Back to Journals » International Journal of Nanomedicine » Volume 14
Self-nanoemulsifying ramipril tablets: a novel delivery system for the enhancement of drug dissolution and stability
Authors Alhasani KF, Kazi M ![]() , Ibrahim MA
, Ibrahim MA ![]() , Shahba AA
, Shahba AA ![]() , Alanazi FK
, Alanazi FK
Received 29 January 2019
Accepted for publication 9 April 2019
Published 18 July 2019 Volume 2019:14 Pages 5435—5448
DOI https://doi.org/10.2147/IJN.S203311
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Thomas Webster
Khalid F Alhasani,1 Mohsin Kazi,1,2 Mohamed Abbas Ibrahim,1 Ahmad A Shahba,1,2 Fars K Alanazi1,2
1Department of Pharmaceutics, College of Pharmacy, King Saud University, Riyadh 11451, Kingdom of Saudi Arabia; 2Kayyali Chair for Pharmaceutical Industries, Department of Pharmaceutics, College of Pharmacy, King Saud University, Riyadh 11451, Kingdom of Saudi Arabia
Background: Ramipril (RMP) suffers from poor aqueous solubility along with sensitivity to mechanical stress, heat, and moisture. The aim of the current study is to improve RMP solubility and stability by designing solid self-nanoemulsifying drug delivery system (S-SNEDDS) as tablet.
Methods: The drug was initially incorporated in different liquid formulations (L-SNEDDS) which were evaluated by equilibrium solubility, droplet size, and zeta potential studies. The optimized formulation was solidified into S-SNEDDS powder by the adsorbent Syloid® and compressed into a self-nanoemulsifying tablet (T-SNEDDS). The optimized tablet was evaluated by drug content uniformity, hardness, friability, disintegration, and dissolution tests. Furthermore, pure RMP, optimized L-SNEDDS, and T-SNEDDS were enrolled in accelerated and long-term stability studies.
Results: Among various liquid formulations, F5 L-SNEDDS [capmul MCM/transcutol/HCO-30 (25/25/50%w/w)] showed relatively high drug solubility, nano-scaled droplet size, and high negative zeta potential value. The optimized SNEDDS solidification with Syloid® at ratio (1:1) resulted in a compressible powder with an excellent flowability. The optimized tablet (T-SNEDDS) showed accepted content uniformity, hardness, friability, and disintegration time (<15 minutes). The optimized L-SNEDDS, S-SNEDDS, and T-SNEDDS showed superior enhancement of RMP dissolution compared to the pure drug. Most importantly, T-SNEDDS showed significant (P<0.05) improvement of RMP stability compared to the pure drug and L-SNEDDS in both accelerated and long-term stability studies.
Conclusion: RMP-loaded T-SNEDDS offers a potential oral dosage form that provides combined improvement of RMP dissolution and chemical stability.
Keywords: solid self-nanoemulsifying drug delivery systems (S-SNEDDS), self-nanoemulsifying tablets, ramipril, dissolution improvement, stability studies
Introduction
Liquid self-nanoemulsifying drug delivery systems (L-SNEDDS) offer high potential in improving the dissolution and bioavailability of poorly-water soluble drugs (PWSDs). However, they have several stability limitations, which are mainly due to their liquid nature. These limitations include rancidity, risk of capsule leakage, incompatibility with capsule shell, and possible drug precipitation during manufacturing and storage.1,2 In addition, some drugs may undergo chemical degradation in the presence of lipids and related excipients.1 This problem has created a window of research on the solidification of lipid-based formulations to create suitable delivery systems for such drugs. This will give the advantage of enhancing PWSD bioavailability by lipid based formulations along with the stabilization benefits of the solid dosage form, such as preventing the loaded drug from precipitation and system stabilization.3
Ramipril (RMP) is an example of such poorly-soluble drugs, and is classified as Class II in the Biopharmaceutics Classification System. RMP, an angiotensin converting enzyme inhibitor, is widely used in controlling major diseases like hypertension and heart failure. Owing to its poor aqueous solubility, RMP experiences poor oral bioavailability of 28–35%.4 In addition, RMP has another critical problem, which is sensitivity to temperature, mechanical stress, and humidity.5 The drug is extensively degraded within acidic and alkaline aqueous systems. Thus, poor water solubility and instability are the two major predicaments limiting effective RMP oral delivery. Solidification of L-SNEDDS is a perfect theoretical technique to overcome these two problems, as SNEDDS will enhance the drug aqueous solubility and the solidification will stabilize the drug within the formulation.
Among various solidification techniques, adsorption using porous silica has become more favorable due to various advantages, such as low cost, ease of application, and high adsorption capacity.3 These adsorbents have succeeded in improving the biopharmaceutical properties of many drugs such as ibuprofen, celecoxib,and lovastatin.6–8 These adsorbents are able to convert L-SNEDDS into free-flowing SNEDDS powder, which can be filled directly into capsules or mixed with suitable excipients before compression into tablets.3 Among various solid dosage forms, tablets show several advantages, such as ease of preparation, convenience, cost effectiveness, and dose accuracy.9 However, a limited number of published research studied the formulation of PWSD into SNEDDS as tablet dosage form, and no study has continued to formulate a successful SNEDDS of RMP into a tablet.10
Accordingly, the aim of the current study is to formulate RMP S-SNEDDS as a tablet to achieve combined enhancement of drug solubility and stability. The drug was initially incorporated in a liquid formulation (L-SNEDDS), which was then solidified into S-SNEDDS powder by the adsorbent Syloid® and finally compressed into a self-nanoemulsifying tablet (T-SNEDDS). The optimized tablet was evaluated by various tablet quality control tests, in vitro dissolution, and stability studies.
Materials and methods
Materials
Ramipril (RMP) was donated by Zydus Cadila Healthcare Ltd. (Ahmedabad, Gujrat, India). Cremercoor® MCT 70/30 (MCT, Caprylic/capric acid medium chain triglycerides, 70% caprylic C8, 30% capric C10) was gratuitously provided by Cremer Oleo GmbH & Co. KG (Hamburg, Germany). Imwitor 988 (I988, Mixture of medium chain mono and diglycerides) was purchased from Sasol, GmbH (Werk Witten, Witten, Germany). Capmul MCM (CMCM, medium chain mono diglycerides c8-10) was donated by Abitec Corporation (Germany). The non-ionic surfactants TO106V (PEG-6 sorbitan oleate water-insoluble, HLB=10) and HCO-30 (Polyoxyethylene hydrogenated castor oil, HLB=11) were donated by Nikko Chemicals Co. (Tokyo, Japan). The co-solvent Transcutol® (TC, highly purified diethylene glycol monoethyl ether) was donated by Gattefossé (Lyon, France). Syloid® (SYL) was gratuitously provided by Grace Gmbh & Co. KG (Worms, Germany). The high purity Milli-Q water was obtained through a Milli-Q Integral Water Purification System (Millipore, Bedford, MA). The Ramipril Sandoz® 2.5 mg tablet was used as a marketed drug. All other reagents were of analytical grade and used without further purification.
