Back to Journals » International Journal of Nanomedicine » Volume 15
Self-Microemulsifying Drug Delivery System for Improved Oral Delivery and Hypnotic Efficacy of Ferulic Acid
Authors Liu CS ![]() , Chen L, Hu YN, Dai JL, Ma B, Tang QF, Tan XM
, Chen L, Hu YN, Dai JL, Ma B, Tang QF, Tan XM
Received 29 November 2019
Accepted for publication 10 March 2020
Published 25 March 2020 Volume 2020:15 Pages 2059—2070
DOI https://doi.org/10.2147/IJN.S240449
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Mian Wang
Chang-Shun Liu,1– 3 Li Chen,4 Yan-Nan Hu,1– 3 Jin-Lian Dai,4 Biao Ma,4 Qing-Fa Tang,1– 3 Xiao-Mei Tan1– 3
1School of Traditional Chinese Medicine, Southern Medical University, Guangzhou 510515, People’s Republic of China; 2Guangdong Provincial Key Laboratory of Chinese Medicine Pharmaceutics, Southern Medical University, Guangzhou 510515, People’s Republic of China; 3Guangdong Provincial Engineering Laboratory of Chinese Medicine Preparation Technology, Southern Medical University, Guangzhou 510515, People’s Republic of China; 4School of Pharmacy, Zunyi Medical University, Zunyi 563000, People’s Republic of China
Correspondence: Li Chen; Xiao-Mei Tan Email [email protected]; [email protected]
Purpose: Ferulic acid (FA) is a natural compound which is used to treat insomnia. However, its use is limited because of its poor oral bioavailability caused by extremely rapid elimination. The current study aimed to develop a self-microemulsifying drug delivery system (SMEDDS) to improve the oral delivery of FA and to enhance its hypnotic efficacy.
Methods: FA-SMEDDS was prepared, and its morphology and storage stability were characterized. The formulation was also subjected to pharmacokinetic and tissue distribution studies in rats. The hypnotic efficacy of FA-SMEDDS was evaluated in p-chlorophenylalanine-induced insomnia mice.
Results: FA-loaded SMEDDS exhibited a small droplet size (15.24 nm) and good stability. Oral administration of FA-SMEDDS yielded relative bioavailability of 185.96%. In the kidney, SMEDDS decreased the distribution percentage of FA from 76.1% to 59.4% and significantly reduced its metabolic conversion, indicating a reduction in renal elimination. Interestingly, FA-SMEDDS showed a higher distribution in the brain and enhanced serotonin levels in the brain, which extended the sleep time by 2-fold in insomnia mice.
Conclusion: This is the first study to show that FA-loaded SMEDDS decreased renal elimination, enhanced oral bioavailability, increased brain distribution, and improved hypnotic efficacy. Thus, we have demonstrated that SMEDDS is a promising carrier which can be employed to improve the oral delivery of FA and facilitate product development for the therapy of insomnia.
Keywords: insomnia, ferulic acid, oral administration, pharmacokinetics, SMEDDS
Introduction
Insomnia is most commonly characterized by difficulty in falling asleep, difficulty in maintaining sleep, or early morning awakening. Insomnia is a major public health issue worldwide with substantial medical, psychiatric, and financial ramifications.1 Epidemiological studies found that between 35% and 70% of all adults suffer from symptoms of insomnia,2 which constitutes a huge social and medical burden. Insomnia leads to an increased risk of mental or medical disorders, including depression,3 hypertension,4 and cardiovascular diseases.5 Extensive research on treatment of insomnia has led to the discovery of new sedative and hypnotic drugs for clinical use. Currently, the main routes for discovery of oral hypnotic agents are chemical synthesis and molecules or natural products derived from medicinal herbs, including traditional Chinese medicinal herbs.6 The development of herbal ingredients as hypnotic candidates has led to safety and efficacy studies of these constituents for the treatment of insomnia.7
Semen Ziziphi Spinosae and Chuanxiong Rhizoma are medicinal herbs possessing sedative and hypnotic activities.8 Ferulic acid (4-hydroxy-3-methoxycinnamic acid, FA, Figure 1A), a natural phenolic compound, is a major constituent of Semen Ziziphi Spinosae and Chuanxiong Rhizoma.8,9 Experimental and clinical evidence indicates that FA exerts its hypnotic effects through its neuroprotective, anti-inflammatory, and anti-oxidant activities.10,11 FA also binds with neurotransmitter receptors of the gamma-aminobutyric acidergic and serotonergic systems.10–12 However, FA shows low gastrointestinal (GI) stability and a poor pharmacokinetic profile, characterized by short plasma half-life and low bioavailability, after oral administration, which limits its clinical use. After oral administration, more than 70% of FA is absorbed from the GI tract within 30 min and is rapidly eliminated.13,14 After absorption into the blood, approximately 6% of FA is metabolized to conjugated forms in plasma and bile, and more than 50% of FA is distributed and eliminated by the kidney.15–17 This extensive and rapid elimination by the kidney leads to a short residence time and low plasma level of FA. Moreover, in spite of its ability to cross the blood-brain barrier, the concentration of FA in the brain is very low,17 which limits its use for the treatment of insomnia. Thus, it is essential to develop a suitable strategy to successfully overcome these challenges so as to decrease its renal elimination, increase its plasma levels and brain distribution, which will improve its oral delivery and hypnotic efficacy.
 |
Figure 1 Excipients screen and formula optimization of SMEDDS for FA. Chemical structure of FA (A). Solubility of FA in the tested excipients (B). Compatibility analysis of formula by pseudo-ternary phase diagram in the blank (C) and drug loading conditions (D). |
Various strategies to overcome the low oral bioavailability of drugs have been investigated, including structural modifications,18 formation of polymer-drug complexes,19,20 and the use of GI absorption enhancers.21 These strategies may improve the oral bioavailability of drugs possessing high GI stability, but are not suitable for the oral delivery of FA as it is rapidly metabolized after entering the GI tract. Hence, loading FA into a microparticle delivery system might be an effective strategy to improve its oral delivery, because the carrier could envelop FA, which would decrease its GI exposure and improve its stability in the body. Recently, several studies have reported enhancement in the oral bioavailability of FA with the use of microparticle delivery systems, such as nanoparticles,22,23 nanostructured lipid carriers,24 and nanofiber carriers.25 Although some of these strategies increase the bioavailability of FA, they mainly focus on enhancing its GI absorption without considering the impact of the carriers on the disposition of FA after absorption, including tissue distribution and renal elimination.
