Back to Journals » Drug Design, Development and Therapy » Volume 9
Single and multiple dose pharmacokinetics and tolerability of HX-1171, a novel antioxidant, in healthy volunteers
Authors Kim YH, Choi HY, Lee SH, Lim H, Miki T, Kang J, Han K, Bae K ![]()
Received 22 December 2014
Accepted for publication 4 February 2015
Published 23 March 2015 Volume 2015:9 Pages 1735—1742
DOI https://doi.org/10.2147/DDDT.S79724
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Shu-Feng Zhou
Yo Han Kim,1 Hee Youn Choi,1 Shi Hyang Lee,1 Hyeong-Seok Lim,1 Tokutaro Miki,2 Jong-Koo Kang,3 Kyoung-Goo Han,4 Kyun-Seop Bae1
1Department of Clinical Pharmacology and Therapeutics, College of Medicine, University of Ulsan, Asan Medical Center, Seoul, Republic of Korea; 2Nippon Hypox Laboratories Inc., Yamanashi, Japan; 3College of Veterinary Medicine, Chungbuk National University, Cheongju, Republic of Korea; 4Biotoxtech Co., Ltd, Cheongju, Republic of Korea
Background: HX-1171 (1-O-hexyl-2,3,5-trimethylhydroquinone) is a promising antioxidant with therapeutic potential for hepatic fibrosis. The aim of this study was to investigate the tolerability and pharmacokinetics of HX-1171 in healthy volunteers.
Methods: A randomized, single-blind, placebo-controlled, dose escalation study was conducted in 83 subjects. In the single ascending dose study, 20, 40, 80, 160, 300, 600, 1,200, 1,500 or 2,000 mg of HX-1171 was administered to 67 subjects. In the multiple ascending dose study, 500 or 1,000 mg was administered to 16 subjects for 14 days. The plasma and urine concentrations of HX-1171 were determined by using a validated liquid chromatography-mass spectrometry method. Pharmacokinetic parameters were obtained by non-compartmental analysis. Tolerability was assessed based on physical examinations, vital signs, clinical laboratory tests, and electrocardiograms.
Results: Adverse events reported in the study were all mild in intensity and resolved without any sequelae. HX-1171 was rapidly and minimally absorbed with a median time at maximal concentration of 0.63–1.50 hours and slowly eliminated with a terminal half-life of 21.12–40.96 hours. Accumulation index ranged from 2.0 to 2.2 after repeated dosing for 14 days. For both the single and multiple doses administrations, urinary concentrations indicated that less than 0.01% of the HX-1171 administered was excreted in urine.
Conclusion: HX-1171 was well tolerated and minimally absorbed in healthy volunteers. The pharmacokinetic profile of HX-1171 was consistent with once-a-day dosing.
Keywords: HX-1171, antioxidant, pharmacokinetics, tolerability, healthy subjects
Introduction
Liver fibrosis is defined as the presence of excess collagen due to new fiber formation and accumulation of extracellular matrix.1,2 The primary causes of liver fibrosis are viral infection,3 non-alcoholic fatty liver disease,4,5 and excessive consumption of alcohol.6 The clinical manifestations of liver fibrosis vary widely, from no symptoms at all, to liver failure. The latest surveillance report showed that liver cirrhosis was the 12th leading cause of death in the United States.7
Oxidative stress (OS) is defined as a disturbance in the balance between the production of reactive oxygen species and antioxidant defenses, leading to potential cellular damage.8 Some experimental data suggest that OS mediates the progression of fibrosis, and that OS-related molecules may act as mediators of liver fibrosis.9–11
HX-1171 (1-O-hexyl-2,3,5-trimethylhydroquinone), which is a novel derivative of vitamin E, is a promising antioxidant with therapeutic potential for liver fibrosis (Figure 1). From an experimental study, HX-1171 showed a similar antioxidative activity to that of d,l-α-tocopherol against micelles, and it also exhibited approximately 4.8-fold higher anti-lipid-peroxidation activity than that of d,l-α-tocopherol against the peroxidation in liposomes.12 In another preclinical study, HX-1171 showed liver fibrosis reduction in CCl4-induced liver cirrhosis in Wistar rats.13 Additionally, HX-1171 improved against dimethylnitrosamine-induced liver fibrosis in Sprague Dawley rats.14
 | Figure 1 The chemical structure of HX-1171 (1-O-hexyl-2,3,5-trimethylhydroquinone). |
These results support the hypothesis that HX-1171 is a potential therapeutic agent for liver fibrosis. Thus, it is necessary to investigate HX-1171 in humans in a first-in-human trial. The objective of this study was to investigate the tolerability and pharmacokinetics of HX-1171 in healthy volunteers.
