresponse, marked by increased IL4 secretion.16 In 2016, Lawlor et al showed PLGA MPs triggered heightened NFκB activity and autophagy-dependent killing of tubercle bacilli in infected macrophages.17
Encapsulation of Ag85B, TB10.4, and TB10.4-Ag85B antigens of Mtb, hemagglutinin of Haemophilus influenzae, and surface antigens (SAG1 and SAG2) of Toxoplasma gondii in PLGA MPs all elicited prominent cellular immunity.18–21 On the other hand, PLGA MPs encapsulating tetanus toxoid, diphtheria toxin, hepatitis B surface antigen, and Yersinia pestis F1 antigen all induced TH2-biased humoral immunity.22–25 Likewise, numerous researchers have investigated the role of PLGA microspheres in selective delivery of antitubercular drugs into alveolar macrophages as part of postexposure therapy against TB.26 However, except for few instances, the role of PLGA as a nanoscale vaccine-delivery system has not been investigated in detail.
For protection against intracellular pathogens like Mtb and Salmonella typhimurium, it is essential to establish a TH1-type cellular immunoresponse.27 In this context, the secretory antigens of Mtb have long been considered potential vaccine candidates, as they are the first to encounter the host immune system. Also, there are reports of a strong correlation between multiplication of Mtb in the host and T cell–proliferation response against its secreted antigens, eg, ESAT6, CFP10, 16 kDa protein, Ag85A, and Ag85B.28,29 The Ag85B and ESAT6 antigens have been identified as key immunogenic targets during TB infection in Lefford’s mouse model, as they are strongly recognized T-cell antigens in the early phase of infection and induce cellular immunity by stimulating memory effector cells.30 Carpenter et al showed that intranasal immunization with early secretory antigen ESAT6-encapsulated polylactic acid MPs induced ESAT6-specific IFNγ- and IL4-secreting cells and strongly elicited T-cell memory and effector response in lungs and mediastinal lymph nodes.31 Ag85B, a mycolyltransferase, has been identified as one of the most potent immunogens of Mtb, stimulating both cellular and humoral immunoresponses in Mtb-infected human and animal models. A recombinant fusion antigen of both Ag85B and ESAT6 (H1) has been employed as a promising subunit vaccine candidate against TB and is currently in the clinical phases.32–34 Immunization of mice with a live, recombinant S. typhimurium strain expressing the Ag85B–ESAT6 fusion protein by the oral route is known to trigger an antigen-specific TH1 response with predominantly higher IgG2a and IFNγ titers.35 Further, immunization with the H1 antigen in combination with IC31 adjuvant to BCG-vaccinated and prior or latently Mtb-infected individuals results in a strong, long-lived, antigen-specific T-cell response, reiterating the potential use of H1 antigen as an ideal TB-vaccine candidate.34
Altogether, a critical approach involving a compliant vaccine-delivery vehicle and the right immunogen would provide the best protective immunity against the TB pandemic. In the present study, we set out to explore the vaccine potential of H1-encapsulated PLGA NPs in terms of eliminating the need for adjuvants, requirement of booster doses, antigen stability, and sustained release. H1 NPs were first characterized for their physicochemical properties, cellular uptake, and internalization by macrophages, and finally their immunomodulatory properties were investigated in a C57BL/6J mice model. Our results showed that a single dose of H1 NPs was able successfully to mount protective immunity, encompassing a balanced TH1 and TH2 immune response, against Mtb infection. The efficacy of this nanoformulation could be further improvised to produce a next-generation Mtb vaccine.
Methods
Bacterial strains and culture conditions
Mtb strain was grown in Middlebrook 7H9 broth (BD Biosciences, San Hose, CA, USA) supplemented with 10% (v:v) albumin, dextrose, and catalase (ADC; BD), 0.2% (v:v) glycerol and 0.05% (v:v) Tween 80 or Middlebrook 7H10 agar plates (BD) containing 10% (v:v) oleic acid and ADC (OADC; BD) enrichment and 0.5% (v:v) glycerol. Escherichia coli (E. coli) recombinant strains were grown under standard conditions in Luria–Bertani broth (BD). All cultures were grown at 37°C with shaking at 180 rpm.
Molecular cloning, expression, and purification of H1 antigen
Ag85B and ESAT6 coding sequences for fbpB and esxA were PCR-amplified using suitable primers and Mtb H37Rv genomic DNA as the template. For constructing the recombinant H1 fusion antigen, the fbpB and esxA genes were fused at a unique KpnI site (offered by fbpB-R and esxA-F primers) by restriction digestion. The resulting H1 fusion DNA fragment was cloned under the isopropyl β-D-1-thiogalactopyranoside (IPTG)-inducible T5 promoter of the pQE30 expression vector (Qiagen) at BamH1 and HindIII sites and expressed in E. coli M15 cells (Qiagen) at 37°C for 4 hours. Expression of His6X-tagged recombinant H1 protein was confirmed by immunoblotting using anti-His monoclonal antibody.
The H1 fusion antigen was purified from the inclusion bodies using Ni–nitrilotriacetic acid (NTA) affinity chromatography. Briefly, 500 mL Luria–Bertani broth was seeded with 1 mL overnight-grown culture of H1-expressing E. coli recombinants and grown at 37°C until OD600 reached ~0.6. H1 expression was then induced using 1 mM IPTG for 5 hours. Cells were harvested and suspended in 20 mL buffer A (25 mM Tris-Cl [pH 8], 25% sucrose, 1 mM benzamidine, and 0.1% Tween 20) and lysed by sonication: four cycles (each at 10 seconds on at 30% amplitude, 20 seconds off, at 4°C) on a Sonics Vibra-Cell digital processor.
The lysate was centrifuged at 12,000 rpm for 30 minutes at 4°C. Inclusion bodies in the pellet were washed with 20 mL buffer B (containing 25 mM Tris-Cl [pH 8], 2 M urea, 200 mM NaCl, 0.1% Triton X-100, and 2 mM EDTA), followed by centrifugation. The inclusion-body pellet obtained thereafter was then washed with 20 mL buffer C (25 mM Tris-Cl [pH 8], 200 mM NaCl, 0.01% Triton X-100, 1 mM EDTA), followed by centrifugation. A final wash with 20 mL buffer D (25 mM Tris-Cl [pH 8]) was performed to remove residual detergent. Purified inclusion bodies were solubilized in 50 mM CAPS buffer (pH 11) containing 1.5% N-lauryl sarcosine and 300 mM NaCl for 2 hours. The soluble protein fraction obtained upon centrifugation (11,000 rpm for 30 minutes) was dialyzed against equilibration buffer E (50 mM Tris-Cl [pH 8.0] containing 300 mM NaCl) and further subjected to Ni-NTA affinity chromatography.
The 6x His-tagged recombinant H1 protein bound to the column was eluted with buffer F (buffer E containing 250 mM imidazole). Sample fractions were analyzed with SDS-PAGE. Fractions containing the recombinant protein were pooled, concentrated to 10 mg/mL using a Microbe Advance centrifugal device (3 kDa cutoff), dialyzed against PBS (pH 7.4), and stored at 4°C for not more than 2 weeks.
Preparation of PLGA-encapsulated H1 NPs
PLGA polymer (Sigma-Aldrich) used for H1 encapsulation had a lactide:glycolide feed ratio of 85:15 (molecular weight 50,000–75,000 Da), inherent viscosity of 0.55–0.75 dL/g at 30°C, and its end groups were deactivated with lauryl alcohol. The double-emulsion method, also known as water–oil–water solvent-evaporation, was adapted for preparation of the H1-loaded PLGA NPs.36 Briefly, 1 mg H1 fusion protein in PBS (100 μL, aqueous phase) was emulsified with 4 mL dichloromethane (DCM; Sigma-Aldrich) containing 50 mg/mL PLGA (organic phase). Optimum emulsification was obtained at 4°C by sonication at 35% amplitude for 1 minute using a 2 mm stepped microtip in a 750 W Sonics Vibra-Cell processor. For preparing blank NPs, PBS was used as the internal aqueous phase. The water–oil emulsion obtained was immediately added dropwise to a second aqueous phase of 1% polyvinyl alcohol (PVA; 87%–89% hydrolyzed, average molecular weight 31,000–50,000 Da) solution (12 mL) to minimize particle aggregation by increasing the distance between emulsified droplets. The water–oil–water emulsion was then resonicated with a 6 mm stepped microtip at 30% amplitude for 110 seconds. The resulting emulsion was stirred for 6 hours at room temperature for DCM evaporation and hardening of NPs. Finally, the H1-PLGA NPs were harvested by centrifugation at 12,000 g for 20 minutes and all remaining impurities removed, with subsequent washing with ice-cold distilled water four times. To prevent phase separation, particles were suspended in 5 mL Milli-Q water, frozen using liquid nitrogen, and subjected to overnight lyophilization at −54°C at 0.003 mbar. Lyophilized NPs were stored at −20°C until further use in small aliquots.