Design of liquid self-nanoemulsifying drug delivery system (L-SNEDDS)
Different ratios of oils, surfactants, and co-solvent were prepared and incorporated with the drug to achieve the optimum L-SNEDDS composition (Table 1).11 Six mixtures were designed with consideration of different lipophilicities and different ratios of each component to provide a wide range and variety of solublization. The prepared formulations were categorized in four different types based on their lipid composition and according to the lipid formulation classification system (LFCS).12 Briefly, Type I formulations contain 100% pure oil (surfactant free systems). Type II formulations contain water insoluble surfactants with different oil proportions in the formulation. Type IIIA formulations (subdivision of Type III) contain water soluble surfactants along with significant proportions of oils, while Type IIIB formulations (subdivision of Type III) are predominantly water soluble components with less oil proportions. Type IV formulations contain only water-soluble surfactant/cosolvent (oil free systems).
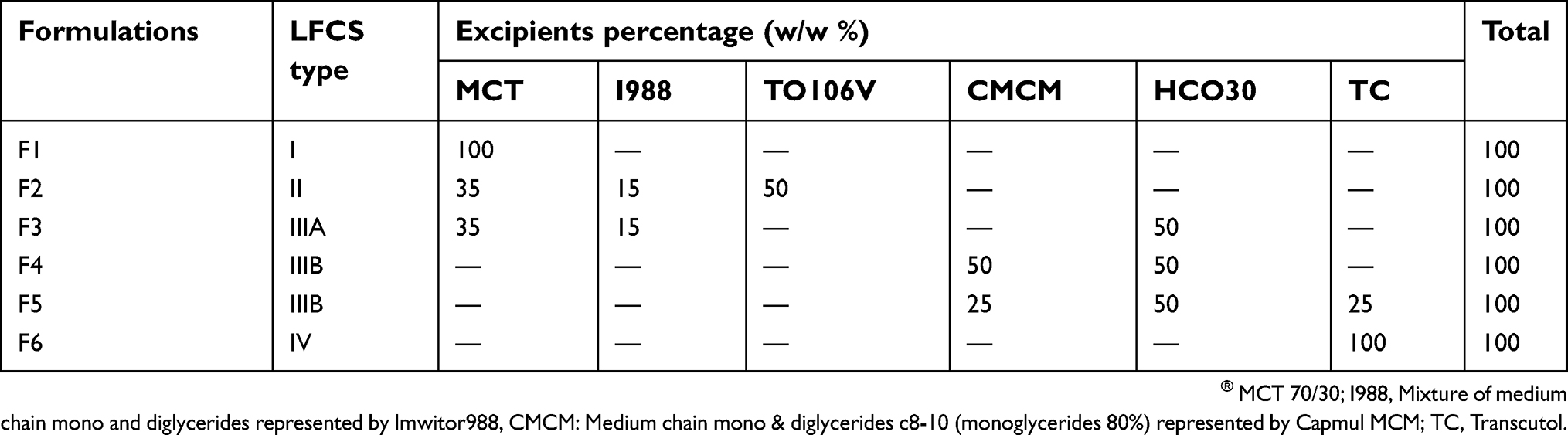 |
Table 1 The compositions of RMP lipid-based formulations |
Equilibrium solubility study
To maximize the drug loading, RMP was incorporated in each lipid-based formulation in excess amounts. After mixing by vortex, the formulations were incubated for 7 days at 37°C to allow for reaching the maximum drug solubility in the formulation. Then, the samples were centrifuged and an aliquot of the supernatant (≈50 mg) was diluted with solvent [acetonitrile: 3% orthophosphoric acid aqueous solution (2:3)] in a 25 mL volumetric flask.13 Three replicates of each sample were analyzed by the adopted analysis method using UHPLC.14
Droplet size and zeta potential
The formulations were diluted at a ratio of 1:1,000 v/v (formulation: distilled water) and mixed for 1 minute before analysis. The droplet size distribution and zeta potential of the diluted formulation were measured using the Zetasizer (Nano ZS, Germany) particle size and zeta potential analyzer.15
Preparation of optimized L-SNEDDS
The optimal L-SNEDDS was prepared by mixing the oil, surfactant, and co-solvent at an optimized ratio. To avoid risk of precipitation, RMP was loaded at ≈85% of its equilibrium solubility in the formulation. Finally, the components were thoroughly mixed to ensure complete drug solubilization and homogenization.2
Solidification of L-SNEDDS by Syloid®
The optimal RMP loaded L-SNEDDS was incorporated into the adsorbent Syloid® (SYL) to convert L-SNEDDS into solid powder (S-SNEDDS). L-SNEDDS was mixed with SYL at ratios 2:1, 1.5:1, 1:1, 1:1.5, and 1:2 (w/w), respectively.16 This step was carried out to screen the influence of L-SNEDDS:SYL ratio on the flowability and processability of the resultant solidified powder and to select the optimal formulation candidate for tablet compression.
Solidified powder (S-SNEDDS) characterization studies
Solidified powders were examined using powder characterization tests such as scanning electron microscopy, differential scanning calorimetry, X-ray diffraction, bulk density, tapped density, and angle of repose.
