Back to Journals » International Journal of Nanomedicine » Volume 18
Self-Nanoemulsifying Drug Delivery System for Enhanced Bioavailability of Madecassic Acid: In vitro and in vivo Evaluation
Authors Lin L, Chen Q, Dai Y, Xia Y
Received 10 February 2023
Accepted for publication 28 April 2023
Published 8 May 2023 Volume 2023:18 Pages 2345—2358
DOI https://doi.org/10.2147/IJN.S408115
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Yan Shen
Li Lin,1,* Qingyong Chen,1,* Yue Dai,2 Yufeng Xia1
1Department of Pharmacognosy, School of Traditional Chinese Pharmacy, China Pharmaceutical University, Nanjing, 211198, People’s Republic of China; 2Department of Traditional Chinese Medicine and Pharmacology, School of Traditional Chinese Pharmacy, China Pharmaceutical University, Nanjing, 211198, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Yufeng Xia, Department of Pharmacognosy, School of Traditional Chinese Pharmacy, China Pharmaceutical University, 639 Longmian Avenue, Jiangning District, Nanjing, 211198, People’s Republic of China, Tel +862583271400, Fax +862585301528, Email [email protected]
Purpose: Madecassic acid (MCA) is a natural triterpenoid isolated from centellae herba that has diverse biological effects, such as anti-inflammatory, antioxidant, and anticancer activities. However, the efficacy of MCA is limited by low oral bioavailability caused by its extremely poor aqueous solubility. This study aimed to develop a self-nanoemulsifying drug delivery system (SNEDDS) for MCA to improve its oral absorption.
Methods: The utilized oil phases, surfactants, and co-surfactants for SNEDDS were selected based on the solubility of MCA and emulsification efficiency. The optimized formulation was characterized for pharmaceutical properties and its pharmacokinetic behavior was examined in rats. Besides, the intestinal absorption property of MCA was investigated using in situ single-pass intestinal perfusion and intestinal lymphatic transport.
Results: The optimized nanoemulsion formula consists of Capryol 90:Labrasol:Kolliphor ELP:Transcutol HP in a weight ratio of 1:2.7:2.7:3.6 (w/w/w/w). MCA-loaded SNEDDS presented a small droplet size (21.52 ± 0.23 nm), with a zeta potential value of − 3.05 ± 0.3 mV. Compared with pure MCA, SNEDDS had a higher effective permeability coefficient and showed 8.47-fold and 4.01-fold of maximum plasma concentration (Cmax) and area under the plasma concentration-time curve (AUC), respectively. Cycloheximide was pretreated before the experiment to evaluate the degree of lymphatic uptake. The results showed that cycloheximide greatly influenced the absorption of SNEDDS, resulting in 82.26% and 76.98% reduction in Cmax and AUC, respectively.
Conclusion: This study reports the MCA-loaded SNEDDS with distinctly enhanced in vitro and in vivo performance compared with pure MCA and concludes that the SNEDDS formulation could be a viable and effective strategy for improving the dissolution rate and bioavailability of poor aqueous-soluble ingredients.
Keywords: madecassic acid, self-nanoemulsifying drug delivery system, pharmacokinetics, intestinal lymphatic transport
Introduction
Madecassic acid (MCA), a typical bioactive pentacyclic triterpenoid isolated from the traditional Chinese medicinal herb Centella asiatica, has a variety of pharmacological activities, including anti-inflammatory,1 antioxidant,2 anti-diabetic,3 neuroprotective,4 and anticancer effects.5,6 MCA exhibited a significant anti-colitis effect in dextran sulfate sodium-induced mice. MCA could decrease the number of gammadeltaT17 cells and attenuate the inflammation via peroxisome proliferator activated receptor gamma-phosphatase and tensin homolog/protein kinase B/glycogen synthase kinase-3beta/nuclear factor of activated T cells pathway,7 and inhibit lipopolysaccharides induced inflammatory stress in RAW 264.7 cells by suppressing nuclear factor kappa-B pathway.8 MCA has a potential for development into a new natural anti-inflammatory drug. MCA contains four hydroxyl groups and one carboxylic group (Figure 1), which was expected to be somewhat soluble in aqueous solutions, but experimentally, saturation is reached at a low concentration (85.0 μg/mL in phosphate buffer solution).9 The calculated partition coefficient (log K) in octanol and water was 3.1 for MCA. The low solubility and high log K of MCA are due to the triterpenoid aglycones mostly comprising hydrophobic regions (pentacyclic skeleton).10 The low aqueous solubility and dissolution rate of MCA are disadvantageous for oral administration and also limit the therapeutic activities. Thus, it is essential to develop a suitable strategy to successfully overcome the challenge to enhance its oral absorption.
 |
Figure 1 The chemical structure of MCA. |
In recent years, nanotechnology has benefited from novel research with resulting innovation in the field of biological sciences. Nanoscaled drug delivery systems have obvious advantages including good pharmacological effectiveness and low toxicity. Diverse formulation methods have been developed to modify the solubility of drugs, such as nanocrystals, micro/nanoemulsion, solid dispersion, and lipid-based drug delivery systems.11 Among them, the self-nanoemulsifying drug delivery system (SNEDDS), a novel isotropic system, consists of an oil phase, a surfactant, and one or more co-solvents or co-surfactants, which offer high potential in ameliorating the solubility, dissolution rate, and oral bioavailability of hydrophobic agents.12 SNEDDS enhances drug delivery by promoting the absorption of drugs via the lymphatic transport pathway, preventing the hepatic first-pass metabolism, reducing the effects of intestinal excretion transport, and avoiding the degradation of drugs in the physiological environment.13,14 The use of SNEDDS has generated much academic and industrial interest as potential formulations for enhancing bioavailability of ingredients. SNEDDS recently proved their advantages in improving oral bioavailability by solubilizing poorly water-soluble compounds such as celastrol, and naringenin (that share some physical properties with MCA).15,16 Moreover, Abushal et al developed a SNEDDS of apremilast by using 15% oil, 60% Smix, and 25% water, which enhanced the relative bioavailability of apremilast by 7.07-fold compared with that of the drug suspension.17 Verma et al also improved the rate of dissolution and oral bioavailability of poorly soluble candesartan by formulating it into SNEDDS.18–21
Accordingly, the aim of the current study is to develop an MCA-loaded SNEDDS formulation to achieve improvement of pharmacokinetic behavior. The selected formulation was evaluated by in vitro dissolution and in vivo studies. In vivo evaluation of the selected formulation was studied in Sprague-Dawley rats, and to assess the relative bioavailability, pharmacokinetic parameters were compared with pure MCA. In addition, the intestinal absorption and lymphatic transport of MCA-loaded SNEDDS were studied to explore the underlying mechanism.
