Back to Journals » Research Reports in Clinical Cardiology » Volume 5
SERCA2a: its role in the development of heart failure and as a potential therapeutic target
Authors Fragoso-Medina J, Zarain-Herzberg A
Received 30 October 2013
Accepted for publication 2 December 2013
Published 11 February 2014 Volume 2014:5 Pages 43—55
DOI https://doi.org/10.2147/RRCC.S38614
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Jorge Fragoso-Medina, Angel Zarain-Herzberg
Department of Biochemistry, School of Medicine, National Autonomous University of Mexico, Mexico City, Mexico
Abstract: The complexity of heart physiology has delayed the implementation of efficient, feasible, and safe therapies to fight against heart diseases for many years. As knowledge of the precise mechanisms governing cardiac hypertrophy and heart failure development increases, the availability of new therapeutic alternatives also grows. Since the cardiomyocyte physiology deeply depends on the correct calcium handling, many efforts to describe accurately the excitation–contraction coupling process in the heart and the proteins involved have been made. Among the proteins participating in calcium handling, sarco/endoplasmic reticulum Ca2+ adenosine triphosphatase-2a (SERCA2a), whose expression and function is decreased in heart failure, stands out because of its critical role regulating Ca2+ concentration in the cardiomyocyte. The importance of SERCA2a has been reflected in numerous studies aimed to describe its expression and function. Recently, gene therapy to deliver SERCA2a has shown promising results in human clinical trials. This paper reviews the current literature knowledge exploring diverse approaches to rescue SERCA2a expression in heart failure. It also discusses some data suggesting other possible therapies that could improve SERCA2a expression and function in cardiac diseases.
Keywords: SERCA2a, cardiac hypertrophy, heart failure, gene therapy, calcium
Introduction
Heart contractile function depends on the correct calcium ion (Ca2+) cycling in the cardiomyocyte accomplished by a tightly synchronized process called excitation–contraction coupling (ECC). In the cardiomyocyte the ECC process starts when a depolarizing stimulus reaches the sarcolemmal voltage-dependent L-type Ca2+ channels also known as dihydropyridine receptors. This allows the entrance of extracellular Ca2+ into the cell sarcoplasm at the triad junction, triggering the opening of the sarcoplasmic reticulum (SR) Ca2+ release channels named ryanodine receptors type-2 (RyR2), which results in a rapid and massive Ca2+ release from the SR lumen to the sarcoplasm and causes cytosolic free calcium concentration ([Ca2+]c) to rise from around 100 nM to 1 μM.1 Then, free Ca2+ binds to the troponin complex, which triggers a conformational change that exposes the actin-binding site for myosin, allowing the cross-bridge between actin and myosin. Upon cross-bridge formation there is a power stroke that moves the actin filament relative to myosin filament and adenosine diphosphate is released, resulting in cardiomyocyte contraction. During relaxation, adenosine triphosphate (ATP) binds to α- and β-myosin heavy chain (MHC) leading to detachment of myosin from actin, and the Ca2+ released from the SR contraction is actively transported from the sarcoplasm into the SR lumen by the cardiac sarco/endoplasmic reticulum Ca2+ ATPase-2a (SERCA2a) pump, varying from around 60% in humans to 90% in mice. The remainder Ca2+ is extruded to the extracellular space mainly by the sodium (Na+)/Ca2+ exchanger, but also by the mitochondrial Ca2+ uniporter and the sarcolemmal Ca2+ ATPase (plasma membrane Ca2+ ATPase) to maintain cardiomyocyte Ca2+ homeostasis.2,3 Inside the SR, Ca2+ binds to the main present protein calsequestrin (CASQ2), which has high Ca2+-binding capacity (40 mol Ca2+/mol protein), moderate affinity for Ca2+ (Kd ~1 mM), and is responsible for storage of Ca2+ but also participates in Ca2+ release by RyR2 during contraction.4–6 The precise control of Ca2+ levels during the contraction–relaxation cycle plays a key role in the normal contractile function of the cardiac myocyte. Altered Ca2+ handling is the main hallmark of the contractile dysfunction observed in heart failure (HF).
This review analyzes the evidence demonstrating alterations in the function of the components regulating Ca2+ handling in cardiac hypertrophy (CH) and HF. It also reviews the current knowledge about the different strategies directed to normalize cardiac SERCA2a activity and/or expression, including pharmacological and gene therapy approaches.
Alterations in calcium handling in the hypertrophic and failing heart
Physiological CH occurs as a result of an adaptation to increased workload, like the one observed in athletes, and is a benign modification of the myocardium. Some of the changes observed are increased heart size, better cardiac performance, enhanced energy utilization, and increased (or unchanged) expression of SERCA2a and α-MHC. In contrast, in pathological CH the increased heart size is accompanied by poor cardiac performance, poor fatty acid utilization, decreased expression of SERCA2a and α-MHC, and activation of a fetal cardiac gene expression program.7 Although adrenergic stimulation has been shown to increase SERCA2a expression,8 a time-course follow-up of hypertrophy has shown that this upregulation occurs in the first stages, but in severe CH the SERCA2 gene is downregulated as occurs in HF.9
Physiological contraction and relaxation parameters are altered in cardiomyocytes of hearts in animal models with CH and in patients with HF.2,10 It has been shown that a decrease in shortening velocity of the cardiomyocyte during contraction correlates with a reduced myofibrillar MHC/ATPase activity,11 as well as with a diminished [Ca2+]c.2,12–14 A decreased rate of Ca2+ removal from the sarcoplasm by the failing cardiomyocyte during myocardial relaxation leads to increased diastolic [Ca2+]c levels, contributing to diastolic dysfunction development present in HF. The observed dysfunction is characterized by increased end-diastolic pressure and ventricular filling abnormalities. The Ca2+ transients of cardiomyocytes from hearts in animal models with CH and from patients with end-stage HF have decreased peak systolic [Ca2+]c and a reduced velocity of Ca2+ transport by the SERCA2a pump into the SR.10 Because of the reduced peak [Ca2+]c derived from reduced Ca2+ load to the SR, there is a reduction of force development by the heart. Due to a low SR Ca2+ content in failing cardiomyocytes,15,16 altered SR Ca2+ handling is thought to be a key component contributing to the decreased contractility and force generation during cardiomyopathy.
Cardiac ECC is controlled by several signaling pathways, including protein kinase A (PKA) and Ca2+/calmodulin-dependent protein kinase II (CaMKII). Among the alterations involved in CH and HF are changes in the expression levels of these regulatory proteins that act as sensors of intracellular Ca2+.17 In HF, increased activity and expression of CaMKII has been demonstrated, which plays a role in decreased contractile properties of the myocardium, uncoupling ECC and possibly leading to sudden death.18 Several studies have shown dilated cardiomyopathy, CH and HF development in models of CaMKII overexpression,5,19 and protective effects against HF development in CaMKII knockout animals.7,20 The alterations include decreased levels of SERCA2a with a subsequent decrease of the recapture of Ca2+ to the SR during diastole, resulting in a smaller amount of Ca2+ available to be released in the following heartbeat.
Closely related to SERCA2a is phospholamban (PLN), a 52 amino acid peptide that is the primary regulator of SERCA2a activity. PLN inhibits the pump associating with SERCA2a, and dissociates from the pump when it is phosphorylated by CaMKII or PKA in response to β-adrenergic stimulation, resulting in increased Ca2+ transport velocity by SERCA2a.21,22 In the failing heart, the SERCA2a/PLN ratio is decreased to a great extent by the reduction of SERCA2a pumps present in the SR membrane. This reduction causes the amount of active SERCA2a to decrease, and therefore decreases its activity and capacity to recapture the Ca2+ released during contraction.23
PLN assembling in pentamers is necessary for fully active SERCA2a inhibition, and equilibrium between phosphorylated (unbound) and dephosphorylated (bound) PLN forms is classically thought to determine SERCA2a activity.24 Although, recent findings25,26 suggest that SERCA activation can occur without dissociation of the SERCA/PLN complex, the decrease in phosphorylated PLN levels causes the SERCA2a activity to decrease. During HF, PLN is hypophosphorylated, which causes a chronic decrease in SERCA2a activity.27 Therefore, reduced SERCA2a activity is not only due to a smaller expression of SERCA2a protein, but also to alterations in the PLN phosphorylation levels as well. Because of increased PKA activity, the inhibitor-1 of the protein phosphatase-1 (PP1) is phosphorylated, releasing PP1 inactivation and resulting in PLN dephosphorylation by PP1 with the consequent activation of PLN.28,29 The SR Ca2+ release channel of the SR (RyR2) has been reported in an unstable state when it is dissociated from the protein calstabin, resulting in an increased probability of channel opening. This results in spontaneous Ca2+ liberations that appear during diastole (diastolic leak) and are associated with ventricular arrhythmias and a decrease in the SR Ca2+ reserve available for the next contraction–relaxation cycle.3,30
CaMKII shares common functional targets with PKA regulating ECC. PLN and RyR2 are phosphorylated by PKA and CaMKII, resulting in an increased SR Ca2+ release. The increased CaMKII activity in HF contributes to the reduced SR calcium content and systolic function, and also causes diastolic SR Ca2+ leak and Ca2+ current changes that may be arrhythmogenic. One of the main causes of sudden death in patients with HF is ventricular arrhythmia. Abnormal RyR2 Ca2+ release from the SR has been linked to arrhythmogenesis; in fact, there is evidence for RyR2 hyperphosphorylation due to PKA and the involvement of phosphodiesterase D in this process.31,32 Recent reports suggest that CaMKII-dependent phosphorylation of RyR2 is involved in enhanced SR diastolic Ca2+ leak and also reduced SR Ca2+ load in HF, contributing to arrhythmias and contractile dysfunction.33 RyR2 phosphorylation by CaMKII at serine 2814 plays a dominant role in arrhythmogenesis and sudden cardiac death in mice with HF. This fact suggests that a destabilization of RyR2 activity due to increased CaMKII phosphorylation results in reduced post-release refractoriness, which is a mechanism involved in arrhythmogenesis and dysfunction in contractility of the failing heart.
