Back to Journals » International Journal of Nanomedicine » Volume 13
Self-nanoemulsifying drug-delivery systems for potentiated anti-inflammatory activity of diacerein
Authors Eltobshi AA, Mohamed EA ![]() , Abdelghani GM, Nouh AT
, Abdelghani GM, Nouh AT
Received 1 July 2018
Accepted for publication 18 August 2018
Published 18 October 2018 Volume 2018:13 Pages 6585—6602
DOI https://doi.org/10.2147/IJN.S178819
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Thomas Webster
Amal Ahmed Eltobshi,1 Elham Abdelmonem Mohamed,2 Galal Mahmoud Abdelghani,2 Ahmed Talaat Nouh3
1Department of Pharmaceutical Technology, Faculty of Pharmacy, Horus University, New Damietta 34517, Egypt; 2Department of Pharmaceutics, Faculty of Pharmacy, Mansoura University, Mansoura 35516, Egypt; 3Department of Pharmaceutics, Faculty of Pharmacy, Delta University for Science and Technology, Gamasa 11152, Egypt
Background: Effective treatment of osteoarthritis necessitates both symptomatic relief and hindrance of joint degeneration progression. Non-steroidal anti-inflammatory drugs permit symptomatic relief only and can cause mucosal injury in the gut. Before absorption, diacerein (Dcn) is converted into rhein that counteracts cartilage degeneration without affecting prostaglandin production. Yet, low solubility and laxative action of unabsorbed rhein in the colon hindered its use. Thus, enhanced Dcn dissolution would allow absorption at the upper gut improving its bioavailability and possibly abolishing the laxative action.
Methods: Therefore, self-nanoemulsifying drug delivery systems (SNEDDSs) with each of gelucire 44/14 (Glc) and d-α-tocopheryl polyethylene glycol 1000 succinate (TPGS) at different drug:carrier weight ratios of 1:1, 1:2, 1:4, 1:6, 1:8 and 1:10 were prepared by melt method and filled into hard gelatin capsules. The optimized binary systems were selected based on solid state characterization, scanning electron microscopy (SEM) and in vitro evaluation of the prepared SNEDDSs in comparison with their corresponding physical mixtures (PMs) and Dcn. The optimized systems were further examined with respect to their morphology, size distribution and ζ-potential. Moreover, the anti-inflammatory activity of the optimized systems against carrageenan-induced paw edema in rats was assessed through estimation of edema and edema inhibition percentages as well as histopathological examination and immunohistochemical localization of tumor necrosis factor-alpha (TNF-α) and caspase-3.
Results: Significantly (P<0.05) enhanced in vitro drug release was recorded for SNEDDSs with either carrier when compared to Dcn and the corresponding PMs. SNEDDSs based on 1:10 Dcn:Glc and 1:8 Dcn:TPGS showed significantly (P<0.05) reduced edema and inflammation as well as expression of TNF-α and caspase-3 relative to positive control and Dcn pretreated groups.
Conclusion: These SNEDDSs can be represented as potential oral drug delivery systems of Dcn for enhanced dissolution and anti-inflammatory activity against carrageenan-induced paw edema.
Keywords: diacerein, gelucire 44/14, d-α-tocopheryl polyethylene glycol 1,000 succinate, self-nanoemulsifying drug-delivery systems, anti-inflammatory activity, paw edema
Introduction
Osteoarthritis is a serious degenerative joint disease affecting 10% of the world population aged >55–60 years.1 Osteoarthritis treatment is challenging, because current therapies, including nonsteroidal anti-inflammatory drugs (NSAIDs) allow symptomatic relief only, without hindering the progression of joint degeneration.2 In addition, NSAIDs cause mucosal injury in the gut by depleting COX1-derived prostaglandins.3 Therefore, an alternative therapy is necessary to downregulate cartilage degeneration without gut injury.
Diacerein (Dcn) is a promising chondroprotective agent that is converted into rhein before absorption. Rhein inhibits the production of IL1β and nitric oxide, which participate in cartilage degeneration.4 Rhein does not affect prostaglandin production; therefore, no gastrointestinal mucosal damage has been reported.5 Consequently, Dcn has been represented as a first-line treatment of osteoarthritis, particularly for patients in whom NSAIDs are contraindicated.6,7 However, this drug is classified according to the Biopharmaceutics Classification System as a class II drug, because it shows low water-solubility. Therefore, its oral bioavailability is low and variable (35%–56%), resulting in a limited therapeutic response.8 Moreover, rhein residue unabsorbed in the colon induces a laxative effect that has been attributed to the enhanced secretion of water and electrolytes in the colon.9 Consequently, different drug-delivery systems (DDSs) have been investigated to improve Dcn dissolution and oral bioavailability. Among these, solid dispersions of Dcn with polyethylene glycol (PEG),10,11 pluronic,12 or sorbitol13 have been reported. Also, self-emulsifying systems of Dcn employing various surfactants, such as Tween, Labrafil, Labrafac, and Labrasol, as well as different oils, such as soy oil, almond oil, olive oil and peanut oil, have been prepared to enhance its bioavailability.14 Moreover, self-emulsifying solid dispersions of Dcn employing PEG 6000, Tween 80, and oleic acid provide increased drug dissolution.15
Self-nanoemulsifying DDSs (SNEDDSs) are isotropic mixtures formed of oil, surfactants, and cosurfactants that produce oil-in-water emulsions into aqueous media upon mild agitation.16 These systems spread readily in the gut, and gastric motility provides the agitation required for self-emulsification.17 SNEDDSs offer some advantages, such as greater stability when compared to solid lipid nanoparticles18 and liposomes.19 Furthermore, they can be put into gelatin capsules, providing solid unit dosage forms to enhance patient convenience, dose accuracy, and drug stability.20 SNEDDSs have been used to potentiate the solubility and oral bioavailability of lipophilic drugs.21 Improved drug dissolution and increased membrane permeability and lymphatic transport have been reported as the main reasons for enhanced absorption of drugs incorporated in SNEDDSs.22,23
Gelucire 44/14 (Glc) is an inert, amphiphilic, semisolid, waxy material with a melting point of 44°C and hydrophilic–lipophilic balance of 14.24 Glc is formed by polyglycolysis of hydrogenated palm-kernel oil using PEG1,500. It is composed of 20% mono-, di-, and triglycerides, 72% mono- and diunsaturated fatty-acid esters of PEG 1500 and 8% free PEG 1500.24 It has a unique composition, since it is formed of different moieties, including surfactants (mono- and diesters of PEG), cosurfactants (monoglycerides), and an oily phase (di- and triglycerides). Consequently, Glc gives an emulsion at body temperature in aqueous fluids. This induces pseudosolubilization of poorly water-soluble drugs, increasing their bioavailability.25 The solubility and therapeutic efficacy of several drugs, including etodolac,26 flurbiprofen,27 and fexofenadine,28 have been improved through the formation of SNEDDS based on Glc. This carrier shows Pgp-inhibitory activity that can reduce drug efflux and enhance absorption.28
Esterification of vitamin E succinate with PEG 1000 produced D-α-tocopheryl PEG 1000 succinate (TPGS), which is a water-soluble derivative of natural vitamin E. This carrier has an average molecular weight of 1,513 Da. It has a lipophilic alkyl tail, hydrophilic polar head, and hydrophilic–lipophilic balance of 13.2.28 TPGS has a relatively low critical micelle concentration of 0.02 wt%, above which it forms a fine emulsion upon contact with an aqueous medium, and thus it can solubilize lipophilic drugs and enhance their absorption.26 It has been reported that TPGS exhibits Pgp-inhibitory activity, and hence it potentiates drug permeability and absorption.28 It is a water-soluble waxy solid possessing a melting-point range of 37°C–41°C. It has been used successfully to enhance the solubility of different water-insoluble compounds, such as paclitaxel,29 celecoxib,30 corticosteroids,31 capuramycin analogue SQ641,32 and propofol.33 SNEDDSs based on TPGS as s single self-emulsifying carrier have been successfully employed to improve the dissolution of etodolac26 and fexofenadine.28 It is stable at a pH range of 4.5–7.5 and <10% is hydrolyzed in neutral aqueous buffer after 3 months, yet it degrades in alkaline media.34 The high stability at lower pH has been also indicated by the degradation of 3.4% of TPGS within 8 hours at pH 1.0. It has been reported that the ester bond between D-α-tocopheryl and succinic acid is stable.35 TPGS has a relatively high oral LD50 (>7 g/kg) for young adult rats of both sexes. It has been approved by the US Food and Drug Administration as a safe pharmaceutical adjuvant.34,36
Therefore, this study aimed to improve Dcn dissolution and anti-inflammatory activity through the preparation of SNEDDSs with Glc and TPGS at different drug:carrier weight ratios of 1:1, 1:2, 1:4, 1:6, 1:8, and 1:10 by melting, to be then out into hard gelatin capsules. Solid-state characterization and in vitro evaluation of the prepared SNEDDSs and their corresponding physical mixtures (PMs) were performed to determine the optimized system with each carrier. The optimized systems were further assessed for morphology, size distribution, ζ-potential and therapeutic efficacy against carrageenan-induced paw edema in rats.
Methods
Materials
Dcn was purchased from Provizer Pharma, India. Glc was obtained as a gift from Gattefossé, Saint-Priest, France. λ-carrageenan, TPGS, and methylcellulose were purchased from Sigma-Aldrich, St Louis, MO, USA. All other chemicals and solvents were of fine analytical grade.
Preparation of SNEDDSs
Dcn SNEDDSs were prepared with Glc and TPGS at drug:carrier weight ratios of 1:1, 1:2, 1:4, 1:6, 1:8, and 1:10 by melting.26,28 Briefly, accurately weighed carrier amounts were placed in a porcelain dish to be melted with continuous stirring in a thermostatically controlled water bath at 50°C±1°C. Accurately weighed drug was added to the molten carrier to be mixed with continuous stirring to obtain a homogeneous melt. The resultant melt was solidified at 4°C, then left at room temperature overnight to attain equilibrium. For comparison, corresponding PMs were prepared by mixing the drug with each carrier using a mortar and a pestle. The prepared systems, each equivalent to 50 mg Dcn, were put into hard gelatin capsules.
