Back to Journals » Clinical, Cosmetic and Investigational Dermatology » Volume 15
Role of HMGB1 in Vitiligo: Current Perceptions and Future Perspectives
Authors Wei G, Pan Y, Wang J, Xiong X ![]() , He Y, Xu J
, He Y, Xu J ![]()
Received 11 July 2022
Accepted for publication 23 September 2022
Published 13 October 2022 Volume 2022:15 Pages 2177—2186
DOI https://doi.org/10.2147/CCID.S381432
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Jeffrey Weinberg
Guangmin Wei,1 Yinghao Pan,2 Jingying Wang,2 Xia Xiong,2 Yuanmin He,2 Jixiang Xu2
1Department of Dermatology, Medical Center Hospital of Qionglai City, Qionglai, Sichuan, People’s Republic of China; 2Department of Dermatology, The Affiliated Hospital of Southwest Medical University, Luzhou, Sichuan, People’s Republic of China
Correspondence: Jixiang Xu, Department of Dermatology, The Affiliated Hospital of Southwest Medical University, No. 25 of Taiping Road, Luzhou, Sichuan, 646000, People’s Republic of China, Email [email protected]
Abstract: Vitiligo is a chronic depigmenting disorder of the skin and mucosa caused by the destruction of epidermal melanocytes. Although the exact mechanism has not been elucidated, studies have shown that oxidative stress plays an important role in the pathogenesis of vitiligo. High mobility group box protein B1 (HMGB1) is a major nonhistone protein and an extracellular proinflammatory or chemotactic molecule that is actively secreted or passively released by necrotic cells. Recent data showed that HMGB1 is overexpressed in both blood and lesional specimens from vitiligo patients. Moreover, oxidative stress triggers the release of HMGB1 from keratinocytes and melanocytes, indicating that HMGB1 may participate in the pathological process of vitiligo. Overall, this review mainly focuses on the role of HMGB1 in the potential mechanisms underlying vitiligo depigmentation under oxidative stress. In this review, we hope to provide new insights into vitiligo pathogenesis and treatment strategies.
Keywords: vitiligo, HMGB1, oxidative stress, melanocytes
Introduction
Vitiligo is a cutaneous depigmentation disorder characterized by the selective loss of melanocytes. Vitiligo affects 0.5% to 2% of the worldwide population, and its incidence continues to increase.1 Since vitiligo usually affects children and teenagers (with almost 50% of the patients developing the disease before the age of 20) and the lesions of chalk-white macules and patches may involve exposed parts such as the face, vitiligo negatively influences patients’ quality of life by decreasing their self-esteem and causing significant psychological distress.2 Although multiple mechanisms, including genetic predisposition, oxidative stress, autoimmune and inflammatory responses and melanocyte detachment, have been implicated in the loss of functional melanocytes in vitiligo,3 the overall pathogenesis of vitiligo is still far from clear, thus rendering the development of effective treatments difficult.4
High mobility group box protein B1 (HMGB1) normally localizes in the nucleus to maintain genomic stabilization and regulate gene transcription.5 Importantly, HMGB1 can be released extracellularly upon exposure to stressful factors such as oxidative stress and function as a damage-associated molecular pattern (DAMP) protein with strong proinflammatory effects. Recent data showed that HMGB1 is overexpressed in both blood and lesional specimens from vitiligo patients. Moreover, oxidative stress triggers the release of HMGB1 from keratinocytes and melanocytes, indicating that HMGB1 may participate and play a crucial role in the pathological process of vitiligo.6 Following this, this review aims to summarize the pathogenic function of HMGB1 in the development of vitiligo, especially in regard to the molecular mechanisms associated with oxidative stress, and to discuss the potential of HMGB1 as a therapeutic target for vitiligo (Figure 1).
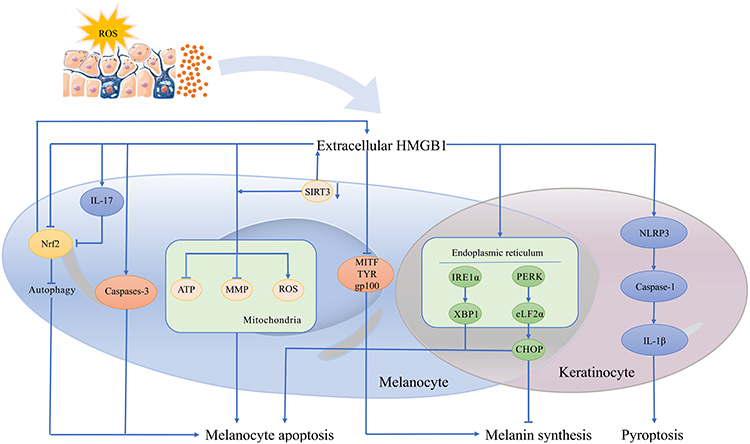 |
Figure 1 The possible pathogenic function of HMGB1 in the development of vitiligo associated with oxidative stress. |
Oxidative Stress-Mediated Destruction of Melanocytes in Vitiligo
Skin is one of the organs that suffers most from oxidative stress due to its direct contact with external environment.7 Among the different types of cutaneous cells, melanocytes especially contain massive amounts of reactive oxygen species (ROS), which are a major source of oxidative stress, because the production of melanin is accompanied by the active production of ROS and melanin itself absorbs ultraviolet that triggers oxidative stress.8 Although ROS act as important secondary messengers, high concentrations of ROS induce the death of melanocytes in various ways and thus lead to the formation of vitiligo.
