Back to Journals » Vascular Health and Risk Management » Volume 10
Serum uric acid and target organ damage in essential hypertension
Received 26 January 2014
Accepted for publication 27 February 2014
Published 2 May 2014 Volume 2014:10 Pages 253—261
DOI https://doi.org/10.2147/VHRM.S61363
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Sandra N Ofori, Osaretin J Odia
Department of Internal Medicine, University of Port Harcourt Teaching Hospital, Port Harcourt, Rivers State, Nigeria
Background: Hypertension is a major risk factor for cardiovascular mortality, as it acts through its effects on target organs, such as the heart and kidneys. Hyperuricemia increases cardiovascular risk in patients with hypertension.
Objective: To assess the relationship between serum uric acid and target organ damage (left ventricular hypertrophy and microalbuminuria) in untreated patients with essential hypertension.
Patients and methods: A cross-sectional study was carried out in 130 (85 females, 45 males) newly diagnosed, untreated patients with essential hypertension. Sixty-five healthy age- and sex-matched non-hypertensive individuals served as controls for comparison. Left ventricular hypertrophy was evaluated by cardiac ultrasound scan, and microalbuminuria was assessed in an early morning midstream urine sample by immunoturbidimetry. Blood samples were collected for assessing uric acid levels.
Results: Mean serum uric acid was significantly higher among the patients with hypertension (379.7±109.2 µmol/L) than in the controls (296.9±89.8 µmol/L; P<0.001), and the prevalence of hyperuricemia was 46.9% among the hypertensive patients and 16.9% among the controls (P<0.001). Among the hypertensive patients, microalbuminuria was present in 54.1% of those with hyperuricemia and in 24.6% of those with normal uric acid levels (P=0.001). Similarly, left ventricular hypertrophy was more common in the hypertensive patients with hyperuricemia (70.5% versus 42.0%, respectively; P=0.001). There was a significant linear relationship between mean uric acid levels and the number of target organ damage (none versus one versus two: P=0.012).
Conclusion: These results indicate that serum uric acid is associated with target organ damage in patients with hypertension, even at the time of diagnosis; thus, it is a reliable marker of cardiovascular damage in our patient population.
Keywords: essential hypertension, serum uric acid, left ventricular hypertrophy, microalbuminuria
Introduction
Hypertension is an important cardiovascular problem worldwide.1 Its prevalence is increasing in developing countries.2 Hypertension has adverse effects on various target organs, thus increasing the risk of stroke, coronary heart disease, and heart failure.3 This leads to high morbidity and mortality in many cases. Elevated blood pressure (BP) is also associated with an accelerated rate of decline in cognitive and renal function.3
In evaluating a patient with hypertension, it is recommended that target organ damage (TOD) be identified in order to better define the individual’s global cardiovascular risk in an effort to guide the decision to begin treatment, and to determine target BP levels.2,3 In less developed parts of the world, the diagnosis of hypertension is often delayed and, as a result, TOD may be present at the time of diagnosis.4 Uric acid (UA) levels tend to be elevated in patients with hypertension.5 Elevated UA is a risk factor for the development of cardiovascular disease, and the European Society of Hypertension–European Society of Cardiology guidelines recommend performing routine laboratory testing for serum UA (SUA) in patients with hypertension.5,6 While it has been shown that SUA is associated with multiple cardiovascular risk factors, including hypertension, metabolic syndrome, diabetes, and renal disease, it remains to be known if UA independently predicts adverse cardiovascular events in patients with hypertension. Various large trials have failed to identify UA as a significant and independent risk factor.7,8
Identifying patients with subclinical TOD provides the best opportunity for preventing progression to overt cardiovascular disease. In clinical practice in limited-resource settings, it may not be practical to screen every newly diagnosed hypertensive individual for subclinical TOD due to limited access to facilities for echocardiography and microalbuminuria; therefore, identification of a relatively inexpensive risk marker, like UA, may help to identify patients who are at higher risk, thereby directing further evaluation.
This study was thus aimed at investigating the relationship between SUA and left ventricular (LV) hypertrophy and microalbuminuria in newly diagnosed, untreated patients with essential hypertension.