Self-microemulsifying drug delivery system (SMEDDS) is a carrier system known for its biocompatibility, stability, controlled release, facilitated absorption, and improved bioavailability.26,27 SMEDDS decreases the elimination and clearance of drugs by altering their distribution in tissues.28,29 Thus, we speculated that SMEDDS may be an effective carrier to improve the pharmacokinetic behavior and anti-insomnia efficacy of FA. In this study, we aimed to 1) prepare SMEDDS as a carrier of FA, 2) characterize it on the basis of its morphology, droplet size, and in vitro release of FA, and 3) examine its impact on the disposition of FA in vivo in terms of its oral bioavailability and tissue distribution in rats and on the hypnotic efficacy of FA in insomnia mice.
Materials and Methods
Materials
FA reference standard (purity 99%) was purchased from the National Institute for the Control of Pharmaceutical and Biological Products (Beijing, China). FA (purity 98%) was purchased from Zelang Medical Technology Co., Ltd (Nanjing, China). Pharmaceutical excipients for FA-SMEDDS, including glyceryl triacetate (Sinopharm Chemical Reagent Co., Ltd; Shanghai, China), OP-10 (Chengdu Jinshan Chemical Reagent Co., Ltd; Chengdu, China), labrasol (Gattefosse China Trading Co., Ltd; Shanghai, China), and polyethylene glycol 400 (PEG 400) (Beijing Solarbio Science and Technology Co., Ltd; Beijing, China), were of analytical grade. p-chlorophenylalanine (PCPA) was purchased from Bide Pharmatech Ltd. (Shanghai, China). Other excipients and chemicals including methanol, acetonitrile, and phosphoric acid were of high-performance liquid chromatographic or analytical grade.
Preparation of SMEDDS for FA
Solubility Determination of FA
Solubility of FA in different media was determined by adding excess amounts of FA to the test media. These samples were continuously stirred at 37 °C for 24 h, centrifuged, and the concentration of FA in the medium was assayed after appropriate dilution.
Pseudo-Ternary Phase Diagram
Pseudo-ternary phase diagrams were constructed using the aqueous titration method to examine the effects of different components of the SMEDDS formulation. Based on the solubility of FA in the media, glyceryl triacetate, a mixture of OP-10 and labrasol (2:1, v/v), and PEG 400 were selected as the oil phase, surfactant, and co-surfactant, respectively. These three phases of SMEDDS were mixed in different proportions (100% = total amount of mixture) and stirred at 37 °C. Pre-microemulsion concentrates (PMCs) were obtained and used for the excipient compatibility assay. A PMC was quickly injected into water while stirring at a rate of 50 rpm at 37 °C to yield a transparent microemulsion. The formulation components would be considered compatible if the self-emulsifying time was < 2 min, self-emulsifying rate was > 90%, droplet size was < 50 nm, and no stratification or precipitate was observed after centrifugation (3000 rpm for 15 min) and incubation for 24 h. The proportions of the PMC components that were found to be compatible were used to draw pseudo-ternary phase diagrams using OriginPro 8.0 software. PMCs containing FA were also assayed in a similar manner to construct pseudo-ternary phase diagrams to examine the effects of FA and different components of the SMEDDS formulations.
Optimization of the SMEDDS Formulation
Box-Behnken design (BBD) was used to optimize the SMEDDS formulation using Design-Expert software (Version 8.0.6; Stat-Ease, Inc., Minneapolis, USA). Three levels each of the oil phase (0.1, 0.2, 0.3), surfactant (0.3, 0.45, 0.6), and co-surfactant (0.1, 0.3, 0.5) were designed as factors for BBD. Saturated drug loading was used as the index to assess the suitability of the formulation component levels obtained by fitting the equation. The SMEDDS formulations were prepared using the results of the BBD, which indicated that the ratio of 0.2: 0.45: 0.3 for oil phase: surfactant: co-surfactant was optimal for the preparation of FA-SMEDDS (Table 1).
 |
Table 1 BBD Assays and Verification Tests for FA-SMEDDS (n = 4) |
Preparation of SMEDDS
The SMEDDS for FA was prepared according to the optimized formula obtained from the BBD analysis. Glyceryl triacetate, a mixture of OP-10 and labrasol (2:1, v/v), and PEG 400 were used as the oil phase, surfactant, and co-surfactant and mixed in the ratio of 0.2: 0.45: 0.3, respectively. SMEDDS containing glyceryl triacetate (0.80 g), mixture of OP-10 and labrasol (1.80 g), and PEG 400 (1.20 g) was prepared. FA (0.30 g) was dissolved in the SMEDDS (3.00 g) to obtain a transparent preparation, FA-SMEDDS.
SMEDDS Characterization
Morphology and Droplet Size
Morphology of FA-SMEDDS was assessed using transmission electron microscopy (H-7650, Hitachi Ltd., Tokyo, Japan). FA-SMEDDS (0.5 g) was dispersed in water (50 mL) and stirred at 37 °C until a microemulsion (1 mg/mL of FA) was obtained. After filtration, 0.2 μL of the microemulsion was deposited on a film-coated copper specimen grid and allowed to stand for 2 min; excess fluid was removed using clean filter paper. The grid was later stained with one drop of 2% (w/v) phosphotungstic acid for 2 min and dried with filter paper. The morphology of the sample was measured using transmission electron microscopy. The droplet size of FA-SMEDDS was measured using a Zetasizer Nano ZS (Zetasizer 3000; Malvern Instrument Ltd., UK) after 100-fold dilution of the microemulsion of FA-SMEDDS (1 mg/mL of FA) with distilled water.
Storage Stability
The storage stability of FA-SMEDDS was assessed by analyzing the appearance of its microemulsion prepared in distilled water (1 mg/mL of FA). The samples were tightly sealed and placed for 1 month at 25 °C and 37 °C, after which the appearance of the microemulsion was assessed, and the concentration of FA was determined.
In vitro Release Experiment
FA released from the SMEDDS was evaluated using the dialysis method. Briefly, FA-SMEDDS (1 g) was dispensed in release media (5 mL) with slight stirring at 37 °C until a microemulsion was obtained. This microemulsion was transferred to a dialysis bag (MWCO 8000–140,000). The dialysis bag was sealed and placed in 1000 mL of release media, namely, distilled water, pH 1.2 hydrochloric acid, or pH 6.8 phosphate buffer, with stirring at the rate of 100 rpm at 37 °C. Samples (5 mL) were taken at 2, 5, 10, 20, 30, 60, 90, and 120 min, and similar volumes of blank release media were added to the bags to replace the loss of volume. The release of FA alone was evaluated and set as the control group. The concentration of FA in the samples was analyzed by liquid chromatography-tandem mass spectrometry (LC-MS/MS), and the cumulative release was calculated as the ratio of FA released to total FA.
Bioavailability and Tissue Distribution Studies
Animal Experiments
Male Wistar rats (200 ± 20 g) were purchased from the Experimental Animal Center of Southern Medical University (Guangzhou, China). All animal experiments in this paper were approved by the Animal Ethics Committee of Southern Medical University. The experimental procedures were in compliance with the National Institutes of Health Guide for Care and Use of Laboratory Animals. Animals were housed in standard cages for 12 h light–dark cycles, provided standard animal chow and tap water daily, and acclimated 3 days before the experiments.