Materials and Methods
Materials and Animals
Madecassic acid (MCA, >98.5%) was provided by Jiangsu Yongjian Pharmaceutical Technology Co., Ltd (Jiangsu, China). Glycyrrhetinic acid (>98%) was procured from China National Institute for the Control of Pharmaceutical and Biological Products (Peking, China). Capryol 90, Labrafil M 1944 CS, Labrafac Lipophile WL 1349, Maisine CC, Peceol, Labrafac PG, Transcutol HP, Lauroglycol FCC, and Labrasol were donated by Guangzhou Tianrun Pharmaceutical Co., Ltd (Guangdong, China). Kolliphor ELP, Kolliphor HS15, and Kolliphor RH40 were gifted from BASF (Ludwigshafen, Germany). Tween 80, ethyl oleate, and oleic acid were acquired from Nanjing Chemical Reagent Co., Ltd (Jiangsu, China). PEG 400 and 1.2-propanediol were acquired from Sinopharm Chemical Reagents Co., Ltd (Shanghai, China). HPLC-grade acetonitrile and methanol were obtained from Merck (Darmstadt, Germany), and HPLC-grade formic acid was purchased from Shanghai Linen Science and Technology Development Co., Ltd (Shanghai, China). Ultrapure water was prepared from a Milli-Q water purification system (Millipore, Milford, MA, USA). All other chemicals were of analytical grade.
Male Sprague-Dawley rats (180–220 g) were supplied by Shanghai SLAC Laboratory Animal Co., Ltd. All animals were housed at controlled temperature (22 ± 2°C) and controlled lighting (12 h dark/light cycle) with access to standard chow and water ad libitum in an SPF environment. All of the experimental procedures were in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals, and were approved by the Institutional Animal Care and Use Committee of China Pharmaceutical University (CPU-2019-06-004).
Design of Self-Nanoemulsifying Drug Delivery System (SNEDDS)
Equilibrium Solubility Determination of MCA
The saturated equilibrium solubility of MCA was examined in diverse oil phases (Labrafac PG, Peceol, Maisine CC, Capryol 90, Labrafil M 1944 CS, Labrafac Lipophile WL 1349, Capryol 90, ethyl oleate and oleic acid), surfactants (Kolliphor RH40, Kolliphor HS15, Kolliphor ELP, Tween 80, Labrasol and Lauroglycol FCC), and co-surfactants (1,2-propanediol, PEG 400, and Transcutol HP). An excess amount of MCA was added to 0.5 mL of different oil phases, surfactants and co-surfactants, the mixture was vortexed for 5 min, ultrasound treatment for 30 min, and placed in a shaker at room temperature for 48 h. Then, the equilibrated samples were centrifuged at 14,000 rpm for 10 min, each supernatant was collected, diluted with methanol, passed through 0.22 μm membrane filter, and analyzed by Agilent 1260 HPLC system (Agilent Technologies, Palo Alto, CA, USA). Chromatographic separation was performed on a Shim-pack CLC-ODS C18 column (4.6 mm × 150 mm, particle size 5 μm, Shimadzu, Kyoto, Japan). The mobile phase consisted of acetonitrile and 0.1% formic acid aqueous solution (70:30, v/v) at flow rate 1.0 mL/min. The column compartment was set at 40°C and detective wavelength was 204 nm. The examination was performed in triplicate and the standard deviation (SD) was calculated.
Emulsification Efficiency Study
The self-emulsification test is used for evaluating the excipients’ miscibility, formulation spontaneity, homogeneity, and appearance after aqueous dilution.22 The apparent spontaneity of emulsion formation against time was mainly measured by visual tests. The formulation was subjected to 1:100 aqueous dilution, and the mixture was stirred for 5 min to provide fast evaluation of formulation appearance and morphology.23 For surfactant screening, surfactant was mixed with the oil phase, and the mixture was reconstituted with distilled water. The spontaneity of the formulation was judged as “A” when a clear or slight blue appearance was visually observed within 1 min. It was judged as “B” when the appearance of the formulation was bluish (semi-clear), within 1 min. When the droplets took 1–2 min to completely spread in water and the appearance was turbid, the formulation could be assigned as “C”. Finally, the formulation was classified as “D” and “E” when the droplets needed within 3 min and more than 3 min to completely spread in water, respectively.13,24,25 Briefly, for co-surfactant selection, the surfactant was mixed with the same amount of co-surfactant, and the oil phase was added, then the mixture was subjected to 1:100 distilled water in a glass beaker.
Pseudo-Ternary Phase Diagram
To identify the region of SNEDDS formulation visually, pseudo-ternary phase diagrams were constructed for selecting oil phases, surfactants, and co-surfactants of different mass ratios. For each phase diagram, the oil phase, surfactant, and co-surfactant at specific ratios were mixed uniformly in various proportions (1:9, 2:8, 3:7, 4:6, 5:5, 6:4, 7:3, 8:2, 9:1).26 Formulations were judged as grade “A” and “B” when the dispersion of droplets in water occurred within 1 min, and clear or semi-clear emulsion with a bluish or bluish tinge color. The types of “A” and “B” formulations were selected for construction of the self-emulsifying region in the phase diagram. Analysis software Origin 8.0 was used to construct the pseudo-ternary phase diagrams.
Droplet Size and Zeta Potential
The formulation was diluted at a ratio of 1:20 v/v (formulation: distilled water) and mixed for 3 min before analysis. The droplet size and zeta potential of the diluted MCA-loaded SNEDDS were determined by a Malvern Zetasizer Nano ZS90 (Malvern Instrument Ltd, UK) at room temperature.