Material and methods
Subjects
Healthy male volunteers aged 20–40 years and with a body mass index of 19–26 kg/m2 were eligible for this study. Volunteers were considered to be in good health based on medical history, physical examinations, vital sign measurements (blood pressure, heart rate, and body temperature), 12-lead electrocardiograms, clinical laboratory tests (hematology, blood chemistry, and urinalysis), serology (hepatitis B surface antigen, hepatitis C virus antibody, and HIV antigen/antibody), and urine drug screening (amphetamine, cocaine, opiate, barbiturate, benzodiazepine, tetrahydrocannabinol, methadone, and methamphetamine) within 4 weeks prior to the first administration of study drug. Subjects who discontinued before completing the study were replaced.
Study design
This first-in-human study was designed as a randomized, single-blind, placebo-controlled, dose escalation clinical trial. In single ascending dose (SAD) study, HX-1171 was administered at single doses of 20, 40, and 80 mg, with six subjects (four active, two placebo) per group, and administered at single doses of 160, 300, 600, 1,200, 1,500, 2,000 mg, with eight subjects (six active, two placebo) per group. In multiple ascending dose (MAD) study, once daily for 14 days at 500 and 1,000 mg with eight subjects (six active, two placebo) per group.
The starting clinical dose was based on results in the rat as most sensitive animal, where an identified no target organ toxicity level of 20 mg/kg was identified in a 13-week good laboratory practice study. When adjusted for the regulatory scaling factor (human equivalent dose of a factor of 0.16 for the rat)15 and applying a safety factor of ten, a maximum safe starting dose would be 0.32 mg/kg or 19.2 mg based on a 60 kg person. The chosen starting dose for this first study was 20 mg.
In SAD study, the subjects were admitted to the Clinical Trial Center (CTC) at Asan Medical Center (AMC) from day −1 through day 3. On day 5 and 7, the subjects revisited the CTC to assess tolerability and pharmacokinetics of HX-1171. Follow-up visit was performed within 12 to 16 days after the drug administration.
In MAD study, the subjects were admitted to the CTC at AMC from day −2 through day 2. From day 3 to day 12, the subjects visited the CTC to assess tolerability and receive study drug. From day 13 to 15, subjects were hospitalized for serial pharmacokinetic urine and blood sampling. On day 14, the last study drug was administered. After discharge, they revisited CTC on 2 consecutive days (day 16 and 17). Follow-up visit was performed within 8 to 12 days after the last drug administration.
The study protocol was approved by the Ministry of Food and Drug Safety and the institutional review board of AMC, Seoul, Republic of Korea (ClinicalTrials.gov identifier: NCT01548391 [SAD]/NCT01889745 [MAD]). The study was conducted at the Clinical Trials Center of AMC from April 2012 to August 2013. All subjects provided written informed consent before screening.
Determination of HX-1171 concentrations
Plasma and urine concentrations of HX-1171 were determined using a validated high performance liquid chromatography coupled with tandem mass spectrometry method. The internal standard was 2,5-di-tert-butylhydroquinone. The sample extracts were analyzed using high performance liquid chromatography (Prominence; Shimadzu, Tokyo, Japan) and an XBridge™ C18 column (3.5 μm, 50 mm× 2.1 mm; Waters, Milford, MA, USA) with mobile phase consisting of distilled water and acetonitrile (35:65, v/v).
The mass spectrometry system (API4000; AB Sciex, Framingham, MA, USA) was operated in positive ion electrospray mode with multiple reaction monitoring. The precursor-to-production reactions monitored were m/z 235.3 → 149.8 for HX-1171 and 221.2 → 164.2 for internal standard. The assay was validated over a range of 0.5–500 ng/mL for plasma (R2>0.995) and 5–500 ng/mL for urine (R2>0.999). Assay accuracy and precision of calibration standard curve ranged from 86.46% to 114.25% and <5.31% in plasma and from 94.90% to 105.21% and <2.51% in urine, respectively.
