Back to Archived Journals » Cell Health and Cytoskeleton » Volume 6
Shelterin complex in telomere protection: recent insights and pathological significance
Received 27 September 2014
Accepted for publication 30 October 2014
Published 3 December 2014 Volume 2014:6 Pages 11—26
DOI https://doi.org/10.2147/CHC.S49687
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 6
Editor who approved publication: Professor Denis Wirtz
Sampada Kalan, Diego Loayza
Department of Biological Sciences, Hunter College, and Graduate Center of the City University of New York, NY, USA
Abstract: Telomeres are essential for chromosome integrity and stability. The telomerase complex is the reverse transcriptase required for the addition of telomeric repeats at chromosome ends and is essential for their maintenance. The enzyme is expressed in over 80% of tumors and, indeed, telomerase is one of the genetic elements required for cellular transformation. In addition, telomeres recruit complexes called shelterin and the CTC1-STN1-TEN1 (CST) complex, which exhibit a high degree of conservation from yeast to mammals. These telomere-associated proteins mediate the roles of telomeres important for chromosome end protection and replication. Recently, some of the known shelterin components and associated telomeric factors have been described as cancer susceptibility genes. Furthermore, following extensive biochemical and genetic dissection of telomere function in a great number of model systems, the past decade has seen great progress in linking specific mutations in telomere-associated proteins with pathologies referred to as “telomeropathies”. These include the dyskeratosis congenita, Hoyeraal–Hreidarsson, and Coats’ plus syndromes, which result from defects in telomere maintenance and protection. We review here the observations and known molecular determinants linking telomere dysfunction to cancer or telomeropathy.
Keywords: telomere, telomerase, POT1, TIN2, RAP1, DNA damage
Introduction: shelterin and telomeres
Telomeres have been the subject of intense scrutiny for several decades, representing now a diverse field for the importance of which E Blackburn, C Greider, and J Szoztak were awarded the 2009 Nobel Prize in Physiology and Medicine. The study of telomere function has gone through many growing steps, from fundamental discoveries all the way to intricate connections with human disease. To name a few, the analysis of terminal sequences in Tetrahymena by E Blackburn and J Gall (1978), the discovery of telomerase activity by C Greider and E Blackburn (1985), the biochemical analysis of telomere binding proteins in ciliates by D Gottschling and T Cech (1984),1 the genetic analysis of telomere function in yeast,2 and the biochemical and genetic characterization of telomere function in mammals,3,4 all contributed in some way to textbook chapters and, more recently, to the description of molecular causes of specific pathologies. Initially, telomeres were the subject of fascination owing to their particular position on chromosomes: How do they terminate? Are there any genes present? Are they made of specific sequences? Are they important elements for the chromosome itself? Over the years, it has been discovered that telomeres are made of short repeated sequences, are bound by a specific protein complex called shelterin, and are essential for chromosome integrity and stability.4 In addition, more recently, it has been discovered that telomere dysfunction is linked to specific pathologies, notably, but not exclusively, cancer.
In this review, we shall outline the structure of telomeres and the proteins that associate specifically with them, and summarize the latest discoveries that implicate telomere integrity in disease (Table 1).
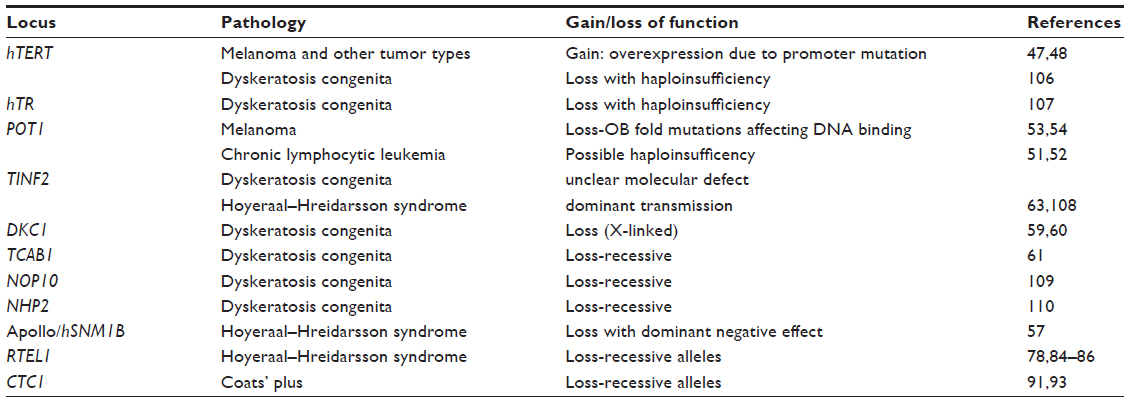 | Table 1 Summary table of loci involved, with related pathologies and nature of mutations, when known |
Telomeres and the shelterin complex
Vertebrate telomeres are composed of the hexanucleotide sequence TTAGGG repeated thousands of times. By nature, the length of the telomere at each chromosome end falls within a range typically between 5 kb and 15 kb in humans, owing to the mechanism of telomere length regulation exerted by the shelterin complex.5 The sequence of telomeres is dictated by the RNA template of the telomerase complex, a dedicated reverse transcriptase able to extend the 3′ end of chromosomes.6 Telomeres terminate with a single-stranded TTAGGG overhang composed of 50–300 nt, which is essential for the formation of the t-loop structure detected in cells, through strand invasion of internal telomere duplex sites.7 The t-loop structure represents a protected telomere, preventing its recognition as a DNA repair substrate.8
Telomeres are bound quantitatively by a six-protein complex called shelterin (Figure 1 and Table 2).9 The paralogues TRF1 and TRF2 are DNA binding proteins which bind the duplex telomeric DNA,10,11 and represent a platform for the association of the rest of the complex. The small molecule TIN2 is able to bind simultaneously TRF1 and TRF2, and recruits the TPP1-POT1 dimer.12,13 POT1 is the telomeric overhang binding protein and, in doing so it represses possible extension of the telomere by telomerase,14 and prevents inappropriate activation of the ataxia-telangiectasia and Rad3-related (ATR) DNA damage response.15 The overhang DNA binding activity of POT1 is mediated by two structural motifs called OB folds, both present in the N-terminal half of the molecule.16,17 The sixth shelterin component is RAP1,18 a TRF2-binding protein, which is critical for the repression of homology-directed repair (HDR) at telomeres.19 TRF2 in turn is important for the inhibition of the ataxia telangiectasia mutated (ATM) response,20 otherwise elicited by unprotected telomeric DNA, which leads to extensive chromosome fusions and apoptosis.21,22
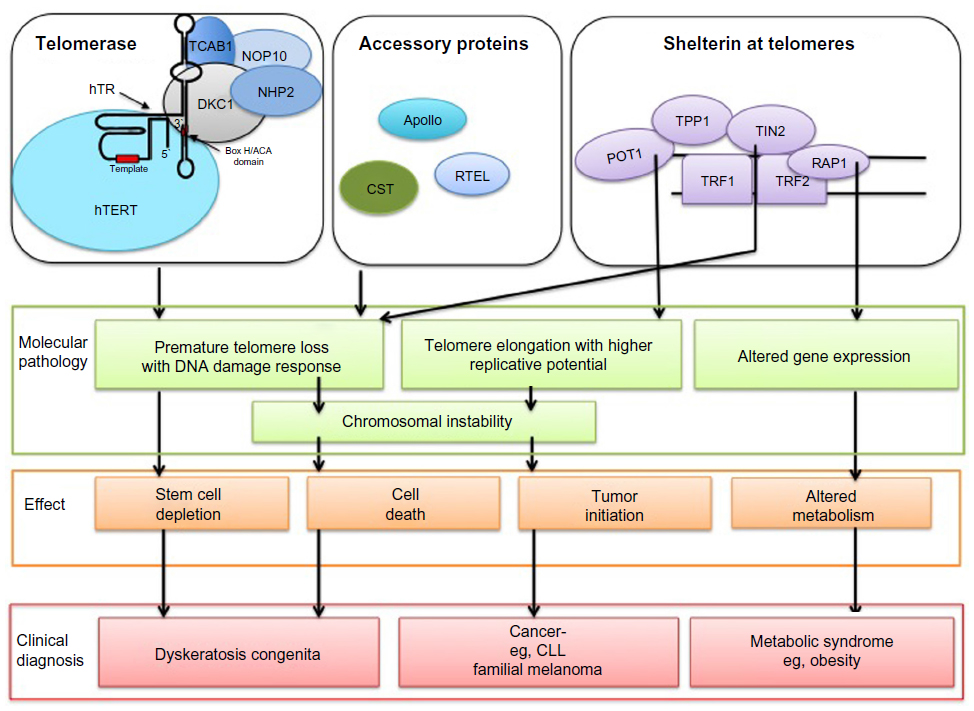 | Figure 1 Mutations in telomerase and shelterin lead to various types of defects and associated diseases. |
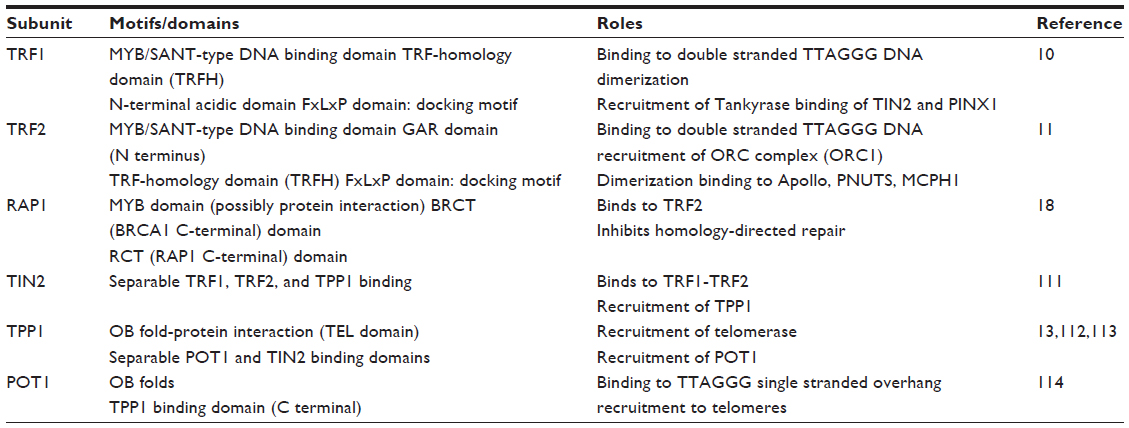 | Table 2 The six subunits of the shelterin complex |
Thus, shelterin is essential for telomere length control and telomere protection in vivo.