In-vitro Release Study
The release of pure MCA and MCA from the optimized formula was studied via dialysis bag technique (MW8000-14000). The dissolution test was carried out in 1% polysorbate 80 in 0.05M phosphate buffer of pH 6.8 ± 0.05 to maintain sink condition, and the temperature was set at 37 ± 0.5 °C.27,28 Pure MCA or MCA-SNEDDS was sealed in a dialysis bag and immersed with 50 mL of fresh release media in a plastic tube. The condition was maintained in an air-bath shaker at a speed of 50 rpm. The samples (0.4 mL) were withdrawn at predetermined time points (0.5, 0.75, 1, 2, 4, 8, 12, 16, 20, 24, 30 and 36 h), and an equal volume of fresh medium was added to the vessel to maintain a constant volume of dissolution medium. Each sample was filtered through a 0.22 μm membrane filter and analyzed for amount of MCA released by HPLC method discussed above. The release studies were carried out in triplicate. The drug release mechanism from the SNEDDS formulation was studied via the application of various kinetic models including zero-order model, first order, Higuchi model, Hixson-Crowell, and Ritger-Peppas model (Origin 8.0).
Bioavailability Studies
The bioavailability of the optimal SNEDDS of MCA (Capryol 90: Labrasol: Kolliphor ELP: Transcutol HP in a weight ratio of 1:2.7:2.7:3.6 (w/w/w/w)) was compared with pure MCA powder on Sprague-Dawley rats. Both MCA-loaded SNEDDS and pure MCA were administered orally at a dose equivalent to 100 mg/kg of MCA. The plasma samples were collected at 0.1, 0.3, 0.5, 0.7, 1, 2, 4, 8, 12, and 24 h after the administration. All plasma samples were centrifuged at 8000 rpm for 6 min, and the supernatant was stored at −20°C until analysis. To 200 μL aliquot of plasma sample, 50 μL internal standard (IS, glycyrrhetinic acid, 400 ng/mL) and 1.1 mL EtOAc were added and vortexed for 6 min to mix in a 1.5 mL polypropylene tube, and were centrifuged at 12,000 rpm for 10 min. The supernatant (1 mL) was evaporated to dryness under vacuum at 40°C. The residue was reconstituted in 150 μL 38:62 methanol/water solution (v/v) and centrifuged at 12,000 rpm for 10 min, and the supernatant was removed into an injection vial, and a 5 μL aliquot was injected into the Agilent 6460 LC-MS/MS system (Agilent Technologies, Palo Alto, CA, USA) for analysis. Separation was performed on an Agilent SB-C18 column (2.1 mm × 50 mm, particle size 1.8 μm, Agilent, USA). The mobile phase consisted of 0.1% aqueous formic acid (FA, v/v, solvent A) and 0.1% FA/ACN (v/v, solvent B). The flow rate was 0.4 mL/min. The gradient was set as follows: 0–2 min, an isocratic elution at 38% B; 2.1–3 min, an isocratic elution at 80% B; 3.1–4 min, an isocratic elution at 38% B. The precursor ions and product ions were m/z 503.3 → 503.3 for MCA, and m/z 469.4 → 425.4 for IS. The fragment was set at 300 V for MCA and 230 V for IS. Collision energy was set at 0 eV for MCA and 40 eV for IS. Quantitation of the drugs in rat plasma was based on the peak area ratio of the cited drug versus that of the IS. Data acquisition and processing were performed using Masshunter software 8.0.
Methods were validated for specificity, linearity, accuracy, precision, extract recovery and stability, according to USA FDA guidelines. Pharmacokinetic parameters including maximum plasma concentration (Cmax), the time to reach Cmax (Tmax), area under the curve (AUC), half-time (T1/2), oral clearance per unit time (CL/F) and mean residence time (MRT) were determined by non-compartmental modeling with PK solver software (issued by China Pharmaceutical University). p value < 0.05 was considered to be statistically significant. All results are expressed as mean ± SD.
Intestinal Permeability Study
The intestinal permeability of the optimal SNEDDS of MCA was evaluated in single-pass intestinal permeability study.29 Firstly, MCA-SNEDDS (containing 200 μg/mL MCA), MCA (200 μg/mL), or MCA (200 μg/mL) with verapamil (0.1 mg/mL) was ultrasonically dispersed in Krebs-Ringer (KR) buffer solution and treated with 37°C water bath to obtain perfusion solution. Then, after abdomen was opened, bile duct was ligated to eliminate the influence of bile. A single-pass constant flow (0.2 mL/min) of solution was perfused into ligated duodenum and jejuna segments (length of ~20 cm). After 30 min, the samples were regularly taken out for 2 h at 15 min intervals and centrifuged. The lengths of the tested sample segments were measured at the end of the perfusion. The supernatants were collected, an equal amount of acetonitrile was added, vortexed for 1 min, and centrifuged at 12,000 rpm for 10 min. The HPLC analysis was the same as “Equilibrium Solubility Determination of MCA”. The effective permeability coefficient was calculated using the following equation.24

Cin is the concentration of MCA in the inlet perfused (μg/mL), Cout is the corrected concentration of MCA in the outlet perfused (μg/mL). Vin and Vout are the entering and exiting volume of the perfusion solution (mL), Q is the flow rate of perfusion solution (mL/min), r is the radius of the rat intestinal (cm), and l is the length of the intestinal segment (cm).
Lymphatic Transport After Oral Administration
Comparing the bioavailability of MCA-SNEDDS after oral dosing with and without pre-treatment with chylomicron flow blocking agent cycloheximide.30 The animals were pre-treated with 3 mg/kg of cycloheximide administered intraperitoneally 1 h before dosing. After the SNEDDS of MCA treatments, plasma samples were collected at 0.1, 0.3, 0.5, 0.7, 1, 2, 4, 8, 12, and 24 h. All plasma samples were centrifuged at 8000 rpm for 6 min, and the supernatant was stored at −20°C until analysis. Sample preparation and HPLC-MS/MS analysis were the same as “Bioavailability Studies”.
Statistical Analysis
All examinations were performed in triplicate and the data were calculated as mean ± standard deviation (SD). Statistical analyses were done using SPSS 22.0 software (IL, USA). Student’s t-test was used to evaluate any significant differences in the experiments, and p value < 0.05 was considered statistically significant.