Determination of H1-encapsulation and -loading efficiency
To evaluate the encapsulation efficiency (EE) and loading efficiency (LE) of H1 NPs, a micro-BCA protein-assay kit (Thermo Fisher Scientific) was used according to the manufacturer’s instructions. Briefly, 10 mg NPs were dissolved in 1 mL acetonitrile (Sigma-Aldrich) for polymer disruption, followed by centrifugation at 12,000 g for 15 minutes. The step was repeated thrice to remove trace amounts of PLGA, and the protein pellet finally obtained was solubilized in 1% SDS. H1 EE was estimated by micro-BCA using BSA standards, dissolved in 1% SDS. Protein integrity was monitored with SDS-PAGE using purified H1 as control upon Coomassie brilliant blue staining. Images were acquired with a Molecular Imager Gel Doc XR system and analyzed using Quantity One software (Bio-Rad). H1-NP EE and LE were calculated:
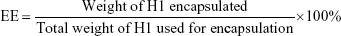
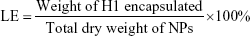
Determination of particle size, polydispersity, and ζ-potential
To measure the size range, polydispersity index, and ζ-potential of H1 NPs, a Zetasizer Nano ZS system (Malvern Instruments Ltd., Worcestershire, UK) with a 633 nm helium–neon laser was employed. Briefly, lyophilized H1 NPs were weighed and suspended in PBS (pH 7.4) to a final concentration of 0.1 mg/mL and introduced into the instrument to read the results. The ζ-potential and particle size were evaluated based upon electrophoretic mobility and laser diffraction, respectively.
Study of nanoparticle morphology by electron microscopy
Surface morphology of the NPs was studied using scanning electron microscopy (EVO40; Carl Zeiss, Jena, Germany). For imaging, lyophilized samples were spread on a carbon tape of an aluminum stub and gold coating applied with sputter coating (Polaron SC7640) at 2 kV for 200 seconds under inert argon, in order to make them conductive. Gold-coated samples were vacuum-dried and then examined at electron high-tension voltage of 20 kV and 50,000× magnification. For transmission electron–microscopy studies, lyophilized NPs were suspended in sterile deionized water and coated on carbon film with 200-mesh copper grids. Imaging was done with electron microscopy (JEM 2100F; Jeol, Tokyo, Japan) under high vacuum and 200 kV voltage.
In vitro release kinetics of H1 NPs
To monitor the in vitro release profile of H1 NPs under physiological conditions, 50 mg H1 NPs were suspended in 1 mL PBS (pH 7.4) and incubated at 37°C with constant stirring at 100 rpm. Supernatants were collected at regular intervals (till 32 days) and the release of H1 antigen estimated by micro-BCA protein estimation. Experiments were done in triplicate. The fractional H1 antigen released in the medium was calculated:
|
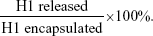
Uptake of nanoparticles by macrophages
To observe the uptake of NPs by macrophages, the H1 protein was fluorescently labeled with fluorescein isothiocyanate (FITC) and used for the preparation of NPs using water–oil–water evaporation. The THP1 human monocytic cell line was obtained from the National Center for Cell Science Repository, Pune, India and maintained in RPMI 1640 medium + 10% FBS up to suboptimal confluence. THP1 monocytes were then differentiated with 10 ng/mL phorbol 12-myristate 13-acetate (Sigma-Aldrich) onto a coverslip already placed at the bottom of a 3 mm–deep culture dish. After cells had adhered on the coverslip, H1-FITC NPs were added to a final concentration of 0.5 mg/mL and incubated at 37°C with 5% CO2 for 4 hours. Cells were then washed thrice with sterile PBS and fixed with 2% paraformaldehyde. The coverslip was then mounted on a glass slide and macrophages stained with DAPI. Untreated cells were taken as negative controls. Confocal images were visualized through both DAPI (blue) and FITC (green) channels under a Nikon Eclipse 80i fluorescent microscope at 600× and 900× magnification, followed by analysis using NIS Elements software. Eventually, a final overlay of both images was done to observe the presence of NPs within the macrophages.
Animal studies and ethical statement
In this study, inbred female C57BL/6J mice 6–8 weeks old were used for immunization. Mice were purchased from the National Centre for Laboratory Animal Sciences, Hyderabad, India. They were housed within microisolator cages using a ventilated animal-caging system in a biosafety level 3 laboratory, maintained under pathogen-free conditions, observed for any sign of disease or sickness, and fed pelleted food and water ad libitum. All studies were approved by the Institutional Animal Ethics Committee and the Institutional Biosafety Committee, Jawaharlal Nehru University, New Delhi, India. Regulations of the former were followed in all mice experiments. For all subsequent studies, mice were immunized by the intraperitoneal route and blood collected by retro-orbital bleeding. During experiments, vaccinated and infected mice were monitored every day. Mice were euthanized by cervical dislocation whenever required.
Mouse-immunization schedule
Figure 1 depicts the complete schedule of mouse immunization, challenge, and protection studies. Briefly, five mouse groups were formed, with 18 mice in each group. All groups were given a single-dose vaccination intraperitoneally, and an adjuvant-free immunization schedule was followed. The first and second groups were vaccinated with antigen H1 alone (50 μg/mouse) and H1-loaded PLGA NPs (50 μg H1 encapsulated in PLGA NPs/mouse), respectively, in a total volume of 0.1 mL per mouse. The second and third groups were immunized with blank NPs and PBS, respectively. Serum was collected at specific time intervals. Six weeks postvaccination, three mice from each group were sacrificed for splenocyte culturing and cytokine profiling. Alongside these, a group of mice were vaccinated with a single dose of BCG (105/mouse, Tubervac, Serum Institute of India) at first immunization as a positive control for protection studies.
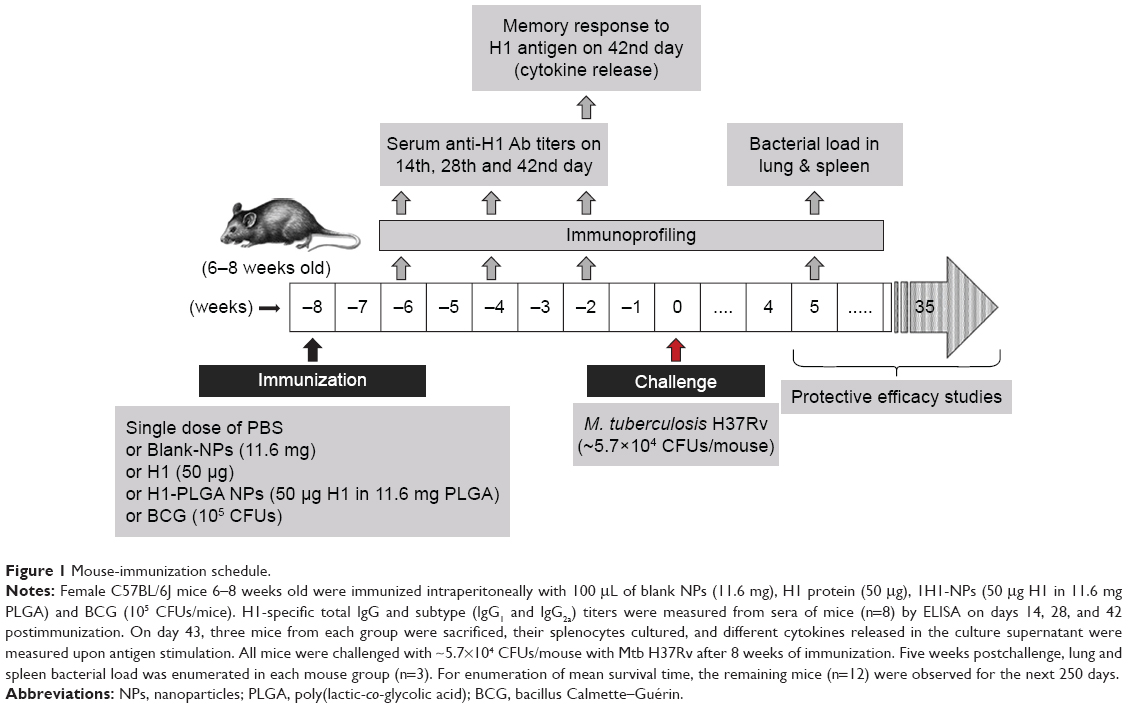 | Figure 1 Mouse-immunization schedule. |
Determination of antigen-specific IgG titers
On days 14, 28, and 42, sera was collected from eight mice, and antigen-specific antibody titers of IgG and its isotypes, IgG1 and IgG2a, were measured by indirect ELISA as previously described, with some modifications.36 ELISAs were performed on 96-well, flat-bottom, high-binding polystyrene microtiter plates (Nunc MaxiSorp). Briefly, wells were precoated with H1 antigen (500 ng/well, diluted in PBS pH 7.4) overnight at 4°C, followed by blocking with 2% BSA and washing thrice with PBST (PBS pH 7.4 containing 0.05% Tween 20). Meanwhile, sera were serially diluted (102–106) and 100 μL of each dilution added to the coated wells in triplicate. After incubation of 2 hours at room temperature, wells were washed five times with PBST and subsequently incubated with either HRP-conjugated goat antimouse IgG (1:10,000 dilution) or its isotypes – IgG1 and IgG2a (1:5,000 dilution) – for 1 hour. Microtiter plates were then washed five times with PBST and finally incubated with OptEIA TMB substrate (BD Bioscience) for 30 minutes in the dark. The reaction was stopped with 1 N HCl and absorbance read at 450 nm using 570 nm as the reference wavelength in a Tecan Sunrise microplate reader. The threshold value for titer determination was taken as the mean absorbance plus three times the SD, obtained at 1:100 dilution of the PBS mouse group (negative control). End point–dilution titers were defined as the inverse of the highest dilution that resulted in an absorbance value (optical density at 450 nm) greater than that of the threshold value.