Patients and methods
Study design
This was a descriptive cross-sectional study carried out in the medical outpatient clinics of the University of Port Harcourt Teaching Hospital, Port Harcourt, Rivers State, Nigeria.
Study population
A total of 130 consecutive, never-treated, newly diagnosed hypertensive subjects aged between 30–70 years of age were included in the study. The study population consisted of an ethnically diverse group of black Nigerians from Rivers State, and the neighboring south–south states of the Niger Delta, involved in the various economic activities in the area, mainly in the oil and gas sector.
The subjects were characterized by: 1) average clinic systolic BP (SBP) values >140 mmHg or diastolic BP (DBP) >90 mmHg, confirmed during two visits at the outpatient clinic; 2) no history or clinical evidence of congestive heart failure, myocardial infarction, arrhythmias, cardiac valve disease, coronary bypass surgery or angioplasty, diabetes mellitus and renal insufficiency, and no treatment with urate-lowering medication; and 3) no clinical evidence of secondary hypertension.
After informed written consent had been obtained, all patients returned to the clinic after an overnight fast and underwent the following procedures: 1) clinic BP measurement; 2) venous blood sample collection for SUA; 3) spot midstream urine collection for microalbuminuria (patients with proteinuria on dipstick testing were excluded); 4) transthoracic echocardiogram; and 5) routine investigations (serum creatinine, fasting lipid profile, and plasma glucose).
Sixty-five non-hypertensive, healthy age- and sex-matched individuals were randomly selected from the hospital staff and patients’ relatives and were classified as controls.
The Ethics Committee of the University of Port Harcourt Teaching Hospital approved the study protocol.
Uric acid
UA was analyzed with the enzymatic colorimetric method using an autoanalyzer. Normal values in the hospital laboratory are <360 μmol/L and <420 μmol/L for women and men, respectively; therefore, individuals who had values above these levels were classified as having hyperuricemia.
Fasting lipid profile
Fasting cholesterol and triglyceride (TG) levels were measured using the enzymatic method with a reagent from Atlas Medical Laboratories (Atlas Development Corporation, Calabasas, CA, USA). Fasting high-density lipoprotein (HDL) was measured with the precipitation method. Low-density lipoprotein (LDL) cholesterol values were calculated using the Friedewald equation when the TG level was <4.0 mmol/L:9

Definition of an abnormal lipid profile:10
- Elevated TG = TG >1.7 mmol/L.
- Hypercholesterolemia = TC >5.2 mmol/L.
- Low HDL cholesterol = HDL cholesterol <1.03 mmol/L.
- Elevated LDL cholesterol = LDL cholesterol >3.0 mmol/L.
Microalbuminuria
Randox immunoturbidimetric assay for urinary albumin (Randox Laboratories, Antrim, UK) was used to determine microalbuminuria in an early morning spot midstream urine sample. Microalbuminuria was defined as an albumin concentration between 20–200 mg/L. This is equivalent to albumin excretion of 30–300 mg/24 hours (in a urine sample collected over 24 hours).11
Blood pressure measurements
BP was measured with a standard (Accoson; AC Cossor and Son [Surgical] Ltd, Essex, UK) mercury sphygmomanometer (with an appropriate cuff size) on the patients’ right arm, as the patients were in the seated position with their feet on the floor after a 5-minute rest. SBP and DBP were taken at Korotkoff phases 1 and 5, respectively to the nearest 2 mmHg. The average of two BP measurements taken 5 minutes apart was used. The presence and severity of hypertension were then defined based on the seventh Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High BP guidelines.1
Echocardiography
Echocardiographic studies for all the patients were carried out with an Aloka Prosound SSD 4000 echocardiography machine (Fair Medical Company Ltd, Matsudo, Japan) equipped with a 2.5 Hz transducer. With the patient in the left lateral decubitus position, targeted echocardiographic estimations were taken. These included the standard two-dimensional oriented motion–mode measurements of interventricular septal thickness in diastole, LV posterior wall thickness in diastole, and LV end diastolic diameter just beyond the tips of the mitral valve leaflets. LV mass (in grams) was automatically calculated with the internal software of the machine.