It is widely known that ROS directly induce cell death by triggering caspase-dependent apoptosis. However, ROS causes melanocyte damage in vitiligo via multiple mechanisms. First, ROS undermine various macromolecules, including lipids, proteins, nucleic acids and carbohydrates, in melanocytes,9 which accelerates the loss of functional melanocytes in vitiligo. Furthermore, chronic exposure to oxidative stress exposes melanocytes to persistent DNA damage, thus leading to senescence and the irreversible stagnation of melanocyte proliferation. Additionally, ROS compromise the function of remaining melanocytes in vitiligo by interfering with the metabolism and differentiation of melanocytes.10
Autophagy is involved in the response to oxidative stress and plays an essential role in protecting cells against oxidative damage. Under oxidative stress, ROS produced in melanocytes prompt autophagy and further inactivate the antioxidant nuclear factor erythroid 2–related factor 2 (Nrf2) pathway, which removes toxic molecules and maintains the redox homeostasis of melanocytes.11 However, the autophagy pathway is impaired in melanocytes in vitiligo, which results in inadequate activation of the Nrf2 pathway in melanocytes exposed to ROS and finally causes premature senescence, decreased proliferation and impaired pigment synthesis in melanocytes.12
Mitochondria are not only the origins of ROS but also victims of oxidative stress, and mitochondrial impairment undoubtedly affects melanocyte survival. ROS-related mitochondrial dysfunction is characterized by mitochondrial outer membrane permeabilization, loss of mitochondrial membrane potential and subsequent melanocyte apoptosis.13 The abnormal lipid composition and compromised intactness of the respiratory chain in melanocyte mitochondria result from ROS-induced mitochondrial impairment.14 Intriguingly, defects in respiratory chain complexes, especially complex I,15 caused by oxidative stress during assembly further induce ROS production, thus forming a vicious feedback loop to destroy melanocytes.
Oxidative stress is a vital factor underlying endoplasmic reticulum (ER) dysfunction in vitiligo. Numerous studies have confirmed that ER subversion by ROS induces melanocyte apoptosis. ER stress is characterized by aberrantly folded proteins in the ER lumen, which elicit the unfolded protein response (UPR),16 and excessive and long-term UPR causes cell death. It has been confirmed that homocysteine (Hcy) induces melanocyte apoptosis by activating ROS-dependent ER stress and UPR activation in vitiligo.17 Moreover, the ROS-induced increase in the expression of chemokine (C-X-C motif) ligand 16 (CXCL16), a chemokine crucial for the migration of cytotoxic T cells (CTLs) in vitiligo, is also due to the activation of the UPR.18
It is worth noting that oxidative stress-mediated melanocyte destruction in vitiligo is affected by the functions of other cutaneous cells. Epidermal keratinocytes secrete cytokines and chemokines under oxidative stress to activate and recruit autoreactive T cells that undermine melanocytes. ROS elevate the Ca2+ levels in vitiligo keratinocytes and then induce the increased release of acetylcholine and the allosteric inhibition of thioredoxin reductase, which eventually leads to the destruction of melanocyte defense and the inhibition of melanogenesis. Langerhans cells (LCs) are activated by inducible heat shock protein 70 (HSP70i), which induces melanocytes under oxidative stress and acts as a warning signal to drive the autoimmune response in vitiligo.19 Dendritic cells (DCs) sense membranal calreticulin (CRT), which is redistributed from the ER lumen to the surface of melanocytes under oxidative stress and then activates a downstream immune response in vitiligo. Moreover, the ROS-induced overexpression of CRT increases the sensitivity of melanocytes to DC-induced immunogenic apoptosis.20
General Profile of HMGB1
HMGB1 has been discovered for four decades and is one of the most abundant and highly conserved proteins in eukaryotic cells. HMGB1 is expressed in almost all eukaryotic cells and is extremely conserved, displaying a homology of 99% between rodents and humans.21 Human HMGB1 consists of 215 amino acid residues and is composed of two DNA-binding domains (A box and B box) and a C-terminal acid tail. HMGB1 is normally distributed in the nucleus but can translocate into the cytoplasm under certain stresses and can even be secreted into the extracellular space in an active or passive manner.22 In the extracellular environment, HMGB1 serves as a proinflammatory DAMP and interacts with downstream receptors, including advanced glycation end products (RAGE), toll-like receptor 2 (TLR2), toll-like receptor 4 (TLR4), toll-like receptor 9 (TLR9), integrin, CD24, chemokine receptor 4 (CXCR4) and N-methyl-D-aspartate receptor.23
HMGB1 is expressed in all types of cells, including keratinocytes and melanocytes, and overexpression of extracellular HMGB1 has been observed in several inflammatory skin conditions, such as psoriasis,24 atopic dermatitis,25 lichen planus,26 cutaneous T-cell lymphoma,27 cutaneous vasculitis,28 systemic lupus,29 Henoch-Schönlein purpura30 and systemic sclerosis.31 Previous studies have reported higher levels of HMGB1 in lesions and plasma samples from patients with vitiligo in the active progressive phase compared with those in the slow progressive phase and healthy people.6,32 Further mechanistic studies indicate that HMGB1 is not only directly involved in ROS-triggered melanocyte apoptosis but also closely associated with the immune disorder caused by oxidative stress in vitiligo.32