Scanning electron microscopy (SEM)
Surface morphology of the samples was visualized by scanning electron microscopy (Jeol JSM 5400LV SEM, Tokyo, Japan) operated at 15 kV. The samples were coated with a thin gold palladium layer (SPI sputter coater, PA19380, USA) under an argon atmosphere in a high vacuum evaporator, and images were then acquired from the system.1,17
Differential scanning calorimetry (DSC)
The samples (3–5 mg) were sealed in aluminum pans and heated at a scanning rate of 10°C min–1 under dry nitrogen flow (30 mLmin–1) over a temperature range of 0–200°C. Indium standard were used to calibrate the DSC temperature and enthalpic scale. Data from the thermal analysis were recorded using the Shimadzu software program in a TA 50I PC system.17
X-ray diffraction (XRD)
Powder crystallinity was assessed by a multipurpose X-ray diffractometer (Ultima IV, Rigaku, Japan). The samples were analyzed using CuKα radiation of wavelength 1.54,056 A, generated at 40 kV voltage, 40 mA current, and receiving slit of 0.3 mm. Analysis were preformed over the 2θ range of 3–60° with an angular increment of 0.5°/min and scan step time of 1.0 second.1,17
Hausner ratio, compressibility index, and angle of repose
The samples (3 gm) were weighted and placed in a graduated cylinder to measure the occupied volume by the powder. Bulk density (BD) was calculated by dividing the weight by the bulk volume (volume before tapping). The same powder was tapped several times from the height of 2 inches and the tapped density (TD) was calculated via dividing the weight by the tapped volume (volume after tapping). BD and TD values were used to calculate the compressibility index (CI, Equation 1) and Hausner ratio (HR, Equation 2) which were used to indicate the powder flowability accordingly.1,18
(1) (2)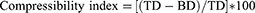
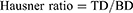
The angle of repose (AR) was studied using the height funnel method.19 The solidified powder was poured from a funnel, at a fixed height, onto the base. The angle of repose was determined as the inverse tangent (arctan) of the maximum cone height to average radius ratio (Equation 3).1
(3)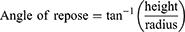
According to USP guidelines, the powder flowability was judged as excellent, good, fair, passable, poor, or very poor, depending on the calculated CI, HR, and AR.18
Tablet pre-formulation and compression
The optimal drug-loaded S-SNEDDS was mixed with Avicel®, magnesium stearate, and Aerosil® at 49.1%, 0.8%, and 0.1% ratios, respectively (Table 2).20 After weighing of the required quantities of ingredients, the powder was mixed for 30 minutes using a turbula mixer (model S2Y, Erweka, Germany). Then, the powder was directly compressed by a single punch tablet compression machine (Korsch EK0, Germany) to produce enough tablets quantity to perform the required tests for evaluation. RMP theoretical content in each tablet was calculated to be 2.5 mg in a total tablet weight of 500 mg (Table 2).
 |
Table 2 The composition of optimized RMP SNEDDS formulations |
Tablets characterization and quality control
Different quality control tests were performed, including drug content uniformity, hardness test, friability, disintegration, and dissolution tests. Depending on each tests requirements, the number of investigated tablets varied from 6–13 for each test.
Drug content uniformity
Each tablet (n=10 tablets) was ground in a mortar and dissolved in 25 mL solvent [acetonitrile: 3% orthophosphoric acid aqueous solution (2:3)]. The samples were analyzed by the developed UHPLC method to check the RMP content.14 According to USP, acceptance criteria for tablet content is 100%±15 and the acceptance value (AV) should be ≤15.21
Hardness test
The hardness of six tablets were evaluated by hardness tester (Erweka TBH 28, Germany) to examine the mechanical strength of the prepared tablets. In addition, this test was also performed frequently during tablet compression as an in-process control test.20
Friability test
The tablet friability was tested using USP-friabilator (Electrolab EF-2 friabilator, India). According to the USP guidelines, the number of tested tablets should be equivalent to 6.5 g.22 Accordingly, 13 tablets were revolved together for 4 minutes at 25 rpm. After the test, the tablets were re-weighed and the weight loss was calculated (Equation 4).1 The percentage weight loss should not exceed 1% to consider the batch accepted.20
(4)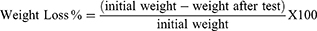
Disintegration test
The disintegration test was carried out according to USP method.23 Six tablets were examined by USP disintegration tester (Electrolab ED-2L) using water as a disintegration medium at 37°C. The disintegration time should not be more than 30 minutes to consider the batch accepted.20
In-vitro dissolution studies
The test was conducted using USP dissolution apparatus II (Model: UDT-804, LOGAN Inst. Corp., USA) coupled with a paddle stirrer at a speed of 50 rpm. In total, 250 mL simulated gastric fluid (with no enzymes, pH =1.2) was used as dissolution medium. The samples were withdrawn at 5, 10, 15, 30, 45, 60, and 120 minutes and filtered using a filter syringe. Thereafter, samples were diluted in appropriate solvent and analyzed by the validated UHPLC method. During the dissolution study, temperature was maintained at 37±0.5°C, and fresh media was added after each withdrawal.24 The dissolution profiles were evaluated using dissolution efficiency,which was calculated from the area under the dissolution curve at time t (determined using the non-linear trapezoidal rule and expressed as a percentage of the area of the trapezoid described by 100% dissolution in the same time).25
Stability study
The chemical and physical stability of L-SNEDDS, T-SNEDDS, and pure RMP were assessed at accelerated and long-term storage conditions, according to the Gulf cooperation council (GCC) guidelines for stability testing.26 The physical appearance of the samples was examined to record any physical changes, while the chemical RMP stability was evaluated based on the percentage of intact RMP remaining in formulation.27
Accelerated stability studies
Pure RMP, L-SNEDDS, and T-SNEDDS samples were stored in climatic stability chambers at 40°C±2°C and relative humidity (RH) of 75%±5%.26,28,29 Samples were withdrawn at predetermined intervals (0, 1, 2, 3, and 6 months) and allowed to equilibrate at room temperature. Samples were evaluated for physical appearance and further assayed by UHPLC to determine the RMP content remaining in the formulations.
Long-term stability study
To gain more information, samples were also stored at long-term storage conditions at 30°C and relative humidity (RH) of 65% (Climatic Zone IV a: Hot/Humid Climate) with predetermined intervals (0, 3,and 6 months).26,28,29 Similar to the accelerated stability studies, samples were evaluated for physical appearance and further analyzed by UHPLC.