Pharmacokinetic assessment and statistical analysis
Sequential blood samples (8 mL) were taken at 0 (pre-dose), 0.25, 0.5, 0.75, 1, 2, 3, 4, 6, 8, 10, 12, 14, 16, 24, 36, 48, 96, and 144 hours after drug administration in SAD study. In MAD study, blood samples were taken before and at 0.25, 0.5, 0.75, 1, 2, 3, 4, 6, 8, 12, and 24 hours after dosing on day 1, just before dosing on day 13, and also at 0.25, 0.5, 0.75, 1, 2, 3, 4, 6, 8, 12, 24, 48, and 72 hours after dosing on day 14. The blood samples were separated by centrifugation at 1,800× g for 8 minutes at 4°C and stored at −70°C until analysis.
Urine samples were collected at predefined intervals of 0–4, 4–8, 8–12, 12–24 and 24–48 hours after dosing in SAD study. In MAD study, urine samples were collected at predefined intervals of 0–6, 6–12 and 12–24 hours after drug administration on day 1 and day 14. Urine volume was measured, and 6 mL urine samples were stored at −70°C before analysis.
The plasma and urine concentration-time profiles of HX-1171 were analyzed by a non-compartmental method using WinNonlin 6.1 (Pharsight Corporation, Mountain View, CA, USA). All analyses were done using actual times of sampling. The measured maximum plasma concentration (Cmax) and time at Cmax (tmax) were determined from the observed values. The terminal elimination rate constant (λz) was estimated by linear regression of the terminal log-linear portion of the plasma concentration-time curves. The area under the plasma concentration-time curve (AUC) from time 0 to the last measurable time (AUClast) and the area under the plasma concentration-time curve within a dosing interval (AUCτ) was calculated by the trapezoidal rule and the AUC extrapolated to infinity (AUCinf) was obtained AUClast + Clast/λz (Clast: the last quantifiable concentration). The terminal half-life (t1/2β) was calculated for each participant as ln(2)/λz.
All statistical analyses were performed using SAS 9.3 (SAS Institute Inc., Cary, NC, USA) and Phoenix WinNonlin 6.1 (Pharsight Corporation). Demographic data and pharmacokinetic parameters were summarized using descriptive statistics. In single dose study, dose proportionality was investigated by evaluating the dose-normalized Cmax and AUC values, using the Kruskal–Wallis test.
Tolerability assessment
Tolerability was assessed throughout the study using vital sign measurements, 12-lead electrocardiograms, clinical laboratory tests (hematology, blood chemistry, and urinalysis), physical examinations, and monitoring of adverse events (AEs). AEs were recorded in terms of symptoms and signs, duration, intensity, relationship to the study drug, action taken, outcome, and seriousness.
Results
Study participants
Sixty-seven healthy male subjects participated and received study drug in the SAD study, and 66 subjects completed the study. One subject withdrew after taking the study drug due to personal reason, and was replaced by another subject. Sixteen male subjects took part in and received at least one dose of study drug in the MAD study, and all subjects completed the study. All subjects were included in the tolerability assessments, and only the subjects who completed the study were included in the pharmacokinetic assessment.
The mean (standard deviation [SD]) age of study participants was 25.9 (4.6) years, the mean (SD) height was 174.4 (5.7) cm, and the mean (SD) weight was 69.5 (6.6) kg in SAD study. In MAD study, the mean (SD) age of study participants was 25.6 (4.0) years, the mean (SD) height was 173.6 (5.0) cm, and the mean (SD) weight was 69.4 (4.3) kg.
Pharmacokinetic analysis
The mean plasma concentration-time profiles of HX-1171 following single-dose administration are shown in Figure 2. In SAD study, pharmacokinetic parameter of some subjects in low dose administered group (20–160 mg) could not be calculated because drug concentration of HX-1171 was below the lower limit of quantification at almost all time points (Table 1). Therefore, pharmacokinetic parameter was compared at 300–2,000 mg dosage group, HX-1171 was rapidly absorbed with tmax 0.63–1.50 hours, and t1/2β was 21.12–40.96 hours (Table 2). There were no statistically significant differences in the dose-normalized Cmax and AUCinf values of the 300–2,000 mg single-dose group, (P-value =0.1365 and 0.1047, respectively) (Figure 3). Until 48 hours after HX-1171 administration, the urine recovery ratio was less than 0.01% at all dose levels. Thus, renal clearance was not evaluated.