Twelve rats were randomly divided into two groups after 12 h of fasting (n=6). Aqueous solutions of FA and FA-SMEDDS were administered orally to the animals; the dose of FA was 40 mg/kg. Blood samples were withdrawn from the caudal vein and placed in heparinized tubes before administration and at 0.17, 0.5, 1, 2, 3, 4, 6, and 8 h after administration. Plasma samples were obtained after centrifugation at 4 °C and 5000 rpm for 15 min. Samples were stored in −20 °C until analysis by LC-MS/MS.
After 12 h of fasting, 48 rats were randomly divided into 8 groups for tissue distribution studies (n=6). Four groups were treated with FA-SMEDDS, and the other four groups were treated with the aqueous solution of FA. Rats were sacrificed by cervical dislocation at 0.5, 2, 4, and 8 h after administration for the collection of heart, liver, spleen, lung, kidney, and brain. Tissue samples were placed in −20 °C until analysis.
Preparation of Samples
Tissue samples were washed with physiological saline, dried, and weighed (0.2 g). Ten microliters of internal standard solution (2 µg/mL of salicylic acid) and 3-fold volumes [v (mL)/w (g)] of methanol were added, and the tissues were homogenized. Samples were centrifuged at 4 °C and 12,000 rpm for 10 min to obtain the supernatant. Plasma and tissue supernatants (100 µL) were extracted with acetonitrile (300 µL) to precipitate protein. This was followed by centrifugation and separation; the supernatant was subsequently evaporated to dryness in a centrifugal evaporator (CVE-3000, Tokyo Rikakikai, Co., Ltd., Tokyo, Japan). The residue was reconstituted in 100 µL of acetonitrile–0.1% formic acid (2:8, v/v) and centrifuged at 12,000 rpm for 10 min at 4 °C. An aliquot of 10 μL was analyzed by LC-MS/MS.
LC-MS/MS Analysis
Samples were analyzed using an LC–MS/MS system consisting of an Agilent 1200 liquid chromatography system and a 6410 triple quadrupole mass spectrometer with an electrospray ionization source (Agilent Technologies Inc., CA, USA). Analytes were eluted through a C18 column (ACE Excel 3, 3 μm, 2.1 mm × 100 mm; Phenomenex Ltd, Macclesfield, UK) guarded with a pre-column at 25 °C. Samples were separated using a mobile phase consisting of acetonitrile (A)-0.1% formic acid (B) and the following gradient: 0–6.0 min with 75–50% B, 6.1–7.4 min with 25% B, and 75–8.0 min with 75% B. The flow rate was 0.4 mL/min, and injection volume was 10 μL. The electrospray ionization source was set in negative ionization mode to analyze FA and its metabolites, ferulic acid 4-O-sulfate (FAS) and ferulic acid 4-O-β-D-glucuronide (FAG). The optimized multiple reaction monitoring parameters of analytes, including precursor-to-product ion transitions (m/z), fragmentor (V), and collision energy (eV), are as follows: FA (193.0–134.1 m/z, 130 V, 10 eV), FAS (273.0–193.0 m/z, 120 V, 7 eV), FAG (369.2–113.0 m/z, 125 V, 7 eV), and internal standard, salicylic acid (137.0–93.2 m/z, 125 V, 12 eV). Method validation assays were carried out for selectivity, linearity, precision, recovery, and stability. Analytes were separated from endogenous materials in release media, blood, and tissues. Separate standard curves were constructed for each experiment (R2 > 0.99). Inter-day and intra-day coefficients of variation in all experiments were < 10%. Recovery of analytes was in the range of 89.21−94.64%, and good stability was obtained within 24 h (relative standard deviation < 10%).
Hypnotic Efficacy Studies
Sixty male Kunming mice (20 ± 2 g) were purchased from the Experimental Animal Center of Southern Medical University (Guangzhou, China) and housed in a controlled environment. Ten mice were injected with physiological saline (0.9% (w/v) NaCl) and considered to be the normal control group (NC). Fifty mice were injected with PCPA (i.p.; 450 mg/kg/d) for two days to establish an insomnia model. Insomnia mice were randomly divided into five groups (n=10): model control (MC), diazepam tablet (DZP; 1 mg/(kg·d)), FA (60 mg/(kg·d)), and low dosage (30 mg/(kg·d)) and high dosage (60 mg/(kg·d)) of FA-SMEDDS. The respective treatments were orally administered to the mice for seven days, and the NC and MC groups were treated with physiological saline. On the seventh day, mice were treated with the respective agents 60 min prior to pentobarbital injection (i.p.; 45 mg/kg). Each mouse was observed for sleep onset: the loss of righting reflex for over 3 min indicated that the mouse was asleep. The time elapsed between the pentobarbital injection and the loss of righting reflex was recorded as sleep latency. The time elapsed between the loss and recovery of righting reflex was recorded as sleeping time. Mice were sacrificed by cervical dislocation for the collection of brain tissue from the hippocampus and hypothalamus. The levels of 5-hydroxytryptamine (5-HT or serotonin) and 5-hydroxyindoleacetic acid (5-HIAA) in the brain tissue were detected using enzyme-linked immunosorbent assay (ELISA) kits (Bioss, Beijing, China) according to the manufacturer’s protocol.
Statistical Analysis
Pharmacokinetic parameters of FA in the plasma and tissues were calculated using non-compartmental analysis of the drug concentration-time profiles using Drug and Statistics (DAS) 3.0 software (Chinese Mathematical Pharmacology Society, Beijing, China). To evaluate the targeting property of FA-SMEDDS, the relative intake efficiency (RE) of FA in each tissue was calculated in terms of the ratio of distribution percentages (DPs) in FA-SMEDDS and free FA groups. All values are presented as mean ± standard deviation (SD) and analyzed by SPSS 20.0 software. Significant differences between the groups were assessed by using one-way analysis of variance (ANOVA) or independent samples’ t-test. Differences were considered statistically significant at p < 0.05.
Results
Preparation of SMEDDS
Solubility
In the tested media, FA demonstrated high solubility in the co-surfactant, compared with its solubility in the surfactant and oil phase (Figure 1B). Among the oil phase components, FA showed the highest solubility in glyceryl triacetate. Among the surfactants, FA dissolved in OP-10 and labrasol. Among the co-surfactants, solubility of FA was higher in PEG 400 and PEG 200 than in glycerol.
Pseudo-Ternary Phase Diagram
The pseudo-ternary phase diagram shows that a large microemulsion region (10%−80%) was obtained for the oil phase and surfactant even with a small proportion of the co-surfactant (Figure 1C). As a result, the solubility of the co-surfactant decreased as the proportion of the oil phase increased. The area of this microemulsion region decreased after the addition of FA (Figure 1D).