Statistical analysis
SPSS 25® software was used to analyze the data. One-way analysis of variance (ANOVA) followed by post-hoc tests (LSD) were used to analyze and compare the results of dissolution and stability study in both conditions. The comparison between dissolution results was performed in terms of dissolution efficiency. P-value<0.05 was considered as significant.13
Results
Equilibrium solubility study
F6 (Type IV), which contained 100% cosolvent, showed the highest RMP solubility of 30.7 mg/g, followed by F5 (Type IIIB), which dissolved 23.2 mg/g (Table 3). On the other hand, F1 (Type I, which contained 100% oil) showed the least RMP solubility of 0.6 mg/g.
 |
Table 3 Effect of liquid formulation composition on equilibrium solubility, droplet size, and zeta potential |
Droplet size and zeta potential
The droplet size and zeta potential of the diluted liquid formulations are presented in Table 3. The aqueous dilution of F1 and F2 presented crude emulsions with larger droplet sizes (43,355 nm and 1,120 nm, respectively), while F3–F6 presented fine emulsions within the nano-metric scale (20–200 nm) and, thus, could be categorized as SNEDDS.30 The zeta potential of the diluted formulations varied from −26.7 to +2.0 mV, where F4 and F5 showed relatively higher negative zeta potential values (−26 to −27 mV). This could give an indication of the potential physical stability of the nanoemulsion generated from these formulations.
Optimization of L-SNEDDS
F5 (CMCM:TC (1:1)/HCO-30 (1/1)) showed relatively high drug loading, low droplet size, and high negative zeta potential. Accordingly, F5 was selected as the optimum RMP L-SNEDDS, and was utilized in subsequent solidification and tablet compression experiments.
Solidification of L-SNEDDS by syloid®
The results from the solidification process revealed that SP1 showed a highly caking, very wet powder with very poor flowability (Table 4). Upon increasing the SYL ratio, the powder caking was reduced along with improved flowability. Hence, SP3 and SP4 showed excellent powder flowability (CI<10%, HR=1.00–1.11, and AR=25–30).18 However, SP3 was preferred to SP4 because it showed better powder properties and possesses a higher SNEDDS ratio (50%w/w), which would lead to higher drug loading within the final formula. Accordingly, SP3 was selected as the optimal solidified SNEDDS (S-SNEDDS) and was utilized in the further tableting experiments.
 |
Table 4 Effect of liquid formulation/adsorbent ratio on the flow properties of solidified powder |
Solidified powder (S-SNEDDS) characterization studies
Scanning electron microscopy (SEM)
The SEM images revealed that pure SYL showed brick-like blocks with small particles on its surface (Figure 1A). On the other hand, the image of S-SNEDDS showed that the adsorbent surface was smoothened after SNEDDS loading without significant changes in the morphology of the adsorbent (Figure 1B).
 |
Figure 1 SEM images of (A) Pure SYL (B) S-SNEDDS (L-SNEDDS:SYL ratio, 1:1).Abbreviations: SEM, scanning electron microscopy; SYL, Syloid; S-SNEDDS, solid self-nanoemulsifying drug delivery system; L-SNEDDS, liquid self-nanoemulsifying drug delivery system. |
Differential scanning calorimetry (DSC)
The chromatograms from the DSC studies showed that SYL has no endothermic peak, while pure RMP exhibited a sharp endothermic peak at 115°C (Figure 2), which confirms the crystalline state of the drug. Upon solidification, the RMP peak was completely disappeared in the case of drug-loaded S-SNEDDS, indicating that RMP was completely solubilized within S-SNEDDS and that the solidification process did not cause any drug precipitation from the formulation.2
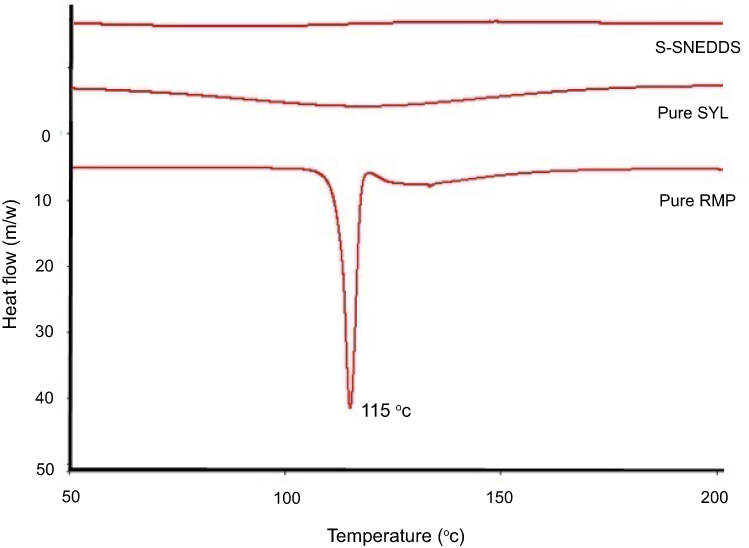 |
Figure 2 DSC chromatograms of pure RMP, pure SYL, and S-SNEDDS.Abbreviations: DSC, Differential scanning calorimetry; RMP, ramipril; SYL, Syloid; S-SNEDDS, solid self-nanoemulsifying drug delivery system. |
X-ray diffraction (XRD)
The XRD findings were in good agreement with the DSC results. Pure RMP exhibited typical X-ray diffraction peaks at 3–30° (2θ) (Figure 3). In contrast, pure SYL and RMP-loaded S-SNEDDS showed complete absence of RMP typical diffraction peaks. These data confirm that RMP was transformed into an amorphous state within the formulation.
 |
Figure 3 XRD of (A) Pure RMP, (B) Pure SYL, and (C) S-SNEDDS.Abbreviations: XRD, X-ray diffraction; RMP, ramipril; SYL, Syloid; S-SNEDDS, solid self-nanoemulsifying drug delivery system. |
Tablet characterization and quality control
During the preparation, it was confirmed that the compressed tablets were ejected easily from the die, showed good appearance, and good mechanical strength with no cracking on the surface.
Tablet content uniformity
The assay mean of tested tablets (X̄) was 98.7%, which was equivalent to 2.46 mg RMP in tablet unit (Table 5). Maximum and minimum assays were 107.3 mg and 92.5 mg, respectively, which were within the acceptable limits (±15%). The acceptance value (AV) for content uniformity was calculated according to USP guidelines (Table 5) and was 12.1, which complies with the acceptance criteria (≤15).21 Accordingly, the compressed tablets showed acceptable content uniformity according to USP guidelines.
 |
Table 5 Content uniformity attributes of RMP T-SNEDDS |
Hardness and friability tests
The hardness mean was 4.95 kp, and the friability test revealed that the weight loss was 0.31% (Table 6) which meets the USP limits for tablet friability test (≤1%).20,22
 |
Table 6 Tablet quality parameters of RMP T-SNEDDS |
Disintegration test
The mean disintegration time for RMP T-SNEDDS was found to be 12.05 minutes (Table 6). The relative delay of disintegration time for these tablets could be due to the presence of lipid-based excipient in the tablet. However, the disintegration time was still falling within the acceptance criteria (≤30 minutes) according to USP.20
In-vitro dissolution studies
Pure RMP showed poor dissolution behavior with a maximum of 59% drug release within the 120 minute run time (Figure 4). On the other hand, all the self-nanoemulsifying formulations (L-SNEDDS, S-SNEDDS, and T-SNEDDS) showed significant enhancement of RMP dissolution efficiency compared to RMP as raw drug (P<0.05). Most importantly, all the self-nanoemulsifying formulations (L-SNEDDS, S-SNEDDS, and T-SNEDDS) achieved >95% drug release at 60 minutes.