Results and Discussion
Equilibrium Solubility Study
Solubility of the drug in excipients plays a significant role in the stability of formulations since numerous formulations experience precipitation prior to encountering in situ solubilization.31 Self-emulsifying nanoemulsions must possess excellent solubility to incorporate drug doses in a minimum volume of the formulation. High drug solubilization is important for improving the efficiency of drug loading into carriers with concomitant advances in oral bioavailability.18 Hence, the selection of media to formulate SNEDDS is critical. The solubility of MCA in diverse oil phases, surfactants, and co-surfactants has been evaluated (Figure 2). The solubilization capacity of oil phase is important to prevent drug precipitation upon dilution, which could affect drug loading efficiency.32 Capryol 90, a medium-chain fatty acid, exhibited good solubilization capacity (14.88 ± 0.86 mg/mL) among the screened oil phases for MCA. Labrasol resulted in the highest solubility (15.00 ± 0.48 mg/mL), followed by Lauroglycol FCC (7.43 ± 0.41 mg/mL). MCA solubility in co-surfactants was higher than its solubility in oil phases and surfactants. Among various co-surfactants screened, Transcutol HP (41.19 ± 2.16 mg/mL) and 1.2-propanediol (16.84 ± 3.81 mg/mL) showed good solubilization of drug.
 |
Figure 2 Solubility of MCA in different oil phases, surfactants, and co-surfactants. Each value shows the mean ± SD (n=3). |
Selection of SNEDDS Components
The emulsification efficiency is a crucial parameter for evaluating the spontaneous emulsification of formulation without the aid of external thermal or mechanical energy. Oil phase is a major component of SNEDDS because of its ability to dissolve large amounts of hydrophobic drugs. Oil phase could control the self-emulsification process and enhance lymphatic transport of drugs. The selection of oil phases mainly depends on solubility of drugs. In comparison with other oil phases, ethyl oleate, Capryol 90 and Labrafil M 1944 CS were found to exhibit good emulsification efficiency with all tested surfactants (Table 1). Therefore, Capryol 90 was selected as an oil phase for SNEDDS formulation and further examinations. Regarding surfactants, emulsification efficiency is the major selection perspective. Various surfactants were also evaluated for their solubilization capacity and emulsification efficiency. Kolliphor RH40, Kolliphor ELP, and Kolliphor HS15 showed comparable emulsification ability for all employed oil phases. The results of emulsification efficiency of Capryol 90 with five surfactants (Lauroglycol FCC, Labrasol, Tween 80, Kolliphor ELP, and Kolliphor RH40) in four different ratios (Km: 1:9, 2:8, 3:7, 4:6, w/w) are shown in Table 2. Labrasol is shown to have the best solubility of screened surfactants, Kolliphor ELP possesses a good capacity to emulsify Capryol 90. However, both dissolution properties and emulsifying abilities are important for surfactants and co-surfactants. Good emulsifying ability is favorable for the spontaneous formation of nanoemulsion. Consequently, both Kolliphor ELP and Labrasol were selected as the cosurfactants of SNEDDS.
 |
Table 1 The Ability of Various Surfactants to Emulsify the Oil Phases |
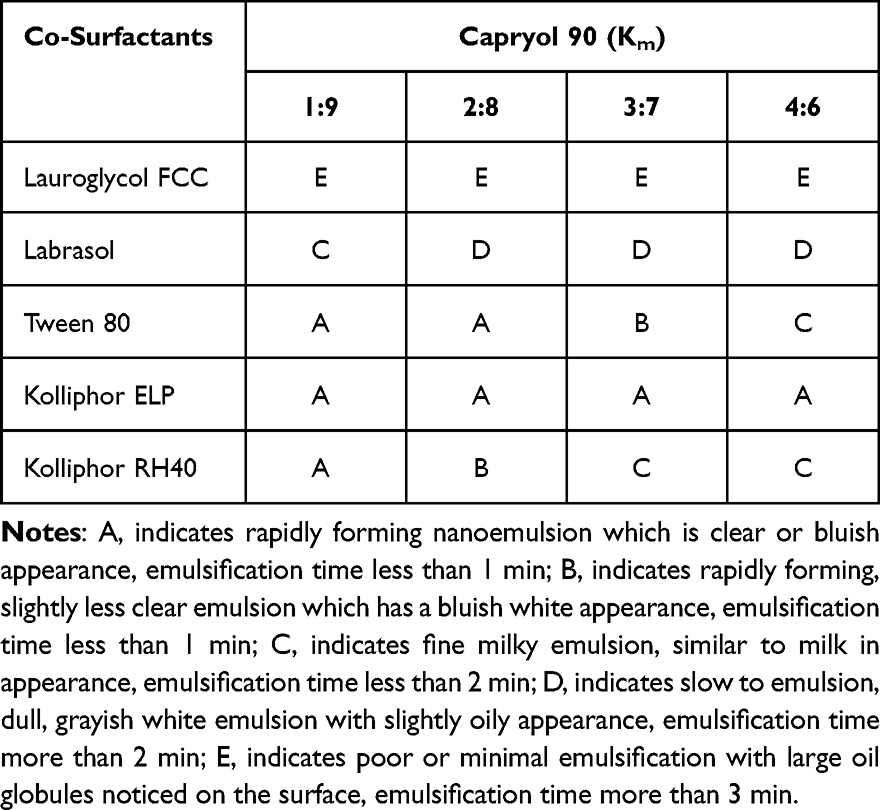 |
Table 2 The Ability of Capryol to Emulsify the Selected Co-Surfactants |
Construction of Pseudo-Ternary Phase Diagrams
The high specificity of the self-emulsification process requires separate selection of oil phases, surfactants, and co-surfactants. The selection of the type and mass ratio of surfactant/co-surfactant is the key factor in the formulation of nanoemulsions. A ternary diagram was established to identify the self-emulsifying region. It helps to select the appropriate excipient ratio for the development of SNEDDS formulations to ensure spontaneous formation of nanoemulsions. Accordingly, to identify the self-nanoemulsifying regions and optimize the concentration of phase ingredients in the SNEDDS, three-phase diagrams were constructed using Caproyl 90 as an oil phase, Kolliphor ELP and Labrasol as surfactants, and Transcutol HP as a co-surfactant. For each selected SNEDDS system, oil phase/surfactant/co-surfactant was prepared at predetermined ratios with 1:9–9:1 (w/w). All components were converted to mass ratios according to different proportions, and the phase diagram was constructed by Origin 8.0 software (Figure 3). This pseudo-ternary phase diagram provided a self-nanoemulsifying region where transparent emulsions were produced. Based on the various combinations, Capryol 90, Kolliphor ELP, Labrasol, Transcutol HP (1:2.7:2.7:3.6) were selected for the formulation of optimized SNEDDS.