Role of HMGB1 in the Oxidative Stress-Mediated Development of Vitiligo
HMGB1 Directly Induces Melanocyte Apoptosis
A previous study claimed that stimulation with ROS or ultraviolet B (UVB) in vitro significantly increased the release of HMGB1 from keratinocytes, which inhibited the expression of melanogenesis-related molecules such as microphthalmia-associated transcription factor (MITF), tyrosinase-related proteins and the gp100 protein in a paracrine manner and finally activate caspase-3 to trigger melanocyte apoptosis.6 More recently, some studies have reported that HMGB1 is also released from lesional melanocytes under oxidative stress in vitiligo and further causes the apoptosis of melanocytes themselves in an autocrine manner.33
HMGB1 is Responsible for Autophagy Damage in Melanocytes Under Oxidative Stress in Vitiligo
As described previously, autophagy defects play a vital role in ROS-induced oxidative damage to melanocytes in vitiligo.34 Defects in autophagy-related protein 7 (ATG7)-dependent autophagy under oxidative stress are accompanied by a decreased growth rate, decreased melanin content, dysfunction of melanogenesis, premature senescence and increased apoptosis of melanocytes, indicating that ATG7-dependent autophagy plays an important role in maintaining the normal biological functions of human melanocytes under oxidative stress.35 Moreover, ATG7-dependent autophagy participates in the dynamic redox balance by regulating the production of ROS as well as the activity of the antioxidant Nrf2 signaling pathway and several antioxidant enzymes.35 Inhibiting the release of HMGB1 can reduce the number of autolysosomes and autophagic flux in human and mouse cells, and HMGB1 acts as an important autophagic sensor in the cellular response to oxidative stress.36 For instance, Petrovićetal found that HMGB1 in hepatocytes is transferred from the nucleus to the extracellular space upon the induction of liver autophagy defects under oxidative stress. The molecular pathway connecting autophagy deficiency and the release of HMGB1 has been shown to be mediated by Nrf2 and inflammasomes. The activation of Nrf2 subsequently leads to the upregulation and activation of caspase-1 and caspase-11, which results in the cleavage of gasdermin D and the formation of gasdermin D pores for the release of HMGB1 from hepatocytes.37 Another study showed that the serum level of HMGB1 in liver-specific Atg7 knockout (L-ATG7KO) mice was increased, and increased HMGB1 expression was observed in ATG7-deficient renal proximal tubules, indicating that the release of HMGB1 is a general event in autophagy-deficient cells regardless of tissue specificity.38 Taken together, these data suggest that dysregulation of ATG7-dependent autophagy could increase the release of HMGB1, which might promote the injury and apoptosis of cells, such as melanocytes, in vitiligo.
The nuclear factor erythroid 2–related factor 2-antioxidant response element (Nrf2-ARE) pathway protects cells from various pro-oxidative stimuli by inducing autophagy.39 Nrf2-p62 is an important mechanism protecting melanocytes from oxidative damage induced by hydrogen peroxide (H2O2).34 During autophagy, intracellular proteins and organelles can bind to autophagosomes by the adaptor protein p62. At the same time, p62/SQSTM1 is the target gene of Nrf2/Keap1 and produces a positive feedback loop between autophagy and Nrf2/Kelch-like ECH-associated protein 1 (Keap1).40 Impairment of the Nrf2/Keap1 pathway suppresses autophagy in melanocytes extracted from perilesional skin from subjects with vitiligo,34 thereby disrupting redox homeostasis and causing premature senescence, decreased proliferation and melanogenesis in melanocytes.35 ROS trigger the release of HMGB1 release from melanocytes in an autocrine manner, leading to the suppression of Nrf2 and downstream antioxidant genes.33 Moreover, knockout of the HMGB1 gene suppresses the expression of the autophagosome marker light chain 3 (LC3) and enhances p62 expression in melanocytes, and the competitive interaction between p62 and Keap1 could promote the activation of Nrf2.33 Therefore, HMGB1 may participate in the ROS-induced destruction of melanocytes in vitiligo by inhibiting both the activation of the Nrf2-p62 pathway and autophagy.
IL-17 is a proinflammatory cytokine that accelerates autophagy. On the one hand, IL-17 induces Bcl-2 phosphorylation and its dissociation from Beclin-1, which activates autophagy.41 On the other hand, interleukin-17 (IL-17) promotes the expression of a number of autophagy-related proteins, such as Beclin-1, LC3-II and ATG5, and enhances the level of autophagy by inhibiting the PI3K signaling pathway.42 Intriguingly, ROS trigger IL-17-mediated autophagic cell apoptosis as a downstream event of autophagy, and IL-17-mediated cell apoptosis is decreased by the inhibition of autophagy via pretreatment with N-acetyl-L-cysteine (NAC).42 The levels of HMGB1 and IL-17 are reported to be increased during liver I/R injury, and HMGB1 can stimulate the production of IL-17 by γδT cells in a TLR4-dependent manner.43 In addition, HMGB1 promotes IL-17 expression, triggers myocardial cell H/R injury and aggravates myocardial apoptosis and autophagy.44 Given that IL-17 is also increased in vitiligo, HMGB1 may cause the autophagic cell apoptosis of melanocytes with the help of IL-17 in subjects with vitiligo.