Cardiac pressure overload, as is seen in hypertensive patients, is the major pathologic stimulus for CH and is the main risk factor for HF development. There are several reports demonstrating that in human pressure overload hypertrophic and failing hearts, as well in CH animal models, there is a marked decrease in SERCA2a content (~50%) of cardiomyocytes.3,34 Since SERCA2a activity is essential for Ca2+ handling and this is altered as a consequence of CH and HF development, efforts to investigate calcium-dependent signaling pathways in this context have been conducted. The best-known two calcium-dependent signaling pathways in cardiomyocytes are the calcineurin/nuclear factor of activated T-cells (NFAT) pathway and the CaMK/myocyte enhancer factor-2 pathway. Both pathways participate in normal heart development and are altered in CH, but the cross-talk between these pathways is still not fully understood.35 One of the common features of CH and HF is downregulation of SERCA2 gene expression, leading to alterations in Ca2+ signaling pathways. Ca2+ signaling fluctuations and contractile function alterations have been demonstrated following specific inhibition of SERCA2a transport activity with thapsigargin, reduction of SERCA2 gene expression by a gene null mutation, and SERCA2 gene silencing with small interfering RNA.36 SERCA2 gene direct transcriptional regulation by NFAT activity has been proposed,37 although the precise molecular mechanisms have not been yet described. Whether NFAT and/or myocyte enhancer factor-2 are involved directly in SERCA2 gene transcriptional regulation is currently under investigation in the authors’ laboratory. The above data highlight the importance of SERCA2 gene expression in the physiopathology of cardiac diseases.
SERCA2a as a therapeutic target
Pharmacological therapies
The current most widely used drugs to treat HF patients include angiotensin-converting enzyme inhibitors, angiotensin II receptor blockers, adrenergic β-blockers, digoxin or digitalis, diuretics, aldosterone antagonists, and inotropes. As a multifactorial clinical syndrome, HF still represents an epidemic threat; therefore, diverse strategies and molecular targets are being investigated. SERCA2a, as a key calcium handling protein, has received special attention in the last 20 years. The following section reviews current literature knowledge positioning SERCA2a as a therapeutic target in the treatment of cardiac diseases. Several drugs that have been recently investigated for their effect on SERCA2a activity and expression are shown in Table 1.
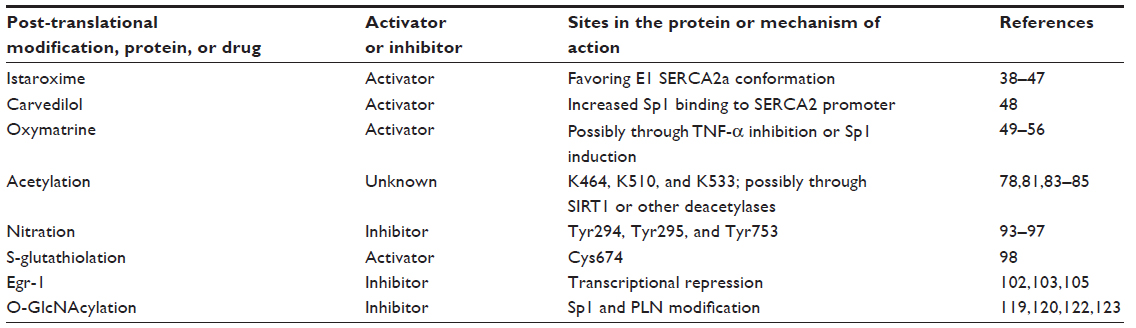 | Table 1 Potential drug therapies targeting SERCA2a for treatment of heart failure |
Istaroxime is a luso-inotropic compound that activates SERCA2a and inhibits the sodium/potassium ATPase.38,39 Due to its double action, istaroxime has been proved to treat acute HF syndrome in guinea pigs and dogs, improving systolic and diastolic function, central hemodynamics, systolic blood pressure, and reducing heart rate.40–42 Istaroxime has also been used in human patients; the Hemodynamic, Echocardiographic, and Neurohormonal Effects of Istaroxime, a Novel Intravenous Inotropic and Lusitropic Agent: a Randomized Controlled Trial in Patients Hospitalized with Heart Failure (HORIZON-HF) trial showed that in addition to its beneficial effects on heart function, istaroxime produces few side effects since there were no neurohormonal or renal changes and no troponin I release, and adverse effects were not life-threatening and only observed at the highest dose.43 Istaroxime effect on SERCA2a is isoform specific since no effect of the drug was observed on SERCA1 from skeletal muscle.40 Recently, the mechanism of istaroxime-driven SERCA2a activation has been addressed and it was observed that this effect is dependent on PLN presence. The SERCA2a/PLN complex was found to dissociate with increasing nanomolar istaroxime concentrations in a similar way to that observed with increasing Ca2+ concentrations. Furthermore, SERCA2a activation was shown to be cyclic adenosine monophosphate (cAMP)/PKA independent since the stimulation was persistent using the PKA inhibitor staurosporine.44 As istaroxime is the only small molecule able to activate SERCA2a activity, and there are some concerns about its specificity, studies have been carried out to design new lusitropic/inotropic compounds with higher effectiveness and lower toxicity. Searching for these molecules has yielded istaroxime analogs with better activity, and even structure–activity relationships have been found.45,46 Further, a high-throughput fluorescence resonance energy transfer assay has been used to find allosteric SERCA2a activators capable of decreasing SERCA2a/PLN-derived fluorescence resonance energy transfer.47 In this study, several compounds previously reported to correct aberrant Ca2+ regulation in HF were shown to activate SERCA2a activity, thus validating the mechanism of action for these drugs.
Carvedilol is a β-adrenergic receptor blocker that elicits its beneficial effects on cardiac function through the restoration of SERCA2a expression in patients with HF. Furthermore, carvedilol prevents oxidative stress-mediated SERCA2 gene downregulation by promoting specificity protein-1 (Sp1) and Sp3 binding to elements located in the proximal promoter, increasing transcription of the gene by this mechanism.48
Among the current therapies to prevent heart disease, traditional medicine has played an especially important role in the People’s Republic of China where herbal medicine is very diverse. Oxymatrine (OMT) is an alkaloid obtained from the roots of the Chinese herb Sophora japonica (Sophora falvescens Ait), also known as ku shen. It has anti-inflammatory, antiviral, immune reaction inhibiting, and anti-hepatic fibrosis protective activities. A few years ago the properties of OMT in treating myocardial infarction in rats were studied, and it was found that a dose of 20 mg/kg reduced infarct size and inhibited lipid peroxidation. Beneficial effects possibly were due to increased endogenous superoxide dismutase activity, increased B-cell lymphoma-2 expression, and a reduction of the apoptotic mediator Fas and [Ca2+]c overload.49 OMT has also been reported to exert antiarrhythmic effects in a rat model of arrhythmia induced by coronary ligation.50 In this study, OMT reduced the action potential duration of cardiomyocytes through reduction of L-type calcium current, enhancement of transient outward potassium current, and inhibition of inward rectifier potassium current. Also, in rats with acute myocardial infarction, this alkaloid was shown to improve hemodynamic parameters and the left ventricle weight/body weight ratio, and to decrease expression of transforming growth factor-β receptor-1 and Smad3 messenger RNAs.51 OMT downregulated the angiotensin-converting enzyme messenger RNA expression and extracellular signal-regulated kinase-1/2, c-Jun N-terminal kinase, and p38 phosphorylation in spontaneous hypertensive rats to levels comparable to those obtained with captopril, a widely used drug to treat HF.52 OMT has been shown to inhibit Janus kinase/signal transducer and activator of transcription protein signaling pathway activation and expression of interleukin-1β and tumor necrosis factor-α;53 the latter being a cytokine that has been associated with SERCA2a downregulation.54 Since the reported effects for OMT include regulation of ionic currents, calcium handling was assessed in a rat model of chronic HF. The Ca2+ transients and SR Ca2+ content were improved by 50 and 100 mg/kg of OMT along with the upregulation of SERCA2a and dihydropyridine receptor expression.55 Furthermore, OMT was shown to induce the Sp1 transcription factor expression in human embryonic kidney 293 cells.56 Taken together, the OMT beneficial effects on cardiac tissue seem to be related to the activation of SERCA2a expression through tumor necrosis factor-α inhibition or Sp1 increased expression. These findings encourage further investigation about OMT and other natural products that could serve as SERCA2a direct or indirect activators.
