Back to Journals » International Journal of Nanomedicine » Volume 9 » Issue 1
Self-assembled or mixed peptide amphiphile micelles from Herpes simplex virus glycoproteins as potential immunomodulatory treatment
Authors Accardo A ![]() , Vitiello MT, Tesauro D, Galdiero M, Finamore E, Martora F, Mansi R, Ringhieri P, Morelli G
, Vitiello MT, Tesauro D, Galdiero M, Finamore E, Martora F, Mansi R, Ringhieri P, Morelli G
Received 15 November 2013
Accepted for publication 15 January 2014
Published 7 May 2014 Volume 2014:9(1) Pages 2137—2148
DOI https://doi.org/10.2147/IJN.S57656
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Antonella Accardo,1 Mariateresa Vitiello,2,3 Diego Tesauro,1 Marilena Galdiero,2 Emiliana Finamore,2 Francesca Martora,2 Rosalba Mansi,1 Paola Ringhieri,1 Giancarlo Morelli1
1Department of Pharmacy, Interuniversitary Centre for Research on Bioactive peptides, CIRPeB, University of Naples "Federico II", Institute of Biostructures and Bioimaging IBB-CNR, Naples, Italy; 2Department of Experimental Medicine, Section of Microbiology and Clinical Microbiology, Second University of Naples, Naples, Italy; 3Department of Clinical Pathology and Transfusion Medicine, University Hospital “Ruggi d'Aragona”, Salerno, Italy
Abstract: The use of micelle aggregates formed from peptide amphiphiles (PAs) as potential synthetic self-adjuvant vaccines to treat Herpes simplex virus (HSV) infection are reported here. The PAs were based on epitopes gB409-505 and gD301-309, selected from HSV envelope glycoprotein B (gB) and glycoprotein D (gD), that had their N-terminus modified with hydrophobic moieties containing two C18 hydrocarbon chains. Pure and mixed micelles of gB and/or gD peptide epitopes were easily prepared after starting with the synthesis of corresponding PAs by solid phase methods. Structural characterization of the aggregates confirmed that they were sufficiently stable and compatible with in vivo use: critical micelle concentration values around 4.0 · 10-7 mol · Kg-1; hydrodynamic radii (RH) between 50–80 nm, and a zeta potential (ζ) around – 40 mV were found for all aggregates. The in vitro results indicate that both peptide epitopes and micelles, at 10 µM, triggered U937 and RAW 264.7 cells to release appreciable levels of cytokines. In particular, interleukin (IL)-23-, IL-6-, IL-8- or macrophage inflammatory protein (MIP)-2-, and tumor necrosis factor (TNF)-α-release increased considerably when cells were treated with the gB-micelles or gD-micelles compared with the production of the same cytokines when the stimulus was the single gB or gD peptide.
Keywords: epitopes, self-adjuvant vaccine, HSV
Introduction
Supramolecular aggregates, such as micelles and liposomes that incorporate drugs and/or contrast agents have been emerging as very promising delivery systems for therapeutic and diagnostic applications in several pathologies. These aggregates, when modified with bioactive molecules (such as peptides or antibodies) on their external surfaces, have been shown to act as target-selective delivery systems in cancer therapy and diagnosis.1–5 Moreover, aggregates obtained from self-assembling peptides have been proposed as vaccines.6–8 Peptides containing the minimal amino acid sequences necessary to stimulate an adaptive immune response are weak immunogens and require either strong adjuvants or their organization into nanometer-sized micelles to be effective for the development of the immune response.9 While the use of adjuvants limits the clinical application of immunogenic peptides (because of their high intrinsic toxicity), the assembling of peptide amphiphiles (PAs) in micelles boosts peptide-specific immune responses without causing any undesirable side effects.7,8 PA micelles have been shown to concentrate the antigen, protect the antigen from degradation, increase uptake and processing by dendritic cells, and induce the production of cytokines that create a robust immune response.10,11 We have studied the effectiveness of micelle aggregates formed from PAs as synthetic self-adjuvants vaccines for the treatment of Herpes simplex Virus (HSV) infections. HSV infections are extremely common and cause a wide range of symptoms, from nonapparent to life-threatening diseases, including genital herpes, orolabial infections, cutaneous and ocular infections, neonatal herpes, herpes encephalitis, disseminated infections, and multiform erythema.12 HSV type 1 (HSV-1) is one of at least eight Herpes viruses that typically cause lifelong recurrent immunopathological diseases in humans.13 The cellular and molecular immune mechanisms underlying recurrent HSV-1 infections are unknown, yet are essential to understand in order to develop efficient immune-prophylactic and immune-therapeutic strategies. Neutralizing antibodies are insufficient for the protection or control of HSV disease. In addition, spontaneous reactivation and shedding of infectious virus occurs at high levels in individuals latently infected with HSV, indicating that the natural immunity acquired following primary infection and subsequent reactivations does not appear to prevent subsequent spontaneous reactivations or shedding of infectious virus capable of spreading the infection. Indeed it is widely considered that a T-helper type 1 (Th1)-driven cell-mediated response is important and sufficient to control the progression of infection. According to the results of a previous study, the replication of HSV-1 in the trigeminal ganglion is controlled by macrophages and T lymphocytes through interferon (IFN)-γ production.14 The expression of IFN-γ has been detected during the acute and latent HSV infection and has been shown to reduce the virus replication in the trigeminal ganglion and brain during acute infection.15