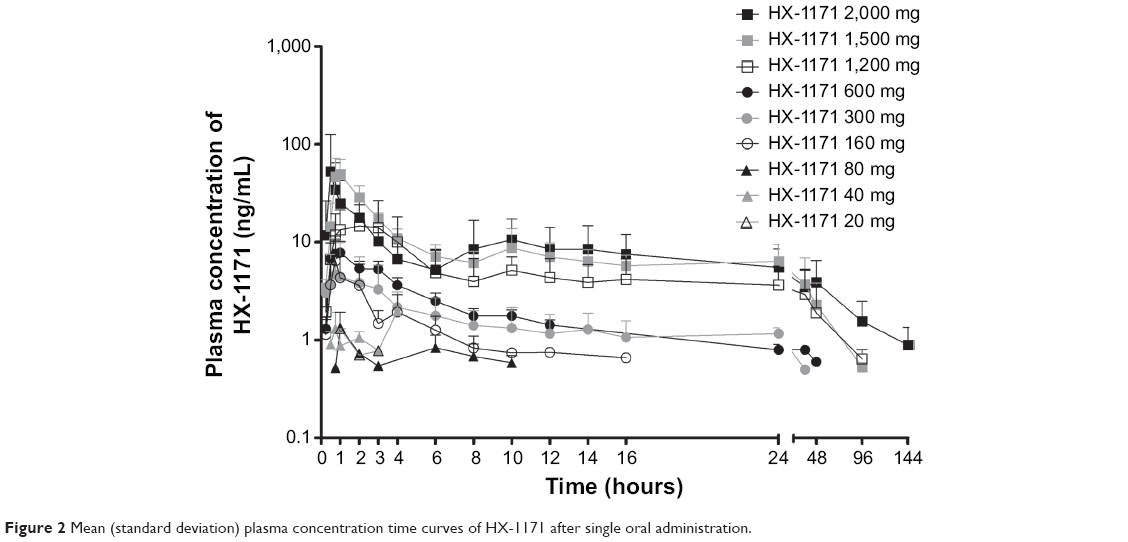 | Figure 2 Mean (standard deviation) plasma concentration time curves of HX-1171 after single oral administration. |
 | Table 1 Pharmacokinetic parameters of HX-1171 in SAD study (20–160 mg) |
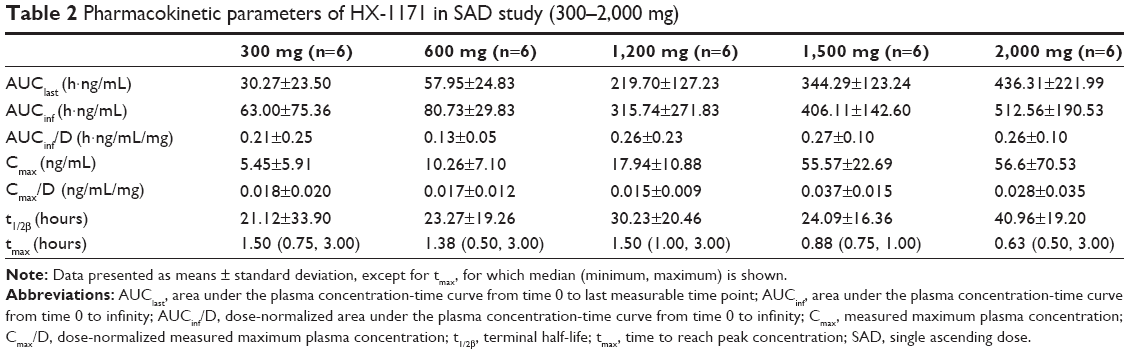 | Table 2 Pharmacokinetic parameters of HX-1171 in SAD study (300–2,000 mg) |
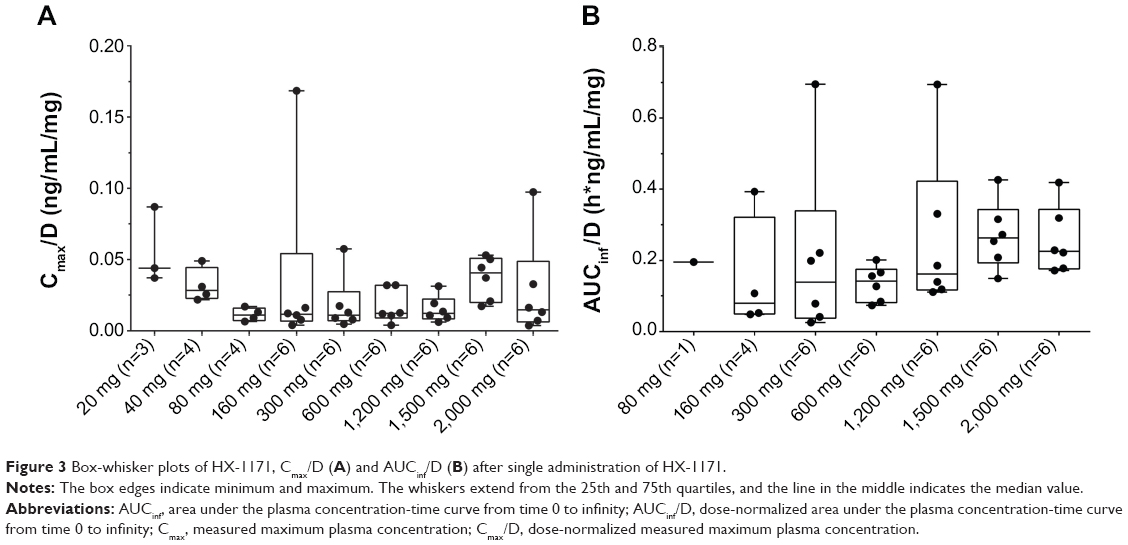 | Figure 3 Box-whisker plots of HX-1171, Cmax/D (A) and AUCinf/D (B) after single administration of HX-1171. |
In MAD study, the mean plasma concentration-time profiles of HX-1171 are shown in Figure 4. Accumulation index ranged from 2.0 to 2.2. It was also rapidly absorbed with tmax 0.88–1.50 hours, and