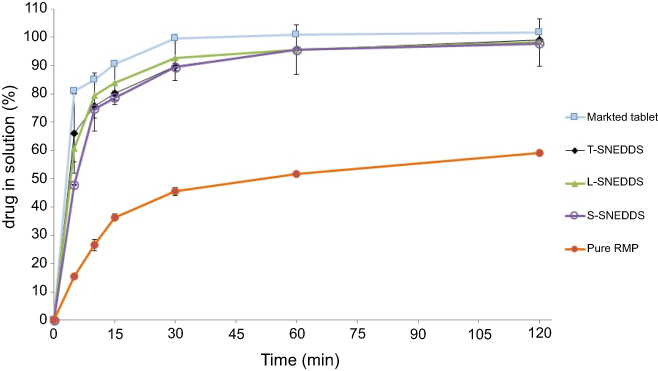 |
Figure 4 In-vitro dissolution studies of RMP formulations using simulated gastric fluid (with no enzymes, pH = 1.2).Abbreviations: RMP, ramipril; T-SNEDDS, self-nanoemulsifying tablet; L-SNEDDS, liquid self-nanoemulsifying drug delivery system; S-SNEDDS, solid self-nanoemulsifying drug delivery system. |
Stability studies
Accelerated stability study
All samples under accelerated conditions were degraded to less than 90% in the first month (Figure 5). L-SNEDDS shows significant (>87%) RMP degradation compared to pure RMP (22%) degradation in the same point (P<0.05). On the other hand, T-SNEDDS (prepared from solidified SNEDDS powder) significantly (P<0.05) reduced RMP degradation to only ≈10% (Figure 5). At the end of the third month, L-SNEDDS maintained only 4% intact RMP in formulation, while T-SNEDDS maintained up to 68% of intact RMP. After 6 months, T-SNEDDS was still able to maintain 62% of the intact drug compared to 27% and 1% in the case of pure RMP and L-SNEDDS, respectively. Regarding physical stability, all samples showed no change in the physical properties and appearances.
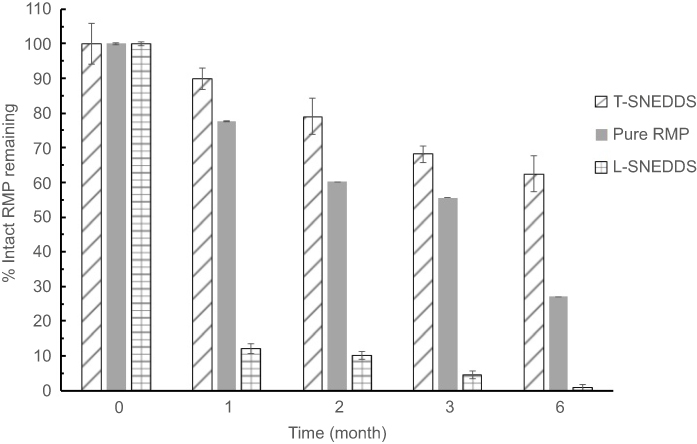 |
Figure 5 Accelerated stability study (40oC/75% RH) for Pure RMP, L-SNEDDS and TSNEDDS.Abbreviations: RMP, ramipril; T-SNEDDS, self-nanoemulsifying tablet; L-SNEDDS, liquid self-nanoemulsifying drug delivery system. |
Long-term stability study
Similar to the accelerated conditions, significant degradation was noticed in the case of L-SNEDDS and pure RMP under the long-term storage conditions (Figure 6). After 6 months of the study, L-SNEDDS and pure RMP maintained 74% and 3% intact RMP, respectively. On the other hand, the T-SNEDDS tablets were able to keep >95% of intact RMP all over the storage period. All samples showed no change in the physical properties and appearances.
 |
Figure 6 Long-term stability study (30oC/65%RH) for Pure RMP, L-SNEDDS and T-SNEDDS.Abbreviations: RMP, ramipril; T-SNEDDS, self-nanoemulsifying tablet; L-SNEDDS, liquid self-nanoemulsifying drug delivery system. |
Discussion
The emerging trends in combinatorial chemistry and drug design led to an increased number of newly developed drug molecules with greater lipophilicity, higher molecular weight, and poor water solubility.31 The poor solubility and slow dissolution can lead to low bioavailability and, hence, suboptimal drug delivery.32 One of the current attractive methods to solve this issue is liquid self-nanoemulsifying drug delivery systems (L-SNEDDS), which potentially improve the solubility of poorly water-soluble drugs (PWSDs) and consequently enhance the drug bioavailability.33 L-SNEDDS is an anhydrous formula of nanoemulsion, which is composed of a liquid homogenous mixture of drug, oil, surfactant, and/or cosurfactant. Upon exposure to GI fluids, these systems would spontaneously self-emulsify to produce enormous drug-loaded micelles (20–200 nm size), leading to enhanced drug solubilization and dissolution within GI aqueous fluids. Small globule size of SNEDDS provides a large interfacial surface area, which improves drug absorption and bioavailability.34 Despite their high potential in improving PWSD bioavailability, L-SNEDDS have several stability problems, which are mainly related to their liquid nature and, thus, could be solved by SNEDDS solidification into powder.