Gene therapy to increase SERCA2a levels
The goal of cardiac gene therapy is to reverse some of the molecular alterations in the failing heart. An obvious approach to restore the SERCA2a protein deficiency caused by HF is to provide more SERCA2a protein. Despite barriers and risks associated with gene therapy, SERCA2a overexpression use for HF shows promising results.66 The first attempts to restore SERCA2a through gene therapy were done using adenoviruses,57,58 since previous studies had shown feasibility for SERCA2a in vivo overexpression.59,60 Lentiviral vectors are also an alternative for gene delivery because they integrate their DNA into host chromosomes, providing a long-term expression of the delivered gene. SERCA2a complementary DNA delivery by a lentiviral vector improved heart function in rat hearts subjected to myocardial infarction.61 Recombinant adeno-associated viruses (AAV) were used in the first gene therapy clinical trial for HF called Calcium Up-Regulation by Percutaneous Administration of Gene Therapy in Cardiac Disease (CUPID).62,63,64 The second phase of this trial used antegrade percutaneous intracoronary artery infusion of the AAV vector AAV1/SERCA2a (MYDICAR®, Celladon Corporation, San Diego, CA, USA), in 39 patients. It began in 2007 and was designed to evaluate the safety profile and biological effects of SERCA2a gene transfer.64–66 After a 3-year follow-up, patients from the CUPID trial showed an acceptable safety profile and improvement in heart function.66 In the trial it was not found to increase adverse effects, laboratory abnormalities, disease-related events, or arrhythmias. The study concluded that after a single intracoronary infusion of AAV1/SERCA2a in patients with advanced HF, the beneficial cardiovascular effects last for several years.
Since the CUPID clinical trial showed promising good results, a clinical trial called CUPID2,67 enrolling 250 patients with HF, has just started in the Cardiovascular Biomedical Research Unit at the Royal Brompton Hospital of England and results will be available in 2015. There are two ongoing gene therapy clinical trials to express SERCA2a in the hearts of patients with HF using AAV1/SERCA2a – one in England and one in France.68 Lastly, another recent trial, named Investigation of the Safety and Feasibility of SERCA Gene Transfer in the Human Failing Heart Using an Adeno-associated Viral Vector, (SERCA/LVAD), started recruitment of patients in the UK in the summer of 2013.69 This trial will test SERCA2 gene therapy in 16 HF patients already fitted with mechanical heart pumps, also known as left ventricular assist devices. These trials will collect information about the effectiveness of the therapy by measuring the amount of the SERCA2 gene and protein that has been introduced into heart muscle (Table 2). Although the above mentioned trials are promising, boosting SERCA2a function has been shown to be counterproductive in the setting of dysregulated RyR2 function in mice.70
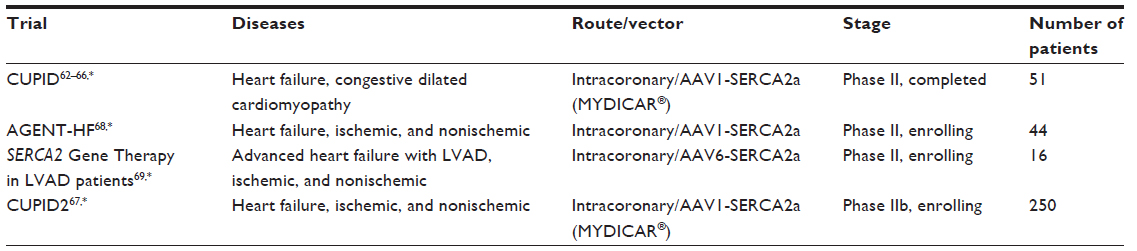 | Table 2 SERCA2 gene therapy clinical trials for heart failure |
Duchenne muscular dystrophy (DMD) is an X-linked recessive disorder characterized by mutations in the dystrophin gene. The vast majority of dystrophin gene mutations lead to loss of dystrophin protein and as a consequence smooth, skeletal, and cardiac muscles of DMD patients show myofiber damage and membrane leakage. Although DMD patients usually die from respiratory failure before their 20s, many men show abnormal electrocardiograms by the age of 18 years. This fact, and the absence of an effective therapy to correct the absence of dystrophin, raises the need for a therapy to correct performance in smooth and cardiac muscle.71 SERCA2a overexpression has been explored to treat DMD heart disease.72 SERCA2a was overexpressed in old female mdx mice, a murine model of DMD cardiomyopathy, finding that the characteristic tachycardia was corrected by the overexpression of SERCA2a. SERCA2a overexpression has also been used to treat skeletal muscle dystrophy. The δ-sarcoglycan-null mice and dystrophin mutant mdx mice subjected to skeletal muscle-specific overexpression of SERCA1 showed dramatically reduced myofiber central nucleation, fibrosis, and serum creatine kinase levels compared with non-treated animals. The loss of exercise capacity and dystrophic phenotype in the gastrocnemius muscle of δ-sarcoglycan-null mice were also rescued. Furthermore, SERCA1 overexpression reduced total cytosolic Ca2+, reversed mitochondria swelling, and reduced calpain activation, suggesting protection from Ca2+-driven necrosis.73
Post-translational SERCA2a modifications as possible therapeutic targets
SERCA2a function in the normal heart is modulated by different means, such as PLN, S100A1, and sarcoglycan interaction, Small ubiquitin-like modifier-1 addition to serine or threonine residues (SUMOylation), glutathiolation, and nitration.74,75 Some of these modifications are presented in Table 3 as possible unexplored therapeutic targets that are under investigation. Increasing SERCA2a activity in cardiomyocytes can be achieved by diminishing the SERCA2a/PLN interaction. Using fluorescent fusion proteins in human embryonic kidney cells and measuring SERCA/PLN interaction by fluorescence resonance energy transfer, PLN mutants with no SERCA inhibitory function competed with wild type for SERCA binding and led to increased SERCA activity.76 Furthermore, it was shown that phosphorylation of PLN induced by forskolin increased SERCA2a/PLN interaction, suggesting that SERCA2 inhibition can be relieved without dissociation of the complex. This strategy raises the possibility of using PLN mutants for gene therapy in heart diseases.
 | Table 3 Possible gene therapies to modify SERCA2a activity or expression |
Since transgenic mice overexpressing CaMKIIδ have increased RyR2 phosphorylation, enhanced SR Ca2+ leak, and lowered SR Ca2+ load, PLN ablation was explored as an alternative to rescue SERCA2 function, ie, SR Ca2+ handling in this animal model.77 Although double mutant mice showed normalized SR Ca2+ loading, SR Ca2+ leak and mitochondrial Ca2+ were enhanced, compromising mice survival. Similarly, in the study by Kalyanasundaram et al70 mentioned in the previous section, the overexpression of SERCA1a in a transgenic model that interferes with RyR2 function was counterproductive. These two studies suggest that increasing SERCA2a function for HF treatment should be used with caution in the setting of dysregulated RyR2 function.
As stated before, a series of post-translational modifications can alter SERCA2a function. SUMOylation is a modification occurring at lysine (Lys) residues of target proteins. It was shown that SERCA2a SUMOylation occurring at Lys480 and Lys585 is essential for the correct ATPase activity and stability of the pump in mouse and human cells.78 Small ubiquitin-like modifier-1 (SUMO-1) and SUMOylated SERCA2a levels are reduced in mouse failing hearts and AAV-mediated gene delivery of SUMO-1 was able to restore cardiac function and SERCA2a protein abundance. Further, SUMO-1 downregulation led to deterioration of cardiac function and decreased SERCA2a function. Although in this study SERCA2a knockdown was not rescued by SUMO-1 overexpression, SERCA2a SUMOylation could serve as a basis for the design of new therapies for HF.