LV mass was indexed to the body surface area using cut-off values of 134 g/m2 and 110 g/m2 for men and women, respectively.12
Relative wall thickness (RWT) was calculated as 2× posterior wall thickness in end diastole/LV end diastolic diameter. A partition value of 0.45 for RWT was used for both men and women.13
Patients with increased LV mass index (LVMI) and increased RWT were considered to have concentric hypertrophy, and those with increased LVMI and normal RWT were considered to have eccentric hypertrophy. Those with normal LVMI and increased or normal RWT were considered to have concentric remodeling or normal geometry, respectively.
Definition of target organ damage
TOD was defined by the presence of microalbuminuria (urinary albumin excretion: 20–200 mg/L) or echocardiographic evidence of LV hypertrophy (LVH) defined as LVMI ≥134 g/m2 in men and ≥110 g/m2 in women.
Definition of degree of target organ damage
The degree was determined by the number of target organs involved (ie, 0= no TOD; 1 TOD = presence of either LVH or microalbuminuria; 2 TOD = presence of both LVH and microalbuminuria).14
Statistical analysis
All data were analyzed using the commercially available Statistical Package for the Social Sciences (SPSS) version 17.0 analytic software (IBM Corporation, Armonk, NY, USA). Data were expressed as mean ± standard deviations, and frequencies as a percentage. Continuous variables were compared with the Students t-test, or one-way analysis of variance, as considered appropriate. Proportions or categorical parameters were compared with the chi-square test. Relations among continuous variables were assessed using Pearson’s correlation coefficient and multiple linear regression analysis. All tests were considered to be statistically significant at the P-value ≤0.05.
Results
There were 130 patients with a female-to-male ratio of 1.9:1. The age range was 31–70 years, with a mean age of 46.8±9.3 years. The age range of the controls was 31–70 years with a mean age of 44.4±9.8 years. There was no statistically significant difference in the mean ages of the cases and controls (P=0.094). The mean body mass index of the cases was 29.0±5.0 kg/m2, which was significantly higher than that of the controls (26.6±3.3 kg/m2; P=0.001). The baseline clinical and laboratory characteristics of the study population are shown in Table 1.
 | Table 1 Clinical and laboratory characteristics of the study population |
LVH was present in 55.4% of the cases and in 10.8% of the controls (P<0.001), and the most common geometric pattern among the cases was concentric hypertrophy, while the majority of the controls had normal LV geometry. The prevalence of microalbuminuria among the cases was 37.7%, while it was 7.7% in the controls (P<0.001). Overall, 67% of the cases had at least one TOD, and females were more affected than males (P=0.045).
The mean SUA was significantly higher in the cases (379.7±109.2 μmol/L) than in the controls (296.9±89.8 μmol/L; P<0.001), and the prevalence of hyperuricemia was 46.9% among the cases and 16.9% among the controls (P<0.001). The mean SUA increased significantly with the severity of hypertension (Figure 1).
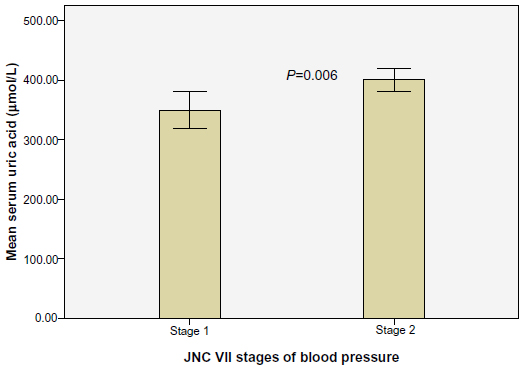 | Figure 1 Relationship between SUA and severity of hypertension. |
Microalbuminuria was present in 54.1% of the hypertensive cases with hyperuricemia, and in 24.6% of the cases with normal UA levels (P=0.001). Similarly LVH was more common among the hypertensive cases with hyperuricemia compared to the hypertensive cases with normal serum uric acid levels (70.5% versus 42.0% respectively, P=0.001). Among the eleven (16.9%) controls that had hyperuricemia, two had LVH and none had microalbuminuria. Table 2 shows the clinical and laboratory characteristics of the 130 cases with and without elevated SUA.