t1/2β was 32.00–33.64 hours (Table 3). On day 1 and 14, the urine recovery ratio was also less than 0.01%.
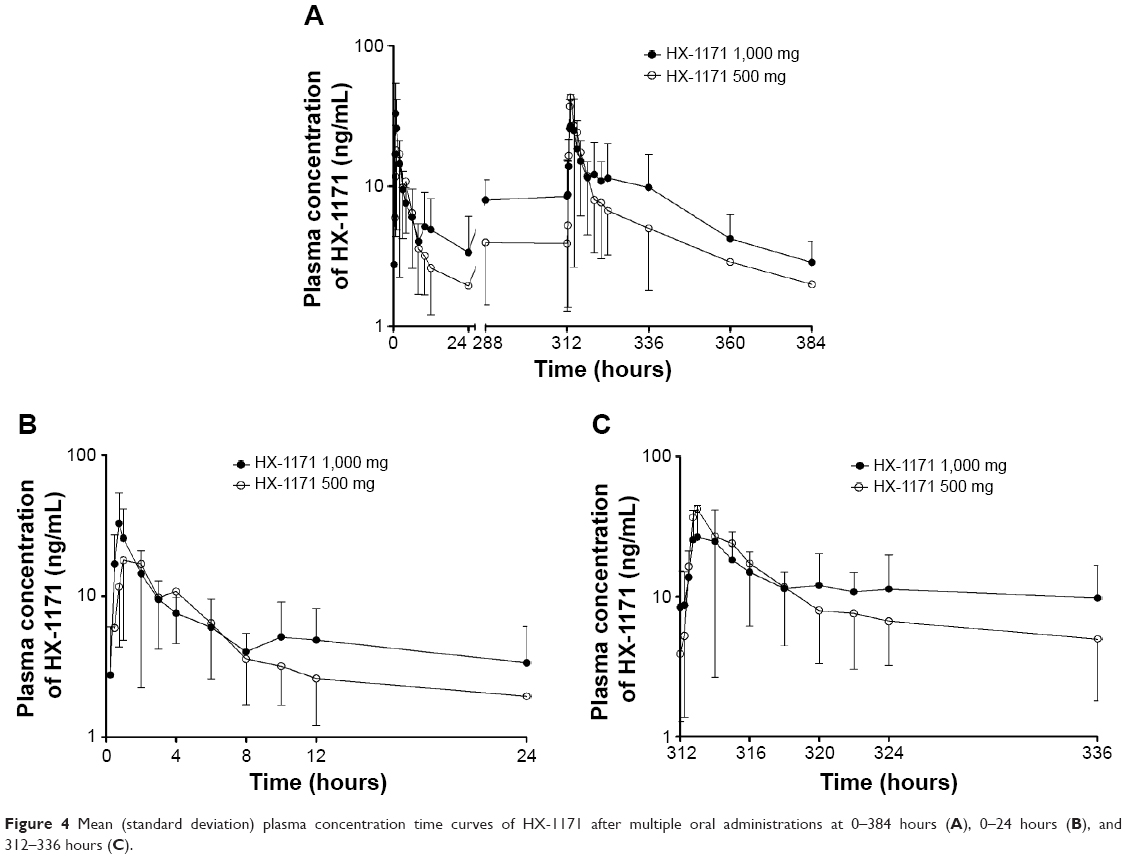 | Figure 4 Mean (standard deviation) plasma concentration time curves of HX-1171 after multiple oral administrations at 0–384 hours (A), 0–24 hours (B), and 312–336 hours (C). |
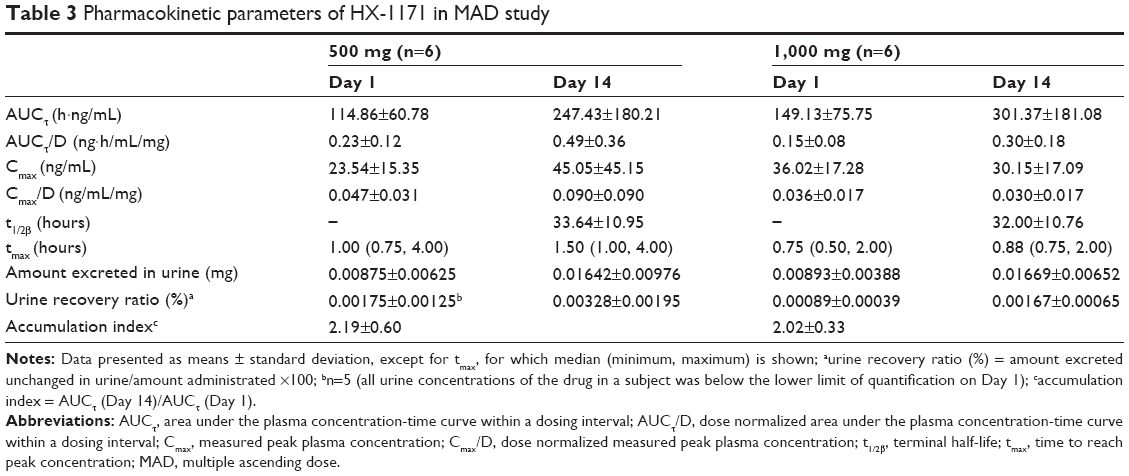 | Table 3 Pharmacokinetic parameters of HX-1171 in MAD study |
Tolerability
Seventeen subjects experienced a total of 23 AEs throughout the SAD study. Three AEs that occurred in three subjects were drug-related. Four subjects experienced a total of four AEs during the MAD study. One AE that occurred in a subject was drug-related. Of the AEs, four were considered to be related to the study drug: abdominal discomfort, conjunctival hyperemia, creatine kinase increased, headache (Table 4). No deaths or serious AEs occurred during the entire course of the study and all AEs were mild in severity. No subject discontinued the study due to AEs, and no clinically significant abnormalities in laboratory tests, physical examinations, vital signs, or electrocardiograms occurred.
 | Table 4 Summary of adverse events (AEs) after single or multiple administration of HX-1171 |
Discussion
The objective of this first-in-human study was to investigate the tolerability and pharmacokinetic characteristics of HX-1171 in healthy volunteers.