Drug content
Drug-content uniformity of the prepared SNEDDSs and PMs was determined by dissolving an accurately weighed amount equivalent to 50 mg of Dcn in a 100 mL 1:1 mixture of acetonitrile:PBS pH 6.8 with sonication for 15 minutes. The resultant mixture was then filtered through a 0.45 μm Millipore filter, suitably diluted, and analyzed spectrophotometrically (Ultraviolet-visible, V-530; Jasco, Tokyo, Japan) at 258 nm against a blank treated the same. All experiments were performed in triplicate to estimate mean values.
Solid-state characterization
Solid-state characterization of pure Dcn and carriers (Glc and TPGS), as well as the prepared SNEDDSs and their corresponding PMs, was performed employing the following techniques.
Fourier-transform infrared spectroscopy
Fourier-transform infrared (FTIR) spectra of the examined samples were obtained (Thermo Fisher Scientific) employing compressed disks of a ground mixture of 2 mg of each sample and 200 mg potassium bromide. Disks were scanned over a wave-number range of 500–4,000 cm−1. The FTIR spectrum of each sample was determined by recording characteristic bands.
Differential scanning calorimetry
Thermal properties of the studied samples were examined (DSC-4; PerkinElmer, Waltham, MA, USA) by heating each sample in crimped aluminum pans at heating rates of 5°C/min over a temperature range of 30°C–400°C under nitrogen gas flow. Indium (99.99% purity, melting point 156.6°C) was used to attain temperature calibration.
X-ray diffractometry
X-ray diffraction (XRD) patterns of the prepared SNEDDSs and corresponding PMs were recorded in comparison with the drug and carriers. XRD patterns were obtained using a diffractometer equipped with CoKα (Diano, Woburn, MA, USA) and operated at 45 kV, 9 mA, and 2θ angle.
Scanning electron microscopy
The morphological characteristics of pure Dcn and its prepared SNEDDSs and respective PMs were examined using scanning electron microscopy (SEM; JSM 5500 LV; JEOL, Tokyo, Japan). The samples were sprinkled and fixed with double-sided adhesive tape on a SEM holder. A layer of gold of 150 °A was used to coat the fixed sample for 2 minutes with a sputter coater (S-150A; Edwards, Crawley, UK) operating in a vacuum (3×10−1 atm) of Argon gas.
In vitro dissolution studies
Dissolution studies of the prepared SNEDDSs and corresponding PMs compared to drug alone were performed using a US Pharmacopeial Convention apparatus I (basket method). Rotation speed was adjusted at 100 rpm. Dissolution medium consisted of 900 mL PBS pH 6.8 and maintained at 37°C±0.5°C. Aliquots of 5 mL were withdrawn at predetermined time intervals of 5, 10, 15, 30, 60, 90, and 120 minutes and replaced with the same volume of a fresh dissolution medium at 37°C±0.5°C. Samples were filtered using 0.45 μm membrane filters, and filtrates obtained were properly diluted with the buffer and measured spectrophotometrically (ultraviolet-visible, V-530) at 258 nm against a blank treated the same. All experiments were carried out in triplicate. Dissolution efficiency (DE) was calculated as percentage of the area under the dissolution curve to time t to that of the rectangle representing 100% dissolution at the same time.37,38 Relative dissolution rate (RDR) was estimated as percentage dissolved Dcn from each binary system either SNEDDS or PM to that of the drug alone at the same time.38,39
Kinetic modeling of dissolution data
In order to elucidate the drug-release mechanism, in vitro dissolution data of the drug from the studied binary systems were analyzed according to zero-order and first-order kinetics,40 as well as diffusion-controlled release mechanisms.41 The Korsmeyer–Peppas kinetic equation (mt/m∞ = ktn) was also employed.42 The model with the highest correlation coefficient was considered to describe Dcn-release mechanism.
Size and ζ-potential measurements
A Ζetasizer Nano ZS (Malvern Instruments, Malvern, UK) utilizing dynamic light scattering (DLS) was used to evaluate size distribution and ζ-potential of the optimized Dcn SNEDDSs with Glc and TPGS. Measurements were done in triplicate for each sample after reconstitution and proper dilution with double-deionized water, to be then sonicated to obtain uniformly distributed particles.
Transmission electron microscopy
The optimized formula of SNEDDSs with either carrier that showed the greatest drug dissolution was further evaluated for its morphology after dispersion into double-deionized water. A drop of a properly diluted SNEDDS was placed on a carbon-coated 300-mesh copper grid to dry slowly in air. Then, the sample was stained with 2% w/v phosphotungstic acid solution for 1 minute at room temperature. The image was taken by transmission electron microscopy (TEM; JEOL 2100) set at an accelerated voltage of 200 kV.
Anti-inflammatory activity against carrageenan-induced paw edema in rats
Animals and treatment
All animal experiments were carried out according to the US National Institutes of Health Guide for the Care and Use of Laboratory Animals (revised 1996). The ethical committee of the Faculty of Pharmacy, Mansoura University, Egypt approved the protocol. Animals were housed under 12 hours’ light–12 hours’ dark cycles at 25°C±1°C and relative humidity of 55%±5%. They were allowed free access to standard laboratory food and water.
Thirty healthy male Wistar rats weighing 180–220 g were divided into five groups of six rats each to assess the anti-inflammatory activity of the optimized SNEDDSs of Dcn with each carrier in comparison with the free drug, as well as normal and positive control groups. Group 1 received normal saline (normal control, 0.1 mL). Rats in group 2 were injected with 0.1 mL of 1% w:v λ-carrageenan in saline into the subplantar region of the right hind paw under light ether anesthesia (positive control). Group 3 orally received Dcn (100 mg/kg) dispersed in 0.5% methylcellulose 30 minutes before λ-carrageenan injection into the right hind paw. Groups 4 and 5 were pretreated orally with optimized SNEDDS with Glc and TPGS, respectively, equivalent to 100 mg/kg Dcn 30 minutes before λ-carrageenan injection into the right hind paw. The investigated SNEDDSs were finely dispersed into double-deionized water before oral administration to rats. Using plethysmography, volume measurements of the injected hind paw were carried out before carrageenan injection and at time intervals of 0.5, 1, 2, 3, 4, 5, 6, 7, 8, 12, and 24 hours following carrageenan injection. Edema and edema inhibition of each group were estimated:43
|
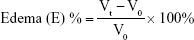
|
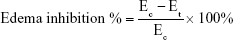
where V0 and Vt are the mean paw volume before carrageenan injection and after carrageenan injection at time t, respectively, and Ec and Et are the edema percentages of control and treated groups, respectively, at the same time interval.
At the end of the study (24 hours), the animals were euthanized then thin and thick skin specimens in paw area of the animals were separated. Specimens were fixed in 10% (v/v) buffered formalin and then routinely processed until paraffin embedding for histopathological examination and immunohistochemical localization of TNF-α and caspase-3.
Histopathological examination
Serial paw-skin sections (thin and thick) of paraffin blocks were obtained and processed to be finally stained with hematoxylin-eosin (H&E) and examined using light microscopy (Leica Microsystems, Wetzlar, Germany).44 Inflammatory response intensity was evaluated in thin- and thick-skin specimens of each animal as 0 = no inflammatory cells, 1 = inflammatory cells <10%, 2 = inflammatory cells 10%–50%, and 3 = inflammatory cells >50%, all with respect to the cell population.45 All readings were performed blindly by a pathologist.
Immunohistochemical localization of TNF-α and caspase-3
Thin and thick specimens of paraffin blocks of rat paws were examined for immunohistochemical localization of TNF-α and caspase-3 following a previously reported procedure.46,47 After paraffin removal with graded xylenes and rehydration in ethanol, blockade of endogenous peroxidase activity was achieved by treatment with 3% hydrogen peroxide (H2O2) for 5 minutes at room temperature. To attain antigen retrieval, paw-skin sections were placed in glass jars containing 0.01 M sodium citrate buffer of pH 6 and boiled in a microwave oven twice for 5 minutes each to permit immunoreactivity. The slides were left to cool to be then rinsed with PBS pH 7.2. The manufacturer’s instructions were followed to perform immunohistochemical staining with ready-to-use polyclonal antibodies for TNFα antibody (1:100 in PBS w:v; Santa Cruz Biotechnology, Dallas, TX, USA) and caspase-3 (1:200 in PBS w:v; Abcam, Cambridge, UK). Detection was performed using a universal kit (Dako, Glostrup, Denmark) by washing slides in PBS for 5 minutes and incubation with secondary antibodies for 30 minutes. After section washing for 5 minutes in PBS, antigen–antibody visualization was attained by 0.04% 3,3′-diaminobenzidine tetrahydrochloride for 1 minute. Sections were washed under running tap water for 10 minutes, then counterstained with Mayer’s hematoxylin. Controls were prepared by staining without primary antibodies. Positively stained cells were observed by a senior pathologist using a digital camera (Olympus) placed on a microscope (Leica). Stain intensity was scored according to 0 = no staining, 1 = week staining, 2 = moderate staining, and 3 = strong staining.48
Statistical analysis
One way ANOVA followed by Tukey–Kramer multiple-comparison tests were employed to analyze the data. GraphPad Prism version 5.00 (GraphPad software, San Diego, CA, USA) was used. At level P<0.05, differences were considered statistically significant.