Therefore, obtaining compounds that conjugate both antiviral and immunomodulatory activities would be very useful for the prevention and/or treatment of these pathologies. The current paradigm in herpes vaccine development is that a highly efficacious vaccine will need to induce a more vigorous and/or different T-cell response than the suboptimal immunity induced by natural infection.16 Decreasing recurrent infections leads to stronger T-cell immunity of both a higher magnitude and wider breadth, or a selective induction of T-cell responses to a specific subset of viral epitope. HSV-1 specifies at least eleven glycoproteins that are expressed in infected cells.17 Most of the research on subunit vaccines has used HSV envelope glycoproteins, specifically glycoprotein B (gB) and/or glycoprotein D (gD), as immunogens since these are the dominant targets for neutralizing antibody production in HSV-infected people. Results from a number of studies indicate that gB and gD, the two major HSV-1/2 antigens, produce some protective immunity against herpes disease in both animal models and humans and are recognized by cluster of differentiation (CD)4+ and CD8+ T cells from both symptomatic and asymptomatic HSV-seropositive humans. gB and gD are attractive choices for subunit vaccines because they are the targets for humoral (neutralizing and antibody-dependent cellular cytotoxicity) and cell-mediated immunity (class I and class II restricted)9,18,19 In addition, gB and gD have high sequence similarity in HSV-1 and HSV-2 and may, therefore, provide protection against both HSV-1 and HSV-2 infections. In the present study, we assembled, in a single micelle, PAs based on gB409−505 and gD301−309 peptides modified with hydrophobic moieties containing two C18 hydrocarbon chains (Figure 1). The structural properties of the pure and mixed micelles and their in vitro immunogenic behavior (ie, interleukin [IL]-6, IL-8, macrophage inflammatory protein [MIP]-2, IL-17, IL-23 and tumor necrosis factor [TNF]-α cytokine-release) is reported. Moreover, the role of the recently described Th1 cytokine in HSV-1 infection, IL-23, was investigated because a previous study demonstrated that IL-23 stimulated the production of IFN-γ and enhanced the activity of cellular immunity.20
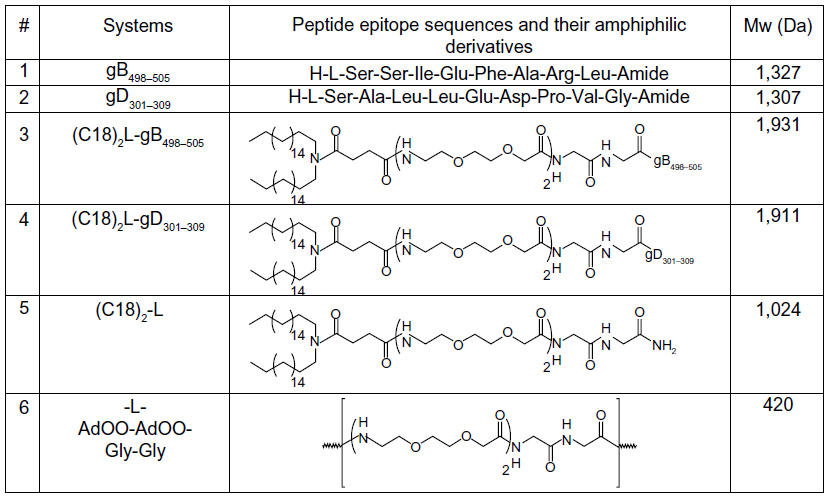 | Figure 1 Amino acid sequences of gB498–505 and gD301–309 peptide epitopes, the schematic representation of the corresponding peptide amphiphilic derivatives and of (C18)2-L adjuvant, and their molecular weights (Mw).
|
Materials and methods
Instrumentation
Protected Nα-fluorenylmethyloxycarbonyl (Fmoc)-amino acid derivatives, coupling reagents, and Rink amide 4-methylbenzhydrylamine (MBHA) resin were Calbiochem®-Novabiochem® branded products (Merck KGaA, Darmstadt, Germany). The Fmoc-8-amino-3,6-dioxaoctanoic acid (Fmoc-AdOO-OH) was purchased from Neosystem SA (Strasbourg, France). N,N-dioctadecyl succinamic acid was prepared according to the experimental procedure reported in literature.21 All other chemicals were generic laboratory-grade products, and were used as received unless otherwise stated. All solutions were prepared by weight, with doubly distilled water. Preparative reversed-phase high-performance liquid chromatography (RP-HPLC) was carried out on a LC8 Shimadzu HPLC system (Shimadzu Corp, Nakagyo-ku, Japan) equipped with a Waters Lambda Max Model 481LC Spectrophotometer (Waters Corp, Milford, MA, USA) ultraviolet (UV) detector, using Phenomenex (Torrance, CA, USA) C18 and C4 (300 Å, 250 × 21.20 mm, 5 m) columns for the peptides (gB498−505 and gD301−309) and the PAs ([C18]2-L-gB498−505 and [C18]2-L-gD301−309), respectively. The elution solvents were a) H2O/0.1% trifluoroacetic acid (TFA) and b) CH3CN/0.1% TFA, from 5% to 70% over 30 minutes at a 20 mL min−1 flow rate. Purity and identity were assessed by analytical liquid chromatography (LC)–mass spectrometry (MS) analyses using a Finnigan™ Surveyor® MSQ™ single quadrupole electrospray ionization column (Thermo Fisher Scientific, Waltham, MA, USA): Phenomenex C18/C4 columns were eluted with an a) H2O/0.1% TFA and b) CH3CN/0.1% TFA, from 5% to 70% over 30 minutes at a 1 mL min−1 flow rate. UV-visible (Vis) spectra were carried out using a UV-Vis Jasco Model 440 spectrophotometer (Jasco Int Co, Ltd, Tokyo, Japan) with a path length of 1 cm. RPMI, Roswell Park Memorial Institute (RPMI) 1640, glutamine, penicillin and streptomycin were Gibco® products purchased from Life Technologies Corp (Carlsbad, CA, USA). Fetal bovine serum (FBS) containing <0.01 endotoxin U/mL was a HyClone brand product (Thermo Fisher Scientific). A cytokine enzyme-linked immunosorbent assay (ELISA) kit was obtained from R&D Systems Inc. (Minneapolis, MN, USA). A Limulus Amebocyte Lysate (LAL) gel-clot assay was purchased from International PBI SpA (Milan, Italy).
Synthesis of peptides and peptide amphiphiles
gD301−309 and gB498−505 peptide epitopes were synthesized using standard solid-phase 9-fluorenylmethoxycarbonyl (Fmoc) procedures.22 The Rink amide MBHA resin (substitution 0.65 mmol · g−1) was used as the solid-phase support, and synthesis was performed on a scale of 0.2 mmol. The resin was swelled in dimethylformamide (DMF) for 30 minutes, and the Fmoc deprotection reaction was performed twice with 30% piperidine in DMF. The amino acid coupling was achieved by adding a twofold molar excess of amino acid, mixed with equimolar amounts of 1-hydroxybenzotriazole, benzotriazol-1-yl-oxy-tris-pyrrolidino-phosphonium (PyBop), and a fourfold molar excess of diisopropylethylamine in DMF. All couplings were performed twice for 1 hour, by using an excess of four equivalents for the single amino acid. To obtain (C18)2-L-gB498−505 and (C18)2-L-gD301−309 PAs and (C18)2-L adjuvant, the lipophilic N,N-dioctadecyl succinamic acid and the Fmoc-AdOO-OH ethoxylic spacer were coupled, as previously described.23 Peptides were fully deprotected and cleaved from the resin with the TFA/triisopropylsilane/ethanedithiol/H2O (93/2/2.5/2.5) mixture, at room temperature. Peptides and PAs were precipitated with ice-cold ethyl ether and water, respectively. Purification of the crude products was carried out by RP-HPLC. Mass spectrometry confirmed the product identity.
Preparation of solutions
Stock solutions of pure or mixed aggregates were prepared by stirring the PA epitopes, at room temperature, in 10 mM tris(hydroxymethyl)aminomethane (TRIS) buffer at pH 8.0, followed by filtering through a 0.45 μm filter.