Telomerase as a genetic element required for cellular transformation
In human cells specifically, the telomerase enzyme is not expressed in most somatic cells, or expressed so poorly that telomeres gradually shorten at each cell division owing to the biochemical properties of DNA replication of linear molecules, referred to as the “end replication problem”. This gradual shortening leads to a permanent state of G1 arrest called “senescence”.23 Therefore, telomeres have been known for a long time as a primordial parameter for the finite replicative potential seen in human cells in culture. Thus, given its impact on limiting cell proliferation, senescence represents an important tumor-suppressive mechanism.24
The bypass of senescence by the establishment of a telomere maintenance system is an essential event in cellular transformation.25 This can be achieved by re-expression of telomerase, likely as a clonal event, leading to long-term telomere maintenance or even telomere elongation.26 Re-expression of telomerase is detected in over 85% of solid tumors in cells.27 However, telomerase-mediated telomere maintenance is not the only mechanism for the bypass of senescence. Another telomere maintenance mechanism, termed alternative lengthening of telomeres (ALT), can be at play, and relies on inter-telomere homology-directed recombination to prevent accumulation of short telomeres, thereby allowing the bypass of senescence.28 It has recently been discovered that reduced expression of ATRX, part of the chromatin remodeling complex ATRX/DAXX and responsible for histone variant H3.3 deposition at telomeres and other genomic sites, is an event associated with ALT in 90% of human ALT cell lines analyzed.29 Although other genetic or epigenetic events are required for establishment of ALT, this pathway stresses that it is telomere maintenance that sustains tumor cell growth, and not telomerase expression by itself.
The importance of telomerase, and specifically the catalytic subunit, in cell transformation has been analyzed by looking at its contribution in the production of cells able to form colonies in soft agar, or in forming aggressive tumors in nude mice.30 By itself, telomerase does not represent an oncogene, as it is unable to induce full transformation of primary human cells. However, the enzyme is sufficient for the bypass of senescence and confers immortality to cells.31 Importantly, such primary cell lines expressing telomerase still possess characteristics of nontransformed cells, such as intact growth control mechanisms, inability to form colonies on soft agar, and failure to produce tumors in nude mice.32,33 In the end, telomere maintenance is necessary, but not sufficient, to produce a transformed phenotype. In addition to telomerase, four other genetic alterations are required for full cellular transformation, which are inactivation of p53, Rb, and PP2A, and oncogenic activation of the MAP kinase signaling pathway, for example, through introduction of activated RAS.30 Importantly, these studies demonstrated the role of telomere maintenance in immortalization and possible cellular transformation and cancer in vivo. Thus, any mutation or condition leading to inappropriate or increased telomere maintenance would be expected to increase cancer predisposition. We shall review below recent published work confirming these ideas.
The shelterin complex is an essential effector of telomere length regulation and telomere protection.4,9 The length of telomeres, and their maintenance, is ultimately dependent on the telomere itself, as it is a regulation exerted in cis by the bound shelterin complexes recruited there.5 One of the many aspects of shelterin is its function as a negative regulator of telomerase-mediated repeat addition at telomeres. Over the years, based on evidence mostly gathered in the yeast and human cell culture systems, it has been established that telomerase is progressively inhibited by increasing amounts of Rap1 (yeast)34 or shelterin (human cells)35 at telomeres, these amounts being ultimately dependent on the length of the individual telomere itself. Thus, a negative-feedback cis regulation is at play, whereby extensive elongation by telomerase leads to increased amounts of shelterin at that specific telomere, resulting in higher inhibition of the enzyme and lower propensity for the telomere to be acted upon. Conversely, significant shortening of a telomere due to the end replication problem leads to lower amounts of shelterin bound, with this relieved inhibition leading to a higher tendency of shorter telomeres to be elongated by telomerase. This simple model is actually not fully reflective of the real situation in that shelterin also possesses a positive influence on telomerase recruitment through its subunit TPP1.36,37 TPP1 possesses an OB fold dedicated to and sufficient for the recruitment of telomerase to telomeres. Since TPP1 requires TIN2 for its own recruitment, depletion or loss of function of TIN2 may have the effect of inhibiting the recruitment of telomerase, resulting in the stalling of the holoenzyme in Cajal bodies where its biogenesis takes place.36 However, the model does account for many observations reported on TRF1, TRF2, and POT1. TRF1 itself is a negative regulator of telomere length by virtue of its ability to significantly reduce mean telomere length when overexpressed.35 A dominant-negative allele of TRF1 lacking the C-terminal MYB-type DNA binding domain and leading to a reduction of telomeric association of TRF1 leads to telomere extension over ~60 doublings before reaching a new, higher setpoint. Analysis of TRF2 wild-type and dominant-negative alleles led to the same conclusion38 that telomere-bound proteins acted as negative regulators of telomerase in cis. Work on POT1 shows that at least part of this regulation occurs as part of the shelterin complex: TRF1 and TRF2 recruit TIN2, TPP1/POT1,13 and POT1 – by virtue of its ability to bind the overhang through the two N-terminal OB fold motifs – prevents extensive telomere elongation in telomerase-positive cells.14 Conceptually, shelterin can be viewed as having dual roles, in telomerase recruitment or in the inhibition of telomere elongation.4 POT1, in particular, is a major player as a negative regulator of telomere length, with its loss of function imparting unregulated telomere elongation in telomerase-positive cells, all other parameters being equal.
Telomere protection and DNA damage responses
By nature, a linear DNA end is highly reactive and can be processed by many DNA repair pathways in cells.39 This means that telomeres are, by virtue of containing the extremities of chromosomes, excellent substrates for DNA repair activities, which can lead to their joining, producing dicentric chromosomes, and resulting in mitotic catastrophe or high genomic instability.40 One machinery performing end-to-end joining is the non-homologous end joining (NHEJ) machinery, which is dependent on the Ku dimer complex Ku70/80, the PIK kinase DNA-PKcs, and ligase IV. These deleterious activities can indeed be highly active at telomeres, for example, upon inactivation of TRF2, in both human and mouse cells.41,42 It has since been discovered that telomeres can elicit many DNA repair activities, which are inhibited by shelterin. Shelterin is therefore essential for a second aspect of telomere function: their protection through inhibition of DNA repair pathways.4
In addition to repressing NHEJ at telomeres, TRF2 exerts a strong inhibition on the ATM pathway, which itself is important for repair through homologous recombination.20 TRF2 was found to associate with ATM and prevent its activation by autophosphorylation. On the other hand, TRF2 deletion or expression of a TRF2 dominant-negative allele leads to immediate activation of ATM and the concomitant induction of p53 and phosphorylation of Checkpoint 2 (Chk2).15,22 Another important DNA repair system that can be activated by telomeres is the ATR pathway. The ATR kinase is known to respond to replication stress and is particularly sensitive to accumulation of single-stranded DNA during DNA replication.43 Its activation results in phosphorylation of targets such as Checkpoint protein 1 (Chk1) and Rad17 and leads to cell cycle delay or apoptosis. Given the presence of a single-stranded overhang at all telomeres, chromosome ends can elicit a potent ATR response. The shelterin subunit POT1 prevents such a response and as such is essential for chromosome integrity.15 The telomeric overhang binding activity of POT1 is essential for the repression of ATR. This has been extensively analyzed in mouse cells deficient for POT1, where an allele deficient in DNA binding, either through deletion of the OB folds or through expression of chimeric molecules, results in the derepression of the ATR response and telomere fusions.44 The extent of the DNA damage response induced by deprotected telomeres has been characterized by studying mouse cells deficient in both TRF1 and TRF2, using a conditional knockout system.45 These cells, as a result of Cre expression, experience removal of the whole shelterin complex from telomeres. In addition to the three pathways mentioned earlier, namely, NHEJ, ATM and ATR, three additional activities are induced: alternative NHEJ, HDR, and a poorly understood 5′ resection process. Of these, the shelterin subunits TRF2 (ATM and NHEJ), POT1 (ATR), and RAP1 (HDR) have been genetically described as inhibitors, with the alt-NHEJ and resection being observed in the absence of Ku or p53BP1, and thus only observed in complex genetic backgrounds.
Overall, the extensive analysis performed on telomere function in mammalian systems has established two major branches impacting disease. First, the specific aspects of telomere replication and maintenance through telomerase represent a limit to the number of divisions a cell can undertake, leading to the tumor suppressor system termed senescence, which has to be overcome in order for cells to be fully transformed. And, second, telomere dysfunction may result in deprotection and the ensuing engagement of multiple DNA repair systems, which can lead to telomere fusions and high genomic instability. The events, in turn, are potentially tumorigenic, and also severely reduce the fitness of the cells experiencing these defects. Below are the recent known examples of pathologies linked to either of the roles ascribed to telomeres.