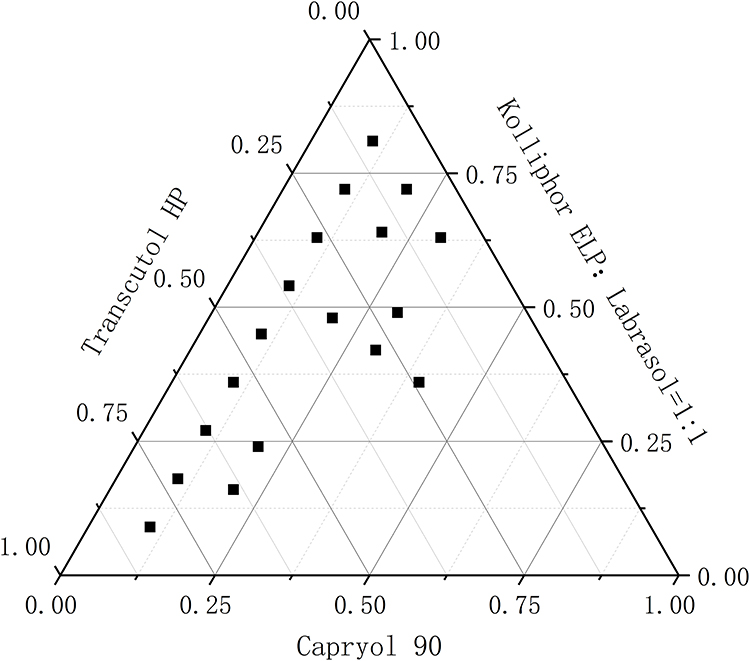 |
Figure 3 Pseudo-ternary phase diagrams for Capryol 90/Labrasol/Kolliphor ELP/Transcutol HP. |
Characterization of SNEDDS
Droplet size is related to the available interfacial area between oil and aqueous phase.33 The droplet size distribution is one of the important characteristics of emulsion stability evaluation and a key step in improving drug bioavailability. The smaller particle size results in an enhanced surface area, thus improving the absorption of drug. The droplet size of optimal MCA-loaded SNEDDS indicated that globules of the emulsion have nanometric range (21.52 ± 0.23 nm) with polydispersity index under 0.16, which demonstrates consistency in distribution of droplet size (Figure 4). The emulsion droplet polarity is also a significant factor in assessing emulsification efficiency. The stability of colloidal dispersions depends on the value of zeta potential. Generally, a stable colloidal dispersion has a zeta potential between −10 and +10 mV, zeta potential values of more than +30 mV or less than −30 mV are considered strongly cationic and strongly anionic, respectively.34 The value of zeta potential was found to be −3.05 ± 0.30 mV, which showed the formulation was negatively charged and gave indication of a stable system.
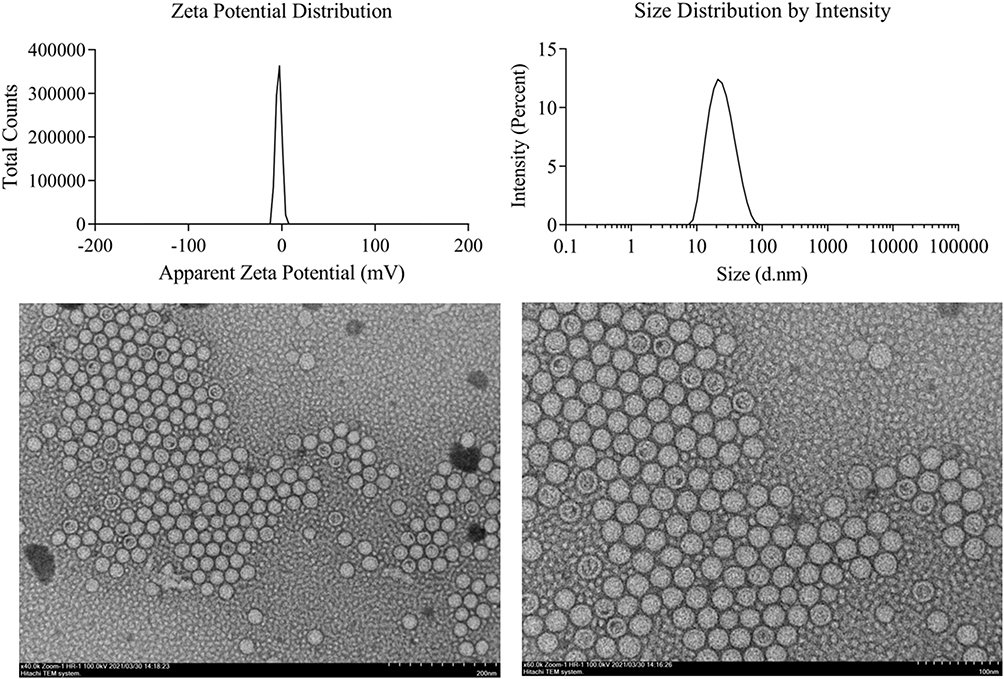 |
Figure 4 Characterization of SNEDDS. |
In-vitro Drug Release Study
The drug release profile of SNEDDS was evaluated in comparison with that of the pure MCA. Performing in vitro release study in a suitable media could predict the bioavailability of drug. As shown in Figure 5, SNEDDS significantly enhanced the rate and extent of MCA release. During the first 4 h of the study, MCA-SNEDDS released more than 15% drug compared with the MCA-suspension that released only 2.59%. At 24 h, the cumulative drug release from SNEDDS and suspension were 68.99% and 23.61%, respectively. About 80% of MCA was released from the SNEDDS formulation at the same period, whereas at the end of the study, the total amount of pure MCA released was 30.63%. The correlation coefficients (R2) and kinetic of drug release from MCA-SNEDDS and MCA suspension are shown in Table 3. The values of R2 suggested the best model fit the drug release pattern from formulation. The best model fit for MCA-SNEDDS was first model with R2 = 0.9912. For MCA-suspension, the best model fitting its drug release kinetic was the zero order with R2 = 0.9804.