 | Table 2 Anthropometric, clinical, and laboratory characteristics of the 130 hypertensive cases with and without elevated SUA |
SUA correlated positively with microalbuminuria among the cases (r=0.529; P<0.001), suggesting that in those with microalbuminuria, urine albumin excretion increased with increasing levels of SUA (Figure 2). This correlation remained significant even after controlling for the following variables: age (r=0.535; P<0.001); body mass index (BMI) (r=0.476; P=0.001); waist circumference (WC) (r=0.489; P<0.001); SBP (r=0.522; P<0.001); and DBP (r=0.537; P<0.001), respectively. The correlation was not significant among the controls (r=0.085; P=0.178). With multiple linear regression analysis, SUA was independently associated with microalbuminuria after adjusting for confounding variables including age, BMI, WC, SBP and DBP. The variables combined explained 31.1% of the variance observed (F=3.154; P=0.012). SUA made a statistically significant and unique contribution to the variance in microalbuminuria (β=0.517; P=0.001).
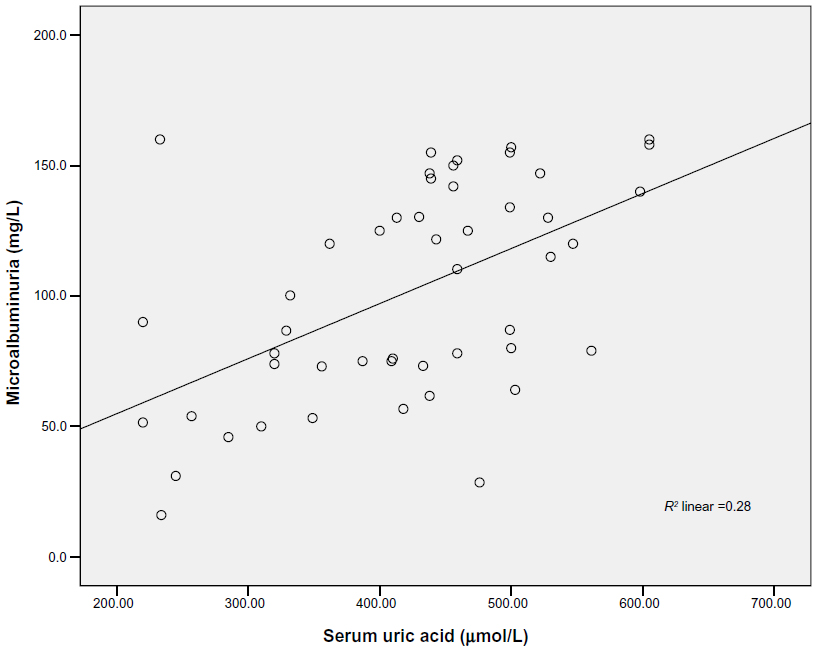 | Figure 2 Correlation between serum uric acid and microalbuminuria among the cases. |
While the correlation between LVMI and SUA among the controls was not significant (r=0.169; P=0.500), it was positive and significant among the cases (r=0.336; P<0.001), with high levels of SUA associated with higher indexed LV mass. The correlation remained significant after controlling for age (r=0.332; P<0.001); BMI (r=0.303; P<0.001); WC (r=0.287; P<0.001); SBP (r=0.308; P<0.001); and DBP (r=0.328; P<0.001). SUA levels were also independently associated with LVMI in regression analysis after adjusting for confounders including age, BMI, WC, SBP, and DBP. The variables combined explained 25.4% of the variance observed in LVMI (P<0.001). SBP made the largest unique contribution (P=0.001), although SUA also made a statistically significant contribution (P=0.003).
There was a significant positive correlation between SUA level and the number of target organs involved (r=0.255; P=0.017). This suggests that the number of TOD increased with increasing levels of SUA. Figure 3 shows that the mean SUA level was higher in the cases with both microalbuminuria and LVH (419.2±135.9 μmol/L), than in those with either of these indices of TOD alone (381.1±93.7 μmol/L), or in those with none (345.8±92.2 μmol/L). This was statistically significant (P=0.012).
 | Figure 3 Mean SUA levels analyzed based on the degree of TOD involvement. |
On further analysis, UA correlated significantly with other cardiovascular risk factors, as shown in Table 3.