BBD for SMEDDS Formulation
BBD analysis yielded the following equation that was used for fitting the experimental data: saturated drug loading = 0.1693–0.1900 × oil phase (w) – 0.0525 × surfactant (w) + 0.1106 × co-surfactant (w). Fitting the equation yielded two formulations (Table 1). Verification tests showed that the relative error in saturated drug loading between verified and predicted values was less than 5%, which indicates good prediction by the fitting equation. However, Formula 1 had a longer self-emulsifying time (> 5 min), whereas this index was shorter for Formula 2 (< 30 s). Hence, Formula 2 was selected as the optimized SMEDDS formulation for FA.
SMEDDS Characterization
Morphology and Droplet Size
The representative image shows spherical microemulsion droplets whose size was approximately 15.24 nm (Figure 2A). The droplet size distribution revealed a Gaussian distribution for the microemulsion, and the average size was 14.89 nm (Figure 2B), which was consistent with the morphological observation.
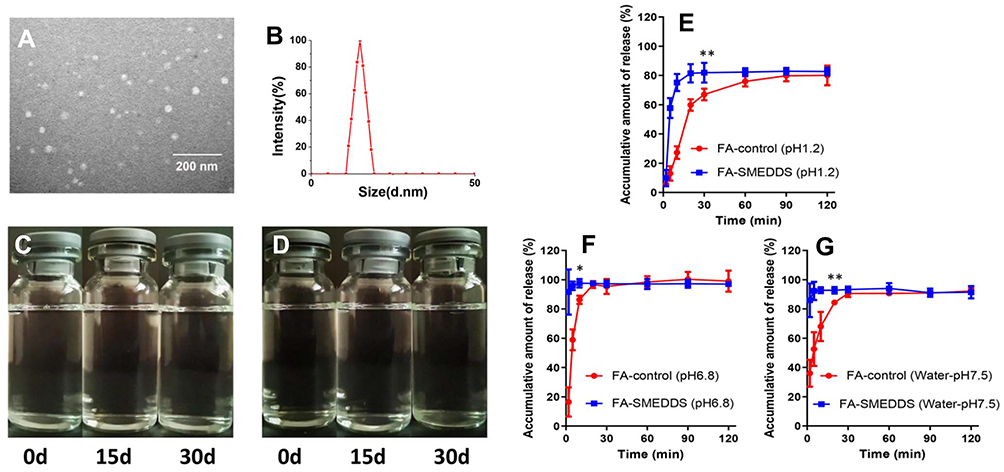 |
Figure 2 Characterization of SMEDDS. FA-SMEDDS showed small droplet size, good stability, and promoted release trait. Morphological feature (× 30,000; (A) and size distribution of FA microemulsion (B). Stability of FA microemulsion evaluated by storing in ambient conditions for 30 days: changes of appearance vs time at 25°C (C) and 37 °C (D). In vitro profiles of FA-SMEDDS and FA in dissolution media, including pH1.2 (E), pH6.8 (F), and water (G). *P < 0.05; **P < 0.01, vs FA-control group, Independent samples t-test. |
Storage Stability
No phase separation or precipitation was found when the microemulsion of FA was stored at 25 °C and 37 °C for 30 days (Figure 2C and D). No remarkable changes were observed in the droplet size or concentration of FA in the microemulsion at the end of the storage period.
In vitro Release Study
The release of FA from the SMEDDS was rapid and achieved saturation at 30 min, indicating that the SMEDDS could facilitate the release of FA (Figure 2E–G). The cumulative release of FA was only approximately 80% in pH 1.2 medium, whereas it was 100% in pH 6.8 medium. Thus, the release of FA was affected by the pH of the environment.
Oral Bioavailability in Rats