TERT and POT1 as cancer susceptibility genes
Given their roles in chromosome stability, it has been proposed that telomere proteins, part of the shelterin complex, and telomerase are excellent candidates for cancer susceptibility genes.25 In addition, it has long been appreciated that telomere maintenance is required for tumor cell proliferation. As such, over 85% of tumors are constituted by cells that are telomerase positive, after re-expression of telomerase, likely resulting from their aberrant gene expression program.27 The remaining 15% possess a telomerase-independent telomere maintenance system termed ALT, which leads to the same ultimate outcome relevant in oncogenesis, namely, cellular immortalization.46 The prediction of telomere-function-associated genes as cancer susceptibility genes has been recently proven correct by a number of studies. First, a mutation linked to overexpression of telomerase has been associated with familial cases of melanoma. Second, it has been found in the past year that POT1 represents a cancer susceptibility gene. We review these two cases below.
TERT in familial and sporadic melanomas
Cutaneous melanoma is an aggressive type of cancer characterized by uncontrolled growth of melanocytes, and accounts for 75% of skin cancer deaths. Familial cutaneous melanoma is a genetic or inherited condition in which 10% of cases have first-degree relative diagnosed with the disease. The BRAF, NRAS, CDKN2A, and CDK4 genes were found to be frequently mutated in these cases.
A specific mutation in the hTERT promoter region has been found by high-throughput sequencing of affected related individuals and is linked to the dominant transmission of the disease in a single family.47 It consists in a T-to-G nucleotide change at position −57 in the TERT promoter, and creates a consensus binding motif for the ETS/TCF class of transcription factors (CCTGAA to CCGGAA). The mutation leads to a 2.2-fold increase in the hTERT promoter activity in luciferase assays. Therefore, it is speculated that the effect of the mutation leads to overexpression of hTERT, and could be the underlying molecular cause of the disease. It is interesting to note that in sporadic melanomas, as well as in melanoma-derived cell lines, similar mutations in the hTERT promoter are frequently encountered. For example, many sporadic melanomas were found to have G-to-A mutations in the −124 to −149 region of the promoter, again creating potential binding sites for ETS/TCF, with the possibility of a similar effect on hTERT transcription. Another study, published on the same issue, found two other mutations located in the hTERT promoter, although at a different location, but present in the vicinity of the transcription start site and each creating an ETS binding sequence as well.48 By whole genome sequencing of malignant melanomas, these two hTERT gene promoter mutations were found to individually correlate with the disease. These were the C–T transitions at dipyrimidines, suggestive of ultraviolet (UV)-induced alterations. As in the previous cases, these were located within 100 bp of the hTERT transcriptional start site, and created an identical sequence corresponding to a consensus binding site for ETS transcription factors. Additional screening of melanomas revealed that either of these mutations could be found in 50 out of 70 tumor samples. The general features are identical to the previous case: dominant transmission, a twofold increase in transcription from the hTERT promoter as measured by luciferase assays, and the presence of this mutation in existing melanoma tissue samples. In addition, the authors could show that the hTERT promoter mutation linked to the emergence of melanoma could also be found in cell lines isolated from other tumor types, such as bladder, thyroid, liver, or lung tumors, among others. Thus, although in vivo overexpression of hTERT is not viewed as oncogenic in itself, it does appear to favor the emergence of tumors.49 The preponderance of melanomas in the tumor profile could be due to the reliance on the RAS pathway for the proliferation of melanocytes upon UV exposure, itself possibly inducing the described hTERT promoter mutations associated with cancer through the production of thymidine dimers in the DNA.47 Therefore, exposure to UV would have a dual impact in inducing the mutation as well as activating RAS in melanocytes. A mutation leading to an increase of expression of telomerase would indeed segregate as a dominant disease if this were confirmed to be the molecular cause. The actual increase in telomerase activity, and possibly of telomere length, in these cells would present important avenues for future work to confirm this model.
POT1 mutations in chronic lymphocytic leukemia
Chronic lymphocytic leukemia (CLL) is a malignant hematological cancer characterized by slow increase in B lymphocytes and is the most frequent leukemia found in adults. CLL might be indolent in some cases or can be aggressive, causing rapid mortality. Detection of the genetic alterations that occur in this disease might help in understanding genetic susceptibility of individuals, disease progression, and clinical outcome. Mutations in the POT1 OB folds, reducing its overhang binding activity, have been found as predispositions to CLL.50 An exome analysis based on the gene size, number of mutations in the gene, and codon composition demonstrated that the shelterin component POT1 was the second most frequently mutated gene in CLL,51 after only SF3B1. Somatic mutations in POT1 were detected in 3.5% of all CLL cases, mostly occurring in wild-type IGHV@ aggressive subtypes (the gene for immunoglobulin heavy chain variable), associated with longer patient survival.51 Twelve recessive somatic mutations were detected, with nine of them localized in the OB folds of POT1, which are critical for the DNA binding activity of POT1. The affected residues were found to be evolutionarily conserved and predicted to reduce the DNA binding ability of POT1. The POT1 mutations were found to be heterozygous with dominant transmission, thus revealing either a dominant-negative effect, or, more likely, a degree of haploinsufficiency for POT1. The expression of exogenous tagged POT1 mutants (Y36N and Y223C affecting the first and second OB, fold respectively) in cell lines showed that the protein was able to localize at telomeres, but could not interact with single-stranded DNA in vitro. Telomere length analysis revealed a significant increase in telomere length with each population doubling without change in telomerase activity,51 thereby likely providing high replicative potential to the cells with bypass of senescence. There was also a marked increase in number of sister telomere fusions and presence of multi-telomeric signals indicative of fragile telomeres, probably caused by stalled replication forks. However, there was no detectable DNA damage at telomeres, suggesting that the main molecular defect was telomere length dysregulation with elongated telomeres, rather than extensive telomere deprotection. In another study, a genome-wide search for susceptibility loci for CLL led to detection of a significant single-nucleotide polymorphism (SNP) located in the 3′UTR of POT1.52 The exact nature of the loss of function remains to be determined, but could be due to destabilization of the mRNA and reduced expression of the protein. This study also reported intriguing associations with hTR and hTERT, both possibly leading to lowered telomerase activity. Although the exact impact on telomerase remains to be confirmed, such mutations could have an effect on tumor incidence through ineffective telomere maintenance and ensuing increased genomic instability, which is itself a known oncogenic process.
Thus, POT1 mutations may initially lead to inappropriate telomere elongation in telomerase-positive lymphocytes, which may drive cancer initiation and progression in CLL in combination with other events inducing genomic instability and full-blown leukemias.50
POT1 mutations in familial melanoma
Genetic screening to identify susceptibility genes that might be altered in melanoma led to the identification of POT1 as a cancer susceptibility gene in several populations in the world.
In one study, melanoma cases from 105 pedigrees from the UK, the Netherlands, and Australia were chosen. The selected cases were unaffected for the already known melanoma predisposition genes CDKN2A and CDK4.53 Variants of POT1 encoding Y89C, Q94E (first OB fold), and R273L (second OB fold) changes were identified in the screen. Interestingly, the Q94E substitution was also detected in some cases of CLL, indicating a possible residue mutated in multiple forms of cancer. The missense mutations were all found to be in the N-terminal OB fold domain essential for the DNA binding activity of POT1. The mutations identified were in highly conserved amino acids within the OB fold as well, suggesting that the variants might prevent binding of POT1 to the overhang. Biochemical gel-shift DNA binding assays showed that all three POT1 mutants analyzed could not interact with single-stranded DNA. Polymerase chain reaction (PCR)-based telomere length analysis of melanoma cases also showed elongated telomere length. In one family, a splice variant between exon 17 and 18 was found, probably resulting in a truncated form of POT1. A C-terminal truncation would not affect DNA binding, but the recruitment of the molecule to telomeres would, which normally occurs through interaction with TPP1. The result would be reduced POT1 recruitment and telomere extension as a loss of function, as is the case for DNA binding defective POT1 mutants, but with a dominant pattern of inheritance as reported, due to haploinsufficiency.
In another genetic study, 101 cases negative for mutations in CDKN2A or CDK4 in 56 unrelated melanoma families from Italy were recruited,54 all displaying dominant inheritance of the disease. Mutation in the second OB fold leading to S270N change, R137H substitution in the α-helix of the first OB fold, and Q623H substitution in the C-terminus containing the TPP1-binding region of POT1 were detected. These residues were also evolutionarily conserved, and the mutant proteins were predicted to be loss-of-function mutations, although this notion awaits biochemical confirmation. Cells from the POT1 variants showed increased telomere lengths and heterogeneity when compared to age-matched controls. There was also a marked increase in average number of fragile telomeres, with no obvious telomeric DNA damage response.
Germline missense mutations causing D224N variant was detected in a single US family. Similar to S270N, the D224N variant was also found in the second OB fold near the DNA-binding domain, likely affecting the binding of POT1 to DNA. Another A532P variant was detected in a family from France; this mutation was near a splice junction and hence could alter normal splicing of POT1 and reduce its expression. Alternatively, these substitutions could affect the interaction with TPP1 and reduce the amounts of POT1 to telomeres. Again, some aspect of haploinsufficiency is to be considered for these predisposing mutations in order to explain their dominant pattern of inheritance.
The detection of POT1 as a frequently mutated gene in a variety of cancer types underscores the importance of telomere maintenance in cancer susceptibility and progression, and provides new insights into cancer detection and treatment. The mutations found could result in excessive telomere lengthening leading to immortalization, or they could induce chromosomal instability due to deprotection and inappropriate induction of DNA repair pathways, either of which could result in tumor initiation. The study of the exact nature molecular defect at play is an important future research endeavor.