Nkx2.5 is a homeobox domain transcription factor essential for heart specification and morphogenesis that has also been shown to be SUMOylated.79 SUMO-1 overexpression in the spontaneously beating HL-1 cell line strongly enhanced Nkx2.5 transcriptional activity and promoted the SUMOylation of undetermined Nkx2.5 cofactors. These data support the role of SUMO-1 as a potential therapy, not only as a SERCA2a activator but also as a complex transcriptional remodeling factor for HF. Lys residues in proteins are as important for SUMOylation as they are for acetylation. Acetylation has been widely reported in histones as an epigenetic mechanism for gene expression control. Recently, acetylation has been investigated as a more extensive post-translational modification in the whole proteome and its role in cancer is a currently addressed topic.80 In this context, acetylation of cardiac proteins has already been reported in the guinea pig acetylome.81 Particularly, SERCA2a can be acetylated in Lys464, Lys510, and Lys533. These three sites lie on the surface of the ATP binding domain, and whether their acetylation affects ATP binding to the pump has not been reported. Sirtuin-1 is a cardioprotective nicotinamide adenine dinucleotide-dependent histone deacetylase82 that has been related to the beneficial effects of resveratrol on cardiac function.83,84 Resveratrol has been shown to rescue SERCA2a expression in the setting of diabetic cardiomyopathy83 and histone deacetylases have been found in sarcomeres.85 Thus, these findings raise the question of whether SERCA2a acetylation may be a prominent modification that would compete or interfere with SERCA2a SUMOylation as proposed by Kho et al,78 and therefore could be of therapeutic interest.
Obesity, diabetes, and hypertension are health problems that have alarmingly grown over the last two decades in the developing world and are major predisposing factors to the development of cardiovascular disease.86 High-fat-fed rats develop obesity that mimics human Western health problems. It has been found that a decreased level of phosphorylated PLN is clearly linked with cardiovascular problems present in this animal model.87,88 Exercise training and diet-induced weight loss have been shown to exert beneficial effects on cardiac performance for both animal HF models and human HF patients.89–92 However, rescued expression of SERCA2a and other calcium handling proteins is not complete, and hence more effective therapies are needed.
A characteristic feature of aging, obesity, and diabetes is oxidative and nitrative stress that alters many protein functions, including SERCA2a activity.93–95 Increased SERCA2a nitration has been shown in human failing hearts.96 This modification has been correlated with increased time to half relaxation in myocytes isolated from control and idiopathic dilated cardiomyopathy hearts. This study also showed in vesicles from porcine hearts that SERCA2a was inactivated by peroxynitrite (ONOO–) exposure and that this inactivation was prevented by PKA pre-treatment. A 60% decrease in SERCA2a activity in the senescent Fischer-344 rat heart relative to that of young adult rat hearts has been reported.97 Although 18% of this decrease was attributed to lower SERCA2a protein abundance, the rest was shown to be due to 3-nitrotyrosine modification of the pump. It was found that there was a more than two-fold increase in age-dependent nitration of SERCA2a and that this nitration was distributed over tyrosine 122 (Tyr122), Tyr130, Tyr497, Tyr586, and Tyr990 in young rat hearts in contrast with senescent rat hearts, where Tyr294, Tyr 295, and Tyr753 were additionally found to be nitrated. Skeletal muscle was also analyzed in this study and full site (1 mol/mol) of Tyr753 nitration was found in young hearts versus additional nitration of Tyr294 and Tyr295 in senescent muscle. The appearance of these latter two nitrotyrosines correlated with diminished ATP utilization by the SERCA2a pump in both skeletal and cardiac muscle under nitrative stress.
Nitric oxide (NO) is one of the signals leading to relaxation in cardiac, skeletal, and smooth muscle, and it performs this function through SERCA2a activation. Using purified protein reconstituted in phospholipid vesicles, it was shown that NO-derived ONOO– directly activated SERCA2a by S-glutathiolation and that this modification was blocked by irreversible oxidation of relevant cysteine thiols. As mutation of SERCA cysteine 674 to serine abolished the pump activity dependence on glutathione and superoxide scavengers decreased S-glutathiolation of SERCA and relaxation by NO, ONOO– was proposed as the mediator. Additionally, atherosclerosis was found to decrease NO-dependent relaxation as well as the S-glutathiolation of SERCA with its consequent activation. Furthermore, a high content of SERCA2a cysteine 674 irreversibly oxidized to sulfonic acid in atherosclerotic aorta was found, confirming a key role for this modification in the diseased tissue.98
Prostaglandin-F2 (PGF2) is a vasoconstrictor involved in inflammatory response that is produced in the myocardium subsequent to infarction or pressure overload.99–101 It was shown that PGF2 stimulates hypertrophic growth of neonatal rat cardiac myocytes, including downregulation of SERCA2a. Interestingly, PGF2 increased early growth response protein 1 (Egr-1) expression and overexpression of Egr-1 decreased transcription of the proximal SERCA2 gene promoter. PGF2 was shown to exert repressive effects on SERCA2 transcription through Egr-1 transcription factor in a process mediated by mitogen-activated protein kinase (MAPK)/p38, Ras, and Rac but not extracellular signal-regulated kinase.102 The MAPK-activated protein kinases 2 and 3 (MK2/3) are known regulators of SERCA2a expression,103 and important players in the cardiac hypertrophic response.104 Recently, it was reported that SERCA2 gene expression was upregulated in MK2/3 double-knockout mice and that this regulation was due to a decreased ratio of transcription factor Egr-1/Sp1.105 It was found that SERCA2 gene promoter activity was directly dependent on MK2 catalytic activity. Also, the MK2/3 double-knockout mice showed enhanced expression of the peroxisome proliferator-activated receptor-γ co-activator-1α gene, which has been shown to be decreased in HF106 and restored after losartan treatment accompanied by exercise training.107 The above data show that Egr-1 transcription factor and its trigger MAPK proteins are important SERCA2a regulators that could potentially be used as therapeutic targets in cardiac diseases alone or combined with other approaches.
O-GlcNAcylation is a modification that has been recently found to play an important role in CH. The β-O-linkage of N-acetylglucosamine (O-GlcNAc) is controlled by two enzymes – the uridine diphospho-N-acetylglucosamine: peptide β-N-acetylglucosaminyltransferase (O-GlcNAc transferase [OGT]) and the O-β-N-acetylglucosaminidase (O-GlcNAcase) in mammalian cells.108 Increased O-GlcNAcylation has been reported in many cardiac dysfunctions,109 but it has also been shown that in vivo ischemic preconditioning increases O-GlcNAc.110 In the latter study, pharmacological augmentation of O-GlcNAc levels was sufficient to reduce myocardial infarct size in mice subjected to acute myocardial infarct. On the other hand, OGT overexpression was reported to reduce post-hypoxic damage in mouse cardiac myocytes and OGT silencing or inhibition to sensitize the formation of mitochondrial permeability transition pore induced by calcium.111,112 O-GlcNAcylation has also been investigated in vascular smooth muscle cells and it was found that increased O-GlcNAc levels mediated by endothelin-1 were abolished by OGT silencing.113 It was reported that pressure overload-induced CH (via transverse aortic constriction) in mice or treatment of neonatal rat cardiac myocytes with phenylephrine increased total O-GlcNAc levels that mirrored the enhanced activated NFAT signaling.114
Among the causes provoking left ventricular dysfunction, the dysregulation of energetic balance plays a substantial role due to the highly energy-consuming process that contraction–relaxation cycling represents. For example, mice with cardiomyocyte-restricted knockout of the insulin receptor show increased mitochondrial dysfunction following myocardial infarction when compared with wild type littermates and this impairment is accompanied by a decrease in several proteins including SERCA2a, peroxisome proliferator-activated receptor-α, and glucose transporter type-4.115 Glucose transporter type-4 knockout mice exhibit deficient contractile performance, CH, fibrosis, and reduced expression of RyR2 and SERCA2a proteins.116 Sp1 is a key transcriptional regulator of SERCA2 gene expression in the normal and hypertrophic heart;117,118 its transcriptional activity is decreased by O-GlcNAcylation.119 High O-GlcNAcylation levels in streptozotocin-mediated diabetic rats have been related to deleterious effects of diabetic cardiomyopathy. In this model, the overexpression of O-GlcNAcase diminished global O-GlcNAcylation level and also diminished SERCA2a and PLN expression, but increased phosphorylated PLN.120 Since one of the main therapies in the setting of diabetes is exercise training, its effects on O-GlcNAcylation interactions with transcriptional co-repressors were studied in a type-2 diabetes mouse model.121 Paradoxically, it was shown that exercise increased O-GlcNAc level. Diabetic hearts showed higher levels of histone deacetylase-1 and -2 activity and lower levels of mammalian switch-independent-3A. In sedentary diabetic hearts, mammalian switch-independent-3A and OGT were shown to be less associated with histone deacetylase-1 and -2, respectively, compared with controls, but exercise removed these differences. In fact, derived from Sp1 O-GlcNAcylation, SERCA2 gene transcriptional repression was also reported in cardiomyocytes exposed to high glucose.122 Moreover, high glucose treatment in cardiomyocytes was recently shown to promote PLN O-GlcNAcylation and it was proposed that this modification could improve PLN-mediated SERCA2a inhibition.123 The Nkx2.5 transcription factor is also downregulated in the heart tissue of streptozotocin-induced diabetic mice.124 Thus, the above data provide a possible mechanism that explains decreased SERCA2a function and expression in diabetic cardiomyopathy.