The plasma concentration–time profiles of FA (Figure 3A) and its pharmacokinetic parameters (Table 2) revealed that the T1/2 and CL/F values were 1.39 h and 2.50 × 105 L/h/kg, respectively, and the AUC0-t was 82.86 h·μg/mL. Compared with free FA alone, T1/2 for the FA-SMEDDS group was delayed to 2.10 h, while the CL/F decreased to 1.42 × 105 L/h/kg (P < 0.05). The AUC0-t of FA was significantly increased to 143.38 h·μg/mL, and the relative bioavailability (Fr) of FA-SMEDDS was 185.96% (P < 0.01). SMEDDS also increased the V/F of FA from 1.22 × 106 L/kg to 3.14 × 106 L/kg, indicating the change in the distribution of FA.
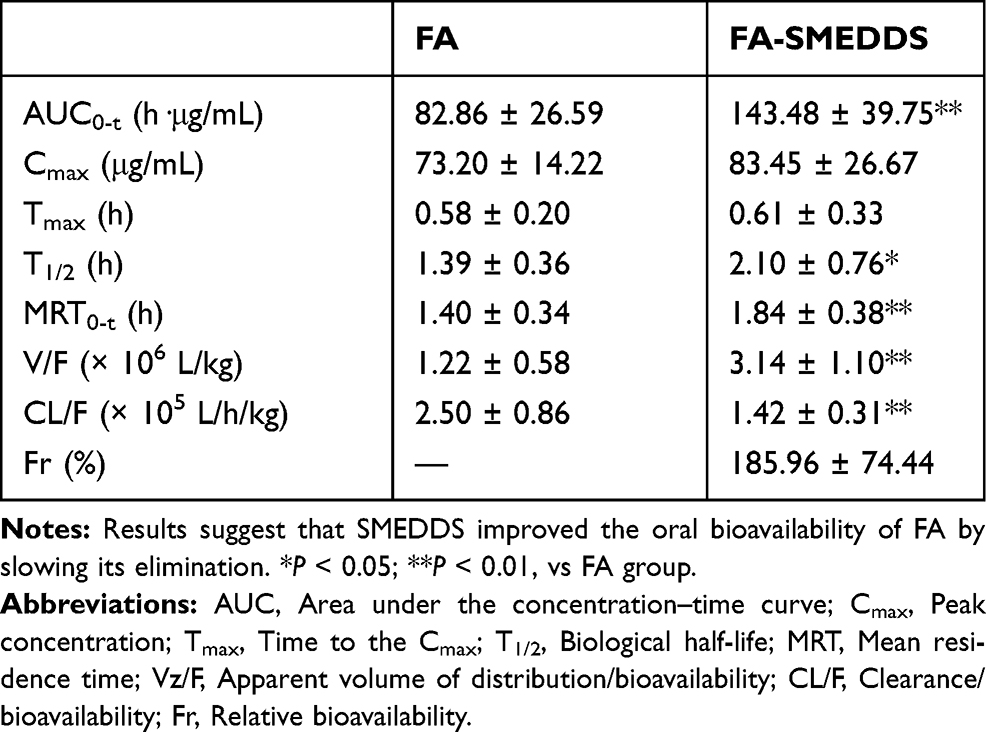 |
Table 2 Main Pharmacokinetic Parameters of FA After Oral Administration in Rats (n=6) |
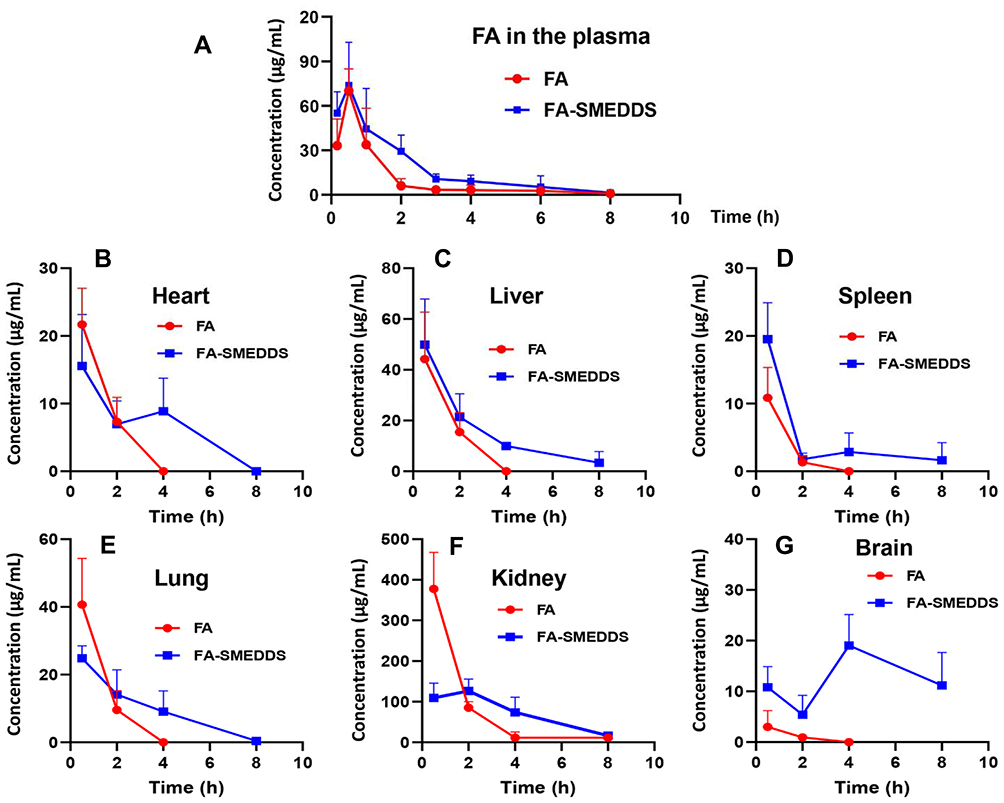 |
Figure 3 Pharmacokinetics and tissue distribution of FA after oral administration in rats. Results suggested that SMEDDS increases plasma exposure of FA, and decreases its renal distribution and enhances its brain accumulation. Plasma concentration–time profiles of FA (A). Concentration–time profiles of FA in the tissues, including heart (B), liver (C), spleen (D), lung (E), kidney (F), and brain (G). |
Tissue Distribution of FA and Its Metabolites in Rats
After oral administration, FA was widely distributed in the body (Figure 3B–G). An analysis of the DP calculations revealed an abundance of FA in the tissues (Table 3) in the following order: kidney (76.1%) > liver (9.3%) > lung (7.6%) > heart (4.6%) > spleen (1.8%) > brain (0.7%). After loading of FA in the SMEDDS, the concentrations of FA in the liver, spleen, and brain were remarkably increased. The distribution of FA was also changed by SMEDDS. DP value of FA in the kidney decreased from 76.1% to 59.4%, whereas the DP in the brain increased from 0.7% to 10.4% (P < 0.01), which yields a high RE value of 14.87.
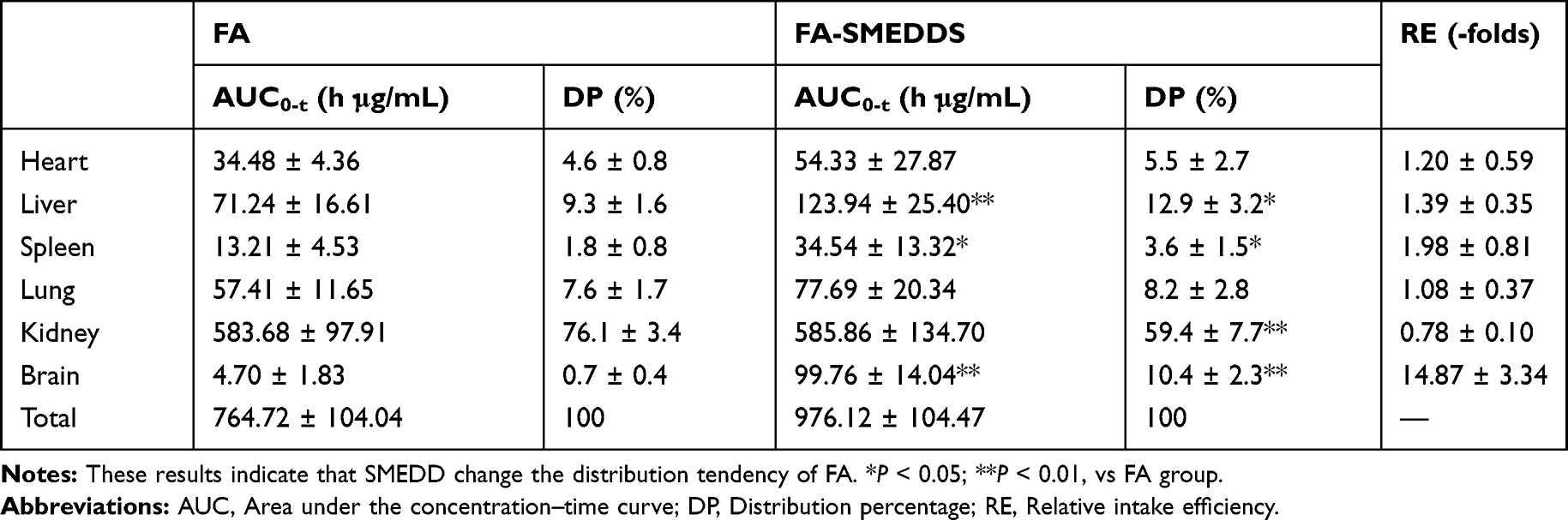 |
Table 3 Analysis of Distributed Tendency of FA in the Tissues. (n=6) |
The metabolites of FA, FAS and FAG, were mainly found in the liver and kidney tissues (Figure 4A–D). The concentrations of FAS in the tissues were higher than those of FAG. SMEDDS significantly changed the levels of FAS and FAG in the kidney (P < 0.05); in particular, it decreased the renal distribution of FAG by approximately 50% (Figure 4E and F).