Dyskeratosis congenita (DC): a telomere disease
DC is a rare disease characterized by the presence of the three symptoms of leukoplakia, nail dystrophy, and reticular skin pigmentation, but also often associated with other medical problems such as bone marrow failure, pulmonary fibrosis, and predisposition to cancer.55,56 The disease is linked to telomere dysfunction, as the molecular defect lies in genes essential for telomere maintenance and protection, mainly TINF2, the telomerase RNA (hTR), and the catalytic subunit (hTERT), as well as many genes involved in the biogenesis of the telomerase complex, namely DKC1, NOP10, and NPH2. The latter three genes are important for the stability of hTR. A severe form of DC, termed Hoyeraal–Hreidarsson Syndrome (HHS), maps to SNM1B/Apollo, a TRF2-associated factor.57 Unlike the involvement of telomerase or shelterin subunit in cancer described above, the disease is believed to be the result of telomere dysfunction and deprotection, and as such its various forms are referred to as “telomeropathies”. In the great majority of the cases, the cells of the affected individuals exhibit extremely short telomeres, specifically in their circulating lymphocytes, and sometimes also in their fibroblasts. The symptoms are at least in part the result of somatic stem cell failure,55 possibly in the tissues where their renewal is most important, and as a consequence, reveal the earlier and most visible functions in the body dependent on stem cell renewal. The tissues reliant on keratinocytes are particularly sensitive, leading to loss of hair and nail dystrophy, and of course, the immune system is highly impacted, resulting in high incidence of infections.
DC and telomerase
As discussed earlier, telomere dysfunction can result from telomere loss, for instance, due to a defect in telomerase, or from deprotection, leading to inappropriate activation of DNA repair pathways. There is evidence for both systems being linked to DC depending on the causative molecular defect. Loss-of-function mutations in the telomerase complex lead to DC, with associated short telomeres but with no obvious signs of telomere deprotection. These can affect the enzyme in many ways: by reducing the activity of the catalytic subunit (hTERT), by mutating the RNA component (hTR), or by affecting components essential for hTR stability (DKC1, NOP10, NHP2) or for the trafficking of the complex itself (TCAB1) (reviewed in Refs 55 and 58). In general, all these defects point to the same molecular cause: a reduction in telomere maintenance, leading to excessive shortening and reduced cellular fitness, either of somatic stem cells or peripheral blood cells. When the reduction in activity is particularly impaired, haploinsufficiency is observed in the case of hTERT and hTR. The disease then follows an autosomal dominant pattern of inheritance. Some cases have been documented as de novo mutations in families in which, when multiple cases arise in different generations, an evident anticipation is observed, whereby the symptoms are manifested at an earlier age in each generation. This phenomenon is compatible with telomere shortening being the culprit in the disease’s etiology, with the anticipation of symptoms arising from shorter telomeres in the germ cells of the affected individuals.
Mutations in DKC1 have been the first to be linked to DC itself.59,60 The gene is present on the X chromosome, and mutations are associated with a strong reduction in hTR amounts in the affected cells. The same is true for mutations in NOP10 and NHP2, and, along with DKC1, these gene products form a complex that binds to hTR and is essential for its stability. The association is mediated by an H/ACA domain in hTR.59 The case of TCAB1 mutations presents a different aspect of telomerase biogenesis in that this component is essential for the incorporation of the telomerase complex in Cajal bodies, themselves known to be important for the association of the holoenzyme with telomeres, and as such, important for telomere maintenance.61 In absent or reduced TCAB function, hTR can be seen to mislocalize outside of Cajal bodies in the nucleus, and accumulates in the nucleolus, causing a defect in telomere repeat addition.61 Indeed, in DC patient cells defective for TCAB1, hTR is not present in the Cajal bodies and the telomeres are significantly shorter, down, to <1 percentile compared to unmatched controls, and shorter than the heterozygous parents or sibling. The disease has been documented in two compound heterozygous patients.61
DC and shelterin: the case of TIN2
Since shelterin inactivation leads to telomere elongation, mutations in its subunits are at first glance not expected to be associated with DC. In reality, it is now accepted that at least some shelterin subunits are important for the recruitment of telomerase. Specifically, TPP1 possesses an OB fold domain which can recruit telomerase through interactions with the TEN and CTE domains of hTERT.36 Although this interaction could be mediated by other proteins, specific mutations in the TPP1 OB fold could theoretically result in DC. Additionally, other shelterin subunits could have a similar effect on telomerase recruitment, or telomerase maintenance or replication. So far, such mutations have been identified in TIN2.62,63 These mutations are dominant and mostly cluster in a 15-residue segment present C-terminally to the TRF1, TRF2, and TPP1 interacting surfaces, and do not impact in an obvious way on their binding.62 The telomeres are extremely short in affected cells, confirming the molecular determinant of the pathogenesis. The exact impact of these mutations remains to be clarified. They could affect the integrity of the complex, the recruitment of telomerase, the stability of TPP1 at telomeres, or other roles in protection. In this context, a mouse model of disease provides informative clues on the importance of TIN2.64 The defect has been reconstituted in the mouse by knocking of the corresponding dominant allele in mouse TINF2, K267E (human K280E). This allele is lethal at homozygocity but, as observed in humans, presents a dominant transmission of the phenotype. The mice develop pancytopenia and reduced fertility, with display of anticipation across generations. Consistent with a telomere-based defect, telomeres shortened over the generations, as well as in mouse embryonic fibroblast (MEF) upon proliferation in culture. A very interesting and informative observation was made on these TINF2+/DC mice, in that significant telomere shortening was observed also in telomerase-deficient mice (mTR−/−), although to a lesser degree than in their wild-type mTR+/+ counterparts. Therefore, the telomere maintenance defect was at least in part due to a telomerase-independent process, possibly related to a replication problem or excessive telomere degradation due to deprotection. Indeed, a significant induction of ATR was seen at these defective telomeres. Thus, the mouse model strongly suggests that the TIN2 defect leading to DC is the result of telomere deprotection, rather than a strictly telomerase-based maintenance defect as described for DKC1, hTR, and hTERT mutations, for example. Another informative mouse model is the POT1b knockout.65 In this organism, there are two POT1 loci which display a degree of functional divergence.66 POT1a is essential and mostly involved in repressing ATR.15 POT1b knockouts are viable but display extensive telomere degradation, due to deprotection of the 5′ end against nucleolytic activities able to catalyze extensive 5′ end resection, thereby producing abnormally long 3′ overhangs.66 The single POT1b−/− knockout exhibits phenotypes compatible with DC disease symptoms: moderate pancytopenia, nail dystrophy, atrophy of the intestinal mucosa, cutaneous hyperpigmentation, and infertility.64 These phenotypes were greatly enhanced in mTR+/− heterozygous mice, with additional bone marrow failure and a severely reduced life span of 4 months. It is important to note that, in the mouse, neither the mTR−/− nor the mTERT−/− knockout displays DC-like phenotypes. Therefore, in the mouse, the disease is due to defects cumulative to a simple telomere maintenance defect, such as protection or replication defects, as uncovered by the POT1b or TINF2 mouse models described above. It is important to keep the POT1 locus in mind for unassigned cases of DC-like symptoms in humans. Specifically, POT1 has been shown to be important for preventing inappropriate 5′ end telomeric processing,67 an activity that could be related to disease in humans other than its importance as a cancer susceptibility locus as described earlier.
DC and telomere associated factors: Apollo, RTEL1, and CTC1
In addition to shelterin, many transiently associated factors participate in telomere function (Figure 1).68 Importantly, a trimolecular complex called the CST complex, composed of CTC1, STN1 (also known as OBFC1), and TEN1, participates in telomere replication.69 Of those additional factors, three have been associated with DC through their importance in telomere function. Strikingly, these mutations are linked to a particularly severe form of the disease, referred to as Coats’ plus syndrome for CTC1, or the previously mentioned HHS for telomere-associated factors Apollo or RTEL1.