Fluorescence studies
The values of critical micellar concentrations (CMC) of (C18)2-L-gB498−505, (C18)2-L-gD301−309, and (C18)2-L pure aggregates; and (C18)2-L-gB498−505/(C18)2-L-gD301−309 (R=50/50) or (C18)2-L/(C18)2-L-gB498−505 (R=80/20 or R=50/50) mixed aggregates were obtained by fluorescence measurements. Fluorescence spectra were recorded at room temperature on a Jasco Model FP-750 spectrofluorophotometer, in a 1.0 cm path length quartz cell. Equal excitation and emission bandwidths were used throughout the experiments, with a recording speed of 125 nm · min−1 and automatic selection of the time constant. The CMC were measured using 8-anilino-1-naphthalene sulfonic acid ammonium salt (ANS) as the fluorescent probe.24,25 Small aliquots of lipopeptide solutions, diluted in 10 mM TRIS buffer at pH 8.0, were added to a fixed volume (1.0 mL) of fluorophore (1 · 10−5 M ANS) dissolved in the same buffer. Final spectra, to be used for calculations, were obtained after blank correction and adjustment for dilution. The intensity was followed as a function of PA molecule concentration. The CMC values were determined by linear least-squares fitting of the fluorescence emission at 480 nm upon excitation at 350 nm versus PA concentration that was lower and higher than the change of slope, as previously reported.26,27
Circular dichroism
Far-UV circular dichroism (CD) spectra of the epitopes or PA epitopes in TRIS buffer at pH 8.0 at 2 · 10−4 M concentration were collected at room temperature on a Jasco J-810 CD Spectropolarimeter, equipped with a Thermo NesLab™ RTE111 Refrigerated Bath/Circulators Digital Controller (Thermo Fisher Scientific), using a 1 mm quartz cell, at 25°C. The spectra were recorded from 260 to 195 nm, with a bandwidth of 3 nm, a time constant of 16 seconds, and a scan rate of 10 nm/min. Other experimental settings were: scan speed =10 nm · min−1; sensitivity =50 mdeg; time constant =16 seconds; and bandwidth =1 nm. Each spectrum was obtained by averaging three scans and by subtracting the contributions from other species in solution. Mean residue ellipticities (MRE), in units of deg × cm2/dmol/res, were calculated using the equation
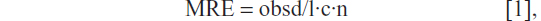
where obsd is the ellipticity measured in millidegrees, l is the length of the cell in centimeters, c is the peptide concentration in moles per liter, and n is the number of amino acid residues in the peptide. Sample spectra of peptides were also recorded at different percentages of trifluoroethanol (TFE) (20%, 40%, 60%, and 80%).
Dynamic light scattering (DLS) characterization
Mean diameter and zeta-potential (ζ) were carried out using a Zetasizer Nano ZS (Malvern Instruments, Malvern, UK) that employed a 173° backscatter detector. Other instrumental settings were the measurement position: 4.65 mm; attenuator: 8; temperature 25°C; and cell: disposable sizing cuvette (Malvern Instruments, Malvern, UK). DLS samples were prepared at the final concentration of 2 · 10−4 M and centrifuged at room temperature at 13,000 rpm for 5 minutes. For each batch, hydrodynamic radii and size distribution were the mean of three measurements, and values were calculated as the mean of three different batches. The ζ values of the aggregate surface were collected as the average of 20 measurements.
Cell lines
U937 monocytes (ATCC CRL-1593.2; American Tissue Type Collection [ATTC], Manassas, VA, USA) were grown at 37°C in 5% CO2 in RPMI 1640 supplemented with 10% heat-inactivated FBS, glutamine (2 mM), penicillin (100 U/mL), and streptomycin (100 U/mL), and differentiated as previously described.28 RAW 264.7 cells, a murine macrophage line, were obtained from ATCC (TIB-71TM). The cells were cultivated in RPMI 1640 supplemented with 10% heat-inactivated FBS, 2 mM L-glutamine, 100 U/mL penicillin, and 0.1 mg/mL streptomycin. The cells were maintained at 37°C, with 5% CO2, in a humidified incubator.29
Endotoxin contamination
All solutions and peptide preparations used in our experiments were tested for the presence of endotoxin, using a LAL assay, as described by Yin et al.30 The lower detection limit of this assay was 0.1 EU/mL.
Cytotoxicity test
To determine the effect of various concentrations on cell viability, the colorimetric assay described by Mosmann was used.31 In this assay, the pale yellow tetrazolium salt 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) is cleaved by active mitochondria to produce a dark blue formazan product. Briefly, cells were seeded onto 96-well culture plates, at a number of 2 × 104 per well. After a 4-hour incubation to allow seeding of the cells, samples were added into each well, at the following concentrations: 1, 5, 10, 20, and 50 μM. The plates were incubated at 37°C, with 5% CO2, for 24 hours. The medium was then discarded and the MTT reagent added. Then, the plates were reincubated at 37°C for an additional 3 hours to allow the development of formazan and then read with an ELISA microplate reader (Model 3550; Bio-Rad Laboratories, Hercules, CA, USA), using a wavelength of 490 nm. Cisplatin (20 μM) was used as a positive control of cell death.
Cytokine evaluations
Culture supernatants for ELISA were collected (3 × 106 cells/mL) at 24 hours postincubation of the U937 and RAW 264.7 cells with stimuli. The optimal concentrations of stimuli (10 μM) and time point of stimulation were selected based on preliminary experiments (data not shown). Lipopolysaccharide was used as a positive control of the efficiency of cytokine assay. After incubation, the samples were centrifuged at 1,800 rpm, at 4°C, for 10 minutes, and the supernatants were collected and stored at −70°C. IL-6, IL-8 or the functional homolog of human IL-8, MIP-2, TNF-α, IL-17, and IL-23 were analyzed (Abcam Inc, Cambridge, UK). Cytokine-release was measured according to the manufacturer’s recommendations. The assay employed a specific antibody for each of the cytokines coated on a 96-well plate. Standards, samples, and biotinylated anti IL-6, IL-8 or MIP-2, TNF-α, IL-17, and IL-23 were pipetted into the wells, and the cytokine present in the samples was captured by the antibody immobilized to the wells and by the biotinylated specific detection antibody. Standard and sample dilutions were added in duplicate wells to each plate. All analyses were performed at least three times for each individual cell-stimulation assay.
Statistical analysis
The results were expressed as the means of three experiments ± standard deviation (SD); differences were considered significant at values of P≤0.01 (Student’s t-test).