 |
Figure 4 Tissue distribution of FAS and FAG after oral administration in rats. These results indicate that SMEDDS decreases the FA transformed into FAS and FAG in the kidney. Distribution of FAS in the liver (A) and kidney (B). Distribution of FAG in the liver (C) and kidney (D). AUC0-t values of FAS (E) and FAG (F). *P < 0.05; **P < 0.01, vs FA group, Independent samples t-test. |
Hypnotic Efficacy in Mice
SMEDDS dose-dependently increased the hypnotic efficacy of FA in insomnia mice. Sleep time and sleep latency were increased and decreased by 105.9% and 69.4%, respectively, by high dosage of FA-SMEDDS, which was higher and lower than these parameters for FA (64.2% and 55.0%, respectively), but comparable with those for DZP (119.1% and 76.3%, respectively) (Figure 5A). Moreover, levels of 5-HT and 5-HIAA were reduced in the hippocampus and hypothalamus regions of insomnia mice, and FA caused a slight reversal in these changes, which was strengthened by SMEDDS. SMEDDS dose-dependently enhanced the levels of 5-HT and 5-HIAA in the brain (P < 0.05; Figure 5B and C), restoring the levels of these neurotransmitters to their normal levels. These results indicate that SMEDDS enhanced the hypnotic effect of FA through the regulation of the serotonergic system.
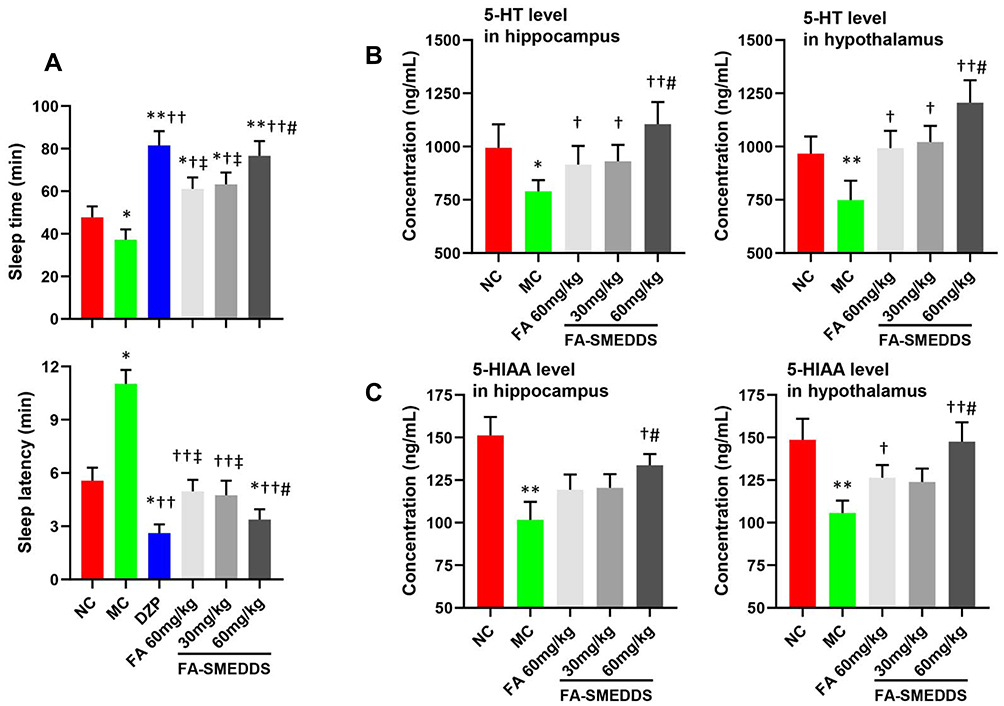 |
Figure 5 Hypnotic efficacy of FA-SMEDDS in PCPA-induced insomnia mice. Findings implied that SMEDDS improved hypnotic efficacy of FA may by regulating serotonergic system. Sleep time and sleep latency (A). The levels of 5-HT (B) and 5-HIAA (C) in the hippocampus and hypothalamus. *P < 0.05; **P < 0.01, vs NC; †P < 0.05; ††P < 0.01, vs MC; ‡P < 0.05, vs DZP; #P < 0.05, vs FA, One-way ANOVA. |
Discussion
In the present study, FA was successfully formulated using SMEDDS and data from solubility and formulation screening experiments using pseudo-ternary phase diagrams and BBD. FA-SMEDDS showed a high self-emulsifying rate, small droplet size, and good stability. After oral administration in rats, SMEDDS remarkably increased the AUC value of FA (P < 0.01), indicating enhanced oral bioavailability of FA.
Our pharmacokinetic analyses revealed that free FA had rapid absorption and elimination in plasma, which was consistent with previous reports.17,30 SMEDDS enhanced the oral bioavailability of FA by slowing its elimination, rather than by enhancing its absorption. SMEDDS did not change the Cmax and Tmax values of FA, which indicates that its absorption was not affected by SMEDDS. These data are consistent with those of in vitro release. Although SMEDDS facilitated the release of FA within 30 min, this improvement could not have had a remarkable impact on its absorption because more than 70% of free FA is absorbed from the GI tract within 30 min.13,14 However, T1/2 and MRT values of FA in the FA-SMEDD group were prolonged, which indicates that SMEDDS reduced the elimination rate of FA and extended its residence time. SMEDDS also decreased the CL/F value of FA, indicating a reduction in clearance. These results demonstrate the decrease in elimination of FA by SMEDDS.
Our pharmacokinetic experiments reveal that SMEDDS increased the apparent volume of distribution of FA (P < 0.01), which implies that SMEDDS facilitated drug distribution in the peripheral tissues. Hence, we investigated the impact of SMEDDS on the tissue distribution of FA. SMEDDS decreased the distribution of FA in the kidney, and as FA is mainly distributed and metabolized in the kidney,15–17 the enhancement in bioavailability of FA by SMEDDS might be due to the reduction in its renal elimination. Absorbed FA is mainly transformed into metabolites, FAS and FAG, in the kidney and liver.15,31 Hence, we explored the impact of SMEDDS on the metabolic profiles of FA by measuring the hepatic and renal concentrations of FAS and FAG. FA was mainly converted into FAS, which is consistent with previous reports.32 In the kidney, AUC values of FAS and FAG in the FA-SMEDDS group were significantly lower than those of the FA alone group, which suggests that the renal metabolism of FA was decreased by SMEDDS. Our findings indicate that SMEDDS reduced the renal distribution of FA and slowed its metabolism, which extended its residence in the body.