Other gene therapies regulating SERCA2a function and expression
As the cardiomyocyte physiology and the SERCA2a interactions are complex, other targets for gene therapy have been tested to override deleterious effects produced by CH (Table 3). Some of them ultimately impact SERCA2a function or expression, highlighting the importance of correct calcium handling in the cardiomyocyte.
Recently, miR-1, a cardiomyocyte-restricted micro RNA, was found to post-transcriptionally directly inhibit the expression of calmodulin and myocyte enhancer factor-2a, and indirectly the expression of GATA4, suggesting that miR-1 is a central regulator of calcium signaling in CH.125 As miR-1 has been shown to inhibit endothelin-1 expression,126 low levels of miR-1 would result in low SERCA2a levels and hence in a poor cardiac performance. A recent publication showed that AAV-mediated restoration of miR-1 expression in male Sprague Dawley rats subjected to ascending aortic stenosis ameliorated adverse effects derived from CH such as fibrosis, MAPK activation, and SERCA2a downregulation.127 The above results show a regulation of SERCA2a expression by miR-1.
As stated above, the phosphorylated/dephosphorylated ratio of PLN derived from phosphorylation by CaMKII or PKA or dephosphorylation by PP1a delimits SERCA2a pump activity. Hajjar et al showed that not only PLN phosphorylation status modifies SERCA2a activity since recombinant adenovirus-mediated PLN overexpression (Ad.RSV.PL) led to a significant prolongation of the relaxation phase, a decrease in peak [Ca2+]c, and an elevation in resting [Ca2+]c in membrane preparations from cardiomyocytes. Ad.RSV.PL also prolonged the time course of shortening in myocytes, but simultaneous SERCA2a overexpression partially reverted these effects.128
S100A1 is an EF-hand calcium-binding sensor protein that has been shown to interact with SERCA2a and activate it, among other functions like regulating RyR2, titin, and mitochondrial ATPase function.129 Due to these functions, S100A1 gene therapy has recently been explored in small and large animal models for HF.130,131 In both rat and domestic pig models, S100A1 transgene expression normalized cardiomyocyte Ca2+ cycling and SR calcium handling, and energy homeostasis resulted from cryoinfarction in rats or balloon occlusion of the left circumflex coronary artery in domestic pigs.132
Conclusion
Cardiovascular diseases are the main cause of morbidity and mortality in the world. Decreased SERCA2a content in the SR of patients with failing hearts is responsible for its decreased contractility. The current goal of the cardiovascular research is to fight back against HF by targeting and reversing some of the critical molecular changes arising in the heart when it fails. During the last decade, several researchers have focused their efforts on trying to correct SERCA2a expression in the hypertrophic and failing heart. Various pharmacologic and genetic approaches aimed to correct SERCA2a expression have been recently explored, shedding new light on the molecular mechanisms responsible for SERCA2a expression and activity. A growing number of approved gene therapy clinical trials have been designed to increase SERCA2a expression in the myocardium of patients with HF. The results published show long lasting results improving cardiac function after a single intracoronary administration of AAV carrying the SERCA2a complementary DNA. The biosafety and efficacy of AAV/SERCA2a vectors has been demonstrated for the treatment of patients with HF, opening the field for testing new targets to modulate cardiac gene expression with more advanced vector systems for cardiovascular gene therapy in the near future.
Acknowledgment
This work was financed by CONACYT grant 164413 to AZH.
Disclosure
The authors report no conflicts of interest in this work.
References
Bers DM. Excitation – Contraction Coupling and Cardiac Contractile Force. 2nd ed. Boston, MA: Kluwer Academic Publishers; 2001. | |
Bers DM. Cardiac excitation – contraction coupling. Nature. 2002; 415(6868):198–205. | |
Bers DM. Calcium cycling and signaling in cardiac myocytes. Annu Rev Physiol. 2008;70:23–49. | |
Yano K, Zarain-Herzberg A. Sarcoplasmic reticulum calsequestrins: structural and functional properties. Mol Cell Biochem. 1994;135(1):61–70. | |
Beard NA, Laver DR, Dulhunty AF. Calsequestrin and the calcium release channel of skeletal and cardiac muscle. Prog Biophys Mol Biol. 2004;85(1):33–69. | |
Beard NA, Wei L, Dulhunty AF. Control of muscle ryanodine receptor calcium release channels by proteins in the sarcoplasmic reticulum lumen. Clin Exp Pharmacol Physiol. 2009;36(3):340–345. | |
Bernardo BC, Weeks KL, Pretorius L, McMullen JR. Molecular distinction between physiological and pathological cardiac hypertrophy: experimental findings and therapeutic strategies. Pharmacol Ther. 2010;128(1):191–227. | |
Anwar A, Taimor G, Korkusuz H, et al. PKC-independent signal transduction pathways increase SERCA2 expression in adult rat cardiomyocytes. J Mol Cell Cardiol. 2005;39(6):911–919. | |
Arai M, Suzuki T, Nagai R. Sarcoplasmic reticulum genes are upregulated in mild cardiac hypertrophy but downregulated in severe cardiac hypertrophy induced by pressure overload. J Mol Cell Cardiol. 1996;28(8):1583–1590. | |
Reyes-Juarez JL, Zarain-Herzberg A. El retículo sarcoplásmico, su función y su papel en las enfermedades cardiacas. [Function and role of the sarcoplasmic reticulum in heart disease]. Arch Cardiol Mex. 2006; 76(Suppl 4):S18–S32. Spanish. | |
Alpert NR, Brosseau C, Federico A, Krenz M, Robbins J, Warshaw DM. Molecular mechanics of mouse cardiac myosin isoforms. Am J Physiol. 2002;283(4):H1446–H1454. | |
Vannier C, Chevassus H, Vassort G. Ca-dependence of isometric force kinetics in single skinned ventricular cardiomyocytes from rats. Cardiovasc Res. 1996;32(3):580–586. | |
Baker AJ, Figueredo VM, Keung EC, Camacho SA. Ca2+ regulates the kinetics of tension development in intact cardiac muscle. Am J Physiol. 1998;275(3 Pt 2):H744–H750. | |
Palmer S, Kentish JC. Roles of Ca2+ and crossbridge kinetics in determining the maximum rates of Ca2+ activation and rela-xation in rat and guinea pig skinned trabeculae. Circ Res. 1998;83(2):179–186. | |
Teucher N, Prestle J, Seidler T, et al. Excessive sarcoplasmic/endoplasmic reticulum Ca2+-ATPase expression causes increased sarcoplasmic reticulum Ca2+ uptake but decreases myocyte shortening. Circulation. 2004;110(23):3553–3559. | |
Bassani JW, Qi M, Samarel AM, Bers DM. Contractile arrest increases sarcoplasmic reticulum calcium uptake and SERCA2 gene expression in cultured neonatal rat heart cells. Circ Res. 1994;74(5):991–997. | |
Kaprielian R, del Monte F, Hajjar RJ. Targeting Ca2+ cycling proteins and the action potential in heart failure by gene transfer. Basic Res Cardiol. 2002;97(Suppl 1):I136–I145. | |
Soltis AR, Saucerman JJ. Synergy between CaMKII substrates and β-adrenergic signaling in regulation of cardiac myocyte Ca(2+) handling. Biophys J. 2010;99(7):2038–2047. | |
Zhang T, Johnson EN, Gu Y, et al. The cardiac-specific nuclear delta(B) isoform of Ca2+/calmodulin-dependent protein kinase II induces hypertrophy and dilated cardiomyopathy associated with increased protein phosphatase 2A activity. J Biol Chem. 2002;277(2):1261–1267. | |