Results and discussion
Drug content
Table 1 illustrates average drug content of the binary systems. According to this table, Dcn content in the prepared SNEDDSs and PMs with either carrier ranged 96.11%±1.04%–104.52%±0.70%, complying with the pharmacopoeial limit of 90%–110%.49
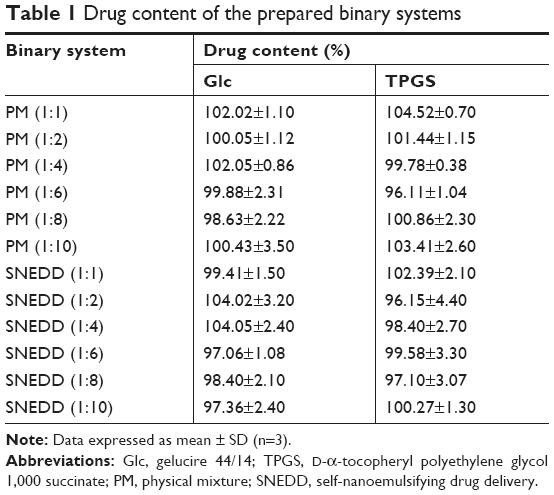 | Table 1 Drug content of the prepared binary systems |
Solid-state characterization
Fourier-transform infrared spectroscopy
FTIR spectra of Dcn and carriers (Glc, TPGS), as well as prepared SNEDDSs and their corresponding PMs, are presented in Figure 1. Characteristic absorption peaks of Dcn were observed at 3,438 cm−1 (−OH stretching band of −COOH), 1,771 cm−1 (C=O stretching band of ester group), 1,685 cm−1 (C=O stretching band of −COOH), 752 cm−1 (benzene), 1,025 cm−1 (C−O, ester stretching), 1,595 cm−1 (C=C, aromatic ring stretching), and 2,935 cm−1 (C−H, aliphatic system stretching).11,50 Pure Glc and TPGS peaks appeared at 1,739 cm−1 (C−O stretching of ester) and 1,112 cm−1 (C=O stretching of alcohol) (primary or secondary; Figure 1).28 The presence of characteristic drug and carrier peaks with no additional peaks in any PMs or SNEDDSs can negate any interaction between the drug and either carrier, and suggests Dcn stability in these systems.
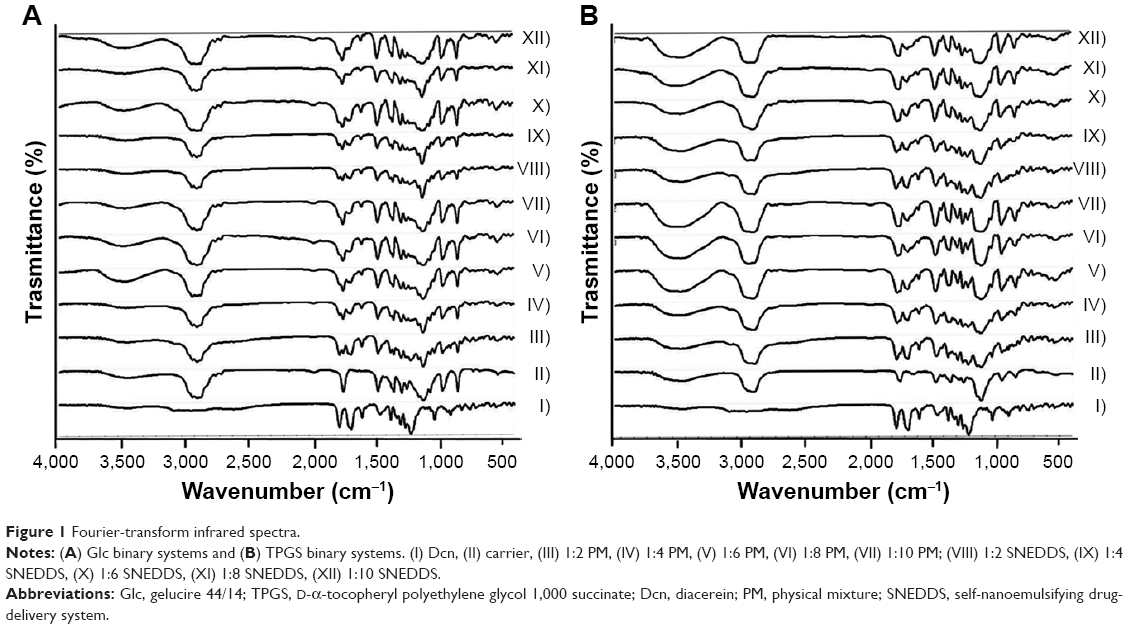 | Figure 1 Fourier-transform infrared spectra. |
Differential scanning calorimetry
Figure 2 illustrates differential scanning calorimetry (DSC) thermograms of Dcn, Glc and TPGS, as well as PMs and SNEDDSs of Dcn with each carrier. In Dcn thermograms, a sharp endothermic peak appeared at 246°C, equivalent to its melting point and indicating its crystalline anhydrous nature.11 DSC thermograms of Glc and TPGS showed respective sharp endothermic peaks at 46°C and 39.5°C, representing the low melting points of these waxy carriers (Figure 2).28 PMs of Dcn and each carrier showed endothermic peaks of both components up to drug:carrier weight ratios of 1:8 and 1:6 in case of Glc and TPGS, respectively. The disappearance of the drug peak beyond these ratios can be attributed to dilution by the higher content of the carrier. Meanwhile, Dcn endothermic peaks disappeared at DSC curves of SNEDDSs with Glc and TPGS at all drug:carrier weight ratios. This may indicate complete conversion of Dcn into the amorphous form or homogeneous dispersion of the partially crystalline drug into the molten carrier. In accordance with FTIR results, no new peaks were noticed in DSC curves of Dcn PMs or SNEDDSs with both carriers, negating the possibility of drug–carrier interaction.
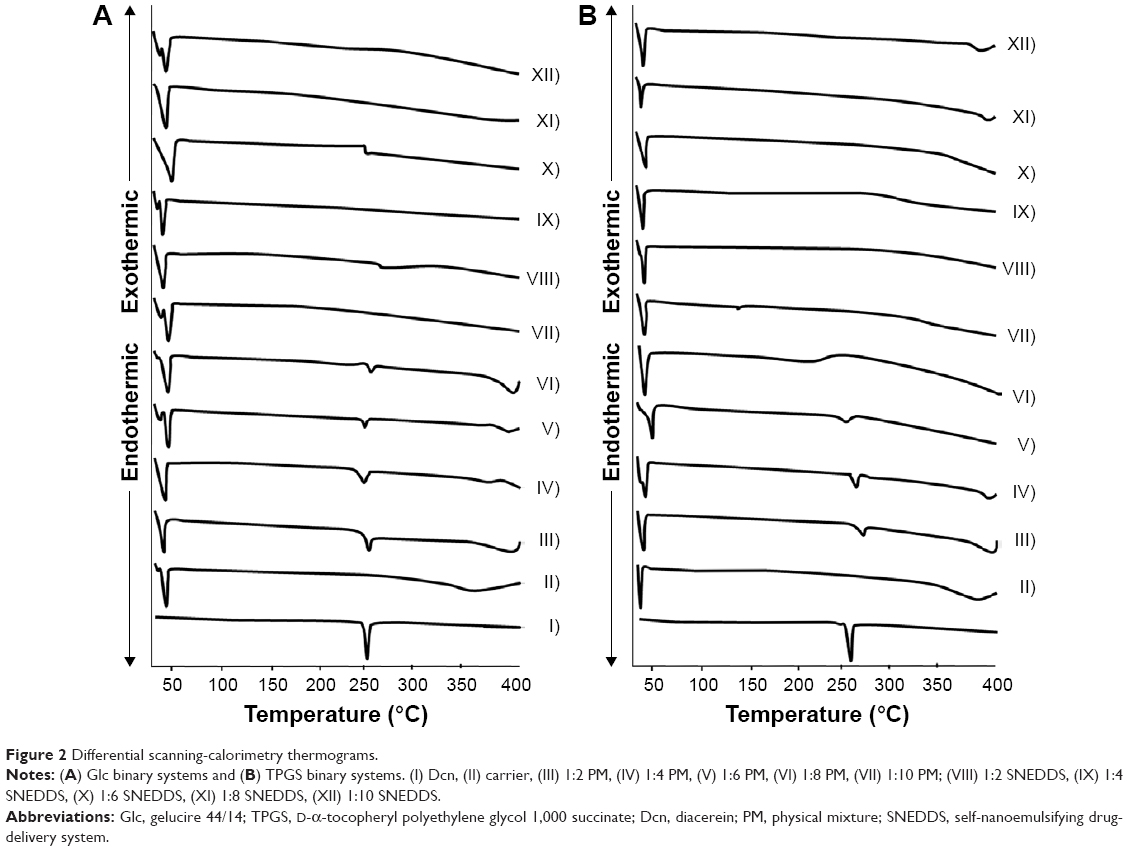 | Figure 2 Differential scanning-calorimetry thermograms. |
X-ray diffractometry
XRD patterns of Dcn exhibited diffraction peaks at 5.18°, 10.51°, 17.35°, 21.06°, 21.44°, 21.90°, 25.06°, and 27.88° (2θ degree), reflecting its crystalline nature (Figure 3). XRD patterns of Glc demonstrated its crystalline structure, as indicated by typical triglyceride peaks of 19.20° and 23.30° at 2θ (Figure 3A).51 Regarding TPGS, predominant peaks were observed at 19.12° and 23.12°, suggesting its crystalline nature (Figure 3B).52 PMs of Dcn and each carrier showed a reduction in intensity of drug peaks, possibly due to dilution by the increased content of the carriers. When compared to the corresponding PMs, diffractograms of SNEDDSs with Glc or TPGS showed either noticeable reduction (particularly with TPGS) or absence of Dcn peaks, possibly indicating reduced crystallinity or amorphization of the drug. In agreement with FTIR and DSC results, no new peaks were observed in the examined PMs and SNEDDSs, indicating the absence of interaction between the drug and the carriers used.