Role of HMGB1 in ER Stress Induction in Vitiligo
The change in ER homeostasis leads to ER stress, which is characterized by the accumulation of unfolded or misfolded proteins in the ER lumen, and ER stress induces the UPR.16 Compared with healthy melanocytes, melanocytes in vitiligo are reported to have more dilated ERs.45 In addition, melanocytes from vitiligo patients have intrinsic defects, which reduce their ability to manage cellular stress, leading to an increase in ROS production and induction of the UPR, which in turn activates congenital inflammation.45,46 X-box-binding protein 1 (XBP-1) is an important regulator of ER stress.47 Genome-wide association studies have shown that XBP1 polymorphisms are associated with an increased risk of suffering from vitiligo. XBP1 regulates plasma cell differentiation and mediates the inflammatory response to ER stress by activating the expression of major histocompatibility complex II (MHC-II) genes.48 In addition, XBP1 increases the expression of immune mediators such as IL-6 and IL-8 as a result of ER stress in melanocytes;49 meanwhile, recent study revealed that XBP1 is involved in the differentiation of Th17 cells and elevated the level of IL-17A in vitiligo.50 Activation of the protein kinase R-like endoplasmic reticulum kinase-eukaryotic translation initiation factor 2α (PERK-eIF2α) and inositol-requiring enzyme 1 alpha-X-box-binding protein 1 (IRE1α-XBP1) pathways is of great importance for the production of CXCL16 induced by H2O2 in keratinocytes.18 Previous studies suggest that HMGB1 promotes the secretion of CXCL16 from keratinocytes by binding to RAGE and activating the endoplasmic reticulum kinase (ERK) and nuclear factor-κB (NF-κB) signaling pathways.32 The increased expression and release of CXCL16 from keratinocytes in response to oxidative stress play a critical role in skin infiltration of CD8+ T cells and dendritic cells in vitiligo.32 As a key regulatory molecule of ER stress, XBP-1 plays an important role in the HMGB1-induced maturation and activation of DCs and upregulation of RAGE on DC surfaces.47 HMGB1 may play an important role in CXCL16 expression in stressed keratinocytes by inducing endoplasmic reticulum stress in vitiligo.
An increased level of Hcy, an amino acid metabolite, has been identified as a circulatory marker of oxidative stress and considered a risk factor for vitiligo.51 Hcy can induce melanocyte apoptosis by activating the ROS and ER stress-associated PERK-eIF2α-CHOP (C/EBP homologous protein) pathways and thereby inhibit melanogenesis.17 HMGB1 induces ER stress in human endothelial progenitor cells (EPCs) and triggers EPC apoptosis via RAGE-mediated activation of the PERK/eIF2α pathway.51 In addition, HMGB1 participates in the UPR system and induces the degradation of proteins and apoptosis associated with Huntington’s disease (HD).52 HMGB1 induces ER stress in endothelial cells in a dose-dependent manner and promotes the apoptosis of macrophages induced by oxLDL through the ERS/CHOP pathway.53 Therefore, HMGB1 may be responsible for the formation of ER stress in vitiligo and needs to be studied further.
HMGB1 Intensifies Oxidative Stress-Induced Mitochondrial Dysfunction, Which Potentiates Vitiligo
Studies have confirmed the ultrastructural disorder of mitochondria in melanocytes in vitiligo,54 especially the disappearance of cristae, which may hamper the assembly of respiratory chain complexes and enhance the production of ROS.55 Therefore, mitochondrial dysfunction is closely related to the vulnerability of melanocytes to oxidative stress. Sirtuin 3 (SIRT3) has been described as a versatile molecule in regulating mitochondrial function and mitochondrial dynamics.56 Excessive oxidative stress could significantly dampen SIRT3 activity and expression,57 and SIRT3 deficiency leads to melanocyte apoptosis by inducing severe mitochondrial dysfunction and the release of cytochrome c into the cytoplasm.58 In addition, excessive carbonylation of SIRT3 dampens PGC1α activation and transcriptional function, leading to decreased expression and functional deficiency of SIRT3 in vitiligo.59 Overexpression of SIRT3 reduces the release of layered double hydroxide (LDH) and HMGB1 and alleviates mitochondrial apoptosis.60 Furthermore, SIRT3 plays a key role in regulating mitochondrial bioenergy, ROS formation and proinflammatory responses in macrophages in acute lung injury (ALI), another disease characterized by exuberant proinflammatory responses and mitochondrial dysfunction. SIRT3 stimulated by vinblastine reduces the severity of ALI induced by lipopolysaccharide (LPS), as shown by decreased neutrophil influx, pulmonary permeability, HMGB1 release and nuclear-binding domain-like receptor 3 (NLRP3) activation.61 Recent studies indicate that the upregulation of SIRT3 expression inhibits the activation of NF-κB, HMGB1, NLRP3 and caspase-1.61 High levels of SIRT3 and superoxide dismutase 2 (SOD2) induced by mesenchymal stem cells (MSCs) suppress the NF-κB/HMGB1/NLRP3/caspase-1 signaling pathway, thereby inhibiting the apoptosis of lung tissue cells. Collectively, these data suggest that SIRT3 deficiency mediates melanocyte apoptosis by aggravating mitochondrial damage in vitiligo, which may be related to suppression of the NF-kB/HMGB1/NLRP3/Caspase-1 pathway.
HMGB1 is closely related to oxidative stress and mitochondrial apoptosis.62 Inhibition of HMGB1 maintains the balance of mitochondrial membrane potential and mitochondrial fusion/division, decreases the levels of mitochondrial ROS and reduces the apoptosis of bone marrow mesenchymal stem cells.63 Mechanical stretching induces excessive release of HMGB1 from mouse pulmonary vascular endothelial cells, which causes mitochondrial oxidative damage and leads to increased mitochondrial ROS generation, decreased mitochondrial membrane potential, decreased intracellular ATP levels and impaired respiratory chain complex activities.64 In addition, HMGB1 leads to increased mitochondrial fission through dynamin-related protein 1 (DRP1) phosphorylation.65 Therefore, HMGB1 may be closely related to mitochondrial dysfunction induced by oxidative stress in melanocytes in vitiligo.