Evaluation of secreted cytokines after in vitro stimulation of splenocytes
To study memory response to H1-antigen recall, three mice from each group were killed on day 43 day postimmunization and their spleens aseptically removed. Splenocytes were spilled off from the spleen-capsule slices using frosted-glass slides in RPMI 1640 medium supplemented with 10% FBS, and the suspension was passed through a 70 μm–cutoff cell strainer to remove contaminating tissue and to get a homogeneous cell suspension. Contaminating red blood cells were lysed using red blood–cell lysis buffer. The cells obtained were washed twice with complete RPMI 1640 medium (containing 10% FBS) and viability assessed by trypan blue exclusion staining. Splenocytes were then seeded in 96-well tissue-culture plates (BD Bioscience) at 106 cells/well and stimulated with either 5 μg/mL of concanavalin A (Sigma Aldrich) as positive control or with 10 μg/mL H1 antigen (test) and incubated at 37°C under a humidified condition of 5% CO2. Unstimulated cells were treated as background control. After 72 hours of incubation, culture supernatants were collected and stored at −80°C until further use.
Levels of IFNγ, TNFα, IL10, and IL4 cytokines were measured in splenocyte-culture supernatant using the OptEIA kit as per the manufacturer’s instructions. A linear regression equation derived by plotting the absorbance of known cytokine standards (provided by the manufacturer) at 450 nm was used to extrapolate test-cytokine concentrations in the culture supernatant.
Mtb-challenge and -protection studies
For protection studies, at 8 weeks after vaccination the remaining vaccinated mouse groups (15 mice/group) were challenged intravenously with Mtb H37Rv through the lateral tail vein (for challenge dose, see Results). All infections were carried out within laminar flow safety enclosures in the BSL3 facility, and infected animals were housed in microisolator cages, as described earlier. For enumeration of lung and spleen bacillary load, three mice from each group were sacrificed at 5 weeks postinfection. Briefly, the lung and spleen from each vaccinated mouse were removed aseptically, homogenized in sterile PBS, serially diluted, and plated on Middlebrook 7H10 agar plates containing OADC supplement. Plates were incubated at 37°C for 3–4 weeks and colonies counted. Results are expressed as log10 of bacterial counts (obtained CFU) in the lung and spleen of the vaccinated mice. For protective-efficacy studies, all remaining challenged mice (n=12/group) were monitored for weight loss, behavioral changes, and survival/death due to infection for the next 250 days.
Statistics and data presentation
The results reported here are means ± SE of the data obtained from each immunized mouse group. The Mtb-challenge experiments were evaluated using Kaplan–Meier survival estimates using GraphPad Prism. For comparison of data obtained from H1 NP–vaccinated mice to those immunized with H1 alone, P-values were calculated using one-way ANOVA, followed by Tukey’s multiple-comparison test. Differences in statistical significance are given in figures and legends. P<0.05 was considered significant.
Results
Cloning, expression, and purification of H1 antigen
Coding sequences corresponding to Ag85B and ESAT6 were fused to create a 1,266 bp–long H1 fusion product. This product was cloned under the IPTG-inducible T5 promoter of the pQE30 vector and expressed in E. coli M15 cells. Expression of H1 fusion antigen was mostly in the inclusion-body fraction, and hence the latter was isolated and purified to homogeneity before solubilization. Recombinant H1 protein was purified by Ni-NTA affinity chromatography of the solubilized fraction, followed by size-exclusion chromatography (as described in Methods). Our preparation yielded ~9.2 mg/L soluble recombinant H1 antigen and was the sole protein band (>99% purity) visible on SDS-PAGE after Coomassie brilliant blue staining (Figure 2B) and immunoblotting using anti-His monoclonal antibody (data not shown).
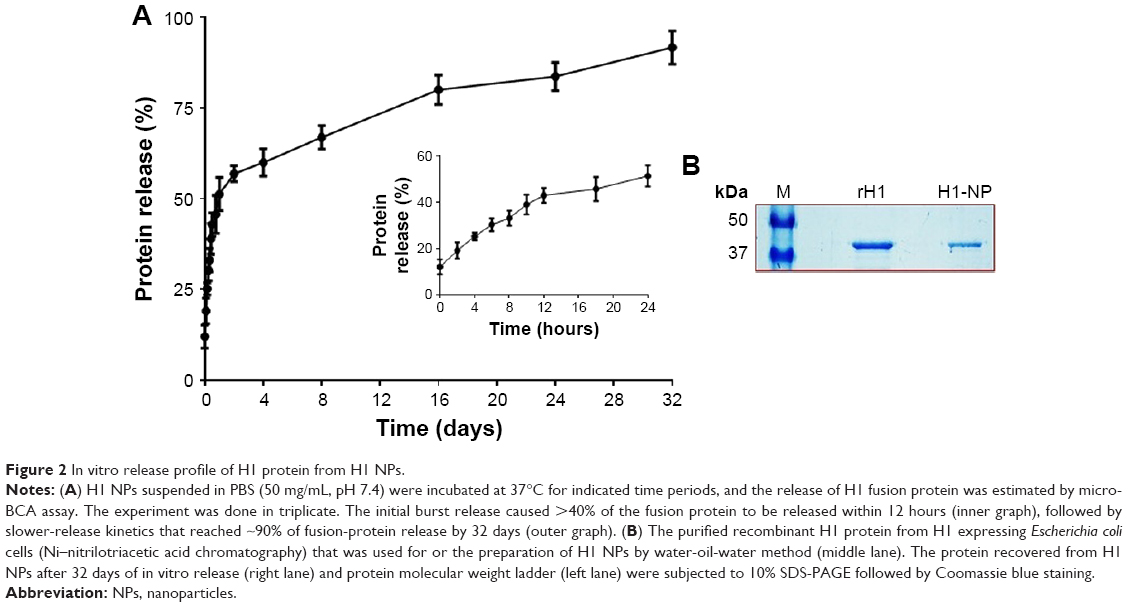 | Figure 2 In vitro release profile of H1 protein from H1 NPs. |
Morphological and physicochemical characterization of H1-PLGA NPs
H1-PLGA NPs were prepared by double-emulsion solvent evaporation. For primary emulsion preparation, the particle size–determining step – the ratio of inner aqueous phase (protein in PBS) to outer organic phase (DCM) – was kept at 1:40. A ratio of 1:12 was then maintained between the organic phase to the outer aqueous phase (ie, 1% PVA), to ensure formation of unaggregated NPs with hardened PVA coating. We obtained H1-antigen EE and LE of 86.18%±3.2% and 0.5%±0.03%, respectively. Scanning electron microscopy of H1-loaded PLGA NPs revealed a uniform, globular, smooth appearance, with no cavities (Figure 3A). Under transmission electron microscopy (Figure 3B), the particles appeared as intact, electron-dense spheres of ~200–300 nm diameter. Dynamic light scattering of an H1-NP suspension on the Zetasizer Nano ZS indicated narrow size distribution and greater colloidal stability (Figure 3C and D). Mean particle diameter was found to be 244.4±32.6 nm in PBS (pH 7.4), with a polydispersity index of <0.2, suggesting negligible heterogeneity in the nanoformulation. The ζ-potential of these particles was found to be −4±0.6 mV, signifying a slight negative charge on the surface of these NPs. These results were reproducible in multiple batches.