 |
Table 3 Mathematical Models of MCA Release from SNEDDS and Suspension |
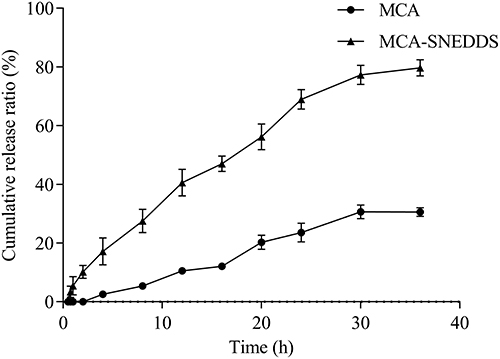 |
Figure 5 In vitro drug release profiles of pure MCA and SNEDDS formulation in phosphate buffer (pH 6.8). Each value represents the mean ± SD (n = 3). Abbreviations: MCA, madecassic acid; MCA-SNEDDS, madecassic acid loaded self-nanoemulsifying drug delivery system. Note: Data represent mean ± SD (n = 5). |
Oral Bioavailability in Rats
The ultimate aim of oral drug delivery is to enhance the bioavailability of compounds. In this section, the oral absorption efficiency of MCA-SNEDDS was investigated in Sprague-Dawley rats when compared with pure MCA. A rapid and accurate HPLC-MS/MS analysis method was developed and validated to determine the plasma concentration of MCA. The plasma concentration-time profiles of MCA (Figure 6) and its pharmacokinetic parameters (Table 4) revealed that the T1/2, Cmax, and CL/F values were 3.36 h, 16.59 ng/mL, and 1.94 mg/ng*mL, respectively, and the AUC was 72.29 h·ng/mL, while Cmax and AUC of MCA-SNEDDS were significantly increased to 140.44 ng/mL and 290.24 h·ng/mL, respectively. The relative bioavailability of MCA-SNEDDS was 401.5%. MCA-SNEDDS showed much higher oral bioavailability than that of pure MCA. The improvement in bioavailability of MCA of the SNEDDS formulation may be due to the decreased particle size and increased solubility of MCA. High drug solubilization capacity and self-emulsifying ability of SNEDDS formulation may have resulted in the increased AUC of MCA. As the results show, formulating MCA into SNEDDS was an efficient strategy to facilitate the oral absorption of MCA.
 |
Table 4 Pharmacokinetic Parameters of MCA and MCA-SNEDDS |
 |
Figure 6 The concentration-time curves of MCA and MCA-SNEDDS. Abbreviations: MCA, madecassic acid; MCA-SNEDDS, madecassic acid loaded self-nanoemulsifying drug delivery system. Note: Data represent mean ± SD (n = 5). |
Intestinal Permeability Study
Permeability in the intestinal tract of any formulation is a crucial factor, which decides the absorption and bioavailability of the drug from that formulation.35 Hence, the intestinal permeability of MCA was evaluated in male Sprague-Dawley rats. The results indicated that the effective permeability coefficient (Peff) of MCA was 1.13×10−5 cm/s (Figure 7). An increase in permeability of MCA was found after co-perfusion with the verapamil (0.1 mg/mL), a P-gp inhibitor (1.30 × 10−5 cm/s, p < 0.01), which indicated that MCA might be a substrate of P-gp. It also suggested that P-gp could efficiently efflux MCA in the gut wall, which might be responsible for the poor oral bioavailability of MCA. The Peff of MCA-SNEDDS formulation was 1.48 times (p < 0.01) that of pure MCA, indicating that MCA permeability could be significantly improved using SNEDDS formulation. Both surfactants Kolliphor ELP and Labrasol can inhibit the efflux transporter P-gp.36 Besides, Labrasol has been reported to play a strong role in improving intestinal permeation.37 Reasonably, the result may also be attributed to lymphatic transport of the drug via the triglyceride core of the chylomicron, which is the main lymphatic transport mechanism of non-dissolvable drugs through lipid carriers such as SNEDDS.24,38 Therefore, the optimal SNEDDS formulations exhibited significantly higher intestinal permeability compared with plain drug solution which might be related to the Labrasol and Kolliphor ELP.
 |
Figure 7 The Peff of MCA and MCA-SNEDDS. |
Lymphatic Transport Studies
Intestinal absorption is a complex process influenced by drug metabolism, transporters, and food. Lymphatic transport is a potential way of enhancing oral bioavailability, preventing live first-pass effect and increasing pharmacodynamic activities in components.39 To access the degree of lymphatic uptake, cycloheximide, a chylomicron flowing blocker, was pretreated prior to the study. The plasma concentration-time profile of MCA-SNEDDS after oral dosing to rats as well as after cycloheximide pre-treatments is shown in Figure 8. The parameters calculated from the pharmacokinetic profiles are presented in Table 5. Cycloheximide pre-treatment significantly decreased both the rate and extent of absorption of MCA as indicated by 5.64-fold Cmax decrease and a 4.34-fold AUC decrease. The result indicated that cycloheximide blocked the lymphatic transport of MCA-SNEDDS formulation, thereby reduced the oral absorption. Lipids and surfactants can promote post-enterocytic lymphatic uptake of drugs and nanocarriers. Labrasol was found to at least partly enter the lymph route, thus bypassing the systemic metabolism of the liver and having higher oral bioavailability compared with a drug suspension.40 Labrasol in MCA-SNEDDS prescription might contribute to promote the absorption of MCA in MCA-SNEDDS via lymphatic transport and increase the bioavailability.
 |
Table 5 Pharmacokinetic Parameters of MCA-SNEDDS with/without Pretreatment with Cycloheximide |
 |
Figure 8 Plasma concentration-time profile of MCA-SNEDDS with/without pretreatment with cycloheximide. Abbreviations: MCA, madecassic acid; MCA-SNEDDS, madecassic acid loaded self-nanoemulsifying drug delivery system. Note: Data represent mean ± SD (n = 5). |
Conclusion
In order to improve the solubility and bioavailability of the poor aqueous-soluble ingredient MCA via oral route, SNEDDS could be a favorable choice. In this study, SNEDDS of various compositions have been evaluated to select the nanocomposites that exhibit optimal characteristics, including solubilization capacity and emulsification efficiency. Moreover, a ternary phase diagram was established to determine the specific amount required for each component. In that, the best formulation was selected with 10% Capryol 90, 27% Kolliphor ELP and 27% Labrasol, and 36% Transcutol HP, as an oil phase, surfactants, and a co-surfactant, respectively. The corresponding zeta potential of −3.05 mV, droplet size (21.52 nm) was less than 200 nm and a PDI value of 0.16 demonstrated the formation of emulsion with nanosized droplets and a uniform distribution. SNEDDS retained the self-emulsifying properties of SNEDDS while contributing to an improved solubilization of the entrapped compound compared with the pure component. Pharmacokinetic data suggest that SNEDDS remarkably improved the oral bioavailability of MCA. Compared with pure MCA, SNEDDS showed a 1.48-fold improved Peff throughout the intestine in the in situ single-pass intestinal permeability study. Furthermore, pretreatment of cycloheximide, a chylomicron flowing blocker, greatly influenced the absorption of SNEDDS. The SNEDDS formulation is a good candidate for enhancing oral absorption of MCA via improved Peff and lymphatic uptake. The representative SNEDDS formulated for MCA in the current research provides collective advantages, such as superior self-emulsification efficiency, high drug loading capacity, improved dissolution, permeation, and enhanced bioavailability of MCA. This work reveals the importance of SNEDDS development for MCA, which may bring about a synergistic effect in treatment along with a lower dose administration. Based on in vivo and in vitro evidence, MCA, a natural pentacyclic triterpenoid, possesses important pharmacological effects, such as wound healing, anti-inflammatory, and antioxidant activities. There is a need for further study to research the applicability and synergistic effect of MCA-SNEDDS to target diseases.