In single dose study, the median tmax of HX-1171 was short (0.63–1.50 hours), reflecting rapid absorption from the proximal small intestine. Secondary peaks indicate the occurrence of enterohepatic recirculation.16 A second peak (about 10 hours after administration) was especially prominent in the 1,200–2,000 mg group. This finding has also been shown in a preclinical study, in which the pharmacokinetics of HX-1171 in rats revealed first peak (tmax) at 0.5 hours and second peak at 8 hours.17 From the dose-normalized Cmax and AUCinf values of the 300–2,000 mg single-dose group, there were no statistically significant differences. However, the dose range and inter-individual variability was large and the tested doses were not evenly spread, making the conclusion debatable.18 In linear regression, the 95% confidence intervals of the slope for log transformed Cmax and AUCinf of the 300–2,000 mg single-dose group were 0.823–1.728 and 1.290–1.539, respectively.
In multiple dose study, the median tmax of HX-1171 on day 1 and 14 were 0.75–1.50 hours. The average half-life on day 14 was 32.00–34.64 hours. These parameters were similar to single dose study, but the coefficient of variations (CVs) were smaller than single dose study (single dose study: 46.87%–160.47% versus multiple dose study: 32.56%–33.64%, Figure 5). The blood sampling was performed 72 hours after the last dose, and %AUCextrapolation (% of extrapolation part of AUCinf) of two subjects was more than 30%. For this reason, more blood sampling was performed after 72 hours after the last dose; it would be expected to characterize the t1/2β more accurately.
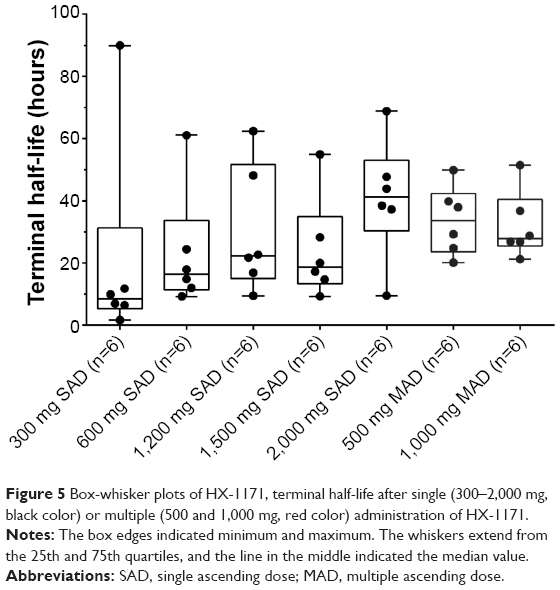 | Figure 5 Box-whisker plots of HX-1171, terminal half-life after single (300–2,000 mg, black color) or multiple (500 and 1,000 mg, red color) administration of HX-1171. |
In this study, the urine recovery ratio at 24 hours (MAD) or 48 hours (SAD) after HX-1171 administration was less than 0.01% of the administered dose. Although urine recovery of HX-1171 is negligible, fecal elimination of HX-1171 identification in another study would be helpful to determine whether the drug was eliminated through feces. In rats, the total amount of unchanged drug recovered from the feces at 24 hours was 45.2%±2.3%.17
The most frequent AE after HX-1171 administration in the study was hypertriglyceridemia. These symptoms were mild and the relationship of study drug was “unlikely related” or “definitely not related”. Three cases of hypertriglyceridemia were reported at follow-up visit (after 13–14 days of drug administration), and two cases of hypertriglyceridemia were reported before drug administration. In addition, there were no statistically significant changes in occurrence rates between the placebo and HX-1171 treatment groups, and the rate was not dose-dependent.
This study had several limitations that should be considered. First, the study was conducted in fasted state only, absorption rate at fed state of HX-1171 was unknown. Second, the results of the present study could not demonstrate any antioxidant effect, because the levels of reliable biomarker were not measured in this study. Third, these results were obtained from healthy male subjects, the pharmacokinetics of HX-1171 may differ in patient population in relation to sex or age. Therefore, future studies are needed to investigate tolerability, efficacy, and pharmacokinetics including effect of food in patients.
In summary, HX-1171 was well tolerated in normal healthy subjects. Although the CVs of pharmacokinetic parameter after administration of HX-1171 were large, the pharmacokinetic profile of HX-1171 was consistent with once-a-day dosing. Based on these findings, the tolerability and efficacy of HX-1171 should be evaluated in further studies involving patients.