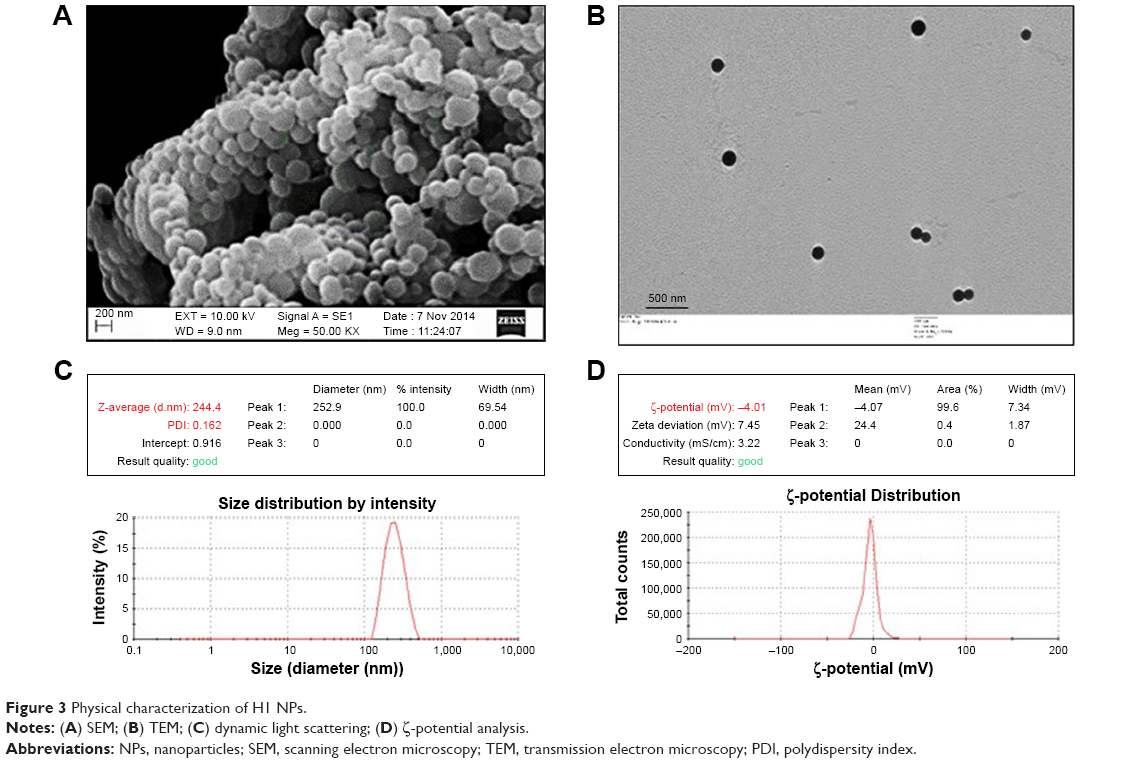 | Figure 3 Physical characterization of H1 NPs. |
In vitro release of protein from PLGA nanoparticles
The rate of H1 release from H1-encapsulated PLGA NPs was estimated with the micro-BCA kit at regular intervals till 32 days under in vitro conditions at physiological pH and temperature (as described in Methods). Figure 2A shows the in vitro release profile of H1 antigen from H1 NPs. Surprisingly, after only 2 hours of incubation, a burst release of ~19.13%±3.5% of the protein was detected in the supernatant. By 12 hours, ~42.8%±3.2% of H1 protein had been released in the supernatant. However, by 24–48 hours, the release profile had reached a plateau and thereupon displayed controlled and sustained release for 32 days. On day 32, slow and sustained release of ~91.5%±4.5% of the total H1 protein was observed, suggesting that under in vivo conditions the H1 antigen would probably have been released by H1 NPs for a longer duration to stimulate the immune system. The integrity of the antigen released after 32 days was further assessed by SDS-PAGE. An intact band of the desired molecular weight was observed (~43 kDa), without any degradation of the protein (Figure 2B), revealing H1 to be a stable antigen. Based on this release profile, the dose of H1 antigen for subsequent mouse immunization was assessed. Briefly, a dose of 50 μg H1 antigen/mouse was selected for vaccination, as nearly 50% of the antigen would be released in a burst within the first 24 hours. The subsequent slow release of the remaining antigens from the NP depot would then act as boosters for the mouse.
Intracellular uptake of H1 NPs by THP1 macrophages
Phagocytosis and antigen presentation to immunoeffector cells is an inherent property of macrophages. Therefore, the ability of THP1 macrophages to phagocytose H1 NPs was examined under ex vivo conditions. Briefly, the human macrophage cell line THP1 was incubated at 37°C with 5% CO2 for 4 hours with FITC-labeled H1 antigen–encapsulated PLGA NPs, and NP internalization was monitored under confocal microscopy. The microscopy confirmed the internalization of FITC-labeled H1 antigens by the macrophages (Figure 4). The H1 NP–release profile suggested that after 4 hours of incubation, ~20% of the antigen would have been released in the medium. However, due to experimental limitations at this stage, it cannot be distinguished whether the observed fluorescence was from FITC-H1 antigens phagocytosed alone or as PLGA encapsulates. H1 NPs were observed at both the periphery and inside the macrophages. These results suggest that once the H1 NPs are injected into an immune system, they will be efficiently taken up by APCs like macrophages.
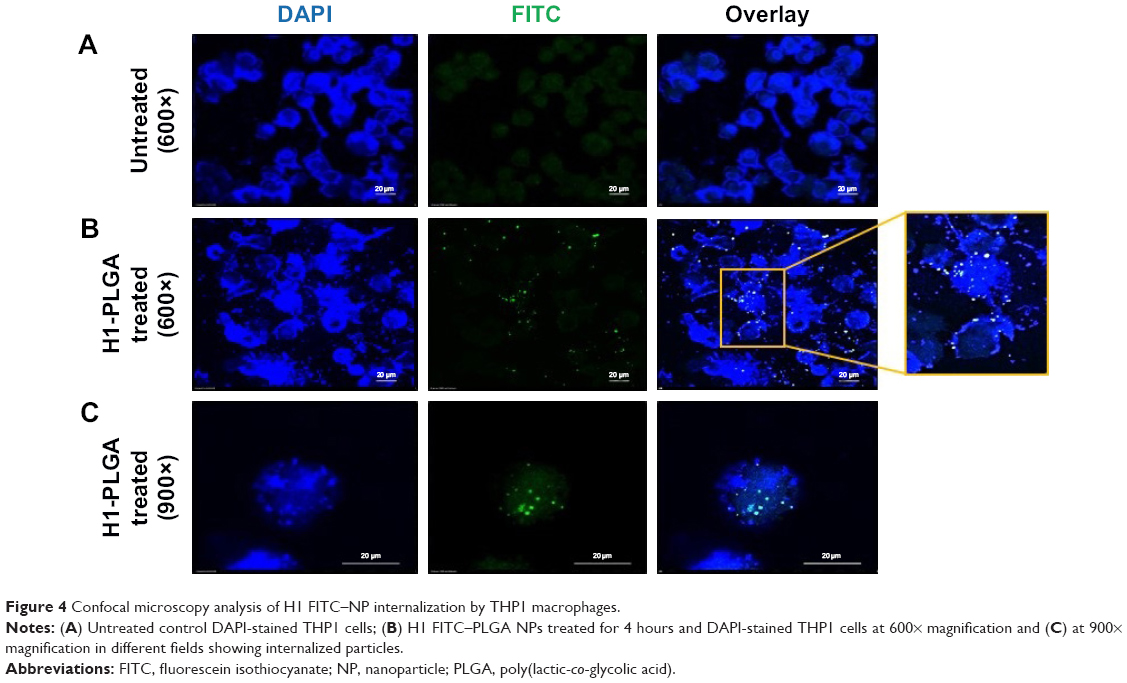 | Figure 4 Confocal microscopy analysis of H1 FITC–NP internalization by THP1 macrophages. |
Single-dose H1 NPs induced heightened humoral immunoresponse
Following a single-dose immunization schedule, serum-antibody titers in different mouse groups were measured on days 14, 28, and 42. As expected, the blank PLGA NPs showed IgG levels similar to the PBS-immunized group (Figure 5), affirming their nonimmunogenic nature. Interestingly, after only 14 days of immunization, H1 NP–immunized mice displayed a significantly higher total IgG titers (P<0.05) than antigen alone (Figure 5A). The end-point total IgG titers of H1 NP-immunized mice were ~6.39- and ~7.9-fold higher on days 28 and 42 postvaccination than the antigen alone–immunized group (P<0.0001 for both, Figure 5B and C).