Ethical Approval and Consent to Participate
The study was approved by the Institutional Animal Care and Use Committee of China Pharmaceutical University (CPU-2019-06-004).
Acknowledgments
This work was supported by the National Natural Science Foundation of China (No. 81874361), and the “Double First-Class” University Project (CPU2022QZ31).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Xu X, Wang Y, Wei Z, et al. Madecassic acid, the contributor to the anti-colitis effect of madecassoside, enhances the shift of Th17 toward Treg cells via the PPARgamma/AMPK/ACC1 pathway. Cell Death Dis. 2017;8:e2723. doi:10.1038/cddis.2017.150
2. Tabassum R, Vaibhav K, Shrivastava P, et al. Centella asiatica attenuates the neurobehavioral, neurochemical and histological changes in transient focal middle cerebral artery occlusion rats. Neurol Sci. 2013;34:925–933. doi:10.1007/s10072-012-1163-1
3. Arancibia-Radich J, Gonzalez-Blazquez R, Alcala M, et al. Beneficial effects of murtilla extract and madecassic acid on insulin sensitivity and endothelial function in a model of diet-induced obesity. Sci Rep. 2019;9:599. doi:10.1038/s41598-018-36555-1
4. Siddiqui S, Khan F, Jamali KS, et al. Madecassic acid reduces fast transient potassium channels and promotes neurite elongation in hippocampal CA1 Neurons. CNS Neurol Disord Drug Targets. 2020;19:12–26. doi:10.2174/1871527318666191111105508
5. Valdeira ASC, Darvishi E, Woldemichael GM, et al. Madecassic acid derivatives as potential anticancer agents: synthesis and cytotoxic evaluation. J Nat Prod. 2019;82:2094–2105. doi:10.1021/acs.jnatprod.8b00864
6. Yun X, Zhang Q, Fang Y, et al. Madecassic acid alleviates colitis-associated colorectal cancer by blocking the recruitment of myeloid-derived suppressor cells via the inhibition of IL-17 expression in gammadeltaT17 cells. Biochem Pharmacol. 2022;202:115138. doi:10.1016/j.bcp.2022.115138
7. Yun X, Fang Y, Lv C, et al. Inhibition of the activation of γδT17 cells through PPARγ–PTEN/Akt/GSK3β/NFAT pathway contributes to the anti-colitis effect of madecassic acid. Cell Death Dis. 2020;11:752. doi:10.1038/s41419-020-02969-x
8. Won JH, Shin JS, Park HJ, et al. Anti-inflammatory effects of madecassic acid via the suppression of NF-kappaB pathway in LPS-induced RAW 264.7 macrophage cells. Planta Med. 2010;76:251–257. doi:10.1055/s-0029-1186142
9. Rafat M, Fong KW, Goldsipe A, et al. Association (micellization) and partitioning of aglycon triterpenoids. J Colloid Interface Sci. 2008;325:324–330. doi:10.1016/j.jcis.2008.05.046
10. Wannasarit S, Puttarak P, Kaewkroek K, et al. Strategies for Improving healing of the gastric epithelium using oral solid dispersions loaded with pentacyclic triterpene-rich centella extract. AAPS PharmSciTech. 2019;20:277. doi:10.1208/s12249-019-1488-7
11. Di Costanzo A, Angelico R. Formulation strategies for enhancing the bioavailability of silymarin: the state of the art. Molecules. 2019;24:2155. doi:10.3390/molecules24112155
12. Verma R, Mittal V, Pandey P, et al. Exploring the role of self-nanoemulsifying systems in drug delivery: challenges, issues, applications and recent advances. Curr Drug Deliv. 2022. doi:10.2174/1567201819666220519125003
13. Shen J, Bi J, Tian H, et al. Preparation and evaluation of a self-nanoemulsifying drug delivery system loaded with Akebia saponin D-phospholipid complex. Int J Nanomedicine. 2016;11:4919–4929. doi:10.2147/IJN.S108765
14. Baral KC, Song JG, Lee SH, et al. Enhanced bioavailability of AC1497, a novel anticancer drug candidate, via a self-nanoemulsifying drug delivery system. Pharmaceutics. 2021;13:1142. doi:10.3390/pharmaceutics13081142
15. Qi X, Qin J, Ma N, et al. Solid self-microemulsifying dispersible tablets of celastrol: formulation development, charaterization and bioavailability evaluation. Int J Pharm. 2014;472:40–47. doi:10.1016/j.ijpharm.2014.06.019
16. Khan AW, Kotta S, Ansari SH, et al. Self-nanoemulsifying drug delivery system (SNEDDS) of the poorly water-soluble grapefruit flavonoid Naringenin: design, characterization, in vitro and in vivo evaluation. Drug Deliv. 2015;22:552–561. doi:10.3109/10717544.2013.878003
17. Abushal AS, Aleanizy FS, Alqahtani FY, et al. Self-Nanoemulsifying Drug Delivery System (SNEDDS) of Apremilast: in vitro Evaluation and Pharmacokinetics Studies. Molecules. 2022;27:3085. doi:10.3390/molecules27103085
18. Verma R, Kaushik D. Design and optimization of candesartan loaded self-nanoemulsifying drug delivery system for improving its dissolution rate and pharmacodynamic potential. Drug Deliv. 2020;27:756–771. doi:10.1080/10717544.2020.1760961
19. Verma R, Kaushik A, Almeer R, et al. Improved pharmacodynamic potential of rosuvastatin by self-nanoemulsifying drug delivery system: an in vitro and in vivo evaluation. Int J Nanomedicine. 2021;16:905–924. doi:10.2147/IJN.S287665
20. Verma R, Kaushik D. Development, optimization, characterization and impact of in vitro lipolysis on drug release of telmisartan loaded SMEDDS. Drug Deliv Lett. 2019;9:330–340. doi:10.2174/2210303109666190614120556