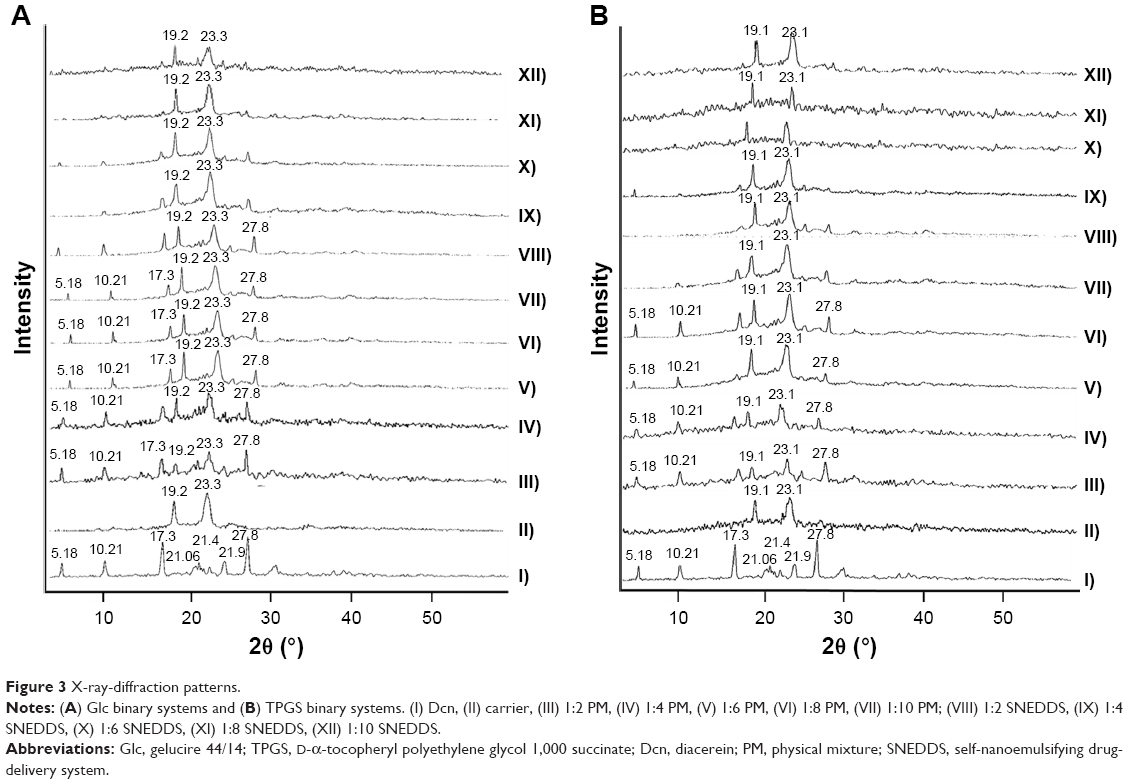 | Figure 3 X-ray-diffraction patterns. |
Scanning electron microscopy
SEM of pure Dcn and its binary systems – either PMs or SNEDDSs with Glc and TPGS – is presented in Figure 4. Pure Dcn appeared as crystals of irregular shape and size. Pure Glc (Figure 4A) and TPGS (Figure 4B) existed as waxy lipids with smooth surfaces. In all PMs, irregular drug crystals were present together with the smooth waxy Glc (Figure 4A) and TPGS (Figure 4B), whereas, SNEDDSs with Glc (Figure 4A) and TPGS (Figure 4B) presented a uniform and homogeneous waxy surface similar to that of the pure carriers, possibly due to the homogeneous drug dispersion into the waxy carriers.28
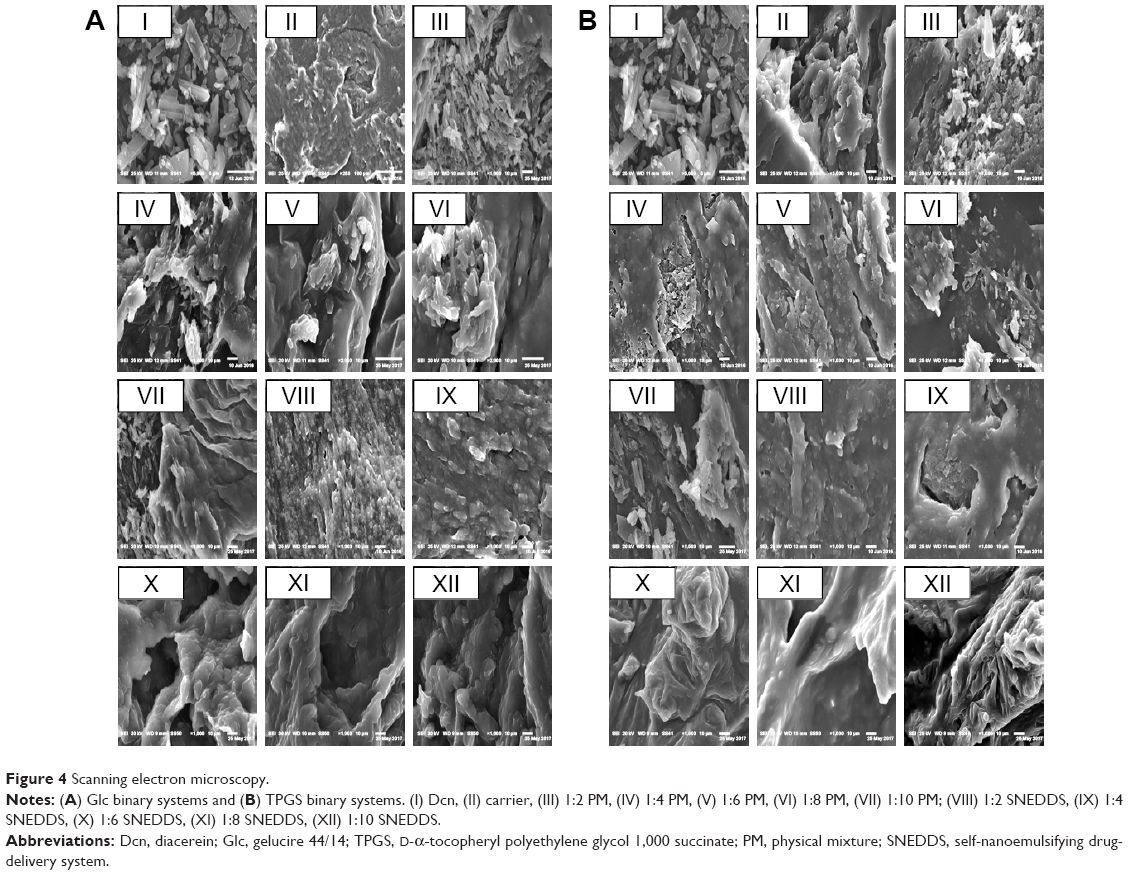 | Figure 4 Scanning electron microscopy. |
In vitro release study
Dcn in vitro release in PBS (pH 6.8) from capsules containing PMs or SNEDDSs with each carrier (Glc and TPGS) compared to that of the drug alone is illustrated in Figure 5. With either carrier, Dcn release from the prepared binary systems (PMs or SNEDDSs) was biphasic. The initial burst release was followed by a gradual-release phase. Such behavior has been reported for these carriers.26,28 Therefore, DE at 30 minutes (DE30) was calculated as a dissolution parameter during the initial burst-release phase to evaluate the significance of effects of binary-system type (PM and SNEDDS) and carrier concentration, as well as carrier nature (Glc or TPGS), if any, on Dcn in vitro release (Table 2). DE has been used as a dissolution parameter to evaluate the effects of SNEDDSs with Glc or TPGS on dissolution of incorporated drugs.26,28
 | Figure 5 In vitro Dcn release in phosphate buffer (pH 6.8) from capsules containing PMs or SNEDDSs with each carrier compared to the drug alone. |
 | Table 2 Dissolution efficiency (DE) and relative dissolution rate (RDR) of Dcn binary systems |
Physical mixing of the drug with Glc or TPGS at all ratios, except 1:1 Dcn:Glc PMs, significantly (P<0.05) enhanced drug release compared to the drug alone (Figure 5A and C, respectively). Formulation of Dcn as SNEDDSs with Glc and TPGS resulted in significantly (P<0.05) higher drug release than that seen with the drug alone and corresponding PMs (Figure 5B and D, respectively). Therefore, it can be said that Dcn in vitro release was dependent on the binary system being significantly (P<0.05) higher in the case of SNEDDSs, as was indicated by the significantly higher average values of DE30 of these systems relative to those of corresponding PMs (Table 2). Calculated values of RDR at the same time (RDR30) reflected such behavior (Table 2). The faster drug dissolution on incorporation of Dcn into SNEDDSs than physical mixing with either carrier may have indicated crystallinity reduction or complete amorphization of the drug, as confirmed by DSC, XRD, and SEM results (Figures 2–4). An amorphous drug is expected to dissolve at a faster rate than a crystalline form, due to its high energy.38,39
With regard to carrier concentration, the effects of the carriers differed. Generally, an increase in Glc concentration from drug:carrier weight ratio 1:1 to 1:10 significantly (P<0.05) enhanced Dcn release, except with 1:2 SNEDDS (Figure 5B), as reflected by mean DE30 values (Table 2), whereas, physical mixing of Dcn with increased concentrations of TPGS from 1:1 to 1:10 drug:carrier ratio resulted in an insignificantly different Dcn release (Figure 5C), as suggested by DE30 estimates in Table 2. Beyond a drug:carrier weight ratio of 1:2, increased TPGS concentration in its SNEDDSs with Dcn caused a significant (P<0.05) improvement in drug release (Figure 5D and Table 2).
There was an insignificant difference regarding carrier-nature effects on Dcn release following physical mixing with Glc or TPGS (Figure 5A and C, respectively). On the other hand, SNEDDSs showed significantly (P<0.05) different drug release up to 1:8 drug:carrier weight ratio. Interestingly, SNEDDSs based on 1:8 Dcn:TPGS showed insignificantly different drug release when compared to 1:10 Dcn:TPGS and 1:10 Dcn:Glc SNEDDSs. DE30 estimates in Table 2 confirmed these results.