Apollo
Apollo is a TRF2-associated protein which possesses a 5′ to 3′ exonuclease domain and a metallo-β-lactamase motif.70,71 Apollo is implicated in interstrand crosslink repair, outside of its roles at telomeres.72 The depletion of Apollo by shRNA leads to telomere dysfunction-induced foci (TIF) formation associated with diminished cell proliferation and premature senescence in primary cells, with induction of p53.71 Studies on mouse knockout cells have established that TIF induction resulting from the loss of Apollo occurs in cells going through S phase, suggesting that the telomere damage observed is related to DNA replication.73–75 The resulting telomere damage leads to induction of ATM and end-to-end telomere fusions through NHEJ, indicative of telomere deprotection. Chromosome orientation fluorescent in vitro hybridization (FISH) on the fused telomeres indicated that Apollo is important to regenerate the 3′ telomeric overhang on the telomere created by leading-strand DNA synthesis.73,74 This model is in accordance with the biochemical activity of the protein, since a 5′ to 3′ exonuclease would be expected to create a 5′ recessed end on a blunt DNA molecule. Thus, Apollo appears to be important for the processing of one of the daughter telomeres, but its absence leads to deleterious fusions and subsequent production of dicentric chromosomes followed by additional breaks and fusion events. Additionally, Apollo deletion in the mouse leads to a significant increase in telomere doublets observed on metaphase chromosomes,74 indicative of the fragile telomeres seen upon replication fork collapse. Apollo, therefore, could contribute to two processes: the formation of the overhang on leading strand telomeres, and the effective replication through the telomeric tract (see also Ref 76). The connection between Apollo and HHS comes from the molecular analysis of cells from a young girl presenting the HHS symptoms of intrauterine growth retardation, microcephaly, lack of B lymphocytes, and progressive aplastic anemia.57 This patient died at 4 years of age due to severe bone marrow failure. Intriguingly, the affected cells present a shorter Apollo transcript previously undetected, resulting from abnormal splicing into the fourth exon, and producing a shorter Apollo gene product termed Apollo-Δ. The production of Apollo-Δ is not the consequence of a mutation in the gene itself, but rather of an unknown alteration elsewhere leading to the aberrant splicing form. Apollo-Δ therefore corresponds to a C-terminal truncation of the protein of otherwise normal sequence. The missing domain corresponds to the TRF2 binding domain, and Apollo-Δ is thus unable to localize to telomeres. Further analysis determined that the Apollo-Δ mutant protein acts as a dominant negative molecule, and the patient cells or cells expressing the mutant Apollo protein display phenotypes in accordance with the pathology initially described. The fibroblasts present a severe growth defect, exhibit premature senescence in culture, and accumulate a significant number of TIFs. The length of the telomeres was not particularly affected, arguing in this case for telomere deprotection rather than shortening being the primary molecular defect. The analysis of the effect of expressing Apollo-Δ in wild-type fibroblasts supported the notion that the mutant protein exerts a dominant-negative effect: the cells underwent premature senescence, exhibited a significant growth defect, and possessed a high degree of chromosomal abnormalities, particularly telomere fusions and doublets. The mutant protein failed to interact with TRF2 and did not localize to telomeres. The dominant effect of this mutation could be due to the titration of another important unknown factor away from telomeres. Overall, this study identified mutations in Apollo as a possible cause for HHS in its important role as a telomere protector.
Regulator of telomere length 1
Regulator of telomere length 1 (RTEL1) is an ATP-dependent 5′ to 3′ helicase part of the Rad3-related DEAH subfamily, with the helicase domain present in the N-terminal half of the protein (reviewed in Ref 77). The molecule also possesses harmonin-N-like domains,78 which are putative protein interaction domains, a PCNA interacting protein (PIP) domain involved in binding to proliferating cell nuclear antigen (PCNA), and a C-terminal RING finger domain with possible ubiquitin ligase activity. The significance of the helicase domain has been well characterized biochemically, and results in the ATP-dependent disassembly of recombination intermediates such as in vitro models of strand invasion substrates, D-loops, t-loops, and G-quadruplex DNA molecules.79 As such, RTEL1 has an anti-recombination activity important for chromosome integrity and efficient DNA repair. The gene was first described as a factor important for telomere length regulation in mice, through crosses between Mus musculus (length >25 kb) and Mus spretus (length 5–15 kb).80 The locus responsible for this difference was identified as RTEL1,81 with a difference in splicing being responsible for expression of a variant in M. spretus possibly exhibiting reduced function. Thus, in mice, RTEL1 acts a positive regulator of telomere length. The role of RTEL1 in the mouse was further refined through genetic and biochemical analysis in knockout embryonic stem cells, the gene being essential for embryonic development between day 10 and day 11.5.81 The RTEL1−/− cells showed significantly shorter telomeres compared to wild-type and heterozygous +/− cells, and high incidence of chromosomal abnormalities consisting of end-to-end intrachromosomal fusions, chromosome ends with no detectable telomeres, and frequent chromosome breaks and fragments. Also very informative was the study of hypomorphic alleles in the Caenorhabditis elegans DOG-1 gene (deletion of guanine-rich DNA), the worm homologue of RTEL1.82 The mutant worms possess a specific mutator phenotype of germline and somatic deletions of extended poly-G tracts throughout the genome. This phenotype is compatible with a role for the RTEL1 helicase in resolving complex secondary structures generated during lagging strand DNA synthesis and, although no telomere deletions were observed in the worm, provides a possible interpretation for the telomere phenotypes observed in mammals. A telomeric role for RTEL1 in the mouse was described as an activity preventing the resolution of the t-loop into a t-circle, where significant loss can occur through the unchecked action of the SLX4 nuclease complex.83 In this context, RTEL1 is proposed to be a t-loop “destabilizer”, thereby preventing excessive production of t-circles leading to rapid deletion of terminal sequences. Thus, in absent or reduced function of RTEL1, a significant telomere loss is observed, consistent with its previously documented role in telomere stability. The proposed model ascribes two important roles for RTEL1 at telomeres: first, in destabilizing t-loops, thereby preventing excessive excision of t-circles; and, second, in the resolution of G4-DNA structures which would otherwise impede lagging-strand DNA replication and result in telomere fragility.83 These two functions would rely on the known helicase activity of the molecule.
In humans, the RTEL1 gene has been implicated as a brain tumor susceptibility gene by genome-wide association studies. Recessive mutations in the gene have also been found as causing HHS, the severe form of DC previously mentioned.84 Specifically, a family with four affected siblings was analyzed by whole exome sequencing in the discovery of two separate mutations in RTEL1, namely M492I and R974X, able to cause HHS in the compound heterozygote.85 The patients’ cells displayed chromosome abnormalities such as fusions and signal-free ends, short telomeres, and a notable growth defect. This study also included the ectopic expression of RTEL1 in affected cells, which complemented the observed phenotype, establishing that RTEL1 was indeed the cause of the defect. Another study focused on three patients in two unrelated families, with recessive transmission of the disease in both cases.86 The telomeres showed a moderate shortening (6.6 kb, 7.0 kb, and 5.6 kb in patients’ blood cells compared to 7.8–9.8 kb range in unaffected parents or siblings). The telomerase activity in patients was not reduced as measured by telomeric repeat amplification protocol (TRAP) assays; however, an ongoing DNA damage response was observed in affected cells, combined with extensive chromosomal abnormalities such as anaphase bridges, chromatid fusions, telomere loss, and, interestingly, telomere-sister exchanges (t-SCE). The t-SCE phenotypes results from sister telomere recombination after replication, and thus appears to be a process regulated by RTEL1 as well. It would be interesting to establish whether t-SCEs occur in other RTEL1-negative cells, and indeed in cells where the causative mutation is in a different locus, to explore whether this type of chromosomal abnormality is common to other DC cases. The two patient siblings P1 and P2 are compound heterozygotes for I669M and C1244R. The I669M substitution is located in the helicase catalytic core, itself located from residues 1–760, proximal to the ATP-binding domain. The C1244R substitution lies in the C-terminal RING domain of the protein. It will be of interest to study the exact biochemical impact of these mutations. The third patient examined possessed a V745M substitution in addition to an intronic mutation possibly affecting splicing. The mutant protein showed severely reduced expression in patient cells. Overall, RTEL1 is important for the control of recombination, DNA replication, and, perhaps, other important DNA repair activities,87 and its loss of function has a profound impact on telomere function, thereby causing HHS, a severe form of DC. The protein may have additional roles to the ones outlined here on telomere function, underlying deficiencies extending from a core set of symptoms described for other less penetrant DC-causing mutations.
CTC1
The CST complex was first identified in budding yeast as a protective activity required to prevent telomere deprotection.69 It is composed of three subunits, CDC13, STN1, and TEN1. All three subunits contain OB folds and the complex was found to bind single-stranded DNA, and in particular, the telomeric overhang. The complex has also been found in higher eukaryotes, first in plants (Arabidopsis thaliana88) and subsequently in mammals, through genetics and sequence homology, and has been discovered biochemically as an activity required for polymerase-α priming activity, and thus termed “AAF” for “alpha associated factor”.89 The complex has an additional activity in counteracting excessive telomerase elongation cycles during S phase.90 The characterization of a syndrome with severe DC-like symptoms has linked CTC1, the largest subunit (1217 residues, 132 kD), to the disease, implicating the CST telomeric complex in pathologies related to those previously described for telomere dysfunction.91 This syndrome, named Coats’ plus, is a rare disorder associated with retinopathy and intracranial calcifications, in addition to DC-like manifestations such as bone marrow failure, alopecia, and nail dystrophy.92 Initially, a search for possible causative mutations for Coats’ plus was performed by whole exome sequencing and revealed that, in nine of the ten unrelated Coats’ plus patients analyzed, biallelic CTC1 mutations were present and linked to the disease.91 Altogether, 14 mutations in CTC1 were observed to correlate with the syndrome, with four altering the first OB fold of the protein (residues 241–287), and three others (between residues 975 and 987) affecting a small region just upstream of the second OB fold of the protein. Leukocyte telomere length was analyzed by flow-FISH and found to be significantly shortened in patient cells in three cases where the parents’ DNA was available as controls. The single heterozygous individuals were found to possess shorter than average telomere length, but not so critically short as to result in the pathology. In two of three cases analyzed, a significant increase in the number of nuclear γH2AX foci was observed in patient cells, indicative of an ongoing DNA damage response in these cells. Thus, a possible telomere protection defect could be responsible for the disease at the cellular level, although more general defects in DNA replication could not be excluded. The correlation between CTC1 mutations and Coats’ plus was established in another independent study, further strengthening the notion that this syndrome is a severe case of DC linked to telomere dysfunction.93 In light of the degree of the clinical symptom overlap between Coats’ plus and severe forms of DC, in particular HHS, 73 patients, previously found to bear no obvious disease-causing mutations in known DC-associated genes, were analyzed for potential mutations in the CST coding genes. Of those, six patients were determined to be compound heterozygous for mutations in the CTC1 gene. No mutations were found so far at the STN1 or TEN1 loci. The mutations affected residues part of the first two OB folds (between positions 221 and 464) as well as a domain immediately N-terminal to the third OB fold (residues 1040–1110), between residues 944 and 987. Telomere length was not severely affected in these cases. The molecular etiology of Coats’ syndrome has been further clarified by an extensive study on the effect of disease-causing mutations in CTC1 on telomere function specifically, in human cultured cells.94 CTC1 mutations led to replication defects and accumulation of single-stranded DNA, in particular at telomeres, likely due to their nature as fragile replication sites. In addition, CTC1 defects led to decreased association with Polα-primase and reduced single-stranded DNA binding and telomere association in vivo. Interestingly, telomere length control was also disrupted in some cases, leading to extensive elongation, in sharp contrast to other DC-causing defects, which led to telomere shortening. Of note, no obvious telomere deprotection or DNA damage response was reported. These alleles all exhibit recessive transmission in families, but display dominant negative phenotypes when overexpressed in a telomerase-positive tumor human cell line. Thus, in at least some cases, CTC1 mutations may cause the pathology through deprotection and replication defects, which may result in inappropriate lengthening of telomeres, consistent with a role in preventing excessive elongation by telomerase.90 Of a panel of eleven disease-associated mutations analyzed, the most common feature was a defect in telomere replication with accumulation of single-stranded DNA.94 This defect could be due to poor interactions with Polα-primase, which is required for efficient lagging strand DNA replication through telomeres, or fill-in synthesis. Importantly, the telomere elongation phenotype in telomerase-positive cells could mask a role in telomere maintenance that could be important in human somatic telomerase-negative cells. Further studies in primary human cells should clarify this issue.