 | Table 3 Pearson’s correlation coefficients of SUA and clinical variables |
Discussion
The main findings in this study were that hyperuricemia was more prevalent among hypertensive individuals than among the normotensive controls, and elevated SUA in the patients with hypertension was accompanied by increased urinary albumin concentrations and increased LVMI. This was similar to the findings by Poudel et al,15 who conducted a cross-sectional study of untreated hypertensive individuals in Nepal. It was found that hyperuricemia was more common among hypertensive patients when compared to the normotensive controls (28.8% versus 13.7%, respectively; P<0.001).15 However, this prevalence was lower than the 46.9% rate found in the present study. This difference may be attributed to the higher BMI and SBP of the patients in this study. A retrospective study of 351 patients with essential hypertension in Northern Nigeria also concluded that hyperuricemia was more common among hypertensive patients compared to normotensive individuals.16 In that population, there was a significant positive correlation between SUA and SBP and DBP. This is in contrast to our study where even though stage 2 (compared to stage 1) hypertension was associated with higher mean SUA, there was no significant correlation between SUA and SBP or DBP, respectively.
Another important finding in the present study was that hypertensive patients who had elevated UA levels had evidence of an increasing number of target organ involvement than those with UA levels in the normal range. Moreover, UA correlated significantly with other cardiovascular risk factors such as increased BMI, increased WC (which is a measure of visceral adiposity), and hypertriglyceridemia. This has also been shown in other studies.17–19 In an interesting commentary published in the Journal of Human Hypertension, Reynolds20 notes that UA can be considered one of the components of the metabolic syndrome, as various studies have shown its association with BP, obesity, TG, and HDL cholesterol.21,22 Although he correctly points out that correlation does not prove causation, a possible pathogenetic mechanism linking these associations includes the release of free fatty acids from visceral adipose tissue, which increases hepatic gluconeogenesis.23 This reduces peripheral tissue glucose uptake, thus causing hyperinsulinemia. In turn, this causes avid renal salt retention, which drives up the BP, as well as increases urate reabsorption.23 This state is also associated with the release of proinflammatory markers including fibrinogen and C-reactive protein, which all act in concert with dyslipidemia to increase the overall cardiovascular risk of an individual.23 The significant positive correlation between microalbuminuria and SUA levels found in this study may be a result of the association between UA and risk factors for renal damage, like systolic hypertension and obesity, or due to their common link with the proinflammatory state.24 This is supported by the recent findings of Dmitriev et al,25 who studied 100 hypertensive patients at moderate and high risk for experiencing cardiovascular events. At SUA levels >319 μmol/L, the hypertensive patients were found to have microalbuminuria, as well as elevated C-reactive protein (a nonspecific marker of inflammation). The latter also correlated with SUA, especially in the patients at high risk.25
The regression analysis in the present study showed that UA was a significant predictor of microalbuminuria, independent of SBP, which also contributed to the variance observed in microalbuminuria. Since one of the major sites of UA production in the cardiovascular system is the vessel wall, especially the endothelium, elevated UA may be a marker of endothelial dysfunction, and microalbuminuria is the renal manifestation of generalized endothelial dysfunction.26 Laboratory testing for SUA in parts of Nigeria is cheaper and far more readily available than that for microalbuminuria, and it can be assessed even in primary care settings to identify hypertensive patients at higher cardiovascular risk.
Hypertension promotes renal dysfunction which, in turn, increases SUA and activates the renin–angiotensin–aldosterone system; angiotensin II is essential in the development of LVH.27 This may partly explain the observation in this study, that UA correlated significantly with LVMI and was a unique predictor of the variance in LV mass. This is consistent with the findings by Iwashima et al,19 where UA not only correlated positively with LVMI, but it also predicted the risk for future cardiovascular events during the follow-up period. However, their study differs from this one in the sense that the patients had longstanding hypertension, and some of them were on diuretics and/or uricosuric agents. The positive correlation in their study persisted even after excluding the effects of drugs on UA levels.19 On the other hand, Tsioufis et al24 did not demonstrate a significant correlation between UA and LVMI in the recently diagnosed hypertensive patients in their study. In their entire study population, the mean duration of hypertension was 5.5 years, and LVMI was within the normal values. The majority (65%) of their patients were also on treatment for hypertension, although they had a 4-week washout period prior to participation in the study. These differences in patient characteristics may partly account for the disparate results.