Collectively with solid characterization and SEM results, in vitro release studies revealed that the optimized systems were 1:8 Dcn:TPGS and 1:10 Dcn:Glc SNEDDSs, which were further evaluated through particle-size and ζ-potential measurements, as well as TEM examination. In addition, these optimized systems were assessed with respect to their influences on anti-inflammatory activity against carrageenan-induced paw edema in rats compared to the drug.
Release kinetics
Due to the biphasic release pattern of Dcn from all prepared binary systems, kinetic analysis of Dcn-release data from these systems was carried out during burst- and gradual-release phases (Tables 3 and 4, respectively). During the burst-release phase, the results revealed that the release mechanism of Dcn from these systems varied between first- and zero-order kinetics (r2≥0.9142), and hence the Korsmeyer–Peppas model was applied to verify the release mechanism. Release exponent (n) values greater than the unity suggested supercase II transport, which refers to the erosion of polymer chain and could explain the initial burst release (Table 3).53 Meanwhile, the Higuchi diffusion model showed drug release from these systems during the gradual-release phase (Table 4). Accordingly, the Fickian mechanism (diffusion-controlled release) during the gradual-release phase was confirmed by n<0.5.42
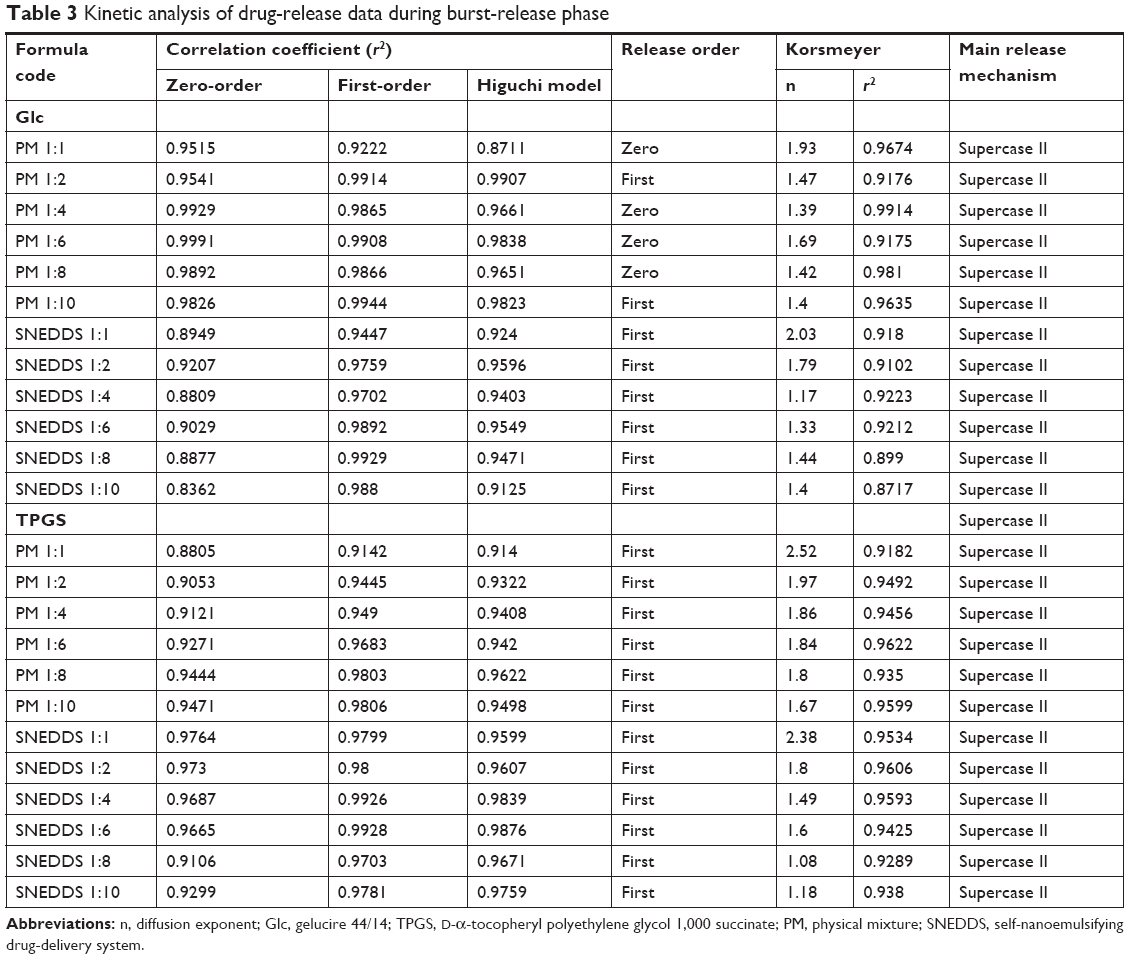 | Table 3 Kinetic analysis of drug-release data during burst-release phase |
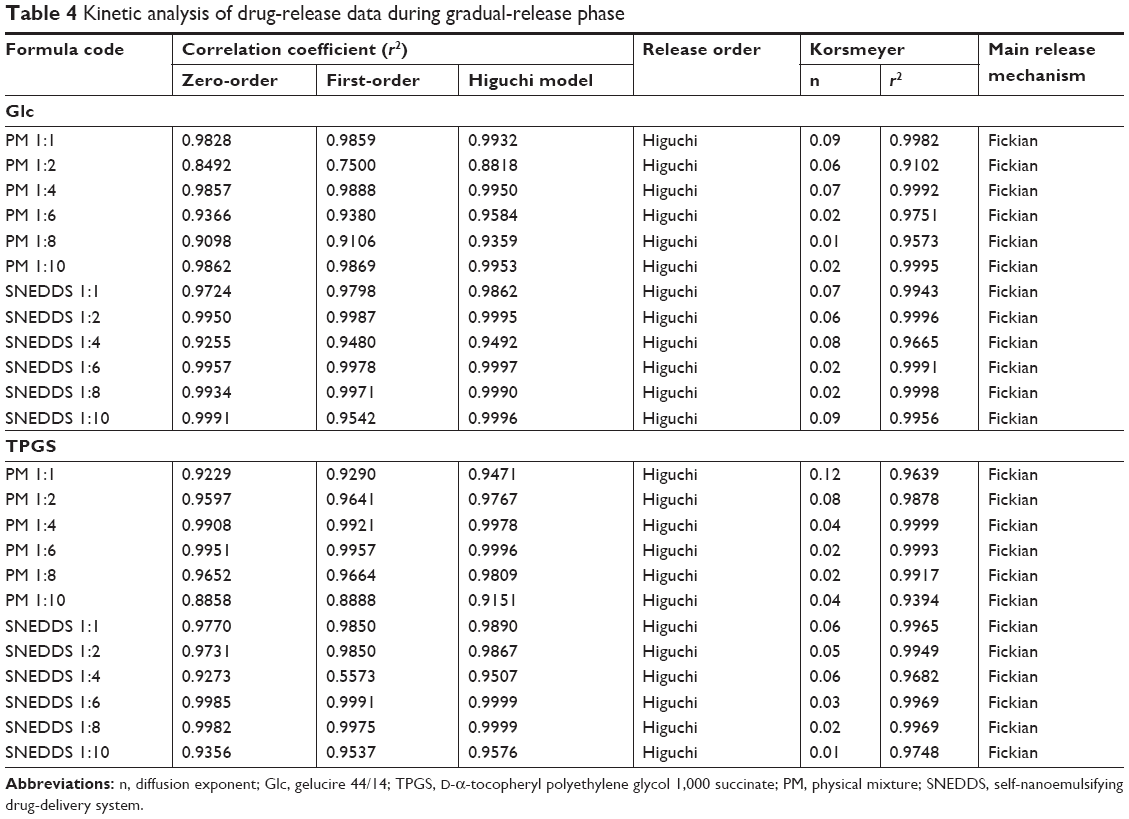 | Table 4 Kinetic analysis of drug-release data during gradual-release phase |
Size and ζ-potential measurements
Optimized SNEDDSs were nanoscopic, with mean diameters of 183±6.55 and 131±6.69 nm for 1:10 Dcn:Glc and 1:8 Dcn:TPGS, respectively. Average respective polydispersity index values were 0.41±0.08 and 0.49±0.08, indicating uniform size distribution. Facilitated access of cells and tissue is expected for nanoparticles of diameter <200 nm, permitting enhanced bioavailability.54 The faster drug absorption in the upper gut may largely diminish the laxative effect, due to unabsorbed rhein in the colon. Mean ζ-potential values were −15.87±0.95 and −23.40±1.35 for 1:10 Dcn:Glc and 1:8 Dcn:TPGS SNEDDSs, respectively, indicating hindered particle aggregation on dispersion into aqueous media, since the charged particles repel one another.55
Transmission electron microscopy
Figure 6 presents TEM images of 1:10 Dcn:Glc and 1:8 Dcn:TPGS SNEDDSs following dispersion into double-deionized water. In accordance with particle-size measurements, this figure shows well-dispersed and nearly spherical particles of diameter <200 nm, though the images show particles with diameters smaller than that determined by DLS. It has been reported that DLS records particles’ hydrodynamic diameter, and hence particles larger than their actual size are observed.56 Moreover, DLS is affected by the dispersion/aggregation behavior of particles in solution.57 The nanoscopic diameter may strongly reflect the possibility of improved absorption and bioavailability of Dcn upon incorporation into SNEDDSs with Glc and TPGS at respective drug:carrier weight ratios of 1:10 and 1:8. This can result in potentiated anti-inflammatory activity and reduced laxative action of this drug.