Results and discussion
Identification of peptide epitopes
The identification of B-cell and T-cell epitopes is crucial for the development of effective vaccines. HSV-1 gB and gD peptide epitopes were chosen on the basis of the literature. Prediction analysis (Chou–Fasman and predicting one-dimension methods) of the primary structure of gD from the HSV-1 and immunogenicity experiments indicated the presence of an immune dominant antigenic determinant in the gD301–309 region (301SALLEDPVG309).32 The gD301–309 peptide epitope was located close to the hydrophobic membrane anchor domain of gD. Conformational analysis of the gD301–309 sequence confirmed the presence of a type I β-turn structure, which included the aspartate-proline-valine-glycine (DPVG) epitope core.33 Moreover, previous studies indicated that the gD301–309 epitope induced gD-specific responses and also elicited an immune response that conferred protection against lethal HSV-1 infection.34 gB493–509 was located in domain II of glycoprotein gB (498SSIGFARL505). HSV gB is the major component of virus infected cell membranes and the virion envelope, and is essential for virus entry and infectiveness. It is the most studied protein for the design of efficient HSV deoxyribonucleic acid (DNA) vaccines.35,36 An vivo study has previously shown that peptide epitope 498–505 of gB, chosen in this study, is one immune-dominant cytotoxic T-lymphocyte (CTL) epitope, targeting around 90% of the CTL response generated by C57BL/6 mice following infection with HSV type 1.37
Peptide synthesis and aggregates formulation
Peptide epitopes gB498−505 and gD301−309 and the corresponding lipophilic derivatives, (C18)2-L- gB498−505 and (C18)2-L- gD301−309, reported in Figure 1, were synthesized by solid-phase methods, using Rink-amide MBHA resin, as a polymeric support, and Fmoc/tert-butyl (tBu) chemistry, according to standard SPPS protocols.22 PAs were obtained by coupling the N,N-dioctadecyl succinamic acid at the free N-terminus directly onto the resin. A linker, formed by two glycine residues and two oxyethylene spacers, was introduced between the peptide moiety and alkyl chains. The linker was introduced in order to distance the peptide epitopes from micelle surface, thus improving peptide bioavailability. The glycine and oxyethylene residues did not alter the net charge of the monomers. After RP-HPLC purification, products were isolated in good yield, and their purity was assessed using analytical HPLC and electrospray ionization mass spectrometry (ESI-MS). Self-assembling and mixed aggregates of gB and gD epitope PAs (as listed in Table 1) were formulated by dissolving the monomers, in the chosen ratio, in 10 mM TRIS buffer at pH 8.0. Aggregates based on (C18)2-L adjuvant, formed by the two alkyl chains and the glycine-glycine-leucine-leucine (GGLL) spacer (Figure 1, row 5) at 100% or in combination with (C18)2-L-gB498−505 at 80/20 and 50/50 molar ratios were also prepared. The latter aggregates were formulated in order to better clarify how crowding of peptide epitopes on the external surface of the supramolecular aggregates influences their immune modulatory activity. All aggregates were studied from the structural and biological point of view. Physicochemical characterization (CMC determination, DLS, and CD measurements) of these aggregates diluted with the adjuvant were also performed, to highlight the structure–activity relationship.
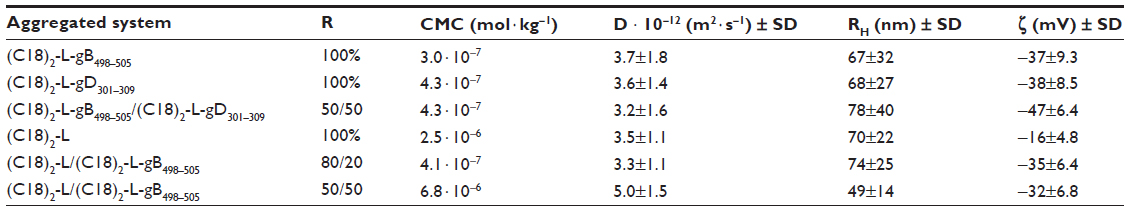 | Table 1 List of the investigated aggregated systems, with the ratio (R) between the amphiphilic monomers, and their structural properties |
Micelle structural characterization
Pure and mixed aggregates of the two lipophilic epitopes were fully characterized by fluorescence spectroscopy and DLS to evaluate their CMC values, RH, and ζ. Aggregates based on (C18)2-L adjuvant at 100% or in combination with (C18)2-L-gB498–505 at 80/20 and 50/50 molar ratios were also structurally characterized. Apparent CMC values were determined by a fluorescence-based method, using ANS as a probe. The fluorescence intensity at 480 nm, corresponding to the maximum of the spectrum, as a function of the PA concentration is reported in Figure 2A, where apparent CMC values can be visualized by the graphical break point. As reported in Table 1, the apparent CMC values of pure and mixed aggregates (≈4.0 × 10−7 mol · Kg−1) were quite similar, thus indicating that the different sequence of the epitope did not influence the CMC values. Instead, the CMC value of the (C18)2-L adjuvant was slightly higher (≈2.5 × 10−6 mol · Kg−1) with respect to that of pure and mixed PA-based aggregates; anyway this value was sufficiently low to assure a good micelle stability.
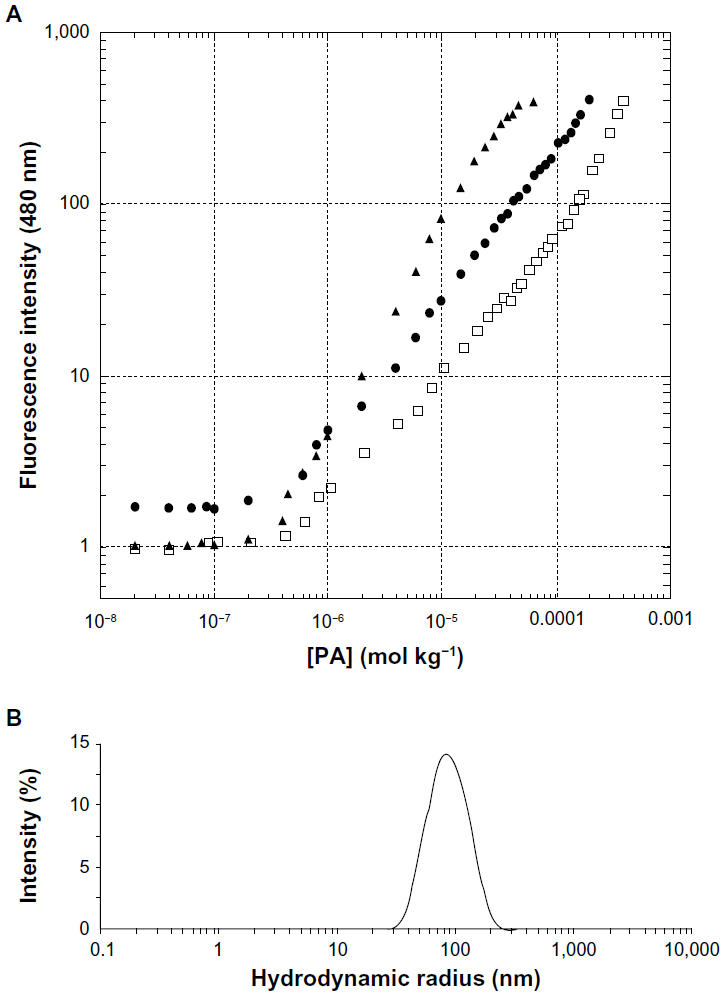 | Figure 2 Structural characterization of the aggregates, by fluorescence and dynamic light scattering.
|
The size and ζ of the aggregates were measured by DLS, at θ¸=173°, on self-assembled PAs at a concentration of 2 × 10−4 M in 10 mM TRIS buffer, at pH 8.0. Results, collected in Table 1, (also, Figure 2B) showed a RH between 50–80 nm for all aggregates. The CMC values and structural data indicated good compatibility for an in vivo use of the obtained aggregates: even when diluted in the bloodstream, they should remain stable in the micelle form and without collapsing, even when in bigger aggregates.