RMP is a potent anti-hypertensive drug, which is widely used in the management of hypertensive disorders. However, oral administration of RMP faces a plight of low aqueous solubility and low bioavailability, which could largely limit its medicinal application in clinical practice. Furthermore, RMP suffers from chemical instability, which is mainly due to its sensitivity to temperature and humidity. Therefore, it is necessary to break these predicaments by exploring a suitable drug delivery system to improve its aqueous solubility as well as chemical stability. Several studies have been carried out to enhance RMP aqueous solubility using liquid self-emulsifying drug delivery systems, but ended with facing various stability issues. Solidification of RMP L-SNEDDS is expected to overcome such limitations, as SNEDDS will enhance the drug aqueous solubility and the solidification will stabilize the drug within the formulation. An earlier study demonstrated that the RMP self-microemulsifying drug delivery system (SMEDDS) has been solidified into powder form using Aersoil 200 as adsorbent in various ratios.35 However, it was found that mixing RMP liquid SMEDDS with the adsorbent formed a damp mass. So, it was necessary to dry the damp solid mixture in the oven at 40°C for 3 hours, which would raise stability issues for such a sensitive drug moiety. In the current study, RMP L-SNEDDS was solidified into free-flowing powder and further compressed into a tablet with no heat or excessive mechanical stress application.
In order to achieve an optimum SNEDDS formula, six formulations were screened for their equilibrium drug solubility, droplet size, and zeta potential. The equilibrium solubility study is an important tool to determine the possible drug content in a single unit dose (such as capsule or tablet). The solubility results revealed that RMP shows limited solubility in pure oils (represented by F1, LFCS-Type I) and maximized solubility in pure co-solvents (represented by F6, LFCS-Type IV) (Table 3). These findings could be correlated to the intermediate value of RMP partition coefficient (log P=3.32),36 which implies low drug solubility in natural lipids and much greater solubility in amphiphilic surfactants, co-surfactants, and co-solvents.37 These data reveal the fact that RMP is a hydrophobic, rather than being a lipophilic, moiety. Hence, it is not a suitable candidate for Type I, Type II, or Type IIIA LFCS systems, which contain a significant amount of lipophilic materials.33
During the design of SNEDDS formulation, it is valuable to determine the droplet size of diluted formulation which is strongly associated with self-emulsification efficiency.15 The large droplet size of F1 and F2 could be explained by the lack of hydrophilic surfactants and predominance of lipophilic oils used in Type I and II formulations.33 The inclusion of hydrophilic excipients (HCO-30 and/or TC) led to a significant reduction of droplet size (<200 nm) and reveals efficient self-emulsification of the formulation (F3–F6, Table 3). On the other hand, the surface charge on the droplets (represented by zeta potential) plays an important role in the physical stability of nanoemulsions. If the droplets have a high negative or positive zeta potential value, they will repel each other, which would improve the nanoemulsion physical stability by lowering the Ostwald ripening rate.38 Both F4 and F5, which contained the non-ionic surfactant HCO-30 at 50% w/w, showed relatively higher negative zeta potential (Table 3). These findings can be attributed to the composition of the oil (free fatty acids + salts), the presence of some anionic impurities in the surfactant (such as free fatty acids), or adsorption of anionic species from the water (such as hydroxyl ions) to the droplet surfaces.39,40
The optimum L-SNEDDS should be selected by keeping a good balance between lower droplet size, higher drug loading, the ability to maintain drug in solution upon aqueous dispersion, and avoidance of drug precipitation during its transit in GIT.2 Accordingly, F5 (CMCM:TC (1:1)/HCO-30 (1/1)) was preferred over F6 (TC 100%) because it contains oil, surfactant, and co-solvent to blend a successful SNEDDS formulation (Table 3). In fact, F6 (LFCS-Type IV formulation) might experience higher risk of drug precipitation upon aqueous dilution compared with F5 (LFCS-Type IIIB formulation).33 In addition, F5 showed a lower droplet size and higher negative zeta potential compared with F6.
In the solidification process, the optimum L-SNEDDS (F5) was mixed with SYL, at five different ratios, to select the optimal formulation for tablet compression. SP3 (SNEDDS: SYL, 1:1 ratio) showed complete solidification, excellent flowability, and relatively higher SNEDDS loading capacity. These findings are in agreement with previous studies and confirm the ultimate characteristics of SYL as adsorbent.41 DSC and XRD data revealed that RMP did not exist in crystalline form within S-SNEDDS. These data can be correlated to complete drug solubilzation within S-SNEDDS and confirm that the solidification process did not cause any drug precipitation.2 The overall characterization results are in agreement with previous studies, which confirmed that the amorphous state is achievable by SYL and was even maintained in accelerated stability conditions for 3 months.42
In the tableting process, avicel served as a binder and diluent to reduce the lipid content in tablet to 25% and, hence, avoid tablet cracking.20 Magnesium stearate served as a lubricant and glidant and was maintained at 0.8% to reduce its adverse effect on tablet disintegration time. Aerosil® was added at 0.1% to compromise for the limited magnesium stearate ratio in the formula. The excellent tablet content and assay results confirm that the tablet manufacturing procedures were appropriate and caused no drug degradation. It was suggested that RMP loading within solidified SNEDDS substantially protected the drug moiety against mechanical stress and possible heat generation during the tableting process.
The superior enhancement of RMP dissolution in the case of all tested SNEDDS formulations is attributed to the ability of RMP-loaded SNEDDS to provide a favorable nanoemulsion environment that maintains RMP solubilized within the nano-sized micelles, formed upon formulation exposure to GI fluids. The self-nanoemulsification process was fast enough to introduce the drug in solution within significantly less time compared to the pure drug. The solidified powder (S-SNEDDS) showed lower drug release, in the initial 30 minutes, compared to L-SNEDDS. This might have been caused by the silica porous carrier SYL as highly effective adsorbent which adhered to drug particles and showed resistance to drug release. Similar results were reported in the case of itraconazole with SYL, and was suggested to be caused by the developed physical bonds between the drug and carrier.43 However, there was no significant difference (P<0.05) in the overall dissolution efficiency between L-SNEDDS and S-SNEDDS. In fact, S-SNEDDS and T-SNEDDS were able to maintain the superior dissolution of L-SNEDDS, which confirms that the solidification and tableting processes had no significant drawbacks on RMP dissolution. Most importantly, T-SNEDDS showed significant enhancement of RMP dissolution and could be compared to the marketed drug Ramipril Sandoz® 2.5 mg, as no significant difference exists between them.