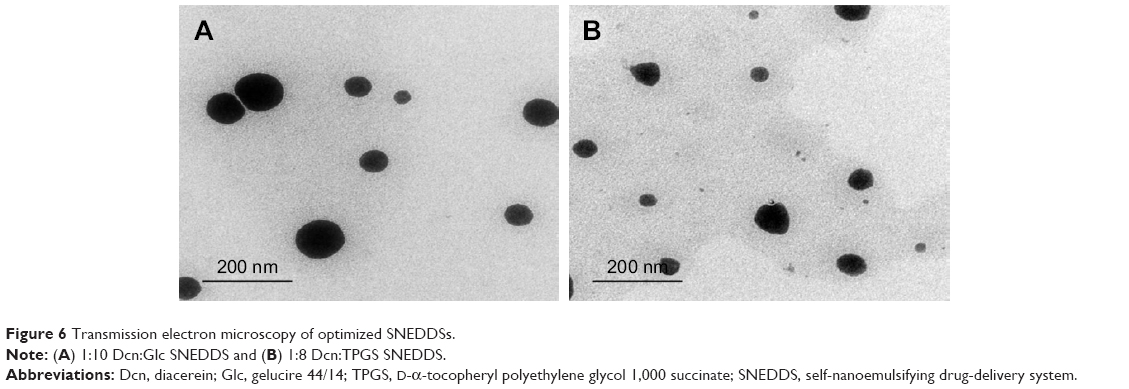 | Figure 6 Transmission electron microscopy of optimized SNEDDSs. |
Anti-inflammatory activity against carrageenan-induced paw edema in rats
Edema inhibition
Figure 7 presents edema up to 24 hours and edema inhibition in groups pretreated orally with 100 mg/kg Dcn or an equivalent dose of the two optimized SNEDDSs in comparison with the positive carrageenan group. Generally, all pretreated groups (III–V) exhibited a significant (P<0.05) reduction in edema when compared to the positive control group (II; Figure 7A). Rats pretreated with optimized SNEDDSs with either carrier (groups IV and V) showed significantly (P<0.05) lower edema than seen with the Dcn group pretreated orally (III).
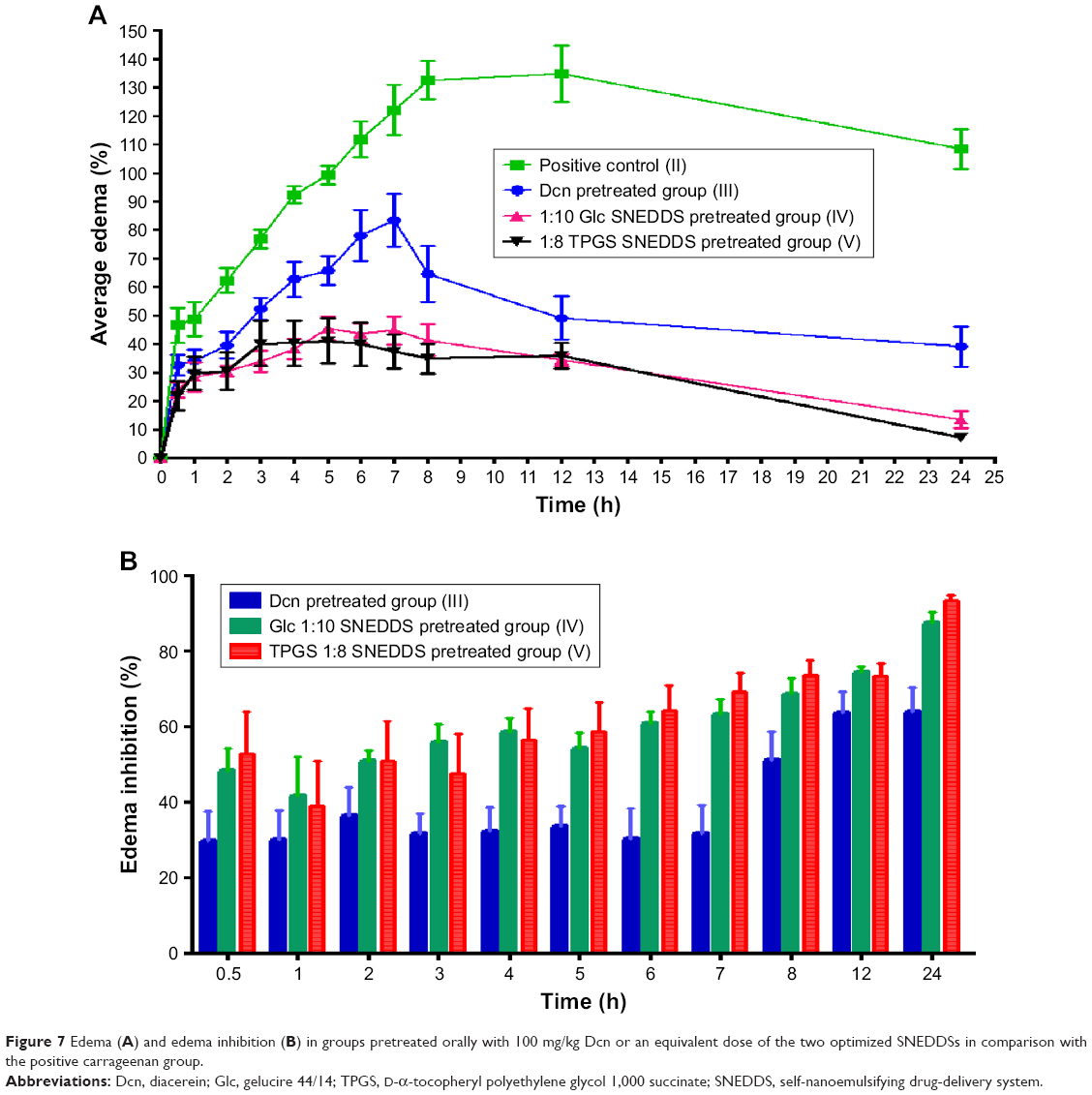 | Figure 7 Edema (A) and edema inhibition (B) in groups pretreated orally with 100 mg/kg Dcn or an equivalent dose of the two optimized SNEDDSs in comparison with the positive carrageenan group. |
In accordance with in vitro release data, pretreatment with 1:10 Dcn:Glc and 1:8 Dcn:TPGS SNEDDSs (groups IV and V, respectively) experienced insignificantly different edema percentages. In agreement, edema inhibition in groups pretreated with either optimized SNEDDS was generally higher than those pretreated with Dcn up to 24 hours (Figure 7B). The superiority of the optimized SNEDDSs over free Dcn regarding anti-inflammatory activity against carrageenan-induced paw edema in rats may be referred to the amorphous nanoscopic Dcn particles permitting enhanced in vitro release, and hence potentiated absorption and bioavailability. Also, facilitated cell and tissue access for nanoparticles of <200 nm diameter can also explain the superiority of the optimized systems.
Histopathological examination
Microscopical examination results of thin and thick paw-skin specimens are depicted in Figure 8. The results revealed normal thin and thick skins in the normal control group (I). Acute inflammation was recognized in dermal layers of thin skin (Figure 8A) and in dermal and muscular layers of thick skin (Figure 8B) in the positive control group (II) treated with carrageenan only. The predominant inflammatory cells were polymorphonuclear cells. Detachment and ulceration were observed in epidermal layers of thin skin in positive control group II and were absent in pretreated groups (III–V). In pretreated groups, less inflammation was noticed in both thin (Figure 8A) and thick skins (Figure 8B).
 | Figure 8 Histological examination (H&E, 100×) of rat-paw skin. |
Results of statistical analysis of inflammation scores for thin and thick skins are shown in Figure 9. The positive control group showed significantly (P<0.05) higher inflammation scores than the normal control. Pretreatment with Dcn, 1:10 Glc-SNEDDS, or 1:8 TPGS-SNEDDS resulted in a significant (P<0.05) reduction in inflammation when compared to the positive control group. In comparison with the Dcn-pretreated group, significantly (P<0.05) lower inflammation scores were observed on pretreatment with the optimized SNEDDSs. When compared to normal control, there were insignificantly different inflammation scores on pretreatment with the optimized SNEDDSs. Drug amorphization confirmed by DSC (Figure 2) and XRD (Figure 3) results and the nanoscopic particles of the optimized SNEDDSs with Glc and TPGS (Figure 6) can be considered major reasons for increased dissolution (Figure 5B and D, respectively) and subsequent enhanced absorption and anti-inflammatory activity.