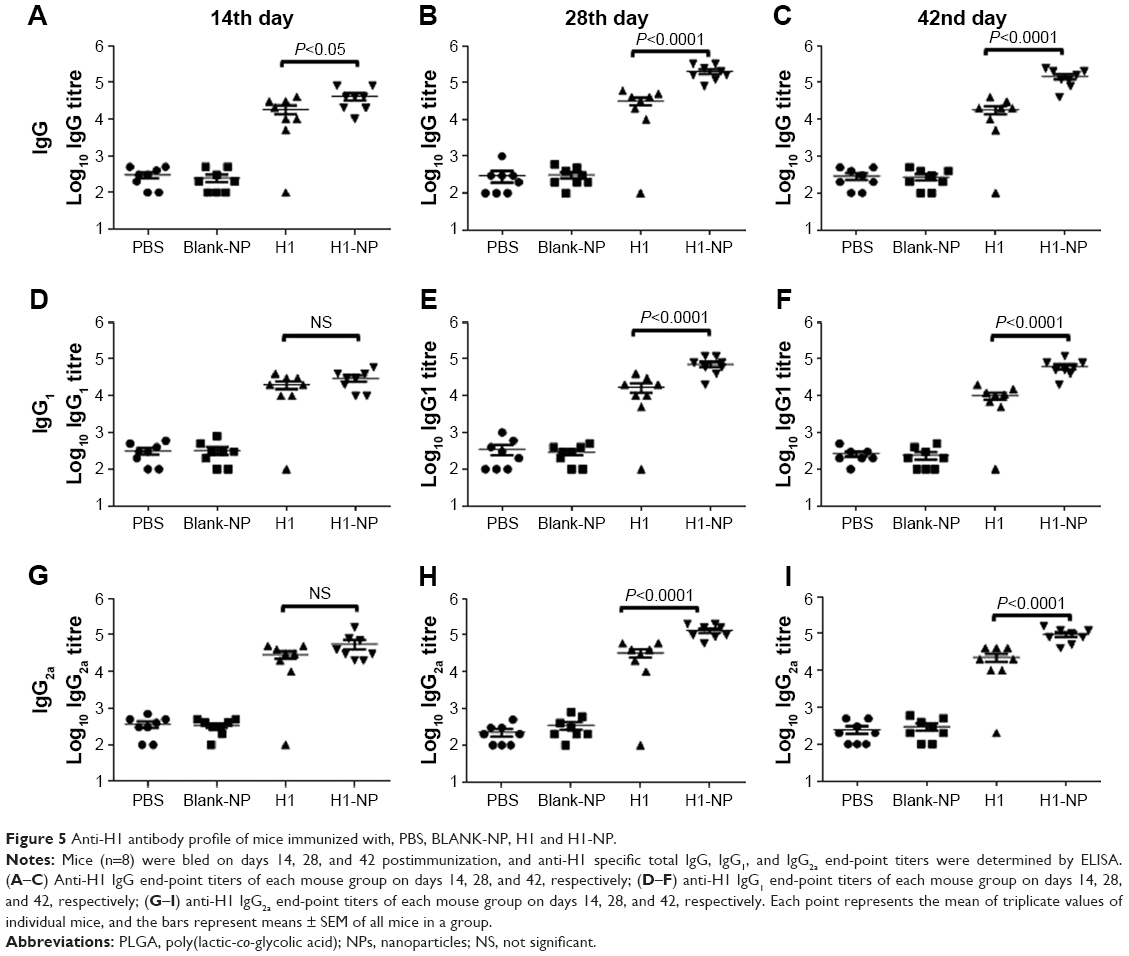 | Figure 5 Anti-H1 antibody profile of mice immunized with, PBS, BLANK-NP, H1 and H1-NP. |
The higher IgG titers induced by H1-NP vaccination illustrated an efficient humoral immunoresponse. Serum titers of IgG2a and IgG1 were also examined to evaluate TH1/TH2 bias of the immunoresponse (Figure 5D–I). For IgG2a, the H1-NP group generated a relatively high end-point titer – ~4.1-fold higher than the H1 alone–treated group (P<0.0001) on day 28 postimmunization (Figure 5H) – though by day 42, mean IgG2a levels in the H1 NP group had decreased marginally over day 28: ~4.1-fold higher than the H1 alone–treated group (Figure 5I). Similarly, on days 28 and 42 day, IgG1 levels of H1 NP–vaccinated mice were ~4.2- and ~6.2-fold higher than H1 alone (Figure 5E and F). Further, the ratio of Ig2a:IgG1 suggested that IgG2a levels were higher than IgG1 in both the H1 alone (~1.9- on day 28 and ~2.2-fold on day 42) and H1 NP (~1.8 on 28th and ~1.5 on 42nd day)–vaccinated mouse groups.
Vaccination with H1 NPs induced TH1-biased immunoresponse
To study the effect of H1 NPs on TH-cell development, memory-recall response of cultured spleen cells from differently immunized mouse groups was evaluated by quantifying cytokines released in the medium after stimulation with the antigen. Briefly, splenocytes from all mice were stimulated with the H1 antigen, and memory-recall response was assessed by quantifying TH1-specific (IFNγ and TNFα) and TH2-specific (IL4 and IL10) cytokine levels in culture supernatant after 72 hours of incubation. In all mouse samples, stimulation of splenocytes by concanavalin A, a positive control, led to secretion of multiple cytokines in the medium (data not shown). As expected, cytokine amounts produced by blank PLGA NP–immunized mice were quite low and similar to the PBS control. H1 NP–vaccinated mice displayed a heightened cytokine response upon antigen stimulation in comparison to the H1 alone–immunized group. Among all the cytokines under investigation, IFNγ levels were ~6.03-fold higher in the H1 NP–treated group (8,174.33 pg/mL) the H1-alone group (1,355.17 pg/mL; Figure 6A). In contrast to robust IFNγ response, levels of TH2-specific IL4 cytokine were much lower: 30.11 pg/mL and 14.89 pg/mL for H1 NP– and H1 alone–immunized mice, respectively (Figure 6C). Levels of TH1-specific TNFα and TH2-specific IL10, were ~2.8-fold (P<0.05) and ~1.6-(P<0.01) fold higher in the H1-NP group than H1 alone–immunized mice (Figure 6B and D). The increased production of TH1-specific IFNγ and TNFα cytokines in the H1 PLGA–vaccinated mice reiterates their immunomodulatory capability in the mouse model and mimics the potentiality of a memory-recall response in cases of mycobacterial infection.
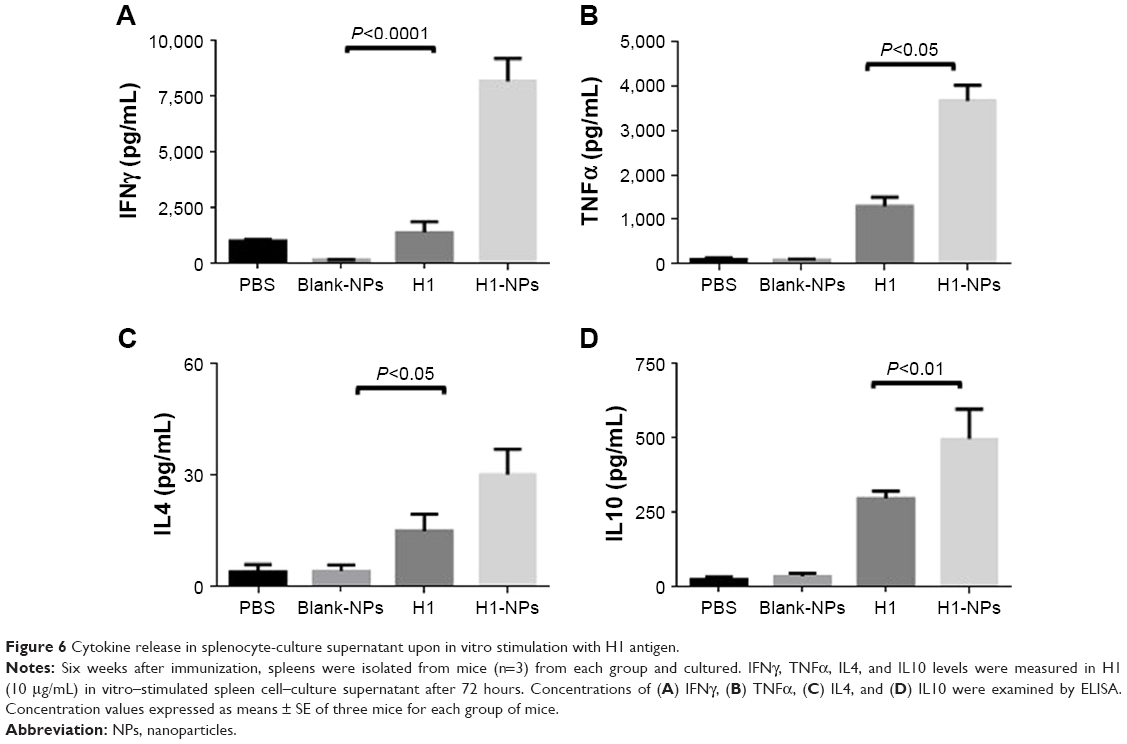 | Figure 6 Cytokine release in splenocyte-culture supernatant upon in vitro stimulation with H1 antigen. |
Protective and survival efficacy of single-dose H1-NP vaccination against tuberculosis
Having established the immunomodulatory properties of H1 NPs, we further evaluated their protective efficacy against Mtb challenge. At 8 weeks after immunization, all mice (15/group) were challenged with ~5.7×104 Mtb H37Rv CFU/mouse (data obtained after serial dilution and plating). Five weeks after Mtb challenge, three mice from each group were sacrificed and the bacterial load in their lungs and spleens enumerated. H1-alone vaccination reduced lung and spleen bacillary load only marginally compared to the PBS (unvaccinated) group, as expected. Compared to antigen alone–immunized mice, the reduction in lung and spleen Mtb load was statistically significant in both H1 NPs and BCG, though to varying degrees: H1 vs H1 NP (P<0.05 for both lung and spleen) and H1 vs BCG (P<0.01 for both lung and spleen; Figure 7). Although BCG vaccination marked the highest decrease in bacillary load in both lung and spleen, statistically it was indifferent to H1-PLGA NPs (Figure 7).