In a more recent study by Yoshimura et al28 of 1,943 hypertensive and normotensive subjects, the relationship between SUA and LVH was significant only among males and not females. Other studies have shown significant relationships among women only.14,29 These sex differences have been attributed to the differences in sex hormones, but they have not been unequivocally proven.14 The link between UA and LV mass might relate to an association of UA with other risk factors, especially renal dysfunction, oxidative stress, severity of hypertension, and obesity.19 Previous reports have shown that UA impaired nitric oxide generation and induced endothelial dysfunction and smooth muscle cell proliferation.30 In experimental and in vitro systems, UA appears to have the ability to induce inflammatory mediators, such as tumor necrosis factor α, and mitogen-activated protein kinases, which are known to induce cardiac hypertrophy.31 UA, by activating the renin–angiotensin–aldosterone system (both at the local and systemic levels), promotes cardiomyocyte growth and interstitial fibrosis, which are the pathologic hallmarks of LVH.32 These discoveries suggest that cardiac hypertrophy may be, at least in part, attributable to an increase in UA itself, via the stimulation of endothelial dysfunction, smooth muscle cell proliferation, and inflammation. Lowering UA has been associated with modest benefits. The LIFE (Losartan Intervention for Endpoint Reduction) study33 demonstrated a 29% reduction in composite cardiovascular outcome (stroke, myocardial infarction, and cardiovascular deaths) in the losartan (an angiotensin receptor blocker) arm of the study, which was attributed to the uricosuric effect of losartan. A recent study demonstrated that in patients with ischemic heart disease, modest LVH regression followed the lowering of SUA with high-dose allopurinol.34 However, as noted by Zoccali and Mallamaci,35 this does not provide sufficient evidence that UA-lowering is the mechanism underlying the benefit of allopurinol and, by implication, other urate-lowering agents, as beyond lowering UA, the inhibition of xanthine oxidase leads to better endothelial responses and modulation of oxidative stress.
Furthermore, the elegant review by Feig et al36 did not find sufficient evidence to conclude that treating asymptomatic hyperuricemia was beneficial, while noting that the adverse effects of a drug like allopurinol can prove fatal.
It was found in this study that SUA increased as the number of TOD went up from none to two. This was similar to the results obtained from 425 untreated essential hypertensive patients (who were a part of the large Microalbuminuria: A Genoa Investigation on Complications trial) where UA was significantly correlated with the presence and number of TOD.14 The findings in this study were also in keeping with the results of a more recent study conducted by Serban et al.37 They found that SUA was significantly higher in hypertensive patients who had more than three indices of TOD compared to those with two or less. Carotid intima–media thickness – a surrogate marker of atherosclerosis – was found to correlate significantly with UA and microalbuminuria in their study. Their study population was, however, not limited to newly diagnosed hypertensive patients; therefore, the independent influence of the duration of hypertension on TOD cannot be ruled out.37
The cases in the present study population were newly diagnosed and largely unaware of their BP status, yet a significant number had evidence of subclinical TOD, which has been shown to herald cardiovascular events.38,39 The significant associations found between SUA and TOD in this study thus suggests that UA is a robust marker for future cardiovascular events and, as such, should be routinely measured in hypertensive individuals as part of their initial cardiovascular work-up at the time of diagnosis.
Limitations of this study include the fact that it is cross-sectional, and thus no causal relationships can be inferred. The limited sample size with the participants recruited from a tertiary health center questions the generalizability of the findings to the average person in the community with hypertension.
The present study was not designed to establish causation; therefore, we cannot comment on the clinical utility of lowering UA to prevent TOD. To firmly establish the causal role of UA in the incidence of TOD among hypertensive patients, large cohort studies are needed. Furthermore a well-designed, randomized, controlled trial is needed to determine the clinical benefits of lowering UA in these patients.
Conclusion
In conclusion, it is the opinion of the authors that in clinical practice in limited-resource settings, a cheap screening test like SUA should be done in all patients presenting with hypertension in order to identify those who are at higher total cardiovascular risk, and who thus warrant more intensive investigations and intervention.