Acknowledgment
The study was funded by Biotoxtech (Cheongju, Republic of Korea), the manufacturer of HX-1171.
Disclosure
The present work was presented as an oral presentation at the 2013 annual meeting of the Korean Society for Clinical Pharmacology and Therapeutics (November 8, 2013, Seoul, Korea). Otherwise, the authors report no conflicts of interest in this work.
References
Anthony PP, Ishak KG, Nayak NC, Poulsen HE, Scheuer PJ, Sobin LH. The morphology of cirrhosis. Recommendations on definition, nomenclature, and classification by a working group sponsored by the World Health Organization. J Clin Pathol. 1978;31(5):395–414. | ||
Friedman SL. Liver fibrosis – from bench to bedside. J Hepatol. 2003;38 Suppl 1:S38–S53. | ||
Lindh M, Horal P, Dhillon AP, Norkrans G. Hepatitis B virus DNA levels, precore mutations, genotypes and histological activity in chronic hepatitis B. J Viral Hepat. 2000;7(4):258–267. | ||
Brunt EM. Nonalcoholic steatohepatitis. Semin Liver Dis. 2004;24(1): 3–20. | ||
Frith J, Day CP, Henderson E, Burt AD, Newton JL. Non-alcoholic fatty liver disease in older people. Gerontology. 2009;55(6):607–613. | ||
Raynard B, Balian A, Fallik D, et al. Risk factors of fibrosis in alcohol-induced liver disease. Hepatology. 2002;35(3):635–638. | ||
Gao B, Bataller R. Alcoholic liver disease: pathogenesis and new therapeutic targets. Gastroenterology. 2011;141(5):1572–1585. | ||
Valko M, Rhodes CJ, Moncol J, Izakovic M, Mazur M. Free radicals, metals and antioxidants in oxidative stress-induced cancer. Chem Biol Interact. 2006;160(1):1–40. | ||
Friedman SL. Mechanisms of hepatic fibrogenesis. Gastroenterology. 2008;134(6):1655–1669. | ||
Ha HL, Shin HJ, Feitelson MA, Yu DY. Oxidative stress and antioxidants in hepatic pathogenesis. World J Gastroenterol. 2010;16(48): 6035–6043. | ||
Sanchez-Valle V, Chavez-Tapia NC, Uribe M, Mendez-Sanchez N. Role of oxidative stress and molecular changes in liver fibrosis: a review. Curr Med Chem. 2012;19(28):4850–4860. | ||
Hino T, Kawanishi S, Yasui H, Oka S, Sakurai H. HTHQ (1-O-hexyl-2,3,5-trimethylhydroquinone), an anti-lipid-peroxidative compound: its chemical and biochemical characterizations. Biochim Biophys Acta. 1998;1425(1):47–60. | ||
An J, Feng GG, Huang L, et al. Effects of 1-O-hexyl-2,3,5-trimethylhydroquinone on carbon tetrachloride-induced hepatic cirrhosis in rats. Hepatol Res. 2010;40(6):613–621. | ||
Jung YR, Lee YJ, Lee NJ, et al. Inhibitory Effect of 1-O-Hexyl-2,3,5-Trimethylhydroquinone on Dimethylnitrosamine-induced Liver Fibrosis in Male SD Rats. Toxicol Res. 2010;26(3):193–201. | ||
US Food and Drug Administration. Guidance for Industry: Estimating the Maximum Safe Starting Dose in Initial Clinical Trials for Therapeutics in Adult Healthy Volunteers. Center for Drug Evaluation and Research; 2005. Available from: http://www.fda.gov/downloads/Drugs/Guidances/UCM078932.pdf. Accessed January 18, 2015. | ||
Roberts MS, Magnusson BM, Burczynski FJ, Weiss M. Enterohepatic circulation: physiological, pharmacokinetic and clinical implications. Clin Pharmacokinet. 2002;41(10):751–790. | ||
HX-1171 (1-O-hexyl-2,3,5,-trimethylhydroquinone). [Investigator’s brochure]. Version 1.0. Biotoxtech Co., Ltd; January, 2012. | ||
Smith BP, Vandenhende FR, DeSante KA, et al. Confidence interval criteria for assessment of dose proportionality. Pharm Res. 2000;17(10):1278–1283. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.