RAP1 and non-telomeric roles: regulation of metabolism and prevention of obesity
Repressor activator protein 1 (RAP1) is an evolutionarily conserved protein from yeast to humans and was first identified in Saccharomyces cerevisiae as a transcriptional regulator. Later, it was found to be a double-stranded telomere binding protein involved in telomere length regulation and in transcriptional silencing of subtelomeric genes through interaction with SIR3 and SIR4 proteins.
In human and mouse cells, RAP1 depends on the interaction with the double-stranded telomere binding protein TRF2 in order to be stable in the nucleus and interact with telomeres.18,95 RAP1 is involved in repression of HDR at telomeres, and hence, plays a role in telomere length maintenance.19 Apart from a telomeric role, RAP1 was shown to impact on subtelomeric gene expression in mice and, surprisingly, to suppress obesity in this organism.96 In human cells, RAP1 was shown to negatively regulate telomere length, presumably by acting in cis.97 Apart from a telomeric role, gene mapping has reveled that RAP1 binds to subtelomeric and extra-telomeric sites and can influence the expression of various genes involved in cell proliferation, metabolism, and neuronal processes in mice.98
A genome-wide gain-of-function screen led to the identification of RAP1 as a modulator of NF-κB signaling.99 Exogenous expression of RAP1 induced NF-κB signaling and RAP1 depletion led to inhibition of its activity.
So far, to our knowledge, no clear evidence of pathology linked to RAP1 was found in humans. However, the mouse phenotype is striking enough to be outlined here as a possible mechanism of disease in humans.
Obesity is a complex problem with adverse effect on health and decreased life expectancy. Excessive body weight and adipose tissue accumulation increase the risk of developing various age-related diseases, such as cardiovascular disease, Type 2 diabetes mellitus, respiratory diseases, and osteoarthritis and also cause predisposition to cancer. In some cases, heredity is responsible, but hormonal, metabolic, and behavioral factors are largely involved.
Studies in humans show that adiposity is directly related to decreased telomere length in leukocytes.100 Telomere length shortening has also been implicated in cellular aging. It was demonstrated that the activation of the DNA damage response due to shortening of telomeres can lead to repression of peroxisome proliferator-activated receptor gamma, coactivator 1 alpha and beta (PGC-1α and PGC-1β) genes.101 These genes are master regulators of mitochondrial physiology, energy homeostasis, lipid and carbohydrate metabolism, and inflammation. Thus a link exists between telomere dysfunction and altered metabolism during aging.
Previously, yeast RAP1 was shown to control the expression of glycolytic enzymes and ribosomal genes, however a role of mammalian RAP1 in metabolism was largely unknown. ChIP-sequence analysis showed that RAP1 was found to bind extra-telomeric sites and RAP1-null mice showed a significant change in gene expression of a variety of genes, as shown by quantitative PCR (qPCR) and microarray analysis.98 Recent studies have now highlighted the role of RAP1 as a transcriptional regulator of genes involved in metabolism. The RAP1 gene was deleted in the mouse and, in contrast to its interaction partner TRF2, was found to be nonessential.19 The lack of RAP1 at telomeres led to strongly increased HDR, resulting in altered telomere length and rampant sister telomere exchanges. RAP1 was not required for other established TRF2 functions, such as inhibition of ATM and restriction of NHEJ between chromosome ends.19 RAP1 was found to bind extra-telomeric sites and RAP1-null mice showed a significant change in gene expression by qPCR and microarray analysis.98,102 Two recent publications have demonstrated the role of RAP1 in cellular metabolism through PGC gene regulation. It was observed that RAP1-null mice showed deregulation of several pathways including cell adhesion and metabolism. A RAP1 mutant that was unable to interact with TRF2 also showed similar effect, confirming that gene regulation was independent of its interaction with TRF2. RAP1 deficiency in mice caused glucose intolerance and insulin resistance, indicating its role in diabetes onset.103 It was also found that RAP1 deficiency affects adipocyte differentiation and causes increased white fat accumulation, altered liver metabolism, and liver steatosis, thus leading to obesity. The effect of RAP1 deletion was more profound in female than male mice, suggesting a hormonal influence. RAP1 was found to be involved in transcriptional activation of the PPARα and PGC1α genes by binding to their promoter regions.102 Although this role for RAP1 is extratelomeric, it is possible that the pool of available protein for binding to telomeres through TRF2 or to promoters is tightly regulated, providing an indirect link between the two types of genomic sites.104 Thus, a telomeric protein is shown to influence metabolic activities, suggestive of an interplay between telomeric homeostasis and metabolism.
Conclusion
Because of the diversity of their roles and regulation, telomeres are active sites of the chromosome where many molecular complexes converge (Figure 1). Mutations in many loci are able to interfere with telomere function. These defects lead to two broad categories of pathologies, namely, cancer through upregulation of telomerase activity and several forms of DC often associated with telomere deprotection (Table 1). The molecular etiology of these pathologies was preceded by great and productive investments in the understanding of the fundamental processes underlying telomere function, both at the biochemical and genetic level, and focusing on a great variety of experimental systems. As a result, the conceptual and knowledge base was primed for the discovery of the loci associated with the related conditions, which has sustained tremendous progress in the past decade with the advent of genomics. We would insist that it is of utmost importance to keep such efforts going in order to understand causes and possible cures for diseases, as is currently happening for pathologies associated with abnormal telomere function.
Note added during revision
During the review of this manuscript, a study linking TPP1 to HHS was published [Kocak H et al (2014), Genes and Development 28:2090, PMID: 25233904], showing that mutations in the OB fold of TPP1 result in the disease. The proband examined displays symptoms of HHS as described here, and mutant cells exhibit short telomere length due to a defect in telomerase recruitment and processivity. Thus, TPP1 represent the second shelterin component, after TIN2, implicated in DC/HHS in humans.
Acknowledgments
The DL laboratory was supported by an SC3 award # 1SC3GM094071-01A1 from the NIGMS. The authors thank Wan Shan Fowler and the DL laboratory for help, advice, and comments on the manuscript, as well as Dr N Ismaili and Dr C Hedvat (NYU Medical Center) for comments and suggestions.
Disclosure
The authors declare no conflicts of interest in this work.