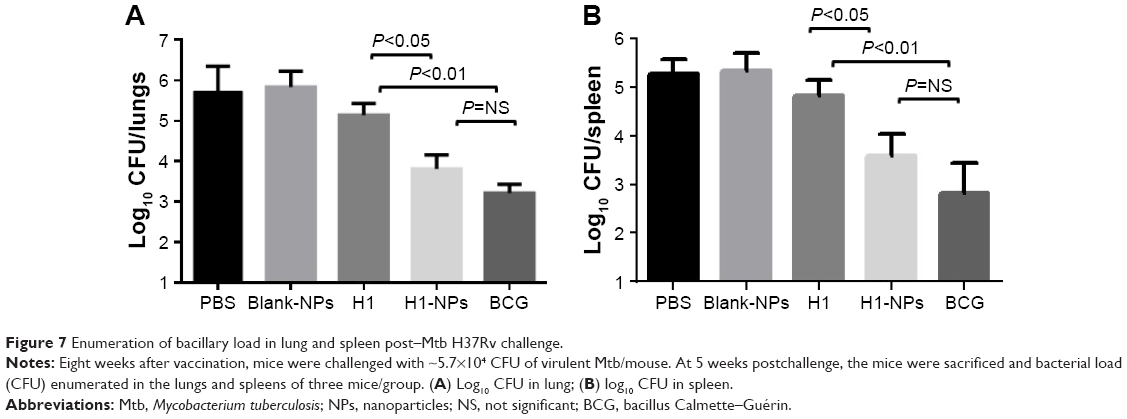 | Figure 7 Enumeration of bacillary load in lung and spleen post–Mtb H37Rv challenge. |
Upon assessing Mtb burden in lung and spleen, we further examined if H1-NP vaccination could also aid in long-term mice survival. Mice were monitored for general behavior and changes in weight and survival for 250 days postchallenge. Lacking subunit vaccination, all mice in PBS-control and blank-NP groups died in <100 days, with a mean survival time (MST) of 50.5 and 58 days, respectively (Figure 8). All mice in the H1 alone–immunized group died of infection, with an MST of 80 days, much earlier than those vaccinated with H1-PLGA NPs (MST 177 days). The MST was doubled upon H1-NP vaccination compared to the H1 alone–treated group (P<0.001, Figure 8). As observed earlier, the highest survival efficacy was observed in BCG-immunized mice, with an MST of 189.5 days. However, again this was statistically insignificant compared to H1 NP–vaccinated mice, reiterating that BCG and H1 NPs delivered similar protection against the Mtb challenge.
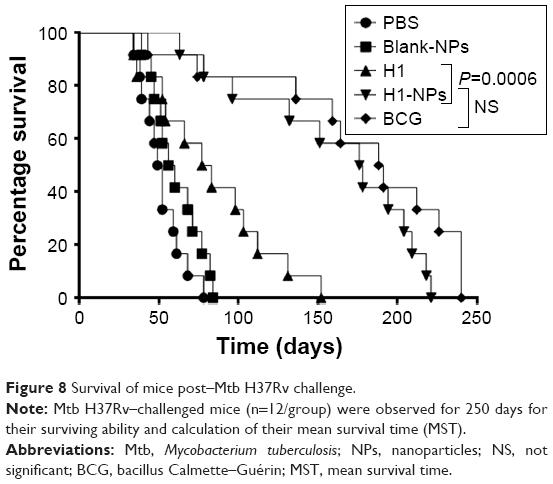 | Figure 8 Survival of mice post–Mtb H37Rv challenge. |
Discussion
The limitations of BCG in protection against pulmonary TB in adults demand new effective vaccination approaches. Vaccines modulating host response toward TH1-cell dependent immunity are in high demand, as they play a key role in attaining long-lasting immunomemory and effective protection against intracellular pathogens like Mtb, Neisseria gonorrhoeae, and Salmonella typhi.37,38 Well-known aluminum salts or squalene-based adjuvants like AS03 and MF59, licensed for human use, are known to be poor inducers of TH1 immunity.39 On the other hand, various adjuvants like CAF01, IC31, and GLA-SE, which induce significant levels of TH1-specific cytokines as a marker of protective immunity against TB, are still in different phases of clinical trials.40,41 In this milieu, NP- or MP-based delivery systems have emerged as exciting next-generation vaccination strategies. Earlier, multiple attempts were made using immunogenic antigens of Mtb, such as ESAT6, Ag85B, and TB10.4, either individually or as bivalent fusion entities (Ag85B–TB10.4 or Ag85B–ESAT6), and the FDA-approved PLGA polymer as the encapsulating carrier. Notably, all these studies had limitations. First, the formulations were tested only in the micrometer range (mean MP size ~3.3 μm),19,42 while PLGA induces superior TH1-response in the nanometer range (size 200–600 nm).43 Secondly, these formulations were investigated under ex vivo conditions or lacked protection studies.19,44 Lastly, most of these formulations were administered by respiratory31 or subcutaneous45 routes, and thus required multiple boosters in comparison to other parenteral routes. As such, in the present study we evaluated the vaccine potential of H1 (bivalent fusion antigen of Ag85B and ESAT6 proteins)-encapsulated PLGA NPs after single-dose immunization and compared it to BCG in protection against tuberculosis. We explicitly opted for the intraperitoneal route for vaccine delivery to facilitate uptake of PLGA NPs preferentially by resident macrophages and to trigger a polarized TH1 response, as reported by Conway et al.46
The competence of an NP-based vaccine-delivery system primarily depends on three key factors: the structural integrity of the encapsulated antigen, its subsequent immunogenicity, and ensuing protective efficacy. In this context, PLGA NPs have been reported to be one of the most stable particles formulated from natural polymers when stored properly.47 Earlier studies have also emphasized PLGA’s role in antigen protection from aspecific proteolysis under in vitro or in vivo conditions.42,48 Indeed, in our in vitro release studies, the H1 antigen released by the PLGA NPs was shown to be intact, with no signs of degradation until the 32nd day. This further suggests that water–oil–water evaporation might not be punitive on the protein structure either. The H1 protein has also been previously stated to be stable in a liposome formulation.49 Typically, PLGA NPs have negative ζ-potential, attributed to their carboxyl groups.50 Contrary to this, H1 NPs carried a weak negative charge (ζ-potential −4±0.6 mV). This relative decrease in intensity of the negative charge on NPs might have eased the interaction with the negatively charged membrane of THP1 cells and hence uptake efficiency.
Besides surface charge, the size of a particle is also crucial for its phagocytosis by APCs.51,52 Several studies have shown the correlation of immunoresponse with the size of particles used for immunization.12,14,53 NPs of 200–300 nm have been reported to generate a strong cellular immunoresponse alongside a heightened humoral response.10,12,36,53 In the present study, H1 NPs were 244.4±32.6 nm, further encouraging their uptake by APCs, owing to their desirable size range. Alongside size, the shape and morphology of particles are also important for scrutiny to assert they are better antigen carriers. Indeed, the H1 NPs met these standards of shape, surface smoothness, and uniform size and showed antigen release after 32 days under physiological conditions. Altogether, it is tempting to conclude that PLGA as a carrier of H1 antigen facilitates endosomal entry into APCs and exerts a “depot effect” by increasing residence time at the site of vaccination and improving antigen-specific immunoresponses. A similar mode of action has been reported for PLGA MPs, immunostimulating complexes, and unbiodegradable polystyrene particles also.