HMGB1 Promotes Keratinocyte-Mediated Melanocyte Loss in Vitiligo
HMGB1 can be released from keratinocytes after exposure to ROS or UVB.66 Constant HMGB1 stimulation of keratinocytes can impair natural barrier formation, and HMGB1 heavily impacts the gene products of pivotal skin barrier differentiation genes, such as filaggrin, involucrin and loricrin. Consequently, HMGB1 has deleterious effects on skin barrier protein expression and epidermal growth.67
Keratinocytes are a vital nexus in the pathomechanism of vitiligo, as they mediate oxidative stress, secrete cytokines to recruit autoreactive T cells and interfere with signal transduction in melanocytes, consequently inducing melanocyte destruction.68 The ROS‐induced release of ATP from keratinocytes might not only stimulate neighboring keratinocytes to generate CXCL9 to chemoattract CD8+ T cells but also trigger Caspase‐3‐mediated melanocyte apoptosis. Treatment with monosodium urate (MSU) crystals induces the release of ATP and HMGB1 from peritoneal macrophages and THP-1 cells. Upon entry of MSU, the cells release ATP, which acts in an autocrine manner on the P2X7 receptor. The cells then release HMGB1 in a manner dependent on Panx-1 and the P2X7 receptor. This DAMP mediates cellular inflammation by activating transcription factors via TLR-4 activation. The activation of the P2X7 receptor induced by extracellular ATP induces the secretion of proinflammatory molecules such as IL-1β and the release of HMGB1 in hepatocytes, which further enhances the inflammatory response mediated by IL-1β.69 ATP also induces the nuclear and cytoplasmic transport and release of HMGB1 in vascular smooth muscle cells. The serum level of IL-1β in patients with vitiligo is increased and related to the activity and severity of the disease. Taken together, these results indicate the involvement of HMGB1 in ATP-mediated melanocyte apoptosis under oxidative stress. Studies have confirmed that UVB irradiation promotes the release of HMGB1 by keratinocytes, and NAC significantly inhibits the release of HMGB1 from UVB-irradiated keratinocytes. Application of recombinant HMGB1 to keratinocytes was shown to reduce the expression of E-cadherin,66 ultimately weakening melanocyte–keratinocyte adhesion and enhancing melanocyte detachment.70 Furthermore, detachment disrupts fibronectin adhesion‐mediated apoptosis suppression, which intensifies melanocyte apoptosis and necrosis.
In keratinocytes, transient receptor potential melastatin 2 (TRPM2) activates the NLRP3 inflammasome by inducing calcium influx into cells and mitochondria, and the NLRP3 inflammasome enhances the functions of CD8+ and CD4+ T cells in vitiligo through the IL-1β/IL-1R pathway.71 In a pathophysiological study of Kawasaki disease, researchers proved that HMGB1 released by immune cells promotes the HMGB1/RAGE/cathepsin B signaling pathway in endothelial cells and subsequently activates endothelial cell pyroptosis via the NLRP3 inflammasome.72 NLRP3 inflammasome activation facilitates the transport of HMGB1 from the nucleus to the cytoplasm and its further secretion from macrophage. The release of HMGB1 from macrophages induced by LPS is dependent on the activation of the NLRP3 inflammasome. Chen et al reported that an adipokine (visfatin) could activate the NLRP3 inflammasome in endothelial cells and promote the production of HMGB1, damaging the intercellular connections in the vascular endothelium.60 HBx increases the secretion of HMGB1 from hepatocytes under H2O2 stimulation, while NAC decreases the secretion of HMGB1 by inhibiting the activation of the NLRP3 inflammasome.73 Therefore, HMGB1 could play an important role in melanocyte destruction mediated by the NLRP3/IL-1β pathway in vitiligo.
To conclude, oxidative stress triggers autocrine-mediated HMGB1 translocation and release by keratinocytes and may promote keratinocyte-potentiated melanocyte loss in vitiligo.
Conclusion and Future Perspectives
We summarized the role of HMGB1 in vitiligo, focusing on its interaction with oxidative stress in the pathogenesis of the disease. Generally, HMGB1 plays a role in not only the direct damage of oxidative stress on melanocytes but also further triggers the autoimmune T-cell response in vitiligo. Given that HMGB1 levels are increased in the sera of patients with vitiligo, especially those in the active progressive stage, the serum HMGB1 level could be a promising biomarker for distinguishing vitiligo from other noninflammatory depigmentation diseases (such as nevus anemicus and amelanotic nevus) and for monitoring vitiligo activity.
The pathogenic role of HMGB1 in the occurrence and development of vitiligo may not be limited to oxidative stress-associated mechanisms. T regulatory cells (Tregs) constitute a key mechanism in maintaining peripheral self-tolerance through the control of autoreactive lymphocyte activation and the prevention of harmful effects of such activation.74 Local Treg deficiency has been observed in the skin of subjects with vitiligo within lesional, nonlesional and perilesional sections, triggering the destruction of melanocytes and the development of vitiligo, and supplementing peripheral skin Tregs can provide protection against depigmentation.75,76 Extracellular HMGB1 exacerbates autoimmune progression and the recurrence of type 1 diabetes by impairing Treg stability and function via regulating RAGE/TLR4-dependent PI3K–Akt–mTOR signaling.77 HMGB1 may play a critical role in immunological self-tolerance breakdown, which leads to aberrant immune responses to melanocytes.