Interestingly, FA-SMEDDS showed a higher distribution in the brain than FA alone. This could be attributed to the following reasons. First, SMEDDS increased the systemic exposure of FA and extended its retention time. Second, SMEDDS increased the apparent volume of distribution of FA, which facilitated the distribution of FA in the peripheral tissues, including the brain. Third, the excipients of SMEDDS, such as labrasol, may facilitate the passage of FA across the blood-brain barrier.33,34 Generally, the hypnotic efficacy of drugs is related to their concentrations in the brain. Therefore, we examined the effects of the high accumulation of FA in the brain on its hypnotic efficacy. We found that FA-SMEDDS significantly prolonged sleep time and shortened sleep latency of insomnia mice, compared with FA alone (P < 0.05), thereby improving the hypnotic efficacy of FA. The serotonergic system is a crucial signaling pathway for FA in the treatment of insomnia.10–12 We hypothesized that SMEDDS strengthens the hypnotic efficacy of FA by regulating the serotonergic system. Compared with FA alone, FA-SMEDDS remarkably increased 5-HT and 5-HIAA levels in the hippocampus and hypothalamus, two important brain regions that regulate sleep. We conclude that SMEDDS facilitated the distribution of FA in the brain and strengthened its regulation of the serotonergic system, thus improving its hypnotic efficacy.
In this study, we found that SMEDDS decreased the distribution of FA in the kidney, reduced its metabolism to FAS and FAG, which extended its residence and increased its oral bioavailability. SMEDDS also facilitated the accumulation of FA in the brain and strengthened the regulatory effects of FA on the serotonergic system, which consequently improved its hypnotic efficacy (Figure 6). However, the exact mechanisms underlying the change in tissue distribution of FA and the passage of FA across the blood-brain barrier, both facilitated by SMEDDS, remain unclear, warranting further study.
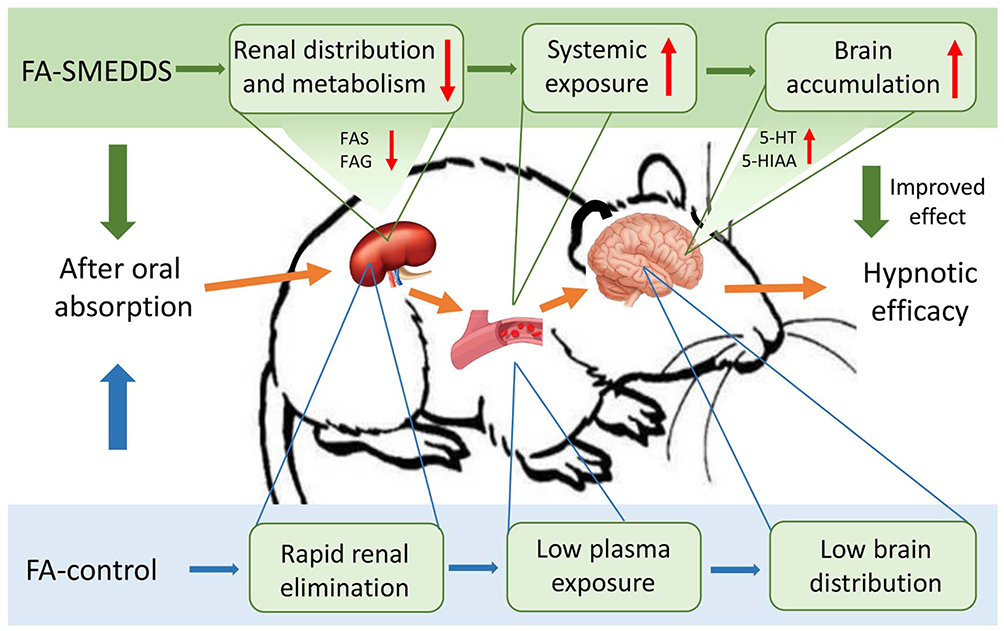 |
Figure 6 Possible mechanism underlining the improved effect of SMEDDS on the oral delivery and hypnotic efficacy of FA. |
Conclusion
A SMEDDS formulation for FA was successfully prepared by screening excipients and optimizing the composition. FA-loaded SMEDDS was evaluated by characterizing its morphology, droplet size, stability, and in vitro release. Pharmacokinetic data reveal that SMEDDS remarkably enhanced the oral bioavailability of FA, decreased its renal distribution, and reduced its metabolism to FAS and FAG in rats. Moreover, SMEDDS improved the distribution of FA in the brain, increased the levels of 5-HT and 5-HIAA, and enhanced the hypnotic efficacy of FA in insomnia mice. We conclude that SMEDDS improved the brain distribution of FA and strengthened its regulation of the serotonergic system, which improved its hypnotic efficacy. Our study is the first to show that SMEDDS improved oral delivery and brain distribution of FA and hence, can be a promising carrier for improved delivery of FA in pharmaceutical applications.
Acknowledgments
This study was supported by the National Key Research and Development Program of China (2018YFC1704503) and China Postdoctoral Science Foundation funded project (2019TQ0140; 2019M663013).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Buysse DJ, Rush AJ, Reynolds CF. Clinical management of insomnia disorder. JAMA. 2017;318(20):1973. doi:10.1001/jama.2017.15683
2. Buysse DJ. Insomnia. JAMA. 2013;309(7):706. doi:10.1001/jama.2013.193
3. Li L, Wu C, Gan Y, et al. Insomnia and the risk of depression: a meta-analysis of prospective cohort studies. BMC Psychiatry. 2016;16(1):375. doi:10.1186/s12888-016-1075-3
4. Meng L, Zheng Y, Hui R. The relationship of sleep duration and insomnia to risk of hypertension incidence: a meta-analysis of prospective cohort studies. Hypertens Res. 2013;36(11):985–995. doi:10.1038/hr.2013.70
5. He Q, Zhang P, Li G, et al. The association between insomnia symptoms and risk of cardio-cerebral vascular events: a meta-analysis of prospective cohort studies. Eur J Prev Cardiol. 2017;24(10):1071–1082. doi:10.1177/2047487317702043
6. Sullivan S. Update on emerging drugs for insomnia. Expert Opin Emerg Dr. 2012;17(3):295–298. doi:10.1517/14728214.2012.693158
7. Singh A, Zhao K. Treatment of insomnia with traditional Chinese herbal medicine. Int Rev Neurobiol. 2017;135:97–115. doi:10.1016/bs.irn.2017.02.006
8. Li M, Wang J, He Q, et al. Compatibility law of Chinese patent medicine of treating insomnia recorded in Pharmacopeia of the People’s Republic of China. J Tradit Chin Med. 2016;57(7):558–562.