Backs J, Backs T, Neef S, et al. The delta isoform of CaM kinase II is required for pathological cardiac hypertrophy and remodeling after pressure overload. Proc Natl Acad Sci U S A. 2009;106(7):2342–2347. | |
Tada M, Kirchberger MA, Repke DI, Katz AM. The stimulation of calcium transport in cardiac sarcoplasmic reticulum by adenosine 3′:5′-monophosphate-dependent protein kinase. J Biol Chem. 1974;249(19):6174–6180. | |
Katz S, Richter B, Eibschutz B. Characterization of calmodulin-dependent and cyclic-AMP-dependent protein kinase stimulation of cardiac sarcoplasmic reticulum calcium transport. Adv Myocardiol. 1985;6:233–247. | |
Hoshijima M, Knoll R, Pashmforoush M, Chien K. Reversal of calcium cycling defects in advanced heart failure toward molecular therapy. J Am Coll Cardiol. 2006;48(9 Suppl 1):A15–A23. | |
Chu G, Li L, Sato Y, et al. Pentameric assembly of phospholamban facilitates inhibition of cardiac function in vivo. J Biol Chem. 1998;273(50):33674–33680. | |
Negash S, Yao Q, Sun H, Li J, Bigelow DJ, Squier TC. Phospholamban remains associated with the Ca2+- and Mg2+-dependent ATPase following phosphorylation by cAMP-dependent protein kinase. Biochem J. 2000;351(Pt 1):195–205. | |
Bidwell P, Blackwell DJ, Hou Z, Zima AV, Robia SL. Phospholamban binds with differential affinity to calcium pump conformers. J Biol Chem. 2011;286(40):35044–35050. | |
Reiken S, Gaburjakova M, Guatimosim S, et al. Protein kinase A phosphorylation of the cardiac calcium release channel (ryanodine receptor) in normal and failing hearts. Role of phosphatases and response to isoproterenol. J Biol Chem. 2003;278(1):444–453. | |
Huang FL, Glinsmann WH. Separation and characterization of two phosphorylase phosphatase inhibitors from rabbit skeletal muscle. Eur J Biochem. 1976;70(2):419–426. | |
Nimmo GA, Cohen P. The regulation of glycogen metabolism. Purification and characterisation of protein phosphatase inhibitor-1 from rabbit skeletal muscle. Eur J Biochem. 1978;87(2):341–351. | |
Braunwald E, Bristow MR. Congestive heart failure: 50 years of progress. Circulation. 2000;102(Suppl 4):IV14–IV23. | |
Lehnart SE, Wehrens XH, Reiken S, et al. Phosphodiesterase 4D deficiency in the ryanodine-receptor complex promotes heart failure and arrhythmias. Cell. 2005;123(1):25–35. | |
Marx SO, Reiken S, Hisamatsu Y, et al. PKA phosphorylation dissociates FKBP12.6 from the calcium release channel (ryanodine receptor): defective regulation in failing hearts. Cell. 2000;101(4):365–376. | |
Ai X, Curran JW, Shannon TR, Bers DM, Pogwizd SM. Ca2+/calmodulin-dependent protein kinase modulates cardiac ryanodine receptor phosphorylation and sarcoplasmic reticulum Ca2+ leak in heart failure. Circ Res. 2005;97(12):1314–1322. | |
Nagai R, Zarain-Herzberg A, Brandl CJ, et al. Regulation of myocardial Ca2+-ATPase and phospholamban mRNA expression in response to pressure overload and thyroid hormone. Proc Natl Acad Sci U S A. 1989;86(8):2966–2970. | |
Zarain-Herzberg A, Fragoso-Medina J, Estrada-Aviles R. Calcium-regulated transcriptional pathways in the normal and pathologic heart. IUBMB Life. 2011;63(10):847–855. | |
Prasad AM, Inesi G. Silencing calcineurin A subunit reduces SERCA2 expression in cardiac myocytes. Am J Physiol Heart Circ Physiol. 2011;300(1):H173–H180. | |
Prasad AM, Inesi G. Regulation and rate limiting mechanisms of Ca2+ ATPase (SERCA2) expression in cardiac myocytes. Mol Cell Biochem. 2012;361(1–2):85–96. | |
Rocchetti M, Besana A, Mostacciuolo G, et al. Modulation of sarcoplasmic reticulum function by Na+/K+ pump inhibitors with different toxicity: digoxin and PST2744 ([E,Z]-3-[[2-aminoethoxy]imino]androstane-6,17-dione hydrochloride). J Pharmacol Exp Ther. 2005;313(1):207–215. | |
Micheletti R, Mattera GG, Rocchetti M, et al. Pharmacological profile of the novel inotropic agent (E,Z)-3-([2-aminoethoxy]imino)androstane-6,17-dione hydrochloride (PST2744). J Pharmacol Exp Ther. 2002;303(2):592–600. | |
Micheletti R, Palazzo F, Barassi P, et al. Istaroxime, a stimulator of sarcoplasmic reticulum calcium adenosine triphosphatase isoform 2a activity, as a novel therapeutic approach to heart failure. Am J Cardiol. 2007;99(2A):24A–32A. | |
Mattera GG, Lo Giudice P, Loi FM, et al. Istaroxime: a new luso-inotropic agent for heart failure. Am J Cardiol. 2007;99(2A):33A–40A. | |
Sabbah HN, Imai M, Cowart D, Amato A, Carminati P, Gheorghiade M. Hemodynamic properties of a new-generation positive luso-inotropic agent for the acute treatment of advanced heart failure. Am J Cardiol. 2007;99(2A):41A–46A. | |
Shah SJ, Blair JE, Filippatos GS, et al. Effects of istaroxime on diastolic stiffness in acute heart failure syndromes: results from the Hemodynamic, Echocardiographic, and Neurohormonal Effects of Istaroxime, a Novel Intravenous Inotropic and Lusitropic Agent: a Randomized Controlled Trial in Patients Hospitalized with Heart Failure (HORIZON-HF) trial. Am Heart J. 2009;157(6):1035–1041. | |
Ferrandi M, Barassi P, Tadini-Buoninsegni F, et al. Istaroxime stimulates SERCA2a and accelerates calcium cycling in heart failure by relieving phospholamban inhibition. Br J Pharmacol. 2013;169(8):1849–1861. | |
Gobbini M, Armaroli S, Banfi L, et al. Novel analogues of istaroxime, a potent inhibitor of Na+, K+-ATPase: synthesis and structure–activity relationship. J Med Chem. 2008;51(15):4601–4608. | |
Gobbini M, Armaroli S, Banfi L, et al. Novel analogues of istaroxime, a potent inhibitor of Na(+), K(+)-ATPase: synthesis, structure–activity relationship and 3D-quantitative structure-activity relationship of derivatives at position 6 on the androstane scaffold. Bioorg Med Chem. 2010;18(12):4275–4299. | |
Cornea RL, Gruber SJ, Lockamy EL, et al. High-throughput FRET assay yields allosteric SERCA activators. J Biomol Screen. 2013;18(1):97–107. | |
Koitabashi N, Arai M, Tomaru K, et al. Carvedilol effectively blocks oxidative stress-mediated downregulation of sarcoplasmic reticulum Ca2+-ATPase 2 gene transcription through modification of Sp1 binding. Biochem Biophys Res Commun. 2005;328(1):116–124. | |
Hong-Li S, Lei L, Lei S, et al. Cardioprotective effects and underlying mechanisms of oxymatrine against ischemic myocardial injuries of rats. Phytother Res. 2008;22(7):985–989. | |
Cao YG, Jing S, Li L, et al. Antiarrhythmic effects and ionic mechanisms of oxymatrine from Sophora flavescens. Phytother Res. 2010;24(12):1844–1849. | |
Shen XC, Yang YP, Xiao TT, Peng J, Liu XD. Protective effect of oxymatrine on myocardial fibrosis induced by acute myocardial infarction in rats involved in TGF-β1-Smads signal pathway. J Asian Nat Prod Res. 2011;13(3):215–224. | |
Huang XY, Chen CX. Effect of oxymatrine, the active component from Radix Sophorae flavescentis (Kushen), on ventricular remodeling in spontaneously hypertensive rats. Phytomedicine. 2013;20(3–4):202–212. | |
Zhang M, Wang X, Wang X, et al. Oxymatrine protects against myocardial injury via inhibition of JAK2/STAT3 signaling in rat septic shock. Mol Med Rep. 2013;7(4):1293–1299. | |
Wu CK, Lee JK, Chiang FT, et al. Plasma levels of tumor necrosis factor-α and interleukin-6 are associated with diastolic heart failure through downregulation of sarcoplasmic reticulum Ca2+ ATPase. Crit Care Med. 2011;39(5):984–992. | |
Hu ST, Tang Y, Shen YF, et al. Protective effect of oxymatrine on chronic rat heart failure. J Physiol Sci. 2011;61(5):363–372. | |