Disclosure
The authors report no conflicts of interest in this work.
References
Chobanian AV, Bakris GL, Black HR, et al; Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure. National Heart, Lung, and Blood Institute; National High Blood Pressure Education Program Coordinating Committee. Seventh report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure. Hypertension. 2003;42(6):1206–1252. | |
Whitworth JA; World Health Organization, International Society of Hypertension Writing Group. 2003 World Health Organization (WHO)/International Society of Hypertension (ISH) statement on management of hypertension. J Hypertens. 2003;21(11):1983–1992. | |
British Cardiac Society; British Hypertension Society; Diabetes UK; HEART UK; Primary Care Cardiovascular Society; Stroke Association. JBS 2: Joint British Societies’ guidelines on prevention of cardiovascular disease in clinical practice. Heart. 2005;91 Suppl 5:v1–v52. | |
Salako BL, Ogah OS, Adebiyi AA, et al. Unexpectedly high prevalence of target-organ damage in newly diagnosed Nigerians with hypertension. Cardiovasc J Afr. 2007;18(2):77–83. | |
Cannon PJ, Stason WB, Demartini FE, Sommers SC, Laragh JH. Hyperuricemia in primary and renal hypertension. N Engl J Med. 1966; 275(9):457–464. | |
European Society of Hypertension-European Society of Cardiology Guidelines Committee. 2003 European Society of Hypertension-European Society of Cardiology guidelines for the management of arterial hypertension. J Hypertens. 2003;21(6):1011–1053. | |
Culleton BF, Larson MG, Kannel WB, Levy D. Serum uric acid and risk for cardiovascular disease and death: the Framingham Heart Study. Ann Intern Med. 1999;131(1):7–13. | |
Duffy WB, Senekjian HO, Knight TF, Weinman EJ. Management of asymptomatic hyperuricemia. JAMA. 1981;246(19):2215–2216. | |
Friedewald WT, Levy RI, Fredrickson DS. Estimation of the concentration of low-density lipoprotein cholesterol in plasma, without use of the preparative ultracentrifuge. Clin Chem. 1972;18(6):499–502. | |
National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III). Third Report of the National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III) final report. Circulation. 2002;106(25):3143–3421. | |
de Jong PE, Curhan GC. Screening, monitoring, and treatment of albuminuria: Public health perspectives. J Am Soc Nephrol. 2006;17(8):2120–2126. | |
Devereux RB. Detection of left ventricular hypertrophy by M-mode echocardiography. Anatomic validation, standardization, and comparison to other methods. Hypertension. 1987;9(2 Pt 2):II19–II26. | |
Ganau A, Devereux RB, Pickering TG, et al. Relation of left ventricular hemodynamic load and contractile performance to left ventricular mass in hypertension. Circulation. 1990;81(1):25–36. | |
Viazzi F, Parodi D, Leoncini G, et al. Serum uric acid and target organ damage in primary hypertension. Hypertension. 2005;45(5):991–996. | |
Poudel B, Yadav BK, Kumar A, Jha B, Raut KB. Serum uric acid level in newly diagnosed essential hypertension in a Nepalese population: a hospital based cross sectional study. Asian Pac J Trop Biomed. 2014;4(1):59–64. | |
Emokpae AM, Abdu A. Serum uric acid levels among Nigerians with essential hypertension. Niger J Physiol Sci. 2013;28(1):41–44. | |
Russo C, Olivieri O, Girelli D, Guarini P, Corrocher R. Relationships between serum uric acid and lipids in healthy subjects. Prev Med. 1996; 25(5):611–616. | |
Nan H, Qiao Q, Dong Y, et al. The prevalence of hyperuricemia in a population of the coastal city of Qingdao, China. J Rheumatol. 2006; 33(7):1346–1350. | |
Iwashima Y, Horio T, Kamide K, Rakugi H, Ogihara T, Kawano Y. Uric acid, left ventricular mass index, and risk of cardiovascular disease in essential hypertension. Hypertension. 2006;47(2):195–202. | |