Autoreactive CD8+ T cells are the major suspects considered to be responsible for the destruction of melanocytes. MSCs suppress the proliferation of CD8+ T cells via the natural killer group 2D (NKG2D) pathway to induce T-cell apoptosis. MSC therapy has also been shown to stimulate the mobilization of healthy melanocytes, leading to the successful repigmentation of skin lesions in patients with vitiligo.78 HMGB1 not only induces the RAGE-dependent recruitment of monocytes and immature dendritic cells, directing cellular responses toward inflammation and an immune response, but also inhibits the hepatocyte growth factor (HGF)-driven migration of MSCs, thereby possibly preventing their immunosuppressive influence at sites of tissue damage when both apoptotic and necrotic cell deaths coincide. Thus, HMGB1 may also be involved in different stages of the autoimmune response to vitiligo, from recognizing autoantigens to adapting to and regulating the immune response.
HMGB1 may be the forefront and an important target for the treatment of vitiligo, bringing new hope for the treatment of vitiligo.
Studies have found that inhibiting the secretion and expression of HMGB1 has protective effects on diabetes and stroke. Blocking the expression, release and translocation of HMGB1 can downregulate the neuroinflammatory response and improve the permeability of the blood–brain barrier, thus alleviating the early brain injury (EBI) induced by subarachnoid hemorrhage (SAH).79 Systemic application of an anti-HMGB1 monoclonal antibody was shown to have a neuroprotective effect on elderly rats undergoing major surgery to prevent memory impairment and anxiety after the operation.80 HMGB1 antagonists and anti-RAGE monoclonal antibodies have also made good progress in the treatment of sepsis. Recent studies have shown that blockade of HMGB1 attenuates disease severity in animal models of skin diseases.81 Meanwhile, the number of HMGB1-oriented pharmacological agents is rapidly expanding, and many of these agents are currently in advanced stages of preclinical and clinical trials and are promising experimental therapeutics. Moreover, these agents have acceptable toxicity and topical application prospects.82 Based on the important role of HMGB1 in the pathogenesis of vitiligo, drugs targeting HMGB1 for the treatment of vitiligo need to be identified. Investigating the various effects of HMGB1 on melanocytes can be extremely valuable for enhancing our understanding of vitiligo and developing new therapeutics.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Kruger C, Schallreuter KU. A review of the worldwide prevalence of vitiligo in children/adolescents and adults. Int J Dermatol. 2012;51(10):1206–1212. doi:10.1111/j.1365-4632.2011.05377.x
2. Hamidizadeh N, Ranjbar S, Ghanizadeh A, Parvizi MM, Jafari P, Handjani F. Evaluating prevalence of depression, anxiety and hopelessness in patients with vitiligo on an Iranian population. Health Qual Life Outcomes. 2020;18(1):20. doi:10.1186/s12955-020-1278-7
3. Bergqvist C, Ezzedine K. Vitiligo: a review. Dermatology. 2020;236(6):571–592. doi:10.1159/000506103
4. Lotti T, Gianfaldoni S, Valle Y, Rovesti M, Feliciano C, Satolli F. Controversial issues in vitiligo patients: a review of old and recent treatments. Dermatol Ther. 2019;32(1):e12745. doi:10.1111/dth.12745
5. Agalave N, Svensson C. Extracellular high-mobility group box 1 protein (HMGB1) as a mediator of persistent pain. Mol Med. 2015;20:569–578. doi:10.2119/molmed.2014.00176
6. Kim J, Lee E, Seo J, Oh S. Impact of high-mobility group box 1 on melanocytic survival and its involvement in the pathogenesis of vitiligo. Br J Dermatol. 2017;176(6):1558–1568. doi:10.1111/bjd.15151
7. Marrot L. Pollution and sun exposure: a deleterious synergy. mechanisms and opportunities for skin protection. Curr Med Chem. 2018;25(40):5469–5486. doi:10.2174/0929867324666170918123907
8. Denat L, Kadekaro A, Marrot L, Leachman S, Abdel-Malek Z. Melanocytes as instigators and victims of oxidative stress. J Invest Dermatol. 2014;134(6):1512–1518. doi:10.1038/jid.2014.65
9. Mitra S, De Sarkar S, Pradhan A, et al. Levels of oxidative damage and proinflammatory cytokines are enhanced in patients with active vitiligo. Free Radic Res. 2017;51:986–994. doi:10.1080/10715762.2017.1402303
10. Guerra L, Dellambra E, Brescia S, Raskovic D. Vitiligo: pathogenetic hypotheses and targets for current therapies. Curr Drug Metab. 2010;11(5):451–467. doi:10.2174/138920010791526105
11. Ma J, Li S, Zhu L, et al. Baicalein protects human vitiligo melanocytes from oxidative stress through activation of NF-E2-related factor2 (Nrf2) signaling pathway. Free Radic Biol Med. 2018;129:492–503. doi:10.1016/j.freeradbiomed.2018.10.421
12. Qiao Z, Wang X, Xiang L, Zhang C. Dysfunction of autophagy: a possible mechanism involved in the pathogenesis of vitiligo by breaking the redox balance of melanocytes. Oxid Med Cell Longev. 2016;2016:3401570. doi:10.1155/2016/3401570
13. Wang Q, Huang L, Yue J. Oxidative stress activates the TRPM2-Ca-CaMKII-ROS signaling loop to induce cell death in cancer cells. Biochim Biophys Acta Mol Cell Res. 2017;1864(6):957–967. doi:10.1016/j.bbamcr.2016.12.014