Finally, it was critical to evaluate the influence of solidification and tablet compression processes on the chemical stability of RMP. In fact, the findings of stability studies revealed the challenging physicochemical properties of RMP as it showed significant degradation in lipid-based systems (L-SNEDDS) and even in pure drug form due to its sensitivity to moisture and heat. However, the solidification into T-SNEDDS led to a significant reduction of RMP degradation within the formulation (P<0.05). It was suggested that RMP loading within solidified SNEDDS substantially protected the drug moiety against negative impacts of temperature and humidity exposure during storage. The overall results established the capacity of T-SNEDDS to protect the drug from critical conditions such as temperature, humidity, and degradation within liquid lipid-based formulations. Accordingly, T-SNEDDS provided combined enhancement of RMP dissolution and stability during manufacturing and storage.
However, RMP degradation was still significant within accelerated storage conditions, which might be caused by drug exposure to elevated humidity environments (RH 75%). Future work should involve further protection of RMP T-SNEDDS against moisture exposure. This can be achieved by moisture resistant coating of T-SNEDDS, which is expected to provide further stabilization of RMP moiety within accelerated storage conditions. Furthermore, in vivo bioavailability studies would be helpful to correlate these in vitro findings and depict the significance of solidified RMP T-SNEDDS.
Conclusion
RMP, an important medication for hypertension and heart failure, was successfully formulated to overcome its poor aqueous solubility, significant degradation in lipid-based formulation, and sensitivity to heat, moisture, and mechanical stress. Six liquid lipid-based formulations were prepared, according to LFCS, to explore the optimal excipients combination to prepare an efficient RMP SNEDDS. F5 (CMCM/TC/HCO 30 [25/25/50]) showed relatively high drug loading, low droplet size, high negative zeta potential, and, hence, was selected as the optimal L-SNEDDS for solidification. Among five tested L-SNEDDS/SYL ratios, the 1:1 ratio resulted in an excellent flowable S-SNEDDS powder and, hence, was selected for further tablet compression experiments. The optimized T-SNEDDS involved using magnesium stearate, avicel, aerosil, along with S-SNEDDS powder. The optimized RMP T-SNEDDS passed all USP requirements for content uniformity, hardness, friability, and disintegration tests. Most importantly, T-SNEDDS provided combined enhancement of RMP dissolution and stability during manufacturing and storage.
Acknowledgments
The authors would like to acknowledge Kayyali Chair for Pharmaceutical Industries for funding this project (Grant no. KM-2019, Research Chair, Deanship of Scientific Research).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Shahba AA-W, Ahmed AR, Alanazi FK, Mohsin K, Abdel-Rahman SI. multi-layer self-nanoemulsifying pellets: an innovative drug delivery system for the poorly water-soluble drug cinnarizine. AAPS PharmSciTech. 2018. doi:10.1208/s12249-018-0990-7
2. Shahba AA, Ahmed AR, Mohsin K, Abdel-Rahman SI, Alanazi FK. Solidification of cinnarizine self-nanoemulsifying drug delivery systems by fluid bed coating: optimization of the process and formulation variables. Pharmazie. 2017;72(3):143–151. doi:10.1691/ph.2017.6089
3. Tang B, Cheng G, Gu JC, Xu CH. Development of solid self-emulsifying drug delivery systems: preparation techniques and dosage forms. Drug Discov Today. 2008;13(13–14):606–612. doi:10.1016/j.drudis.2008.04.006
4. Ekambaram P, Abdul HSA. Formulation and evaluation of solid lipid nanoparticles of. J Young Pharm. 2011;3(3):216–220. doi:10.4103/0975-1483.83765
5. Shafiq S, Shakeel F, Khar RK. Enhanced stability of ramipril in nanoemulsion containing cremophor-EL: a technical note. AAPS PharmSciTech. 2008;9(4):1097–1101. doi:10.1208/s12249-008-9151-8
6. Rao S, Tan A, Boyd BJ, Prestidge CA. Synergistic role of self-emulsifying lipids and nanostructured porous silica particles in optimizing the oral delivery of lovastatin. Nanomedicine (Lond). 2014;9(18):2745–2759. doi:10.2217/nnm.14.37
7. Nguyen TH, Tan A, Santos L, et al. Silica-lipid hybrid (SLH) formulations enhance the oral bioavailability and efficacy of celecoxib: an in vivo evaluation. J Control Release. 2013;167(1):85–91. doi:10.1016/j.jconrel.2013.01.012
8. Tan A, Eskandar NG, Rao S, Prestidge CA. First in man bioavailability and tolerability studies of a silica-lipid hybrid (Lipoceramic) formulation: a phase I study with ibuprofen. Drug Deliv Transl Res. 2014;4(3):212–221. doi:10.1007/s13346-013-0172-9
9. Neeraj BAK, Abhilash C, Rubia C, Rajn B. A review on immediate release drug delivery system. Irjpas. 2014;4(1):78–87.
10. Gumaste SG, Dalrymple DM, Serajuddin ATM. Development of solid SEDDS, V: compaction and drug release properties of tablets prepared by adsorbing lipid-based formulations onto neusilin® US2. Pharm Res. 2013;30(12):3186–3199. doi:10.1007/s11095-013-1106-4
11. Mohsin K, Pouton C. The influence of the ratio of lipid to surfactant and the presence of cosolvent on phase behaviour during aqueous dilution of lipid-based drug delivery systems. J Drug Deliv Sci Technol. 2012;22(6):531–540. doi:10.1016/S1773-2247(12)50092-4
12. Pouton CW. Lipid formulations for oral administration of drugs: non-emulsifying, self-emulsifying and ‘self-microemulsifying’ drug delivery systems. Eur J Pharm Sci. 2000;11(Suppl 2):S93–98.
13. Shahba AA, Mohsin K, Alanazi FK. Novel self-nanoemulsifying drug delivery systems (SNEDDS) for oral delivery of cinnarizine: design, optimization, and in-vitro assessment. AAPS PharmSciTech. 2012;13(3):967–977. doi:10.1208/s12249-012-9821-4
14. Alhasani KF, Mohsin K, ShakEEL F, Alanazi F. Development and validation of stability-indicating ultra high-performance liquid chromatography for ramipril analysis in pharmaceutical dosage forms and its application in lipid-based formulations. Orient J Chem. 2018;34(1):64–74. doi:10.13005/ojc
15. Shahba AA-W, Mohsin K, Alanazi FK, Abdel-Rahman SI. Optimization of self-nanoemulsifying formulations for weakly basic lipophilic drugs: role of acidification and experimental design. Braz J Pharm Sci. 2016;52(4):653–667. doi:10.1590/s1984-82502016000400009
16. Kazi M, Al-Qarni H, Alanazi FK. Development of oral solid self-emulsifying lipid formulations of risperidone with improved in vitro dissolution and digestion. Eur J Pharm Biopharm. 2017. doi:10.1016/j.ejpb.2017.01.015
17. Shazly G, Mohsin K. Dissolution improvement of solid self-emulsifying drug delivery systems of fenofibrate using an inorganic high surface adsorption material. Acta Pharm. 2015;65(1):29–42. doi:10.1515/acph-2015-0003
18. United States Pharmacopeial Convention. Powder flow. In: USP35 NF30, U. S. Pharmacopeia National Formulary. Rockville (MD): United States Pharmacopeial; 2011:801–804.