Reynolds TM. Serum uric acid and new-onset hypertension: a possible therapeutic avenue? J Hum Hypertens. Epub January 16, 2014. | |
Sundstrom J, Sullivan L, D’Agostino RB, et al. Relations of serum uric acid to longitudinal blood pressure tracking and hypertension incidence. Hypertension.2005;45:28–33. | |
Tsouli SG, Liberopoulos EN, Mikhaildis DP, et al. Elevated serum uric acid levels in metabolic syndrome: an active component or an innocent bystander. Metabolism. 2006;55(10):1293–12301. | |
Johnson RJ, Kang DH, Feig D, et al. Is there a pathogenetic role for uric acid in hypertension and cardiovascular and renal disease? Hypertension. 2003;41(6):1183–1190. | |
Tsioufis C, Chatzis D, Vezali E, et al. The controversial role of serum uric acid in essential hypertension: relationships with indices of target organ damage. J Hum Hypertens. 2005;19(3):211–217. | |
Dmitriev VA, Oshchepkova EV, Titov BN, et al. [Is there an association of uric acid level with preclinical target organ damage in moderate- and high-risk hypertensive patients?]. Ter Arkh. 2013;85(9):52–57. Russian. | |
Niskanen LK, Laaksonen DE, Nyyssönen K, et al. Uric acid level as a risk factor for cardiovascular and all-cause mortality in middle-aged men: a prospective cohort study. Arch Intern Med. 2004;164(14):1546–1551. | |
Lorell BH, Carabello BA. Left ventricular hypertrophy: pathogenesis, detection, and prognosis. Circulation. 2000;102(4):470–479. | |
Yoshimura A, Adachi H, Hirai Y, et al. Serum uric acid is associated with the left ventricular mass index in males of a general population. Int Heart J. 2014;55(1):65–70. | |
Matsumura K, Ohtsubo T, Oniki H, Fujii K, Iida M. Gender-related association of serum uric acid and left ventricular hypertrophy in hypertension. Circ J. 2006;70(7):885–888. | |
Rao GN, Corson MA, Berk BC. Uric acid stimulates vascular smooth muscle cell proliferation by increasing platelet-derived growth factor A-chain expression. J Biol Chem. 1991;266(13):8604–8608. | |
Yokoyama T, Nakano M, Bednarczyk JL, McIntyre BW, Entman M, Mann DL. Tumor necrosis factor-alpha provokes a hypertrophic growth response in adult cardiac myocytes. Circulation. 1997;95(5):1247–1252. | |
Watanabe S, Kang DH, Feng L, et al. Uric acid, hominoid evolution, and the pathogenesis of salt-sensitivity. Hypertension. 2002;40(3):355–360. | |
Kjeldsen SE, Dahlöf B, Devereux RB, et al; LIFE (Losartan Intervention for Endpoint Reduction) Study Group. Effects of losartan on cardiovascular morbidity and mortality in patients with isolated systolic hypertension and left ventricular hypertrophy: a Losartan Intervention for Endpoint Reduction (LIFE) substudy. JAMA. 2002;288(12):1491–1498. | |
Rekhraj S, Gandy SJ, Szwejkowski BR, et al. High-dose allopurinol reduces left ventricular mass in patients with ischemic heart disease. J Am Coll Cardiol. 2013;61(9):926–932. | |
Zoccali C, Mallamaci F. Uric acid, hypertension, and cardiovascular and renal complications. Curr Hypertens Rep. 2013;15(6):531–537. | |
Feig DI, Kang DH, Johnson RJ. Uric acid and cardiovascular risk. N Engl J Med. 2008;359(17):1811–1821. | |
Serban C, Dragan S, Christodorescu R, et al. Association of serum uric levels and carotid intima thickness with the numbers of organ damaged in hypertensive patients. International Journal of Collaborative Research on Internal Medicine and Public Health. 2011;3(1):8–16. | |
Klausen KP, Scharling H, Jensen G, et al. New Definition of Microalbuminuria in Hypertensive subjects: Association with Incident Coronary Heart Disease and Death. Hypertension. 2005;46:33–37. | |
Agabiti RE, Muiesan ML. Pathophysiology and treatment of hypertensive left ventricular hypertrophy. Dialogues in Cardiovascular Medicine. 2005;10(1):3–18. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.