14. Dell’ Anna M, Ottaviani M, Bellei B, et al. Membrane lipid defects are responsible for the generation of reactive oxygen species in peripheral blood mononuclear cells from vitiligo patients. J Cell Physiol. 2010;223(1):187–193. doi:10.1002/jcp.22027
15. Sahoo A, Lee B, Boniface K, et al. MicroRNA-211 regulates oxidative phosphorylation and energy metabolism in human vitiligo. J Invest Dermatol. 2017;137(9):1965–1974. doi:10.1016/j.jid.2017.04.025
16. Park K, Lee S, Shin K, Uchida Y. Insights into the role of endoplasmic reticulum stress in skin function and associated diseases. FEBS J. 2019;286(2):413–425. doi:10.1111/febs.14739
17. Chen J, Zhuang T, Chen J, et al. Homocysteine induces melanocytes apoptosis via PERK-eIF2α-CHOP pathway in vitiligo. Clin Sci. 2020;134(10):1127–1141. doi:10.1042/CS20200218
18. Li S, Zhu G, Yang Y, et al. Oxidative stress drives CD8 T-cell skin trafficking in patients with vitiligo through CXCL16 upregulation by activating the unfolded protein response in keratinocytes. J Allergy Clin Immunol. 2017;140(1):177–189.e9. doi:10.1016/j.jaci.2016.10.013
19. Mosenson J, Eby J, Hernandez C, Le Poole I. A central role for inducible heat-shock protein 70 in autoimmune vitiligo. Exp Dermatol. 2013;22(9):566–569. doi:10.1111/exd.12183
20. Zhang Y, Liu L, Jin L, et al. Oxidative stress-induced calreticulin expression and translocation: new insights into the destruction of melanocytes. J Invest Dermatol. 2014;134(1):183–191. doi:10.1038/jid.2013.268
21. Sessa L, Bianchi M. The evolution of High Mobility Group Box (HMGB) chromatin proteins in multicellular animals. Gene. 2007;387:133–140. doi:10.1016/j.gene.2006.08.034
22. Yang H, Wang H, Andersson U. Targeting inflammation driven by HMGB1. Front Immunol. 2020;11:484. doi:10.3389/fimmu.2020.00484
23. Zhao Z, Hu Z, Zeng R, Yao Y. HMGB1 in kidney diseases. Life Sci. 2020;259:118203. doi:10.1016/j.lfs.2020.118203
24. Zhang W, Guo S, Li B, et al. Proinflammatory effect of high-mobility group protein B1 on keratinocytes: an autocrine mechanism underlying psoriasis development. J Pathol. 2017;241(3):392–404. doi:10.1002/path.4848
25. Wang Y, Zhang Y, Peng G, Han X. Glycyrrhizin ameliorates atopic dermatitis-like symptoms through inhibition of HMGB1. Int Immunopharmacol. 2018;60:9–17. doi:10.1016/j.intimp.2018.04.029
26. de Carvalho G, Hirata F, Domingues R, et al. Up-regulation of HMGB1 and TLR4 in skin lesions of lichen planus. Arch Dermatol Res. 2018;310(6):523–528. doi:10.1007/s00403-018-1837-5
27. Senda N, Miyagaki T, Kamijo H, et al. Increased HMGB1 levels in lesional skin and sera in patients with cutaneous T-cell lymphoma. Eur J Dermatol. 2018;28(5):621–627. doi:10.1684/ejd.2018.3400
28. Wang J, Fu L, Yang H, Cao K, Sun Q, Chen T. The anti-inflammatory effects of HMGB1 blockades in a mouse model of cutaneous vasculitis. Front Immunol. 2020;11:2032. doi:10.3389/fimmu.2020.02032
29. Abdulahad D, Westra J, Limburg P, Kallenberg C, Bijl M. HMGB1 in systemic lupus Erythematosus: its role in cutaneous lesions development. Autoimmun Rev. 2010;9(10):661–665. doi:10.1016/j.autrev.2010.05.015
30. Chen T, Guo Z, Wang W, Qin S, Cao N, Li M. Increased serum HMGB1 levels in patients with Henoch-Schönlein purpura. Exp Dermatol. 2014;23(6):419–423. doi:10.1111/exd.12422
31. Yoshizaki A, Komura K, Iwata Y, et al. Clinical significance of serum HMGB-1 and sRAGE levels in systemic sclerosis: association with disease severity. J Clin Immunol. 2009;29(2):180–189. doi:10.1007/s10875-008-9252-x
32. Cui T, Zhang W, Li S, et al. Oxidative stress-induced HMGB1 release from melanocytes: a paracrine mechanism underlying the cutaneous inflammation in vitiligo. J Invest Dermatol. 2019;139(10):2174–2184.e4. doi:10.1016/j.jid.2019.03.1148
33. Mou K, Liu W, Miao Y, Cao F, Li P. HMGB1 deficiency reduces HO-induced oxidative damage in human melanocytes via the Nrf2 pathway. J Cell Mol Med. 2018;22(12):6148–6156. doi:10.1111/jcmm.13895
34. He Y, Li S, Zhang W, et al. Dysregulated autophagy increased melanocyte sensitivity to HO-induced oxidative stress in vitiligo. Sci Rep. 2017;7:42394. doi:10.1038/srep42394
35. Qiao Z, Xu Z, Xiao Q, et al. Dysfunction of ATG7-dependent autophagy dysregulates the antioxidant response and contributes to oxidative stress-induced biological impairments in human epidermal melanocytes. Cell Death Discov. 2020;6:31. doi:10.1038/s41420-020-0266-3
36. Yu Y, Tang D, Kang R. Oxidative stress-mediated HMGB1 biology. Front Physiol. 2015;6:93. doi:10.3389/fphys.2015.00093
37. Khambu B, Huda N, Chen X, et al. HMGB1 promotes ductular reaction and tumorigenesis in autophagy-deficient livers. J Clin Invest. 2019;129(5):2163. doi:10.1172/JCI129234
38. Yang H, Ni H, Ding W, Li S, Zhu G, Yang Y. Emerging players in autophagy deficiency-induced liver injury and tumorigenesis. Gene Expr. 2019;19(3):229–234. doi:10.3727/105221619X15486875608177
39. Zhang L, Li J, Ma J, et al. The relevance of Nrf2 pathway and autophagy in pancreatic cancer cells upon stimulation of reactive oxygen species. Oxid Med Cell Longev. 2016;2016:3897250.