References
Blackburn EH. A history of telomere biology. In: de Lange T, Lundblad V, Blackburn EH, editors. Telomeres. 2nd ed. Huntington, NY: Cold Spring Harbor Laboratory Press; 2006:1–19. | |
Lundblad V. Budding yeast telomeres. In: de Lange T, Lundblad V, Blackburn EH, editors. Telomeres. 2nd ed. Huntington, NY: Cold Spring Harbor Laboratory Press; 2006:345–386. | |
de Lange T. Lasker laurels for telomerase. Cell. 2006;126(6):1017–1020. | |
Palm W, de Lange T. How shelterin protects mammalian telomeres. Annu Rev Genet. 2008;42:301–334. | |
Smogorzewska A, de Lange T. Regulation of telomerase by telomeric proteins. Annu Rev Biochem. 2004;73:177–208. | |
Cristofari G, Lingner J. The telomerase ribonucleoprotein particle. In: de Lange T, Lundblad V, Blackburn EH, editors. Telomeres. 2nd ed. Huntington, NY: Cold Spring Harbor Laboratory Press; 2006:21–47. | |
Griffith JD, Comeau L, Rosenfield S, et al. Mammalian telomeres end in a large duplex loop. Cell. 1999;97(4):503–514. | |
de Lange T. How telomeres solve the end-replication problem. Science. 2009;326:948. | |
de Lange T. Shelterin: the protein complex that shapes and safeguards human telomeres. Genes Dev. 2005;19(18):2100–2110. | |
Chong L, van Steensel B, Broccoli D, et al. A human telomeric protein. Science. 1995;270(5242):1663–1667. | |
Broccoli D, Smogorzewska A, Chong L, de Lange T. Human telomeres contain two distinct Myb-related proteins, TRF1 and TRF2. Nat Genet. 1997;17(2):231–235. | |
Ye JZ, Donigian JR, van Overbeek M, et al. TIN2 binds TRF1 and TRF2 simultaneously and stabilizes the TRF2 complex on telomeres. J Biol Chem. 2004;279(45):47264–47271. | |
Ye JZ, Hockemeyer D, Krutchinsky AN, et al. POT1-interacting protein PIP1: a telomere length regulator that recruits POT1 to the TIN2/TRF1 complex. Genes Dev. 2004;18(14):1649–1654. | |
Loayza D, de Lange T. POT1 as a terminal transducer of TRF1 telomere length control. Nature. 2003;424(6943):1013–1018. | |
Lazzerini Denchi E, de Lange T. Protection of telomeres through independent control of ATM and ATR by TRF2 and POT1. Nature. 2007;448:1068–1071. | |
Loayza D, Parsons H, Donigian J, Hoke K, de Lange T. DNA binding features of human POT1: a nonamer 5′-TAGGGTTAG-3′ minimal binding site, sequence specificity, and internal binding to multimeric sites. J Biol Chem. 2004;279(13):13241–13248. | |
Lei M, Podell ER, Cech TR. Structure of human POT1 bound to telomeric single-stranded DNA provides a model for chromosome end-protection. Nat Struct Mol Biol. 2004;11(12):1223–1229. | |
Li B, Oestreich S, de Lange T. Identification of human Rap1: implications for telomere evolution. Cell. 2000;101(5):471–483. | |
Sfeir A, Kabir S, van Overbeek M, Celli GB, de Lange T. Loss of Rap1 induces telomere recombination in the absence of NHEJ or a DNA damage signal. Science. 2010;327(5973):1657–1661. | |
Karlseder J, Hoke K, Mirzoeva OK, et al. The telomeric protein TRF2 binds the ATM kinase and can inhibit the ATM-dependent DNA damage response. PLoS Biol. 2004;2(8):E240. | |
van Steensel B, Smogorzewska A, de Lange T. TRF2 protects human telomeres from end-to-end fusions. Cell. 1998;92(3):401–413. | |
Karlseder J, Broccoli D, Dai Y, Hardy S, de Lange T. p53- and ATM-dependent apoptosis induced by telomeres lacking TRF2. Science. 1999;283(5406):1321–1325. | |
Shay JW, Wright WE. Telomeres and telomerase in normal and cancer stem cells. FEBS Lett. 2010;584(17):3819–3825. | |
Sedivy JM. Telomeres limit cancer growth by inducing senescence: long-sought in vivo evidence obtained. Cancer Cell. 2007;11(5):389–391. | |
Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–674. | |
Weinberg RA. Telomeres: bumps on the road to immortality. Nature. 1998;396(6706):23–24. | |
Shay JW, Wright WE. Telomerase and human cancer. In: de Lange T, Lundblad V, Blackburn EH, editors. Telomeres. 2nd ed. Huntington, NY: Cold Spring Harbor Laboratory Press; 2006:81–108. | |
Dunham MA, Neumann AA, Fasching CL, Reddel RR. Telomere maintenance by recombination in human cells. Nat Genet. 2000;26:447–450. | |
Lovejoy CA, Li W, Reisenweber S, Thongthip S, et al; ALT Starr Cancer Consortium. Loss of ATRX, genome instability, and an altered DNA damage response are hallmarks of the alternative lengthening of telomeres pathway. PLoS Genet. 2012;8(7):e1002772. | |
Hahn WC, Counter CM, Lundberg AS, Beijersbergen RL, Brooks MW, Weinberg RA. Creation of human tumour cells with defined genetic elements. Nature. 1999;400(6743):448–464. | |
Bodnar AG, Ouellette M, Frolkis M, et al. Extension of life-span by introduction of telomerase into normal human cells. Science. 1998; 279(5349):349–352. | |
Jiang XR, Jimenez G, Chang E, et al. Telomerase expression in human somatic cells does not induce changes associated with a transformed phenotype. Nat Genet. 1999;21(1):111–114. | |
Morales CP, Holt SE, Ouellette M, et al. Absence of cancer-associated changes in human fibroblasts immortalized with telomerase. Nat Genet. 1999;21(1):115–118. | |
Marcand S, Gilson E, Shore D. A protein-counting mechanism for telomere length regulation in yeast. Science. 1997;275(5302):986. | |
van Steensel B, de Lange T. Control of telomere length by the human telomeric protein TRF1. Nature. 1997;385(6618):740–743. | |
Zhong FL, Batista LF, Freund A, Pech MF, Venteicher AS, Artandi SE. TPP1 OB-fold domain controls telomere maintenance by recruiting telomerase to chromosome ends. Cell. 2012;150(3):481–494. | |
Noel JF, Wellinger RJ. Exposing secrets of telomere-telomerase encounters. Cell. 2012;150(3):453–454. | |
Takai KK, Hooper S, Blackwood S, Gandhi R, de Lange T. In vivo stoichiometry of shelterin components. J Biol Chem. 2010;285(2):1457–1467. | |
Ciccia A, Elledge SJ. The DNA damage response: making it safe to play with knives. Mol Cell. 2010;40(2):179–204. | |
de Lange T. Protection of mammalian telomeres. Oncogene. 2002;21(4):532–540. | |
Smogorzewska A, Karlseder J, Holtgreve-Grez H, Jauch A, de Lange T. DNA ligase IV-dependent NHEJ of deprotected mammalian telomeres in G1 and G2. Curr Biol. 2002;12(19):1635. | |
Smogorzewska A, de Lange T. Different telomere damage signaling pathways in human and mouse cells. EMBO J. 2002;21(16):4338–4348. | |
Zeman MK, Cimprich KA. Causes and consequences of replication stress. Nat Cell Biol. 2014;16(1):2–9. | |
Palm W, Hockemeyer D, Kibe T, de Lange T. Functional dissection of human and mouse POT1 proteins. Mol Cell Biol. 2009;29(2):471–482. | |
Sfeir A, de Lange T. Removal of shelterin reveals the telomere end-protection problem. Science. 2012;336(6081):593–597. | |
Reddel RR. Alternative lengthening of telomeres, telomerase, and cancer. Cancer Lett. 2003;194(2):155–162. | |
Horn S, Figl A, Rachakonda PS, et al. TERT promoter mutations in familial and sporadic melanoma. Science. 2013;339(6122):959–961. | |
Huang FW, Hodis E, Xu MJ, Kryukov GV, Chin L, Garraway LA. Highly recurrent TERT promoter mutations in human melanoma. Science. 2013;339(6122):957–959. | |
Patton EE, Harrington L. Cancer: trouble upstream. Nature. 2013; 495(7441):320–321. | |
Chang S. Cancer chromosomes going to POT1. Nat Genet. 2013;45(5):473–475. | |
Ramsay AJ, Quesada V, Foronda M, et al. POT1 mutations cause telomere dysfunction in chronic lymphocytic leukemia. Nat Genet. 2013;45(5):526–530. | |
Speedy HE, Di Bernardo MC, Sava GP, et al. A genome-wide association study identifies multiple susceptibility loci for chronic lymphocytic leukemia. Nat Genet. 2014;46(1):56–60. | |
Robles-Espinoza CD, Harland M, Ramsay AJ, et al. POT1 loss-of-function variants predispose to familial melanoma. Nat Genet. 2014;46(5):478–481. | |
Shi J, Yang XR, Ballew B, et al; NCI DCEG Cancer Sequencing Working Group, NCI DCEG Cancer Genomics Research Laboratory, French Familial Melanoma Study Group. Rare missense variants in POT1 predispose to familial cutaneous malignant melanoma. Nat Genet. 2014;46(5):482–486. | |
Kirwan M, Dokal I. Dyskeratosis congenita, stem cells and telomeres. Biochim Biophys Acta. 2009;1792(4):371–379. | |
Nelson ND, Bertuch AA. Dyskeratosis congenita as a disorder of telomere maintenance. Mutat Res. 2012;730(1–2):43–51. | |
Touzot F, Callebaut I, Soulier J, et al. Function of Apollo (SNM1B) at telomere highlighted by a splice variant identified in a patient with Hoyeraal-Hreidarsson syndrome. Proc Natl Acad Sci U S A. 2010;107:10097–10102. | |
Shtessel L, Ahmed S. Telomere dysfunction in human bone marrow failure syndromes. Nucleus. 2011;2(1):24–29. | |
Mitchell JR, Wood E, Collins K. A telomerase component is defective in the human disease dyskeratosis congenita. Nature. 1999;402(6761):551–555. | |
Heiss NS, Knight SW, Vulliamy TJ, et al. X-linked dyskeratosis congenita is caused by mutations in a highly conserved gene with putative nucleolar functions. Nat Genet. 1998;19(1):32–38. | |
Zhong F, Savage SA, Shkreli M, et al. Disruption of telomerase trafficking by TCAB1 mutation causes dyskeratosis congenita. Genes Dev. 2011;25(1):11–16. | |