Circular dichroism
The CD spectra of the gB498–505 and gD301–309 peptides, in 10 mM TRIS buffer at pH 8.0 and in the water/TFE mixtures (80/20, 60/40, 40/60, and 20/80 v/v), were recorded between 190 and 260 nm. For both peptides, the shape of the CD spectra in water solution showed a negative band between 197 and 201 nm, thus confirming the expected unordered peptide structure (Figure 3A shows the gD301−309 peptide). The addition of TFE induced a conformational rearrangement from a random coil to α-helix. Helix formation can be better appreciated by difference spectra, which showed the growth of a positive and two negative bands centered around 195 nm, and 208 nm and 222 nm, respectively, as typical of α-helical rearrangement. However, the low helical content gained by the peptide, even in TFE, supports its high flexibility. The CD spectra of the PAs were recorded at concentration above the CMC values, to ensure the presence of aggregates in solution. Both self-assembling and mixed aggregates showed a spectrum typical of a random coil conformation (Figure 3B), thus indicating that any structural transition occurred when the gB and gD epitopes were anchored to the aggregate surface. The same behavior was also observed in mixed aggregates of the gB-PA with the adjuvant at the studied molar ratios (data not shown). Usually, a CD spectrum with a large negative maximum at 218 nm, typical of a β-sheet structural motif, is observed when PAs form nanostructures, such as nanofibers.38 The formation of nanofibers is driven by interactions of the hydrophobic chains in the inner core and by the peptide side chains in the hydrophilic shell. gB498–505 and gD301–309 peptide epitopes have a theoretical isoelectric point of 6.0 and 3.7, respectively, both lower than the pH of TRIS buffer solution (pH =8.0). Hence, the Glu501 residue in the gB epitope and Glu305 and Asp306 residues were deprotonated (COO−) in the experimental conditions studied, and a positive charge was expected on the peptide sequence. Moreover, both peptide sequences had a very high aliphatic index and hydropathicity. All these conditions do not favor interaction between peptide side chains and nanofiber formation. As expected, the CD spectra of the mixed micelles based on (C18)2-L adjuvant in combination with (C18)2-L-gB498−505 at 80/20 and 50/50 molar ratios did not show any structuration of the peptide moiety with respect to the pure gB or gD micelles (data not shown).
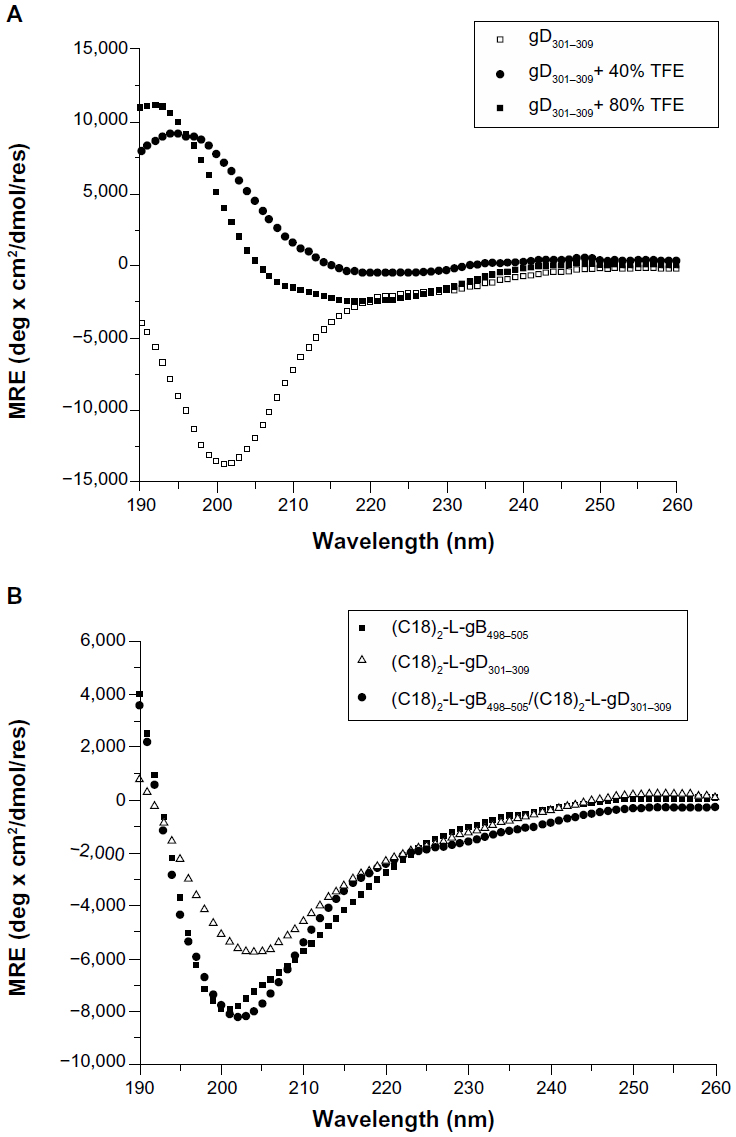 | Figure 3 Far-UV CD spectra of: (A) gD301–309 peptide at 0, 40%, and 80% of TFE in TRIS buffer solution, after subtraction of gD301–309 spectrum at 0% TFE; similar spectra were observed for the gB498–505 peptide; (B) pure and mixed (50/50) amphiphilic epitopes at 2 × 10−4 M concentration, well above the CMC values to ensure the presence of aggregates in solution. |
Cytotoxicity test
The culture media, reagents, and peptide solutions tested for the presence of endotoxin, by LAL tests, were found to contain lower than 0.1 EU of endotoxin per mL (data not shown). The cytotoxic effect of the pure and mixed aggregates of the two gB and gD peptide epitopes, toward human U937 (Figure 4A) and mouse RAW 264.7 macrophage cells (Figure 4B), were evaluated using an MTT assay, in order to determine the concentrations to be used for subsequent assays. Results demonstrated that pure and self-assembling aggregates of gB and gD epitope displayed different, dose-dependent cytotoxicity, in both cell culture systems employed. The various treatments performed were not toxic for the cells; in fact, cytotoxicity (up to a concentration of 50 μM) was not obvious after 24 hours of incubation. Therefore, only the concentration of 10 μM was selected for the subsequent experiments. Cisplatin (20 μM), a platinum-containing anticancer drug, was used as a positive control of cell death, and results showed an average 20% viability of cells after treatment, therefore, as expected, it proved to be quite toxic in the cell cultures.