Zhang Y, Dong Z, Jin L, et al. Arsenic trioxide-induced hERG K(+) channel deficiency can be rescued by matrine and oxymatrine through up-regulating transcription factor Sp1 expression. Biochem Pharmacol. 2013;85(1):59–68. | |
Davia K, Bernobich E, Ranu HK, et al. SERCA2A overexpression decreases the incidence of aftercontractions in adult rabbit ventricular myocytes. J Mol Cell Cardiol. 2001;33(5):1005–1015. | |
del Monte F, Harding SE, Schmidt U, et al. Restoration of contractile function in isolated cardiomyocytes from failing human hearts by gene transfer of SERCA2a. Circulation. 1999;100(23):2308–2311. | |
Hajjar RJ, Kang JX, Gwathmey JK, Rosenzweig A. Physiological effects of adenoviral gene transfer of sarcoplasmic reticulum calcium ATPase in isolated rat myocytes. Circulation. 1997;95(2):423–429. | |
He H, Giordano FJ, Hilal-Dandan R, et al. Overexpression of the rat sarcoplasmic reticulum Ca2+ ATPase gene in the heart of transgenic mice accelerates calcium transients and cardiac relaxation. J Clin Invest. 1997;100(2):380–389. | |
Niwano K, Arai M, Koitabashi N, et al. Lentiviral vector-mediated SERCA2 gene transfer protects against heart failure and left ventricular remodeling after myocardial infarction in rats. Mol Ther. 2008;16(6):1026–1032. | |
Celladon Corporation. Efficacy and Safety Study of Genetically Targeted Enzyme Replacement Therapy for Advanced Heart Failure. Available from http://clinicaltrials.gov/ct2/show/study/NCT00454818. Accessed January 9, 2014. | |
Jaski BE, Jessup ML, Mancini DM, et al. Calcium Upregulation by Percutaneous Administration of Gene Therapy in Cardiac Disease (CUPID trial), a first-in-human Phase I/II clinical trial. J Card Fail. 2009;15(3):171–181. | |
Hajjar RJ, Zsebo K, Deckelbaum L, et al. Design of a Phase I/II trial of intracoronary administration of AAV1/SERCA2a in patients with heart failure. J Card Fail. 2008;14(5):355–367. | |
Jessup M, Greenberg B, Mancini D, et al. Calcium Upregulation by Percutaneous Administration of Gene Therapy in Cardiac Disease (CUPID): a Phase II trial of intracoronary gene therapy of sarcoplasmic reticulum Ca2+-ATPase in patients with advanced heart failure. Circulation. 2011;124(3):304–313. | |
Zsebo KM, Yaroshinsky A, Rudy JJ, et al. Long term effects of AAV1/SERCA2a gene transfer in patients with severe heart failure: analysis of recurrent cardiovascular events and mortality. Circ Res. Epub September 24, 2013. | |
Celladon Corporation. A Study of Genetically Targeted Enzyme Replacement Therapy for Advanced Heart Failure (CUPID-2b). Available from http://clinicaltrials.gov/ct2/show/record/NCT01643330. Accessed January 9, 2014. | |
Assistance Publique - Hôpitaux de Paris. AAV1-CMV-Serca2a GENe Therapy Trial in Heart Failure (AGENT-HF). Available from: http://clinicaltrials.gov/ct2/show/NCT01966887. Accessed January 9, 2014. | |
Imperial College London. The recruitment status of this study is unknown because the information has not been verified recently. Available from: http://clinicaltrials.gov/show/NCT00534703. Accessed January 9, 2014. | |
Kalyanasundaram A, Lacombe VA, Belevych AE, et al. Up-regulation of sarcoplasmic reticulum Ca(2+) uptake leads to cardiac hypertrophy, contractile dysfunction and early mortality in mice deficient in CASQ2. Cardiovasc Res. 2013;98(2):297–306. | |
Nowak KJ, Davies KE. Duchenne muscular dystrophy and dystrophin: pathogenesis and opportunities for treatment. EMBO Rep. 2004;5(9):872–876. | |
Shin JH, Bostick B, Yue Y, Hajjar R, Duan D. SERCA2a gene transfer improves electrocardiographic performance in aged mdx mice. J Transl Med. 2011;9:132. | |
Goonasekera SA, Lam CK, Millay DP, et al. Mitigation of muscular dystrophy in mice by SERCA overexpression in skeletal muscle. J Clin Invest. 2011;121(3):1044–1052. | |
Vandecaetsbeek I, Raeymaekers L, Wuytack F, Vangheluwe P. Factors controlling the activity of the SERCA2a pump in the normal and failing heart. Biofactors. 2009;35(6):484–499. | |
Brini M, Carafoli E. Calcium pumps in health and disease. Physiol Rev. 2009;89(4):1341–1378. | |
Gruber SJ, Haydon S, Thomas DD. Phospholamban mutants compete with wild type for SERCA binding in living cells. Biochem Biophys Res Commun. 2012;420(2):236–240. | |
Zhang T, Guo T, Mishra S, et al. Phospholamban ablation rescues sarcoplasmic reticulum Ca(2+) handling but exacerbates cardiac dysfunction in CaMKIIδ(C) transgenic mice. Circ Res. 2010;106(2):354–362. | |
Kho C, Lee A, Jeong D, et al. SUMO1-dependent modulation of SERCA2a in heart failure. Nature. 2011;477(7366):601–605. | |
Costa MW, Lee S, Furtado MB, et al. Complex SUMO-1 regulation of cardiac transcription factor Nkx2-5. PLoS One. 2011;6(9):e24812. | |
Choudhary C, Kumar C, Gnad F, et al. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science. 2009;325(5942):834–840. | |
Foster DB, Liu T, Rucker J, et al. The cardiac acetyl-lysine proteome. PLoS One. 2013;8(7):e67513. | |
Sundaresan NR, Pillai VB, Gupta MP. Emerging roles of SIRT1 deacetylase in regulating cardiomyocyte survival and hypertrophy. J Mol Cell Cardiol. 2011;51(4):614–618. | |
Sulaiman M, Matta MJ, Sunderesan NR, Gupta MP, Periasamy M, Gupta M. Resveratrol, an activator of SIRT1, upregulates sarcoplasmic calcium ATPase and improves cardiac function in diabetic cardiomyopathy. Am J Physiol. 2010;298(3):H833–H843. | |
Rimbaud S, Ruiz M, Piquereau J, et al. Resveratrol improves survival, hemodynamics and energetics in a rat model of hypertension leading to heart failure. PLoS One. 2011;6(10):e26391. | |
Gupta MP, Samant SA, Smith SH, Shroff SG. HDAC4 and PCAF bind to cardiac sarcomeres and play a role in regulating myofilament contractile activity. J Biol Chem. 2008;283(15):10135–10146. | |
Hossain P, Kawar B, El Nahas M. Obesity and diabetes in the developing world – a growing challenge. N Engl J Med. 2007;356(3):213–215. | |
Relling DP, Esberg LB, Fang CX, et al. High-fat diet-induced juvenile obesity leads to cardiomyocyte dysfunction and upregulation of Foxo3a transcription factor independent of lipotoxicity and apoptosis. J Hypertens. 2006;24(3):549–561. | |
Ouwens DM, Boer C, Fodor M, et al. Cardiac dysfunction induced by high-fat diet is associated with altered myocardial insulin signalling in rats. Diabetologia. 2005;48(6):1229–1237. | |
Alves AJ, Ribeiro F, Goldhammer E, et al. Exercise training improves diastolic function in heart failure patients. Med Sci Sports Exerc. 2012;44(5):776–785. | |
de las Fuentes L, Waggoner AD, Mohammed BS, et al. Effect of moderate diet-induced weight loss and weight regain on cardiovascular structure and function. J Am Coll Cardiol. 2009;54(25):2376–2381. | |
Rolim NP, Medeiros A, Rosa KT, et al. Exercise training improves the net balance of cardiac Ca2+ handling protein expression in heart failure. Physiol Genomics. 2007;29(3):246–252. | |
Paulino EC, Ferreira JC, Bechara LR, et al. Exercise training and caloric restriction prevent reduction in cardiac Ca2+-handling protein profile in obese rats. Hypertension. 2010;56(4):629–635. | |
Assayag P, Charlemagne D, de Leiris J, et al. Senescent heart compared with pressure overload-induced hypertrophy. Hypertension. 1997;29(1 Pt 1):15–21. | |