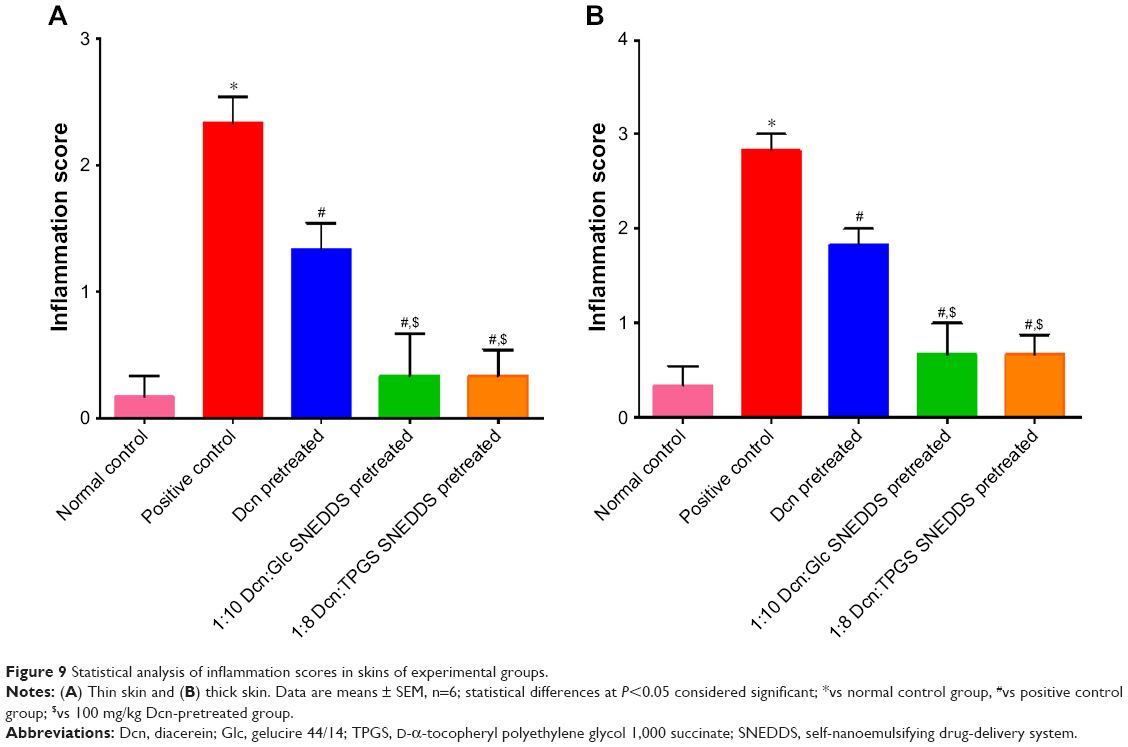 | Figure 9 Statistical analysis of inflammation scores in skins of experimental groups. |
Immunohistochemical localization of TNF-α and caspase-3
Figures 10 and 11 illustrate immunohistochemical evaluation of TNF-α and caspase-3 expression, respectively, in thin and thick rat-paw skin. Positive signals of TNF-α and caspase-3 antibodies were absent in skin of the normal control group (I), strong in skin of the positive control group (II), moderate in rats pretreated with Dcn (III), and mild in groups IV and V of rats pretreated with the optimized SNEDDSs with Glc and TPGS, respectively.
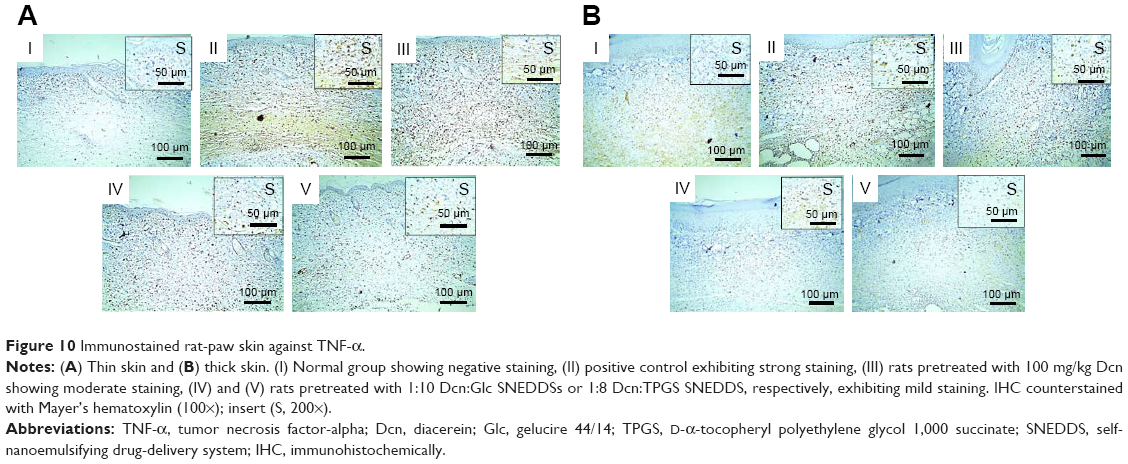 | Figure 10 Immunostained rat-paw skin against TNF-α. |
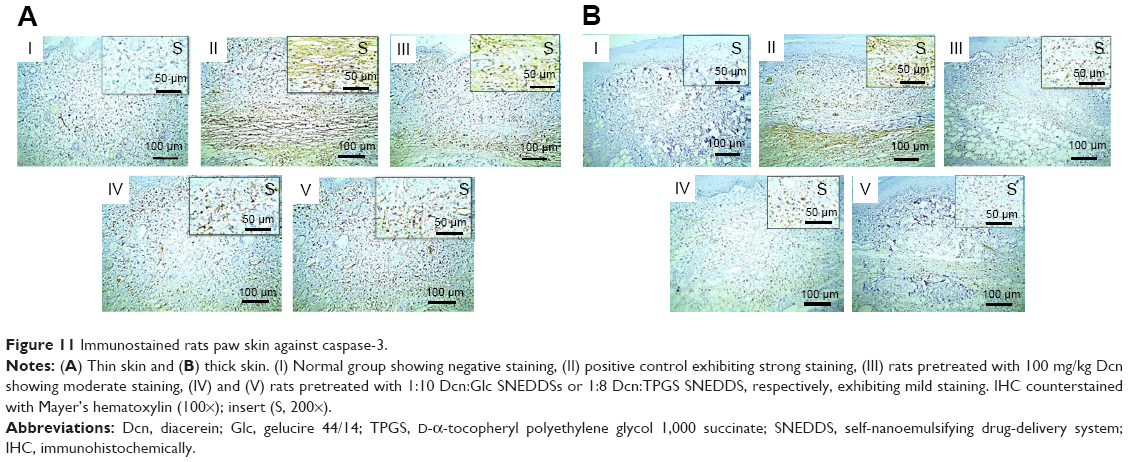 | Figure 11 Immunostained rats paw skin against caspase-3. |
Results of statistical analysis of immunohistochemical intensity scores in examined thin and thick skins are presented in Figure 12. This figure clarified that there was a significant (P<0.05) elevation in TNF-α and caspase-3 expression in thin and thick paw skins of positive control rats (II) relative to normal rats (I). In contrast, a significant (P<0.05) lowering in TNF-α and caspase-3 expression in thin and thick paw skins of rats pretreated with either Dcn (III) or the optimized SNEDDSs (IV and V) was observed when compared to those of normal control (I). In accordance with statistical analysis of inflammation scores, insignificantly different TNF-α and caspase-3 expression intensity in thin and thick paw skins was recorded in rats pretreated with the optimized SNEDDSs (IV and V). Dcn amorphization and nanosized particles encountered with the optimized SNEDDSs can still explain the potentiated anti-inflammatory activity of this drug against carrageenan-induced paw edema in rats.
 | Figure 12 Statistical analysis of IHC intensity scores of rats paw skin. |
Conclusion
In comparison with Dcn and the corresponding PMs, significantly (P<0.05) enhanced in vitro drug release was recorded for SNEDDSs with either Glc or TPGS. In vitro Dcn release was dependent on the binary system type, carrier type, and nature, particularly with SNEDDSs. The optimal systems were 1:10 Dcn:Glc and 1:8 Dcn:TPGS SNEDDSs, since they showed the highest drug release among the binary systems investigated. The crystallinity reduction or amorphization of Dcn and the nanoscopic diameter exhibited by the optimized SNEDDSs, in addition to the Pgp-inhibitory activity of both carriers may explain their superiority regarding in vitro drug release and anti-inflammatory activity against carrageenan-induced paw edema.
Acknowledgment
The authors would like to thank Dr Walaa Awadin, associate professor, Department of Pathology, Faculty of Veterinary Medicine, Mansoura University for her technical support and specimen examination during histopathological examination and immunohistochemical localization of TNF-α and caspase-3.
Disclosure
The authors report no conflicts of interest in this work.