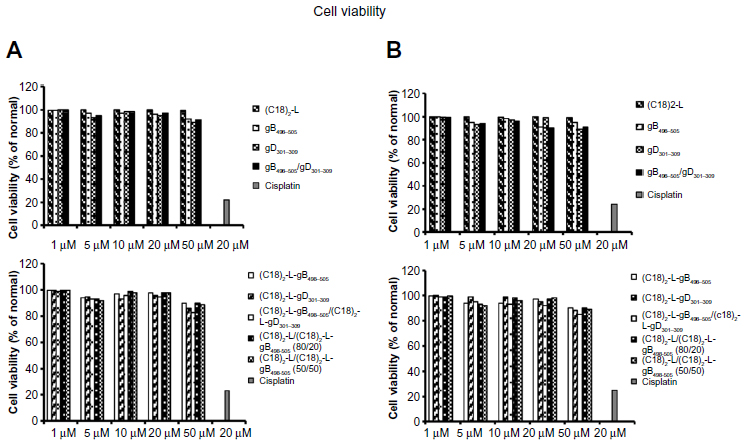 | Figure 4 Effects of PAs (gB, gD, gB/gD) and PA-micelles on cell viability. |
Effects of pure and self-assembling aggregates of gB498–505 and gD301–309 epitope on cytokine production
The efficient elimination of infectious agents, such as viruses, generally requires a combination of both innate and adaptive immunity, where also a proinflammatory host response can contribute to an effective viral clearance.39 The type 1 immunity response is characterized by the activation of mononuclear cells and production of proinflammatory cytokines, such as IFN, TNF, and interleukins, which may or not may be interrelated but which cooperate to regulate the inflammatory and immune responses, inducing an antiviral state in the cells (via the IFNs) or destroying virus-infected cells (via TNF-α). Recently, a novel IFN-γ-inducing cytokine, IL-23, has been discovered40 and seems to be involved in the cross talk of immune cells during viral infection.41 IL-23 is produced by dendritic cells and macrophages in response to pathogens, including certain bacteria and viruses and/or their components. In particular, IL-23 is essential for the survival, activation, and differentiation of Th17 cells in vivo, promoting the production of other proinflammatory mediators, such as IL-6, IL-8, IL-17, TNF-α, and MCP-1.42 However, IL-23, per se, cannot induce the differentiation of naïve CD4+ T cells into Th17 in vitro.43 The effects of gB and gD epitopes and mixed PA micelles on cytokine production (IL-6, IL-8/MIP-2, TNF-α, IL-17, and IL-23) in U937 and RAW 264.7 cells are shown in Figures 5 and 6. Specifically, we tested the release of IL-6, IL-8 or MIP-2, TNF-α, IL-17, and IL-23 by immune-responsive cells, in the form of isolated U937 and RAW 264.7 cells. The supernatant of the U937 and RAW 264.7 cell cultures was tested by ELISA for the presence of secreted cytokines in response to different peptide-micelles constructs and compared with that induced by the core system alone. We also compared the release of cytokines after stimulation with gB498–505 and gD301–309 peptides, with the release obtained when stimulating cells with the PA micelles containing the same peptides. For the stimulation experiments, a U937 human monocytic cell line and RAW 264.7 murine macrophage cell line were seeded at 3 × 106 cells per mL density and then treated for a 24-hour period. In vitro results indicated that the gB498–505 and gD301–309 epitopes and the pure and mixed micelles of epitopes (C18)2-L-gB498−505, (C18)2-L-gD301–309, and (C18)2-L-gB498−505/(C18)2-L- gD301−309) at the concentration of 10 μM triggered U937 and RAW 264.7 cells to release appreciable levels of IL-23, IL-6, IL-8/MIP-2, and TNF-α, whereas very low amounts of IL-17 production were found (Figure 5). In particular, the IL-23-, IL-6-, IL-8- or MIP-2-, and TNF-α-release increased considerably when cells were treated with gB-micelles or gD-micelles compared with the production of the same cytokines when the stimulus was represented by the single gB or gD peptides. The U937 cells were more reactive to such stimulation and reached higher levels of cytokine production compared with the RAW 264.7 cells. Any considerable difference was observed in cytokine production between the gB or gD pure micelles; only a higher efficacy of gB aggregates compared with gD micelles was detected in IL-23-release. Surprisingly, cytokine-release by the gB/gD mixed aggregates was very similar or was slightly lower compared with the effect shown by the gB or gD pure aggregates. Thus, the expected cooperative effect between the two peptide epitopes was not observed. Finally, cytokine-release of the mixed micelles (C18)2-L/(C18)2-L-gB498−505 (at 80/20 and 50/50 molar ratios) was also studied. From the inspection of Figures 5 and 6, it was observed that the dilution of (C18)2-L-gB498−505 with the adjuvant (C18)2-L did not affect the cytokine-release. This result indicated that in the pure micelles, peptide epitopes were constrained in very close packaging on the external surface of the supramolecular aggregate, but this did not influence their immunomodulatory activity.