9. Li Y, Bi K. Study on the therapeutic material basis of traditional Chinese medicinal preparation Suanzaoren decoction. Chem Pharm Bull. 2006;54(6):847–851. doi:10.1248/cpb.54.847
10. Tu Y, Cheng S, Sun H, et al. Ferulic acid potentiates pentobarbital-induced sleep via the serotonergic system. Neurosci Lett. 2012;525(2):95–99. doi:10.1016/j.neulet.2012.07.068
11. Zhou Q, Zhou X, Xu M, et al. Suanzaoren formulae for insomnia: updated clinical evidence and possible mechanisms. Front Pharmacol. 2018;9:76. doi:10.3389/fphar.2018.00076
12. Gao J, Wang Q, Huang Y, et al. In silico study of anti-insomnia mechanism for Suanzaoren prescription. Front Pharmacol. 2019;10:925. doi:10.3389/fphar.2019.00925
13. Zhao Z, Egashira Y, Sanada H. Digestion and absorption of ferulic acid sugar esters in rat gastrointestinal tract. J Agr Food Chem. 2003;51(18):5534–5539. doi:10.1021/jf034455u
14. Zhao Z, Egashira Y, Sanada H. Ferulic acid is quickly absorbed from rat stomach as the free form and then conjugated mainly in liver. J Nutr. 2004;134(11):3083–3088. doi:10.1093/jn/134.11.3083
15. Zhao Z, Egashira Y, Sanada H. Ferulic acid sugar esters are recovered in rat plasma and urine mainly as the sulfoglucuronide of ferulic acid. J Nutr. 2003;133(5):1355–1361. doi:10.1093/jn/133.5.1355
16. Adam A, Crespy V, Levrat-Verny M, et al. The bioavailability of ferulic acid is governed primarily by the food matrix rather than its metabolism in intestine and liver in rats. J Nutr. 2002;132(7):1962–1968. doi:10.1093/jn/132.7.1962
17. Zhang C, Ma W, Zhang Y, et al. Pharmacokinetics, bioavailability, and tissue distribution study of angoroside C and its metabolite ferulic acid in rat using UPLC-MS/MS. Front Pharmacol. 2018;9:1186. doi:10.3389/fphar.2018.01186
18. Subbaiah M AM, Mandlekar S, Desikan S, et al. Design, synthesis, and pharmacokinetic evaluation of phosphate and amino acid ester prodrugs for improving the oral bioavailability of the HIV-1 protease inhibitor atazanavir. J Med Chem. 2019;62(7):3553–3574. doi:10.1021/acs.jmedchem.9b00002
19. Kesharwani SS, Ahmad R, Bakkari MA, et al. Site-directed non-covalent polymer-drug complexes for inflammatory bowel disease (IBD): formulation development, characterization and pharmacological evaluation. J Control Release. 2018;290:165–179. doi:10.1016/j.jconrel.2018.08.004
20. Kumar S, Kesharwani SS, Mathur H, et al. Molecular complexation of curcumin with pH sensitive cationic copolymer enhances the aqueous solubility, stability and bioavailability of curcumin. Eur J Pharm Sci. 2016;82:86–96. doi:10.1016/j.ejps.2015.11.010
21. Chen W, Miao YQ, Fan DJ, et al. Bioavailability study of berberine and the enhancing effects of TPGS on intestinal absorption in rats. AAPS PharmSciTech. 2011;12(2):705–711. doi:10.1208/s12249-011-9632-z
22. Panwar R, Raghuwanshi N, Srivastava AK, et al. In-vivo sustained release of nanoencapsulated ferulic acid and its impact in induced diabetes. Mater Sci Eng C. 2018;92:381–392. doi:10.1016/j.msec.2018.06.055
23. Zhang Y, Zhang K, Li Z, et al. Ethyl oleate-containing nanostructured lipid carriers improve oral bioavailability of trans-ferulic acid ascompared with conventional solid lipid nanoparticles. Int J Pharmaceut. 2016;511(1):57–64. doi:10.1016/j.ijpharm.2016.06.131
24. Hassanzadeh P, Arbabi E, Atyabi F, et al. Ferulic acid-loaded nanostructured lipid carriers: a promising nanoformulation against the ischemic neural injuries. Life Sci. 2018;193:64–76. doi:10.1016/j.lfs.2017.11.046
25. Vashisth P, Sharma M, Nikhil K, et al. Antiproliferative activity of ferulic acid-encapsulated electrospun PLGA/PEO nanofibers against MCF-7 human breast carcinoma cells. 3 Biotech. 2015;5(3):303–315. doi:10.1007/s13205-014-0229-6
26. Bala V, Rao S, Bateman E, et al. Enabling oral SN38-based chemotherapy with a combined lipophilic prodrug and self-microemulsifying drug delivery system. Mol Pharm. 2016;13(10):3518–3525. doi:10.1021/acs.molpharmaceut.6b00591
27. Ding W, Hou X, Cong S, et al. Co-delivery of honokiol, a constituent of Magnolia species, in a self-microemulsifying drug delivery system for improved oral transport of lipophilic sirolimus. Drug Deliv. 2016;23(7):2513–2523. doi:10.3109/10717544.2015.1020119
28. Lu YY, Wang XW, Wang X, et al. Profile of disposition, tissue distribution and excretion of the novel anti-human immunodeficiency virus (HIV) agent W-1 in rats. Arch Pharm Res. 2016;39(7):970–977. doi:10.1007/s12272-016-0727-7
29. Chen L, Liu CS, Chen QZ, et al. Characterization, pharmacokinetics and tissue distribution of chlorogenic acid-loaded self-microemulsifying drug delivery system. Eur J Pharm Sci. 2017;100:102–108. doi:10.1016/j.ejps.2017.01.011
30. Wu Y, Ai Y, Wang F, et al. Determination of ferulic acid in rat plasma by UPLC-MS/MS and its application in a comparative pharmacokinetic. J Shenyang Pharm Univ. 2016;33(5):396–401.
31. Rondini L, Peyrat-Maillard M, Marsset-Baglieri A, et al. Sulfated ferulic acid Is the main in vivo metabolite found after short-term ingestion of free ferulic acid in rats. J Agr Food Chem. 2002;50(10):3037–3041. doi:10.1021/jf011295i
32. Kokumai T, Ito J, Kobayashi E, et al. Comparison of blood profiles of γ-oryzanol and ferulic acid in rats after oral intake of γ-oryzanol. Nutrients. 2019;11(5):1174. doi:10.3390/nu11051174
33. Wang G, Wang J, Li F, To ST. Development and evaluation of a novel drug delivery: pluronics/SDS mixed micelle loaded with myricetin in vitro and in vivo. J Pharm Sci-Us. 2016;105(4):1535–1543. doi:10.1016/j.xphs.2016.01.016
34. Kaur P, Kim K. Pharmacokinetics and brain uptake of diazepam after intravenous and intranasal administration in rats and rabbits. Int J Pharmaceut. 2008;364(1):27–35. doi:10.1016/j.ijpharm.2008.07.030
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.