The ability of the H1 antigen to provoke a heightened antibody response has been well investigated in multiple studies.5,7,32,54 In the present study, augmentation of antigen-specific immunoresponse by H1 NPs led to induction of antigen-specific TH1-type IgG2a and TH2-type IgG1 antibodies, indicating effective presentation of antigens with major histocompatibility–complex class II molecules and subsequent activation of both TH1 and TH2 arms of immunity. Even 6 weeks after single-dose vaccination with H1 NPs, higher IgG and IgG2a production was observed with enhanced production of TH1-dependent IFNγ and TNFα cytokines upon antigen recall. This steady and heightened humoral response can be attributed to sustained release of H1 from PLGA NPs, which act as antigen depots inside host mice, resulting in long-term stimulation of immunoeffector cells and thus negate the need for frequent booster doses. H1 NP–treated mice alongside an amplified antibody response exhibited robust cytokine release as a memory response to antigen recall. Significantly higher levels of TH1-specific IFNγ and TNFα were detected in culture supernatants of splenocytes extracted from H1 NP–vaccinated mice upon stimulation. Although titers of TH2-specific IL4 and IL10 cytokines were also significantly elevated in the H1 NP–vaccinated group in comparison to the control groups, they were much lower than TH1-specific cytokines. Also, IFNγ has been studied extensively in TB and shown to be essential for activation of macrophages during infection and eventually to contain the infection.55 H1 NP–immunized mice also demonstrated elevated levels of TNFα, which was in line with earlier findings of recombinant BCG–expressing Ag85B and ESAT6 substantially increasing TNFα levels in mice.56 This has been attributed to the potential role played by PLGA NPs in NFκB translocation to the cell nucleus, which in turn increases the production of inflammatory cytokine TNFα.57 The neutralization potential of IgG2a is better than IgG1, and hence the strong IgG2a response generated by H1 NPs might play a crucial role in clearing Mtb infection. As levels of both IgG2a and IFNγ were high until day 42, this demonstrates that the response observed was completely TH1-biased in both H1 NP– and H1 alone–treated groups, which is in accordance with other reports stating H1 to be a TH1 antigen.56,58 A TH1-biased modulation of host immunoresponse has also been reported with PLGA NPs conjugated to a protein or peptide that is already recognized for its TH2 bias.24
The efficacy of a vaccine is judged mainly by the degree of protection it provides the recipient. The ability of H1-PLGA to modulate protective efficacy was observed at different levels of host response, from generation of a repertoire of antibodies to activation of TH1-specific cytokines and reduction of Mtb burden in lungs and spleens of mice. Enumeration of the latter illustrated that H1 NP–vaccinated mice were able to reduce bacterial load significantly more in comparison to the H1 alone–immunized control group. Interestingly, compared to H1 alone, the significance of vaccination with H1 NPs is reflected by statistically insignificant bacterial lung/spleen load and extension of MST, equivalent to BCG vaccination. Altogether, our study forms the groundwork and provides data demonstrating H1-encapsulated PLGA NPs as a promising TB-vaccine candidate. For its acceptance as a next-generation vaccine, it needs to be subjected to a higher level of scrutiny on immunologic and toxicological parameters.
Disclosure
The authors report no conflicts of interest in this work.
References
Global Tuberculosis Report 2017. World Health Organization (WHO): Available from: https://www.who.int/tb/publications/global_report/gtbr2017_main_text.pdf. Accessed June 21, 2018. | ||
Luca S, Mihaescu T. History of BCG Vaccine: Amaltea Medical. Bucharest: Editura Magister; 2013. | ||
Fine PE. The BCG story: lessons from the past and implications for the future. Rev Infect Dis. 1989;11(Supplement_2):S353–S359. | ||
Fine PE. Variation in protection by BCG: implications of and for heterologous immunity. The Lancet. 1995;346(8986):1339–1345. doi:10.1016/S0140-6736(95)92348-9 | ||
Ottenhoff TH, Kaufmann SH. Vaccines against tuberculosis: where are we and where do we need to go? PLoS Pathog. 2012;8(5):e1002607. doi:10.1371/journal.ppat.1002607 | ||
Tameris MD, Hatherill M, Landry BS, et al. Safety and efficacy of MVA85A, a new tuberculosis vaccine, in infants previously vaccinated with BCG: a randomised, placebo-controlled phase 2b trial. The Lancet. 2013;381(9871):1021–1028. doi:10.1016/S0140-6736(13)60177-4 | ||
Junqueira-Kipnis AP, Neto LMM, Kipnis A. Role of fused Mycobacterium tuberculosis immunogens and adjuvants in modern tuberculosis vaccines. Front Immunol. 2014;5:188. doi:10.3389/fimmu.2014.00188 | ||
Flynn JL, Chan J. Immune evasion by Mycobacterium tuberculosis: living with the enemy. Curr Opin Immunol. 2003;15(4):450–455. | ||
Gregory AE, Titball R, Williamson D. Vaccine delivery using nanoparticles. Front Cell Infect Microbiol. 2013;3:13. doi:10.3389/fcimb.2013.00072 | ||
Oyewumi MO, Kumar A, Cui Z. Nano-microparticles as immune adjuvants: correlating particle sizes and the resultant immune responses. Expert Rev Vaccines. 2010;9(9):1095–1107. doi:10.1586/erv.10.89 | ||
Silva A, Soema P, Slütter B, Ossendorp F, Jiskoot W. PLGA particulate delivery systems for subunit vaccines: linking particle properties to immunogenicity. Hum Vaccin Immunother. 2016;12(4):1056–1069. doi:10.1080/21645515.2015.1117714 | ||
Akagi T, Baba M, Akashi M. Biodegradable nanoparticles as vaccine adjuvants and delivery systems: regulation of immune responses by nanoparticle-based vaccine. In: Polymers in Nanomedicine. Heidelberg: Springer; 2011:31–64. | ||
Makadia HK, Siegel SJ. Poly lactic-co-glycolic acid (PLGA) as biodegradable controlled drug delivery carrier. Polymers. 2011;3(3):1377–1397. doi:10.3390/polym3031377 | ||
Anderson JM, Shive MS. Biodegradation and biocompatibility of PLA and PLGA microspheres. Adv Drug Deliv Rev. 2012;64:72–82. doi:10.1016/j.addr.2012.09.004 | ||
Oh N, Park J-H. Endocytosis and exocytosis of nanoparticles in mammalian cells. Int J Nanomedicine. 2014;9(Suppl 1):51. | ||
Kanchan V, Panda AK. Interactions of antigen-loaded polylactide particles with macrophages and their correlation with the immune response. Biomaterials. 2007;28(35):5344–5357. doi:10.1016/j.biomaterials.2007.08.015 | ||
Lawlor C, O’Connor G, O’Leary S, et al. Treatment of mycobacterium tuberculosis-infected macrophages with poly (lactic-co-glycolic acid) microparticles drives NFκB and autophagy dependent bacillary killing. PLoS One. 2016;11(2):e0149167. doi:10.1371/journal.pone.0149167 | ||
Kasturi SP, Skountzou I, Albrecht RA, et al. Programming the magnitude and persistence of antibody responses with innate immunity. Nature. 2011;470(7335):543. doi:10.1038/nature09771 | ||
Shi S, Hickey AJ. PLGA microparticles in respirable sizes enhance an in vitro T cell response to recombinant Mycobacterium tuberculosis antigen TB10. 4-Ag85B. Pharm Res. 2010;27(2):350–360. doi:10.1007/s11095-009-0028-7 | ||
Keijzer C, Slütter B, Van Der Zee R, Jiskoot W, Van Eden W, Broere F. PLGA, PLGA-TMC and TMC-TPP nanoparticles differentially modulate the outcome of nasal vaccination by inducing tolerance or enhancing humoral immunity. PLoS One. 2011;6(11):e26684. doi:10.1371/journal.pone.0026684 | ||
Allahyari M, Mohabati R, Amiri S, et al. Synergistic effect of rSAG1 and rGRA2 antigens formulated in PLGA microspheres in eliciting immune protection against Toxoplasama gondii. Exp Parasitol. 2016;170:236–246. doi:10.1016/j.exppara.2016.09.008 | ||
Men Y, Thomasin C, Merkle HP, Gander B, Corradin G. A single administration of tetanus toxoid in biodegradable microspheres elicits T cell and antibody responses similar or superior to those obtained with aluminum hydroxide. Vaccine. 1995;13(7):683–689. | ||