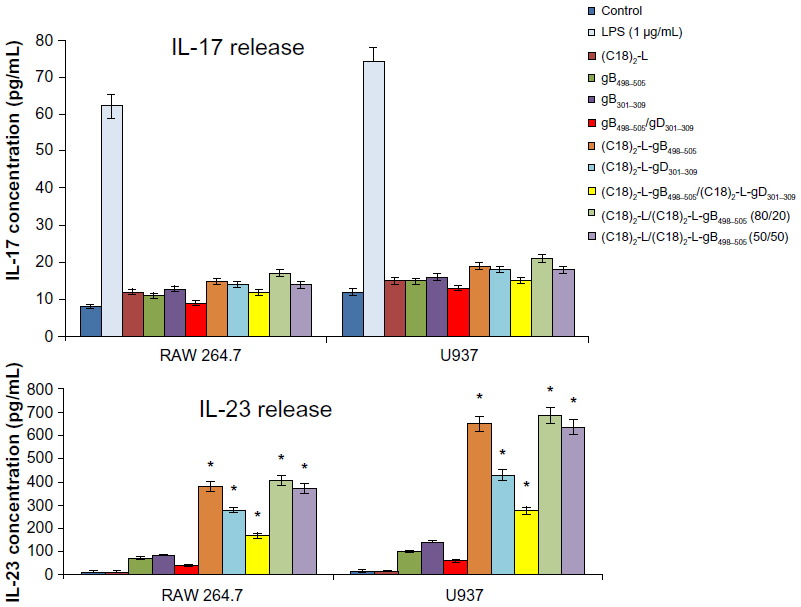 | Figure 5 IL-17- and IL-23-release after treatment with 10 μM of each peptide and peptide-micelles for 24 hours. |
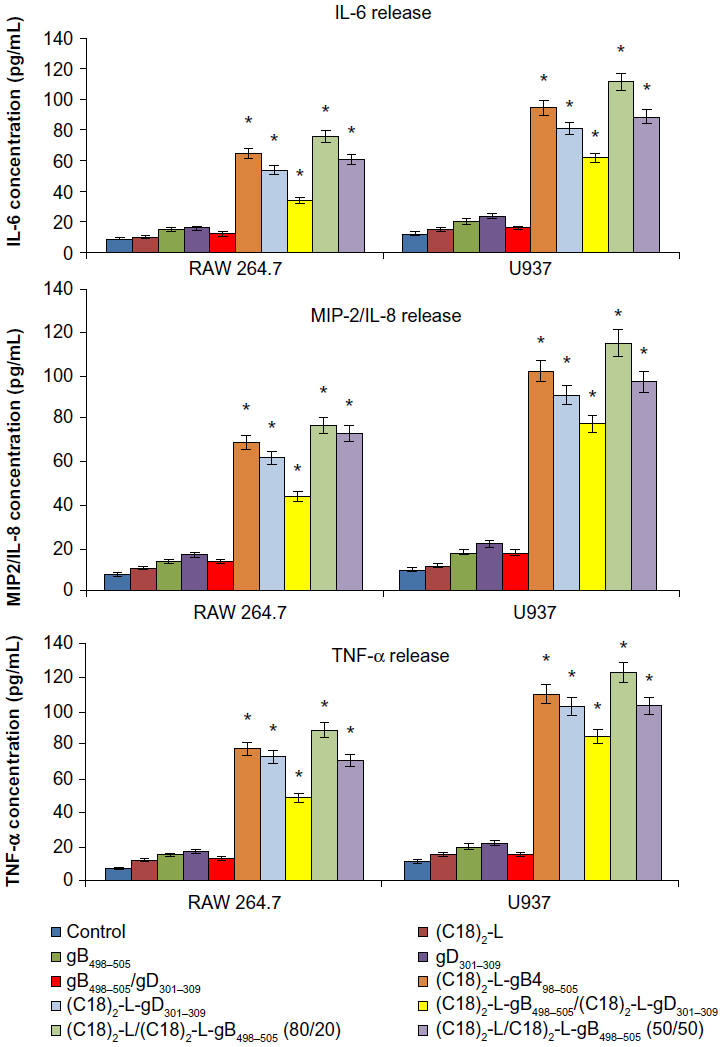 | Figure 6 IL-6-, IL-8- or MIP-2-, and TNF-α-release after treatment with 10 μM of each peptide and peptide-micelles, for 24 hours.
|
Conclusion
HSV-1 is a ubiquitous human virus, and its seroprevalence has been over 70% in most studied populations over the world.17 The aim of this study was to elucidate the structural characterization and immune-modulation capacity of self-assembled or mixed PA micelles derived from HSV-1 glycoproteins gB and gD for the development of a synthetic, multiepitopic HSV-1 vaccine. Pure and mixed micelles of gB and/or gD peptide epitopes were easily prepared, starting from the corresponding PAs that were synthesized by solid-phase methods. Structural characterization of the aggregates confirmed that they were sufficiently stable and thus compatible for following in vivo use. The effects of gB and gD epitopes and mixed PA micelles on cytokine production (IL-6, IL-8/MIP-2, TNF-α, IL-17, and IL-23) in human U937 and mouse RAW 264.7 macrophage cells was investigated. In vitro results indicated that both peptide epitopes and micelles, at 10 μM, triggered U937 and RAW 264.7 cells to release appreciable levels of IL-23, IL-6, IL-8/MIP-2, and TNF-α, whereas very low amounts of IL-17 production were found. In particular, the cytokine-release of gB-micelles or gD-micelles increased considerably with respect to the production triggered by the single gB or gD peptides. gB or gD pure micelles stimulated cytokine production with similar efficacy, whereas, unexpectedly, a cooperative effect between the two peptide epitopes in the mixed micelles was not observed. The cytotoxic effect of pure and mixed aggregates of gB and gD peptide epitopes toward human and mouse macrophage cell lines was determined to exclude eventual cellular toxicity, a priority requirement for any compound considered for in vivo use.The results showed that pure and self-assembling aggregates of gB and gD epitope did not display appreciable signs of cellular toxicity. In conclusion, this work has relevance to studies of the significance of immune modulatory treatments in regard to HSV infections. The experimental data presented indicate that micelle aggregates formed from PAs, as effective synthetic self-adjuvant vaccines, can exert significant immune modulatory effects on macrophage activation, leading to the production of predominantly IL-23 and other proinflammatory cytokines. Therefore, taken together, our results could support a strategy of using these innovative compounds for therapeutic applications targeting protective T-cell responses during viral infection.
Acknowledgments
This work was supported by grants from the Italian Minister of Research (MIUR): grant number FIRB RBRN07BMCT and grant number PRIN E61J11000300001. The authors are grateful to Leopoldo Zona for technical assistance with capture of the NMR spectra.
Disclosure
The authors report no conflicts of interest in this work.