Azhar G, Zhang X, Wang S, Zhong Y, Quick CM, Wei JY. Maintaining serum response factor activity in the older heart equal to that of the young adult is associated with better cardiac response to isoproterenol stress. Basic Res Cardiol. 2007;102(3):233–244. | |
Kaplan P, Jurkovicova D, Babusikova E, et al. Effect of aging on the expression of intracellular Ca(2+) transport proteins in a rat heart. Mol Cell Biochem. 2007;301(1–2):219–226. | |
Lokuta AJ, Maertz NA, Meethal SV, et al. Increased nitration of sarcoplasmic reticulum Ca2+-ATPase in human heart failure. Circulation. 2005;111(8):988–995. | |
Knyushko TV, Sharov VS, Williams TD, Schoneich C, Bigelow DJ. 3-Nitrotyrosine modification of SERCA2a in the aging heart: a distinct signature of the cellular redox environment. Biochemistry. 2005;44(39):13071–13081. | |
Adachi T, Weisbrod RM, Pimentel DR, et al. S-glutathiolation by peroxynitrite activates SERCA during arterial relaxation by nitric oxide. Nat Med. 2004;10(11):1200–1207. | |
Berger HJ, Zaret BL, Speroff L, Cohen LS, Wolfson S. Regional cardiac prostaglandin release during myocardial ischemia in anesthetized dogs. Circ Res. 1976;38(6):566–571. | |
Lai J, Jin H, Yang R, et al. Prostaglandin F2α induces cardiac myocyte hypertrophy in vitro and cardiac growth in vivo. Am J Physiol. 1996;271(6 Pt 2):H2197–H2208. | |
Chazov EI, Pomoinetsky VD, Geling NG, Orlova TR, Nekrasova AA, Smirnov VN. Heart adaptation to acute pressure overload: an involvement of endogenous prostaglandins. Circ Res. 1979;45(2):205–211. | |
Hara S, Arai M, Tomaru K, et al. Prostaglandin F2α inhibits SERCA2 gene transcription through an induction of Egr-1 in cultured neonatal rat cardiac myocytes. Int Heart J. 2008;49(3):329–342. | |
Andrews C, Ho PD, Dillmann WH, Glembotski CC, McDonough PM. The MKK6–p38 MAPK pathway prolongs the cardiac contractile calcium transient, downregulates SERCA2, and activates NF-AT. Cardiovasc Res. 2003;59(1):46–56. | |
Molkentin JD. Calcineurin–NFAT signaling regulates the cardiac hypertrophic response in coordination with the MAPKs. Cardiovasc Res. 2004;63(3):467–475. | |
Scharf M, Neef S, Freund R, et al. Mitogen-activated protein kinase-activated protein kinases 2 and 3 regulate SERCA2a expression and fiber type composition to modulate skeletal muscle and cardiomyocyte function. Mol Cell Biol. 2013;33(13):2586–2602. | |
Rowe GC, Jiang A, Arany Z. PGC-1 coactivators in cardiac development and disease. Circ Res. 2010;107(7):825–838. | |
Kemi OJ, Hoydal MA, Haram PM, et al. Exercise training restores aerobic capacity and energy transfer systems in heart failure treated with losartan. Cardiovasc Res. 2007;76(1):91–99. | |
Ngoh GA, Facundo HT, Zafir A, Jones SP. O-GlcNAc signaling in the cardiovascular system. Circ Res. 2010;107(2):171–185. | |
Lunde IG, Aronsen JM, Kvaloy H, et al. Cardiac O-GlcNAc signaling is increased in hypertrophy and heart failure. Physiol Genomics. 2012;44(2):162–172. | |
Jones SP, Zachara NE, Ngoh GA, et al. Cardioprotection by N-acetylglucosamine linkage to cellular proteins. Circulation. 2008;117(9):1172–1182. | |
Ngoh GA, Watson LJ, Facundo HT, Dillmann W, Jones SP. Non-canonical glycosyltransferase modulates post-hypoxic cardiac myocyte death and mitochondrial permeability transition. J Mol Cell Cardiol. 2008;45(2):313–325. | |
Hirose K, Tsutsumi YM, Tsutsumi R, et al. Role of the O-linked β-N-acetylglucosamine in the cardioprotection induced by isoflurane. Anesthesiology. 2011;115(5):955–962. | |
Lima VV, Giachini FR, Carneiro FS, et al. O-GlcNAcylation contributes to the vascular effects of ET-1 via activation of the RhoA/Rho-kinase pathway. Cardiovasc Res. 2011;89(3):614–622. | |
Facundo HT, Brainard RE, Watson LJ, et al. O-GlcNAc signaling is essential for NFAT-mediated transcriptional reprogramming during cardiomyocyte hypertrophy. Am J Physiol Heart Circ Physiol. 2012;302(10):H2122–H2130. | |
Sena S, Hu P, Zhang D, et al. Impaired insulin signaling accelerates cardiac mitochondrial dysfunction after myocardial infarction. J Mol Cell Cardiol. 2009;46(6):910–918. | |
Domenighetti AA, Danes VR, Curl CL, Favaloro JM, Proietto J, Delbridge LM. Targeted GLUT-4 deficiency in the heart induces cardiomyocyte hypertrophy and impaired contractility linked with Ca(2+) and proton flux dysregulation. J Mol Cell Cardiol. 2010;48(4):663–672. | |
Brady M, Koban MU, Dellow KA, Yacoub M, Boheler KR, Fuller SJ. Sp1 and Sp3 transcription factors are required for trans-activation of the human SERCA2 promoter in cardiomyocytes. Cardiovasc Res. 2003;60(2):347–354. | |
Takizawa T, Arai M, Tomaru K, et al. Transcription factor Sp1 regulates SERCA2 gene expression in pressure-overloaded hearts: a study using in vivo direct gene transfer into living myocardium. J Mol Cell Cardiol. 2003;35(7):777–783. | |
Yang X, Su K, Roos MD, Chang Q, Paterson AJ, Kudlow JE. O-linkage of N-acetylglucosamine to Sp1 activation domain inhibits its transcriptional capability. Proc Natl Acad Sci U S A. 2001;98(12):6611–6616. | |
Hu Y, Belke D, Suarez J, et al. Adenovirus-mediated overexpression of O-GlcNAcase improves contractile function in the diabetic heart. Circ Res. 2005;96(9):1006–1013. | |
Cox EJ, Marsh SA. Exercise and diabetes have opposite effects on the assembly and O-GlcNAc modification of the mSin3A/HDAC1/2 complex in the heart. Cardiovasc Diabetol. 2013;12(1):101. | |
Clark RJ, McDonough PM, Swanson E, et al. Diabetes and the accompanying hyperglycemia impairs cardiomyocyte calcium cycling through increased nuclear O-GlcNAcylation. J Biol Chem. 2003;278(45):44230–44237. | |
Yokoe S, Asahi M, Takeda T, et al. Inhibition of phospholamban phosphorylation by O-GlcNAcylation: implications for diabetic cardiomyopathy. Glycobiology. 2010;20(10):1217–1226. | |
Kim HS, Woo JS, Joo HJ, Moon WK. Cardiac transcription factor Nkx2.5 is downregulated under excessive O-GlcNAcylation condition. PLoS One. 2012;7(6):e38053. | |
Ikeda S, He A, Kong SW, et al. MicroRNA-1 negatively regulates expression of the hypertrophy-associated calmodulin and Mef2a genes. Mol Cell Biol. 2009;29(8):2193–2204. | |
Li D, He B, Zhang H, et al. The inhibitory effect of miRNA-1 on ET-1 gene expression. FEBS Lett. 2012;586(7):1014–1021. | |
Karakikes I, Chaanine AH, Kang S, et al. Therapeutic cardiac-targeted delivery of miR-1 reverses pressure overload-induced cardiac hypertrophy and attenuates pathological remodeling. J Am Heart Assoc. 2013;2(2):e000078. | |
Hajjar RJ, Schmidt U, Kang JX, Matsui T, Rosenzweig A. Adenoviral gene transfer of phospholamban in isolated rat cardiomyocytes. Rescue effects by concomitant gene transfer of sarcoplasmic reticulum Ca(2+)-ATPase. Circ Res. 1997;81(2):145–153. | |
Most P, Remppis A, Pleger ST, Katus HA, Koch WJ. S100A1: a novel inotropic regulator of cardiac performance. Transition from molecular physiology to pathophysiological relevance. Am J Physiol Regul Integr Comp Physiol. 2007;293(2):R568–R577. | |
Pleger ST, Shan C, Ksienzyk J, et al. Cardiac AAV9-S100A1 gene therapy rescues post-ischemic heart failure in a preclinical large animal model. Sci Transl Med. 2011;3(92):92ra64. | |
Pleger ST, Most P, Boucher M, et al. Stable myocardial-specific AAV6-S100A1 gene therapy results in chronic functional heart failure rescue. Circulation. 2007;115(19):2506–2515. | |
Ritterhoff J, Most P. Targeting S100A1 in heart failure. Gene Ther. 2012;19(6):613–621. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.