References
Alcaraz MJ, Megías J, García-Arnandis I, Clérigues V, Guillén MI. New molecular targets for the treatment of osteoarthritis. Biochem Pharmacol. 2010;80(1):13–21. | ||
Berenbaum F. Targeted therapies in osteoarthritis: a systematic review of the trials on www.clinicaltrials.gov. Best Pract Res Clin Rheumatol. 2010;24(1):107–119. | ||
Sostres C, Gargallo CJ, Arroyo MT, Lanas A. Adverse effects of non-steroidal anti-inflammatory drugs (NSAIDs, aspirin and coxibs) on upper gastrointestinal tract. Best Pract Res Clin Gastroenterol. 2010;24(2):121–132. | ||
Jain A, Singh R, Singh S, Singh S. Diacerein protects against iodoacetate-induced osteoarthritis in the femorotibial joints of rats. J Biomed Res. 2015;29(5):405–413. | ||
Dougados M, Nguyen M, Berdah L, Maziéres B, Vignon E, Lequesne M. ECHODIAH investigators study group. evaluation of the structure-modifying effects of diacerein in hip osteoarthritis: ECHODIAH, a three-year, placebo-controlled trial. Arthritis Rheum. 2001;44(11):2539–2547. | ||
Bartels EM, Bliddal H, Schøndorff PK, Altman RD, Zhang W, Christensen R. Symptomatic efficacy and safety of diacerein in the treatment of osteoarthritis: a meta-analysis of randomized placebo-controlled trials. Osteoarthritis Cartilage. 2010;18(3):289–296. | ||
Pavelka K, Bruyère O, Cooper C, et al. Diacerein: benefits, risks and place in the management of osteoarthritis. An opinion-based report from the ESCEO. Drugs Aging. 2016;33(2):75–85. | ||
Elsayed I, Abdelbary AA, Elshafeey AH. Nanosizing of a poorly soluble drug: technique optimization, factorial analysis, and pharmacokinetic study in healthy human volunteers. Int J Nanomedicine. 2014;9:2943–2953. | ||
Yamauchi K, Yagi T, Kuwano S. Suppression of the purgative action of rhein anthrone, the active metabolite of sennosides A and B, by calcium channel blockers, calmodulin antagonists and indomethacin. Pharmacology. 1993;47 Suppl 1(1):22–31. | ||
Aggarwal AK, Singh S. Physicochemical characterization and dissolution study of solid dispersions of diacerein with polyethylene glycol 6000. Drug Dev Ind Pharm. 2011;37(10):1181–1191. | ||
Zaki RM, Ali AA, El Menshawi SF, Bary AA. Effect of binary and ternary solid dispersions prepared by fusion method on the dissolution of poorly water-soluble diacerein. Int J Drug Del. 2013;5(1):99–109. | ||
Patil SB, Shete DK, Narade SB, et al. Improvement in the dissolution profile of diacerein using a surfactant-based solid dispersion technique. Drug Discov Ther. 2010;4(6):435–441. | ||
Garthe OM, Kothawade PS, Mahajan VR. Solubility Enhancement of diacerein by solid dispersion technique. Int J Pharm Res Allied Sci. 2013;2(2):47–55. | ||
Nakhat P, Mandaogade P, Jain GK, No TMUS. 20100303902A1, Washington, DC: U. S. Patent and Trademark Office. 2010. | ||
Aggarwal AK, Singh S. Fast-dissolving and high-drug-loaded, Fatty Acid-based self-emulsifying solid dispersions of diacerein. PDA J Pharm Sci Technol. 2012;66(3):201–213. | ||
Nazzal S, Smalyukh II, Lavrentovich OD, Khan MA. Preparation and in vitro characterization of a eutectic based semisolid self-nanoemulsified drug delivery system (SNEDDS) of ubiquinone: mechanism and progress of emulsion formation. Int J Pharm. 2002;235(1–2):247–265. | ||
Pouton CW. Formulation of poorly water-soluble drugs for oral administration: physicochemical and physiological issues and the lipid formulation classification system. Eur J Pharm Sci. 2006;29(3–4):278–287. | ||
Sharma A, Sharma US. Liposomes in drug delivery: Progress and limitations. Int J Pharm. 1997;154(2):123–140. | ||
Müller RH, Mäder K, Gohla S. Solid lipid nanoparticles (SLN) for controlled drug delivery–a review of the state of the art. Eur J Pharm Biopharm. 2000;50(1):161–177. | ||
Mahmoud EA, Bendas ER, Mohamed MI. Preparation and evaluation of self-nanoemulsifying tablets of carvedilol. AAPS PharmSciTech. 2009;10(1):183–192. | ||
Pouton CW. Lipid formulations for oral administration of drugs: non-emulsifying, self-emulsifying and ‘self-microemulsifying’ drug delivery systems. Eur J Pharm Sci. 2000;11 Suppl 2(2):S93–S98. | ||
O’Driscoll CM. Lipid-based formulations for intestinal lymphatic delivery. Eur J Pharm Sci. 2002;15(5):405–415. | ||
Porter CJ, Trevaskis NL, Charman WN. Lipids and lipid-based formulations: optimizing the oral delivery of lipophilic drugs. Nat Rev Drug Discov. 2007;6(3):231–248. | ||
Chambin O, Jannin V. Interest of multifunctional lipid excipients: case of Gelucire 44/14. Drug Dev Ind Pharm. 2005;31(6):527–534. | ||
Jannin V, Musakhanian J, Marchaud D. Approaches for the development of solid and semi-solid lipid-based formulations. Adv Drug Deliv Rev. 2008;60(6):734–746. | ||
Barakat NS. Etodolac-liquid-filled dispersion into hard gelatin capsules: an approach to improve dissolution and stability of etodolac formulation. Drug Dev Ind Pharm. 2006;32(7):865–876. | ||
Soliman MS, Khan MA. Preparation and in vitro characterization of a semi-solid dispersion of flurbiprofen with Gelucire 44/14 and Labrasol. Pharmazie. 2005;60(4):288–293. | ||
Eedara BB, Veerareddy PR, Jukanti R, Bandari S. Improved oral bioavailability of fexofenadine hydrochloride using lipid surfactants: ex vivo, in situ and in vivo studies. Drug Dev Ind Pharm. 2014;40(8):1030–1043. | ||
Varma MV, Panchagnula R. Enhanced oral paclitaxel absorption with vitamin E-TPGS: effect on solubility and permeability in vitro, in situ and in vivo. Eur J Pharm Sci. 2005;25(4–5):445–453. | ||
Fulzele SV, Chatterjee A, Shaik MS, Jackson T, Singh M. Inhalation delivery and anti-tumor activity of celecoxib in human orthotopic non-small cell lung cancer xenograft model. Pharm Res. 2006;23(9):2094–2106. | ||
Saidi Z, Klyashchitsky B. U.S. Patent No. 6,241,969 B1. Washington, DC: U.S. Patent and Trademark Office. 2001. | ||
Nikonenko BV, Reddy VM, Protopopova M, Bogatcheva E, Einck L, Nacy CA. Activity of SQ641, a capuramycin analog, in a murine model of tuberculosis. Antimicrob Agents Chemother. 2009;53(7):3138–3139. | ||
Momot KI, Kuchel PW, Chapman BE, Deo P, Whittaker D. NMR Study of the Association of Propofol with Nonionic Surfactants. Langmuir. 2003;19(6):2088–2095. | ||
Guo Y, Luo J, Tan S, Otieno BO, Zhang Z. The applications of Vitamin E TPGS in drug delivery. Eur J Pharm Sci. 2013;49(2):175–186. | ||
Christiansen A, Backensfeld T, Kühn S, Weitschies W. Investigating the stability of the nonionic surfactants tocopheryl polyethylene glycol succinate and sucrose laurate by HPLC-MS, DAD, and CAD. J Pharm Sci. 2011;100(5):1773–1782. | ||
Zhang Z, Tan S, Feng SS. Vitamin E TPGS as a molecular biomaterial for drug delivery. Biomaterials. 2012;33(19):4889–4906. | ||
Ahuja N, Katare OP, Singh B. Studies on dissolution enhancement and mathematical modeling of drug release of a poorly water-soluble drug using water-soluble carriers. Eur J Pharm Biopharm. 2007;65(1):26–38. | ||
Mohamed EA, Meshali MM, Foda AM, Borg TM. Improvement of dissolution and hypoglycemic efficacy of glimepiride by different carriers. AAPS PharmSciTech. 2012;13(3):1013–1023. | ||
Abdelkader H, Abdallah OY, Salem HS. Comparison of the effect of tromethamine and polyvinylpyrrolidone on dissolution properties and analgesic effect of nimesulide. AAPS PharmSciTech. 2007;8(3):E110–E117. | ||
Martin AN, Swarbrick J, Cammarata A. Physical Pharmacy: Physical Chemical Principles in The Pharmaceutical Sciences. 4th Lea, Febiger, editors. Philadelphia; 1993. | ||
Higuchi T. Mechanismof of sustained-action medication. theoretical analysis of rate of release of solid drugs dispersed in solid matrices. J Pharm Sci. 1963;52(12):1145–1149. | ||
Ritger PL, Peppas NA. A simple equation for description of solute release I. Fickian and non-fickian release from non-swellable devices in the form of slabs, spheres, cylinders or discs. Journal of Controlled Release. 1987;5(1):23–36. | ||
Mei Z, Li X, Wu Q, Hu S, Yang X. The research on the anti-inflammatory activity and hepatotoxicity of triptolide-loaded solid lipid nanoparticle. Pharmacol Res. 2005;51(4):345–351. | ||
Bancroft JD, Gamble M. Theory and Practice of Histological Techniques. 5th ed. Livingstone Churchill, London UK, 2007:125–138. | ||
Riella KR, Marinho RR, Santos JS, et al. Anti-inflammatory and cicatrizing activities of thymol, a monoterpene of the essential oil from Lippia gracilis, in rodents. J Ethnopharmacol. 2012;143(2):656–663. | ||
El-Sheakh AR, Ghoneim HA, Suddek GM, Ammar El-Sm. Antioxidant and anti-inflammatory effects of flavocoxid in high-cholesterol-fed rabbits. Naunyn Schmiedebergs Arch Pharmacol. 2015;388(12):1333–1344. | ||
Mohamed EA, Abu Hashim II, Yusif RM, et al. Polymeric micelles for potentiated antiulcer and anticancer activities of naringin. Int J Nanomedicine. 2018;13:1009–1027. | ||
Fisher ER, Anderson S, Dean S, et al. Solving the dilemma of the immunohistochemical and other methods used for scoring estrogen receptor and progesterone receptor in patients with invasive breast carcinoma. Cancer. 2005;103(1):164–173. | ||
B. P. British Pharmacopeia, Vol. III, 6th ed. London: The Council of Europe, The Stationary Office; 2010; pp. 3155–3157. | ||
Silverstein RM, Bassler GC, Morrill TC. Spectroscopic Identification of Organic Compounds. 5th Wiley John, Sons Inc., NY USA, editors; 1991. | ||
Faisal W, Ruane-O’Hora T, O’Driscoll CM, Griffin BT. A novel lipid-based solid dispersion for enhancing oral bioavailability of Lycopene – in vivo evaluation using a pig model. Int J Pharm. 2013;453(2):307–314. | ||
Shin SC, Kim J. Physicochemical characterization of solid dispersion of furosemide with TPGS. Int J Pharm. 2003;251(1–2):79–84. | ||
Ofokansi KC, Adikwu MU, Okore VC. Preparation and evaluation of mucin-gelatin mucoadhesive microspheres for rectal delivery of ceftriaxone sodium. Drug Dev Ind Pharm. 2007;33(6):691–700. | ||
Allen C, Maysinger D, Eisenberg A. Nano-engineering block copolymer aggregates for drug delivery. Colloids Surf B Biointerfaces. 1999;16(1–4):3–27. | ||
Bayindir ZS, Yuksel N. Characterization of niosomes prepared with various nonionic surfactants for paclitaxel oral delivery. J Pharm Sci. 2010;99(4):2049–2060. | ||
Fissan H, Ristig S, Kaminski H, Asbach C, Epple M. Comparison of different characterization methods for nanoparticle dispersions before and after aerosolization. Analytical Methods. 2014;6(18):7324–7334. | ||
Eaton P, Quaresma P, Soares C, et al. A direct comparison of experimental methods to measure dimensions of synthetic nanoparticles. Ultramicroscopy. 2017;182:179–190. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.