19. Beakawi Al-Hashemi HM, Baghabra Al-Amoudi OS. A review on the angle of repose of granular materials. Powder Technol. 2018;330:397–417. doi:10.1016/j.powtec.2018.02.003
20. Abdel-Rahman AA, Alanazi FK. Basis of Industrial Pharmacy.
21. United States Pharmacopeial Convention. Uniformity of the dosage forms. In: USP35 NF30, U. S. Pharmacopeia National Formulary. Rockville (MD): United States Pharmacopeial; 2011:420–423.
22. United States Pharmacopeial Convention. Tablet friability. In: USP35 NF30, U. S. Pharmacopeia National Formulary. Rockville (MD): United States Pharmacopeial; 2011:867–868.
23. United States Pharmacopeial Convention. Disintegration. In: USP35 NF30, U. S. Pharmacopeia National Formulary. Rockville (MD): United States Pharmacopeial; 2011:293–295.
24. Aulton M. Aulton’s Pharmaceutics: The Design and Manufacture of Medicines.
25. El Maghraby GM, Elzayat EM, Alanazi FK. Development of modified in situ gelling oral liquid sustained release formulation of dextromethorphan. Drug Dev Ind Pharm. 2012;38(8):971–978. doi:10.3109/03639045.2011.634811
26. The GCC Guidelines for Stability Testing of Active Pharmaceutical Ingredients (Apis) and Finished Pharmaceutical Products (Fpps) [Internet]; 2011.
27. Shahba AA, Alanazi FK, Abdel-Rahman SI. Stabilization benefits of single and multi-layer self-nanoemulsifying pellets: A poorly-water soluble model drug with hydrolytic susceptibility. PLoS One. 2018;13(7):e0198469. doi:10.1371/journal.pone.0198469
28. ICH. ICH Steering Committee, ICH Harmonized Tripartite Guidlines, Q1C, Stability Testing for New Dosage Forms; 1996.
29. ICH. ICH Steering Committee, ICH Harmonized Tripartite Guidlines, Q1A(R2), Stability Testing of New Drug Subtances and Products; 2003.
30. Date AA, Desai N, Dixit R, Nagarsenker M. Self-nanoemulsifying drug delivery systems: formulation insights, applications and advances. Nanomedicine (Lond). 2010;5(10):1595–1616. doi:10.2217/nnm.10.126
31. Lipinski C. Poor aqueous solubility-an industry wide problem in drug discovery. Am Pharm Rev. 2002;5(3):82–85.
32. Müllertz A, Ogbonna A, Ren S, Rades T. New perspectives on lipid and surfactant based drug delivery systems for oral delivery of poorly soluble drugs. J Pharm Pharmacol. 2010;62(11):1622–1636. doi:10.1111/j.2042-7158.2010.01107.x
33. Pouton CW, Porter CJ. Formulation of lipid-based delivery systems for oral administration: materials, methods and strategies. Adv Drug Deliv Rev. 2008;60(6):625–637. doi:10.1016/j.addr.2007.10.010
34. Dou YX, Zhou JT, Wang TT, et al. Self-nanoemulsifying drug delivery system of bruceine D: a new approach for anti-ulcerative colitis. Int J Nanomedicine. 2018;13:5887–5907. doi:10.2147/IJN.S174146
35. Bandivadekar MM, Pancholi SS, Shelke N. Preparation and characterization of solid SMEDDS by adsorbent techniques to improve dissolution profile of poorly aqueous soluble drug ramipril. Int Res J Pharm. 2011;2(6):85–90.
36. Odovic JV, Markovic BD, Trbojevic-Stankovic J, Vladimirov SM, Karljikovic-Rajic KD. Evaluation of ACE inhibitors lipophilicity using in silico and chromatographically obtained hydrophobicity parameters/Procena lipofilnosti ace inhibitora primenom in silico i hromatografski dobijenih parametara hidrofobnosti. Hemijska Industrija. 2013;67(2):209–217. doi:10.2298/HEMIND120522078O
37. Porter C, Pouton C, Cuine J, Charman W. Enhancing intestinal drug solubilisation using lipid-based delivery systems. Adv Drug Deliv Rev. 2008;60(6):673–691. doi:10.1016/j.addr.2007.10.014
38. Saifee M, Zarekar S, Rao VU, Zaheer Z, Soni R, Burande S. Formulation and in vitro evaluation of solid-self-emulsifying drug delivery system (SEDDS) of glibenclamide. Am J Adv Drug Delivery. 2013;1(3):323–340.
39. Silva HD, Cerqueira MA, Vicente AA. Influence of surfactant and processing conditions in the stability of oil-in-water nanoemulsions. J Food Eng. 2015;167:89–98. doi:10.1016/j.jfoodeng.2015.07.037
40. Tian Y, Chen L, Zhang W. Influence of ionic surfactants on the properties of nanoemulsions emulsified by nonionic surfactants span 80/tween 80. J Dispers Sci Technol. 2016;37(10):1511–1517. doi:10.1080/01932691.2015.1048806
41. Patel VI. To Improve the Solubility of Poorly Water Soluble Drugs Using Porous Material. Brooklyn (NY): Industrial pharmacy, faculty of the arnold & marie schwartz college of Pharmacy and health sciences, Long Island University; 2009.
42. Kinnari P, Makila E, Heikkila T, Salonen J, Hirvonen J, Santos HA. Comparison of mesoporous silicon and non-ordered mesoporous silica materials as drug carriers for itraconazole. Int J Pharm. 2011;414(1–2):148–156. doi:10.1016/j.ijpharm.2011.05.021
43. Limnell T, Santos HA, Makila E, et al. Drug delivery formulations of ordered and nonordered mesoporous silica: comparison of three drug loading methods. J Pharm Sci. 2011;100(8):3294–3306. doi:10.1002/jps.22577
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.