References
Accardo A, Tesauro D, Morelli G. Peptide-based targeting strategies for simultaneous imaging and therapy with nanovectors. Polym J. 2013;45:481−493. | |
Yu MK, Park J, Jon S. Targeting strategies for multifunctional nanoparticles in cancer imaging and therapy. Theranostics. 2012;2(1):3–44. | |
Lammers T, Aime S, Hennink WE, Storm G, Kiessling F. Theranostic nanomedicine. Acc Chem Res. 2011;44(10):1029−1038. | |
Accardo A, Salsano G, Morisco A, et al. Peptide-modified liposomes for selective targeting of bombesin receptors overexpressed by cancer cells: a potential theranostic agent. Intern J Nanomed. 2012;7:2007−2017. | |
Kieler-Ferguson HM, Fréchet JM, Szoka FC Jr. Clinical developments of chemotherapeutic nanomedicines: polymers and liposomes for delivery of camptothecins and platinum (II) drugs. Wiley Interdiscip Rev Nanomed Nanobiotechnol. 2013;5(2):130–138. | |
Black M, Trent A, Kostenko Y, et al. Self-assembled peptide amphiphile micelles containing a cytotoxic T-cell epitope promote a protective immune response in vivo. Adv Mater. 2012;24(28):3845−3849. | |
Moyle PM, Toth I. Self-adjuvanting lipopeptide vaccines. Curr Med Chem. 2008;15(5):506–516. | |
Eriksson EM, Jackson DC. Recent advances with TLR2-targeting lipopeptide-based vaccines. Curr Protein Pept Sci. 2007;8(4):412–417. | |
Galdiero S, Vitiello M, Finamore E, et al. Activation of monocytic cells by immunostimulatory lipids conjugated to peptide antigens. Mol BioSyst. 2012;8(12):3166−3177. | |
Lee KC, Carlson PA, Goldstein AS, Yager P, Gelb MH. Protection of a decapeptide from proteolytic cleavage by lipidation and self-assembly into high-axial ratio microstructures: a kinetic and structural study. Langmuir. 1999;15(17):5500–5508. | |
Missirlis D, Khant H, Tirrell M. Mechanism of peptide amphiphile internalization by SJSA-1 cells in vitro. Biochemistry. 2009;48(15):3304–3314. | |
Aymard M. [Current epidemiology of herpes]. Pathol Biol (Paris). 2002;50(7):425–435. French. | |
Whitley RJ, Kimberlin DW, Roizman B. Herpes simplex viruses. Clin Infect Dis. 1998;26(3):541−553. | |
Kodukula P, Liu T, Rooijen NV, Jager MJ, Hendricks RL. Macrophage control of herpes simplex virus type 1 replication in the peripheral nervous system. J Immunol. 1999;162(5):2895−2905. | |
Cantin EM, Hinton DR, Chen J, Openshaw H. Gamma interferon expression during acute and latent nervous system infection by herpes simplex virus type 1. J Virol. 1995;69(8):4898−4905. | |
Koelle DM. Vaccines for herpes simplex virus infections. Curr Opin Investig Drugs. 2006;7(2):136−141. | |
Whitley R. Herpes simplex viruses. In: Knipe DM, Howley PM, Griffen DE, et al. Field’s Virology. 3rd ed. Philadelphia: Lippincott-Raven Publishers; 2006. | |
Liu K, Jiang D, Zhang L, et al. Identification of B- and T-cell epitopes from glycoprotein B of herpes simplex virus 2 and evaluation of their immunogenicity and protection efficacy. Vaccine. 2012;30(19):3034–3041. | |
Kim M, Taylor J, Sidney J, et al. Immunodominant epitopes in herpes simplex virus type 2 glycoprotein D are recognized by CD4 lymphocytes from both HSV-1 and HSV-2 seropositive subjects. J Immunol. 2008;181(9):6604−3615. | |
Lankford CS, Frucht DM. A unique role for IL-23 in promoting cellular immunity. J Leukoc Biol. 2003;73(1):49–56. | |
Schmitt L, Dietrich C, Tampe R. Synthesis and characterization of chelator-lipids for reversible immobilization of engineered proteins at self-assembled lipid interfaces. J Am Chem Soc. 1994;116(19):8485−8491. | |
Chan WC, White PD. Fmoc Solid Phase Peptide Synthesis: A Practical Approach. Oxford: Oxford University Press; 2000. | |
Accardo A, Mansi R, Morisco A, et al. Peptide modified nanocarriers for selective targeting of bombesin receptors. Mol Biosys. 2010;6(5):878−887. | |
Accardo A, Tesauro D, Mangiapia G, Pedone C, Morelli G. Nanostructures by self-assembling peptide amphiphile as potential selective drug carriers. Biopolymers. 2007;88(2):115−121. | |
Morisco A, Accardo A, Gianolio E, et al. Micelles derivatized with octreotide as potential target-selective contrast agents in MRI. J Pept Sci. 2009;15(3):242−250. | |
Birdi KS, Singh HN, Dalsager SU. Interaction of ionic micelles with the hydrophobic fluorescent-probe 1-anilino-8-naphthalenesulfonate. J Phys Chem. 1979;83(21):2733−2737. | |
De Vendittis E, Palumbo G, Parlato G, Bocchini V. A fluorimetric method for the estimation of the critical micelle concentration of surfactants. Anal Biochem. 1981;115(2):278–286. | |
Galdiero M, Tortora A, Damiano N, Vitiello M, Longanella A, Galdiero E. Induction of cytokine mRNA expression in U937 cells by Salmonella typhimurium porins is regulated by different phosphorylation pathways. Med Microbiol Immunol. 2005;194(1–2):13–23. | |
Taylor MF, Paulauskis JD, Weller DD, Kobzik L. In vitro efficacy of morpholino-modified antisense oligomers directed against tumor necrosis factor-alpha mRNA. J Biol Chem. 1996;271(29):17445−17452. | |
Yin ET, Galanos C, Kinsky S, et al. Picogram-sensitive assay for endotoxin: gelation of Limulus polyphemus blood cell lysate induced by purified lipopolysaccharides and lipid A from gram-negative bacteria. Biochim Biophys Acta. 1972;261(1):284−289. | |
Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods. 1983;65(1–2):55–63. | |
Hudecz F, Hilbert á, Mezö G, et al. Epitope mapping of the 273–284 region of HSV glycoprotein D by synthetic branched polypeptide carrier conjugates. Pept Res. 1993;6(5):263−271. | |
Bettahi I, Nesburn AB, Yoon S, et al. Protective immunity against ocular herpes infection and disease induced by highly immunogenic self-adjuvanting glycoprotein D lipopeptide vaccines. Invest Ophthalmol Vis Sci. 2007;48(10):4643–4653. | |
Nesburn AB, Ramos TV, Zhu X, Asgarzadeh H, Nguyen V, BenMohamed L. Local and systemic B cell and Th1 responses induced following ocular mucosal delivery of multiple epitopes of herpes simplex virus type 1 glycoprotein D together with cytosine-phosphate-guanine adjuvant. Vaccine. 2005;23(7):873–883. | |
Stanberry LR, Spruance SL, Cunningham AL, et al; GlaxoSmithKline Herpes Vaccine Efficacy Study Group. Glycoprotein D adjuvant vaccine to prevent gential herpes. N Engl J Med. 2002;347(21):1652−1661. | |
Koelle DM, Corey L. Recent progress in herpes simplex virus immunobiology and vaccine research. Clin Microbiol Rev. 2003;16(1):96–113. | |
Wallace ME, Keating R, Heath WR, Carbone FR. The cytotoxic T-cell response to herpes simplex virus type 1 infection of C57BL/6 mice is almost entirely directed against a single immunodominant determinant. J Virol. 1999;73(9):7619−7623. | |
Behanna HA, Donners JJ, Gordon AC, Stupp SI. Coassembly of amphiphiles with opposite peptide polarities into nanofibers. J Am Chem Soc. 2005;127(4):1193–1200. | |
Romagnani S. Th1/Th2 cells. Inflamm Bowel Dis. 1999;5(4):285–294. | |
Oppmann B, Lesley R, Blom B, et al. Novel p19 protein engages IL-12p40 to form a cytokine, IL-23, with biological activities similar as well as distinct from IL-12. Immunity. 2000;13(5):715–725. | |
Pirhonen J, Matikainen S, Julkunen I. Regulation of virus-induced IL-12 and IL-23 expression in human macrophages. J Immunol. 2002;169(10):5673–5678. | |
Boniface K, Blom B, Liu YJ, de Waal Malefyt R. From interleukin-23 to T-helper 17 cells: human T-helper cell differentiation revisited. Immunol Rev. 2008;226:132−146. | |
Veldhoen M, Hocking RJ, Atkins CJ, Locksley RM, Stockinger B. TGFβ in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity. 2006;24(2):179−189. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.