Walne AJ, Vulliamy T, Beswick R, Kirwan M, Dokal I. TINF2 mutations result in very short telomeres: analysis of a large cohort of patients with dyskeratosis congenita and related bone marrow failure syndromes. Blood. 2008;112(9):3594–3600. | |
Sasa GS, Ribes-Zamora A, Nelson ND, Bertuch AA. Three novel truncating TINF2 mutations causing severe dyskeratosis congenita in early childhood. Clin Genet. 2012;81(5):470–478. | |
Frescas D, de Lange TA. TIN2 dyskeratosis congenita mutation causes telomerase-independent telomere shortening in mice. Genes Dev. 2014;28(2):153–166. | |
Hockemeyer D, Palm W, Wang RC, Couto SS, de Lange T. Engineered telomere degradation models dyskeratosis congenita. Genes Dev. 2008;22(13):1773–1785. | |
Hockemeyer D, Daniels JP, Takai H, de Lange T. Recent expansion of the telomeric complex in rodents: two distinct POT1 proteins protect mouse telomeres. Cell. 2006;126(1):63–77. | |
Hockemeyer D, Sfeir AJ, Shay JW, Wright WE, de Lange T. POT1 protects telomeres from a transient DNA damage response and determines how human chromosomes end. EMBO J. 2005;24:2667–2678. | |
Diotti R, Loayza D. Shelterin complex and associated factors at human telomeres. Nucleus. 2011;2:1–17. | |
Price CM, Boltz KA, Chaiken MF, Stewart JA, Beilstein MA, Shippen DE. Evolution of CST function in telomere maintenance. Cell Cycle. 2010;9(16):3157–3165. | |
Lenain C, Bauwens S, Amiard S, Brunori M, Giraud-Panis MJ, Gilson E. The Apollo 5′ exonuclease functions together with TRF2 to protect telomeres from DNA repair. Curr Biol. 2006;16(13):1303–1310. | |
van Overbeek M, de Lange T. Apollo, an Artemis-related nuclease, interacts with TRF2 and protects human telomeres in S phase. Curr Biol. 2006;16(13):1295–1302. | |
Bae JB, Mukhopadhyay SS, Liu L, et al. Snm1B/Apollo mediates replication fork collapse and S Phase checkpoint activation in response to DNA interstrand cross-links. Oncogene. 2008;27(37):5045–5056. | |
Lam YC, Akhter S, Gu P, et al. SNMIB/Apollo protects leading-strand telomeres against NHEJ-mediated repair. EMBO J. 2010;29:2230–2241. | |
Wu P, van Overbeek M, Rooney S, de Lange T. Apollo contributes to G overhang maintenance and protects leading-end telomeres. Mol Cell. 2010;39(4):606–617. | |
Ye J, Lenain C, Bauwens S, et al. TRF2 and Apollo cooperate with topoisomerase 2alpha to protect human telomeres from replicative damage. Cell. 2010;142(2):230–242. | |
Sarthy JF, Baumann P. Apollo-taking the lead in telomere protection. Mol Cell. 2010;39(4):489–491. | |
Vannier JB, Sarek G, Boulton SJ. RTEL1: functions of a disease-associated helicase. Trends Cell Biol. 2014;24(7):416–425. | |
Faure G, Revy P, Schertzer M, Londono-Vallejo A, Callebaut I. The C-terminal extension of human RTEL1, mutated in Hoyeraal-Hreidarsson syndrome, contains harmonin-N-like domains. Proteins. 2014;82(6):897–903. | |
Barber LJ, Youds JL, Ward JD, et al. RTEL1 maintains genomic stability by suppressing homologous recombination. Cell. 2008;135(2):261–271. | |
Zhu L, Hathcock KS, Hande P, Lansdorp PM, Seldin MF, Hodes RJ. Telomere length regulation in mice is linked to a novel chromosome locus. Proc Natl Acad Sci U S A. 1998;95(15):8648–8653. | |
Ding H, Schertzer M, Wu X, et al. Regulation of murine telomere length by Rtel: an essential gene encoding a helicase-like protein. Cell. 2004;117(7):873–886. | |
Cheung I, Schertzer M, Rose A, Lansdorp PM. Disruption of dog-1 in Caenorhabditis elegans triggers deletions upstream of guanine-rich DNA. Nat Genet. 2002;31(4):405–409. | |
Vannier JB, Pavicic-Kaltenbrunner V, Petalcorin MI, Ding H, Boulton SJ. RTEL1 dismantles T loops and counteracts telomeric G4-DNA to maintain telomere integrity. Cell. 2012;149(4):795–806. | |
Lamm N, Ordan E, Shponkin R, Richler C, Aker M, Tzfati Y. Diminished telomeric 3′ overhangs are associated with telomere dysfunction in Hoyeraal-Hreidarsson syndrome. PLoS One. 2009; 4(5):e5666. | |
Deng Z, Glousker G, Molczan A, et al. Inherited mutations in the helicase RTEL1 cause telomere dysfunction and Hoyeraal-Hreidarsson syndrome. Proc Natl Acad Sci U S A. 2013;110(36):E3408–E3416. | |
Le Guen T, Jullien L, Touzot F, et al. Human RTEL1 deficiency causes Hoyeraal-Hreidarsson syndrome with short telomeres and genome instability. Hum Mol Genet. 2013;22(16):3239–3249. | |
Uringa EJ, Youds JL, Lisaingo K, Lansdorp PM, Boulton SJ. RTEL1: an essential helicase for telomere maintenance and the regulation of homologous recombination. Nucleic Acids Res. 2010;39:1647–1655. | |
Surovtseva YV, Churikov D, Boltz KA, et al. Conserved telomere maintenance component 1 interacts with STN1 and maintains chromosome ends in higher eukaryotes. Mol Cell. 2009;36(2):207–218. | |
Casteel DE, Zhuang S, Zeng Y, et al. A DNA polymerase-α primase cofactor with homology to replication protein A-32 regulates DNA replication in mammalian cells. J Biol Chem. 2009;284(9):5807–5818. | |
Chen LY, Redon S, Lingner J. The human CST complex is a terminator of telomerase activity. Nature. 2012;488(7412):540–544. | |
Anderson BH, Kasher PR, Mayer J, et al. Mutations in CTC1, encoding conserved telomere maintenance component 1, cause coats plus. Nat Genet. 2012;44(3):338–342. | |
Savage SA. Connecting complex disorders through biology. Nat Genet. 2012;44(3):238–240. | |
Walne AJ, Bhagat T, Kirwan M, et al. Mutations in the telomere capping complex in bone marrow failure and related syndromes. Haematologica. 2013;98(3):334–338. | |
Chen LY, Majerska J, Lingner J. Molecular basis of telomere syndrome caused by CTC1 mutations. Genes Dev. 2013;27(19):2099–2108. | |
Celli GB, de Lange T. DNA processing is not required for ATM-mediated telomere damage response after TRF2 deletion. Nat Cell Biol. 2005;7(7):712–718. | |
Duong MT, Sahin E. RAP1: protector of telomeres, defender against obesity. Cell Rep. 2013;3(6):1757–1758. | |
Li B, de Lange T. Rap1 affects the length and heterogeneity of human telomeres. Mol Biol Cell. 2003;14(12):5060–5068. | |
Martinez P, Thanasoula M, Carlos AR, et al. Mammalian Rap1 controls telomere function and gene expression through binding to telomeric and extratelomeric sites. Nat Cell Biol. 2010;12(8):768–780. | |
Teo H, Ghosh S, Luesch H, et al. Telomere-independent Rap1 is an IKK adaptor and regulates NF-kappaB-dependent gene expression. Nat Cell Biol. 2010;12(8):758–767. | |
Lee M, Martin H, Firpo MA, Demerath EW. Inverse association between adiposity and telomere length: the Fels longitudinal study. Am J Hum Biol. 2011;23(1):100–106. | |
Sahin E, Colla S, Liesa M, et al. Telomere dysfunction induces metabolic and mitochondrial compromise. Nature. 2011;470(7334):359–365. | |
Martínez P, Gómez-López G, García F, et al. RAP1 protects from obesity through its extratelomeric role regulating gene expression. Cell Rep. 2013;3(6):2059–2074. | |
Yeung F, Ramírez CM, Mateos-Gomez PA, et al. Nontelomeric role for Rap1 in regulating metabolism and protecting against obesity. Cell Rep. 2013;3(6):1847–1856. | |
Crabbe L, Karlseder J. Mammalian Rap1 widens its impact. Nat Cell Biol. 2010;12(8):733–735. | |
Chen JL, Blasco MA, Greider CW. Secondary structure of vertebrate telomerase RNA. Cell. 2000;100(5):503–514. | |
Armanios M, Chen JL, Chang YP, et al. Haploinsufficiency of telomerase reverse transcriptase leads to anticipation in autosomal dominant dyskeratosis congenita. Proc Natl Acad Sci U S A. 2005;102(44):15960–15964. | |
Vulliamy T, Marrone A, Goldman F, et al. The RNA component of telomerase is mutated in autosomal dominant dyskeratosis congenita. Nature. 2001;413(6854):432–435. | |
Savage SA, Giri N, Baerlocher GM, Orr N, Lansdorp PM, Alter BP. TINF2, a component of the shelterin telomere protection complex, is mutated in dyskeratosis congenita. Am J Hum Genet. 2008;82(2):501–509. | |
Walne AJ, Vulliamy T, Marrone A, et al. Genetic heterogeneity in autosomal recessive dyskeratosis congenita with one subtype due to mutations in the telomerase-associated protein NOP10. Hum Mol Genet. 2007;16(13):1619–1629. | |
Vulliamy T, Beswick R, Kirwan M, et al. Mutations in the telomerase component NHP2 cause the premature ageing syndrome dyskeratosis congenita. Proc Natl Acad Sci U S A. 2008;105(23):8073–8078. | |
Kim SH, Kaminker P, Campisi J. TIN2, a new regulator of telomere length in human cells. Nat Genet. 1999;23(4):405–412. | |
Houghtaling BR, Cuttonaro L, Chang W, Smith S. A dynamic molecular link between the telomere length regulator TRF1 and the chromosome end protector TRF2. Curr Biol. 2004;14(18):1621–1631. | |
Liu D, Safari A, O’Connor MS, et al. PTOP interacts with POT1 and regulates its localization to telomeres. Nat Cell Biol. 2004;6:673–680. | |
Baumann P, Cech TR. Pot1, the putative telomere end-binding protein in fission yeast and humans. Science. 2001;292(5519):1171–1175. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.