Chudina T, Labyntsev A, Manoilo K, Kolybo D, Komisarenko S. Cellobiose-coated poly (lactideco-glycolide) particles loaded with diphtheria toxoid for per os immunization. Croat Med J. 2015;56(2):85–93. | ||
Lutsiak M, Kwon GS, Samuel J. Biodegradable nanoparticle delivery of a TH2-biased peptide for induction of TH1 immune responses. J Pharm Pharmacol. 2006;58(6):739–747. doi:10.1211/jpp.58.6.0004 | ||
Huang S-S, Li I-H, Po-da Hong M-KY. Development of Yersinia pestis F1 antigen-loaded microspheres vaccine against plague. Int J Nanomedicine. 2014;9:813. | ||
Yoshida A, Matumoto M, Hshizume H, et al. Selective delivery of rifampicin incorporated into poly (dl-lactic-co-glycolic) acid microspheres after phagocytotic uptake by alveolar macrophages, and the killing effect against intracellular Mycobacterium bovis Calmette–guérin. Microbes Infect. 2006;8(9):2484–2491. doi:10.1016/j.micinf.2006.06.004 | ||
Khan N, Gowthaman U, Pahari S, Agrewala JN. Manipulation of costimulatory molecules by intracellular pathogens: veni, vidi, vici!! PLoS Pathog. 2012;8(6):e1002676. doi:10.1371/journal.ppat.1002676 | ||
Andersen P, Askgaard D, Ljungqvist L, Bentzon MW, Heron I. T-cell proliferative response to antigens secreted by Mycobacterium tuberculosis. Infect Immun. 1991;59(4):1558–1563. | ||
Hasløv K, Andersen Å, Nagai S, Gottschau A, Sørensen T, Andersen P. Guinea pig cellular immune responses to proteins secreted by Mycobacterium tuberculosis. Infect Immun. 1995;63(3):804–810. | ||
Brandt L, Oettinger T, Holm A, Andersen AB, Andersen P. Key epitopes on the ESAT-6 antigen recognized in mice during the recall of protective immunity to Mycobacterium tuberculosis. J Immunol. 1996;157(8):3527–3533. | ||
Carpenter ZK, Williamson ED, Eyles JE. Mucosal delivery of microparticle encapsulated ESAT-6 induces robust cell-mediated responses in the lung milieu. J Control Release. 2005;104(1):67–77. doi:10.1016/j.jconrel.2005.01.014 | ||
Dietrich J, Andersen C, Rappuoli R, Doherty TM, Jensen CG, Andersen P. Mucosal administration of Ag85B-ESAT-6 protects against infection with Mycobacterium tuberculosis and boosts prior bacillus Calmette-Guerin immunity. J Immunol. 2006;177(9):6353–6360. | ||
van Dissel JT, Arend SM, Prins C, et al. Ag85B–ESAT-6 adjuvanted with IC31 promotes strong and long-lived Mycobacterium tuberculosis specific T cell responses in naïve human volunteers. Vaccine. 2010;28(20):3571–3581. doi:10.1016/j.vaccine.2010.02.094 | ||
Van Dissel JT, Soonawala D, Joosten SA, et al. Ag85B–ESAT-6 adjuvanted with IC31 promotes strong and long-lived Mycobacterium tuberculosis specific T cell responses in volunteers with previous BCG vaccination or tuberculosis infection. Vaccine. 2011;29(11):2100–2109. doi:10.1016/j.vaccine.2010.12.135 | ||
Hall LJ, Clare S, Pickard D, et al. Characterisation of a live Salmonella vaccine stably expressing the Mycobacterium tuberculosis Ag85B–ESAT6 fusion protein. Vaccine. 2009;27(49):6894–6904. doi:10.1016/j.vaccine.2009.09.007 | ||
Manish M, Rahi A, Kaur M, Bhatnagar R, Singh S. A single-dose PLGA encapsulated protective antigen domain 4 nanoformulation protects mice against Bacillus anthracis spore challenge. PLoS One. 2013;8(4):e61885. doi:10.1371/journal.pone.0061885 | ||
Igietseme JU, Eko FO, He Q, Black CM. Antibody regulation of T-cell immunity: implications for vaccine strategies against intracellular pathogens. Expert Rev Vaccines. 2004;3(1):23–34. doi:10.1586/14760584.3.1.23 | ||
Ebensen T, Guzmán CA. Immune modulators with defined molecular targets: cornerstone to optimize rational vaccine design. Hum Vaccin. 2008;4(1):13–22. | ||
Lee S, Nguyen MT. Recent advances of vaccine adjuvants for infectious diseases. Immune Netw. 2015;15(2):51–57. doi:10.4110/in.2015.15.2.51 | ||
Agger EM. Novel adjuvant formulations for delivery of anti-tuberculosis vaccine candidates. Adv Drug Deliv Rev. 2016;102:73–82. doi:10.1016/j.addr.2015.11.012 | ||
Zhu B, Dockrell HM, Ottenhoff TH, Evans TG, Zhang Y. Tuberculosis vaccines: opportunities and challenges. Respirology. 2018;23(4):359–368. doi:10.1111/resp.13245 | ||
Allahyari M, Mohit E. Peptide/protein vaccine delivery system based on PLGA particles. Hum Vaccin Immunother. 2016;12(3):806–828. doi:10.1080/21645515.2015.1102804 | ||
Joshi VB, Geary SM, Salem AK. Biodegradable particles as vaccine delivery systems: size matters. Aaps J. 2013;15(1):85–94. doi:10.1208/s12248-012-9418-6 | ||
Kirby DJ, Rosenkrands I, Agger EM, Andersen P, Coombes AG, Perrie Y. PLGA microspheres for the delivery of a novel subunit TB vaccine. J Drug Target. 2008;16(4):282–293. doi:10.1080/10611860801900462 | ||
Dhiman N, Khuller G. Protective efficacy of mycobacterial 71-kDa cell wall associated protein using poly (DL-lactide-co-glycolide) microparticles as carrier vehicles. FEMS Immunol Med Microbiol. 1998;21(1):19–28. | ||
Conway MA, Madrigal-Estebas L, McClean S, Brayden DJ, Mills KH. Protection against Bordetella pertussis infection following parenteral or oral immunization with antigens entrapped in biodegradable particles: effect of formulation and route of immunization on induction of TH1 and TH2 cells. Vaccine. 2001;19(15–16):1940–1950. | ||
De S, Robinson DH. Particle size and temperature effect on the physical stability of PLGA nanospheres and microspheres containing Bodipy. AAPS PharmSciTech. 2004;5(4):18–24. doi:10.1208/pt050453 | ||
Qi C, Chen Y, Jing Q-Z, Wang X-G. Preparation and characterization of catalase-loaded solid lipid nanoparticles protecting enzyme against proteolysis. Int J Mol Sci. 2011;12(7):4282–4293. doi:10.3390/ijms12074282 | ||
Hamborg M, Kramer R, Schanté CE, et al. The physical stability of the recombinant tuberculosis fusion antigens h1 and h56. J Pharm Sci. 2013;102(10):3567–3578. doi:10.1002/jps.23669 | ||
Wang Y, Li P, Kong L. Chitosan-modified PLGA nanoparticles with versatile surface for improved drug delivery. AAPS PharmSciTech. 2013;14(2):585–592. doi:10.1208/s12249-013-9943-3 | ||
Foged C, Brodin B, Frokjaer S, Sundblad A. Particle size and surface charge affect particle uptake by human dendritic cells in an in vitro model. Int J Pharm. 2005;298(2):315–322. doi:10.1016/j.ijpharm.2005.03.035 | ||
Champion JA, Walker A, Mitragotri S. Role of particle size in phagocytosis of polymeric microspheres. Pharm Res. 2008;25(8):1815–1821. doi:10.1007/s11095-008-9562-y | ||
Li X, Sloat BR, Yanasarn N, Cui Z. Relationship between the size of nanoparticles and their adjuvant activity: data from a study with an improved experimental design. Eur J Pharm Biopharm. 2011;78(1):107–116. doi:10.1016/j.ejpb.2010.12.017 | ||
Olsen AW, van Pinxteren LA, Okkels LM, Rasmussen PB, Andersen P. Protection of mice with a tuberculosis subunit vaccine based on a fusion protein of antigen 85b and esat-6. Infect Immun. 2001;69(5):2773–2778. doi:10.1128/IAI.69.5.2773-2778.2001 | ||
Ellner JJ, Hirsch CS, Whalen CC. Correlates of protective immunity to Mycobacterium tuberculosis in humans. Clin Infect Dis. 2000;30(Supplement_3):S279–S282. doi:10.1086/313874 | ||
Xu Y, Liu W, Shen H, Yan J, Qu D, Wang H. Recombinant Mycobacterium bovis BCG expressing the chimeric protein of antigen 85B and ESAT-6 enhances the TH1 cell-mediated response. Clin Vacc Immunol. 2009;16(8):1121–1126. doi:10.1128/CVI.00112-09 | ||
Nicolete R, Dos Santos DF, Faccioli LH. The uptake of PLGA micro or nanoparticles by macrophages provokes distinct in vitro inflammatory response. Int Immunopharmacol. 2011;11(10):1557–1563. doi:10.1016/j.intimp.2011.05.014 | ||
Shi C, Wang X, Zhang H, Xu Z, Li Y, Yuan L. Immune responses and protective efficacy induced by 85B antigen and early secreted antigenic target-6 kDa antigen fusion protein secreted by recombinant bacille Calmette–Guérin. Acta Biochim Biophys Sin (Shanghai). 2007;39(4):290–296. |
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.