21. Corrie L, Gulati M, Kaur J, et al. Quality by Design-based RP-HPLC method for estimation of curcumin in rat plasma and fecal microbiota extract-based solid self-nano emulsifying drug delivery system. Curr Drug Res Rev. 2023. doi:10.2174/2589977515666230120140543
22. Shahba AA, Sherif AY, Elzayat EM, et al. Combined Ramipril and Black Seed Oil Dosage Forms Using Bioactive Self-Nanoemulsifying Drug Delivery Systems (BIO-SNEDDSs). Pharmaceuticals. 2022;15:1120. doi:10.3390/ph15091120
23. Kazi M, Al-Swairi M, Ahmad A, et al. Evaluation of Self-nanoemulsifying drug delivery systems (SNEDDS) for poorly water-soluble talinolol: preparation, in vitro and in vivo assessment. Front Pharmacol. 2019;10:459. doi:10.3389/fphar.2019.00459
24. Yin HF, Yin CM, Ouyang T, et al. Self-nanoemulsifying drug delivery system of genkwanin: a novel approach for anti-colitis-associated colorectal cancer. Drug Des Devel Ther. 2021;15:557–576. doi:10.2147/DDDT.S292417
25. Abdel-All SR, Shakour ZTA, Abouhussein DMN, et al. Phytochemical and biological evaluation of a newly designed nutraceutical self-nanoemulsifying self-nanosuspension for protection and treatment of cisplatin induced testicular toxicity in male rats. Molecules. 2021;26:408. doi:10.3390/molecules26020408
26. Panigrahi KC, Patra CN, Rao MEB. Quality by design enabled development of oral self-nanoemulsifying drug delivery system of a novel calcimimetic cinacalcet hcl using a porous carrier: in vitro and in vivo characterisation. AAPS PharmSciTech. 2019;20:216. doi:10.1208/s12249-019-1411-2
27. Nair AB, Singh B, Shah J, et al. Formulation and evaluation of self-nanoemulsifying drug delivery system derived tablet containing sertraline. Pharmaceutics. 2022;14:336. doi:10.3390/pharmaceutics14020336
28. Liu CS, Chen L, Hu YN, et al. Self-microemulsifying drug delivery system for improved oral delivery and hypnotic efficacy of ferulic acid. Int J Nanomedicine. 2020;15:2059–2070. doi:10.2147/IJN.S240449
29. Dahlgren D, Roos C, Peters K, et al. Evaluation of drug permeability calculation based on luminal disappearance and plasma appearance in the rat single-pass intestinal perfusion model. Eur J Pharm Biopharm. 2019;142:31–37. doi:10.1016/j.ejpb.2019.06.011
30. Lind ML, Jacobsen J, Holm R, et al. Intestinal lymphatic transport of halofantrine in rats assessed using a chylomicron flow blocking approach: the influence of polysorbate 60 and 80. Eur J Pharm Sci. 2008;35:211–218. doi:10.1016/j.ejps.2008.07.003
31. Parmar N, Singla N, Amin S, et al. Study of cosurfactant effect on nanoemulsifying area and development of lercanidipine loaded (SNEDDS) self nanoemulsifying drug delivery system. Colloids Surf B Biointerfaces. 2011;86:327–338. doi:10.1016/j.colsurfb.2011.04.016
32. Michaelsen MH, Wasan KM, Sivak O, et al. The effect of digestion and drug load on halofantrine absorption from self-nanoemulsifying drug delivery system (SNEDDS). AAPS J. 2016;18:180–186. doi:10.1208/s12248-015-9832-7
33. Eleftheriadis GK, Mantelou P, Karavasili C, et al. Development and characterization of a self-nanoemulsifying drug delivery system comprised of rice bran oil for poorly soluble drugs. AAPS PharmSciTech. 2019;20:78. doi:10.1208/s12249-018-1274-y
34. Qureshi KA, Mohammed SAA, Khan O, et al. Cinnamaldehyde-Based Self-Nanoemulsion (CA-SNEDDS) accelerates wound healing and exerts antimicrobial, antioxidant, and anti-inflammatory effects in rats’ skin burn model. Molecules. 2022;27:5225. doi:10.3390/molecules27165225
35. Kamble PR, Shaikh KS. Optimization and Evaluation of Self-nanoemulsifying Drug Delivery System for Enhanced Bioavailability of Plumbagin. Planta Med. 2022;88:79–90. doi:10.1055/a-1332-2037
36. Seljak KB, Berginc K, Trontelj J, et al. A self-microemulsifying drug delivery system to overcome intestinal resveratrol toxicity and presystemic metabolism. J Pharm Sci. 2014;103:3491–3500. doi:10.1002/jps.24114
37. McCartney F, Jannin V, Chevrier S, et al. Labrasol® is an efficacious intestinal permeation enhancer across rat intestine: ex vivo and in vivo rat studies. J Control Release. 2019;310:115–126. doi:10.1016/j.jconrel.2019.08.008
38. Sun M, Zhai X, Xue K, et al. Intestinal absorption and intestinal lymphatic transport of sirolimus from self-microemulsifying drug delivery systems assessed using the single-pass intestinal perfusion (SPIP) technique and a chylomicron flow blocking approach: linear correlation with oral bioavailabilities in rats. Eur J Pharm Sci. 2011;43:132–140. doi:10.1016/j.ejps.2011.04.011
39. Rysanek P, Grus T, Lukac P, et al. Validity of cycloheximide chylomicron flow blocking method for the evaluation of lymphatic transport of drugs. Br J Pharmacol. 2021;178:4663–4674. doi:10.1111/bph.15644
40. Ghassemi S, Haeri A, Shahhosseini S, et al. Labrasol-enriched nanoliposomal formulation: novel approach to improve oral absorption of water-insoluble drug, carvedilol. AAPS PharmSciTech. 2018;19:2961–2970. doi:10.1208/s12249-018-1118-9
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.