40. Jain A, Lamark T, Sjøttem E, et al. p62/SQSTM1 is a target gene for transcription factor NRF2 and creates a positive feedback loop by inducing antioxidant response element-driven gene transcription. J Biol Chem. 2010;285(29):22576–22591. doi:10.1074/jbc.M110.118976
41. Kang R, Zeh H, Lotze M, Tang D, Beclin T. 1 network regulates autophagy and apoptosis. Cell Death Differ. 2011;18(4):571–580. doi:10.1038/cdd.2010.191
42. Zhou J, An X, Dong J, et al. IL-17 induces cellular stress microenvironment of melanocytes to promote autophagic cell apoptosis in vitiligo. FASEB J. 2018;32(9):4899–4916. doi:10.1096/fj.201701242RR
43. Wang X, Sun R, Wei H, Tian Z. High-mobility group box 1 (HMGB1)-Toll-like receptor (TLR) 4-interleukin (IL)-23-IL-17A axis in drug-induced damage-associated lethal hepatitis: interaction of γδ T cells with macrophages. Hepatology. 2013;57(1):373–384. doi:10.1002/hep.25982
44. Hu X, Zhang K, Chen Z, Jiang H, Xu W. The HMGB1‑IL‑17A axis contributes to hypoxia/reoxygenation injury via regulation of cardiomyocyte apoptosis and autophagy. Mol Med Rep. 2018;17(1):336–341. doi:10.3892/mmr.2017.7839
45. Jadeja S, Mayatra J, Vaishnav J, Shukla N, Begum R, Concise A. Review on the role of endoplasmic reticulum stress in the development of autoimmunity in vitiligo pathogenesis. Front Immunol. 2020;11:624566. doi:10.3389/fimmu.2020.624566
46. Shoab Mansuri M, Singh M. Could ER stress be a major link between oxidative stress and autoimmunity in vitiligo? J Pigment Disord. 2014;01(03). doi:10.4172/2376-0427.1000123
47. Zhu X, Yao F, Yao Y, Dong N, Yu Y, Sheng Z. Endoplasmic reticulum stress and its regulator XBP-1 contributes to dendritic cell maturation and activation induced by high mobility group box-1 protein. Int J Biochem Cell Biol. 2012;44(7):1097–1105. doi:10.1016/j.biocel.2012.03.018
48. Ren Y, Yang S, Xu S, et al. Genetic variation of promoter sequence modulates XBP1 expression and genetic risk for vitiligo. PLoS Genet. 2009;5(6):e1000523. doi:10.1371/journal.pgen.1000523
49. Toosi S, Orlow S, Manga P. Vitiligo-inducing phenols activate the unfolded protein response in melanocytes resulting in upregulation of IL6 and IL8. J Invest Dermatol. 2012;132(11):2601–2609. doi:10.1038/jid.2012.181
50. Jadeja S, Vaishnav J, Bharti A, Begum R. Elevated X-box binding protein1 splicing and interleukin-17A expression are associated with active generalized vitiligo in Gujarat population. Front Immunol. 2021;12:801724. doi:10.3389/fimmu.2021.801724
51. Huang Q, Yang Z, Zhou J, Luo Y. HMGB1 induces endothelial progenitor cells apoptosis via RAGE-dependent PERK/eIF2α pathway. Mol Cell Biochem. 2017;431:67–74. doi:10.1007/s11010-017-2976-2
52. Angelopoulou E, Paudel Y, Piperi C. Exploring the role of high-mobility group box 1 (HMGB1) protein in the pathogenesis of Huntington’s disease. J Mol Med. 2020;98(3):325–334.
53. Wu H, Chen Z, Chen J, et al. High Mobility Group B-1 (HMGB-1) promotes apoptosis of macrophage-derived foam cells by inducing endoplasmic reticulum stress. Cell Physiol Biochem. 2018;48(3):1019–1029. doi:10.1159/000491970
54. Dell’ Anna M, Ottaviani M, Kovacs D, et al. Energetic mitochondrial failing in vitiligo and possible rescue by cardiolipin. Sci Rep. 2017;7(1):13663. doi:10.1038/s41598-017-13961-5
55. Li J, Huang Q, Long X, et al. Mitochondrial elongation-mediated glucose metabolism reprogramming is essential for tumour cell survival during energy stress. Oncogene. 2017;36(34):4901–4912. doi:10.1038/onc.2017.98
56. Yang W, Nagasawa K, Münch C, et al. Mitochondrial sirtuin network reveals dynamic SIRT3-dependent deacetylation in response to membrane depolarization. Cell. 2016;167(4):985–1000.e21. doi:10.1016/j.cell.2016.10.016
57. Song Y, Li S, Geng W, et al. Sirtuin 3-dependent mitochondrial redox homeostasis protects against AGEs-induced intervertebral disc degeneration. Redox Biol. 2018;19:339–353. doi:10.1016/j.redox.2018.09.006
58. Yi X, Guo W, Shi Q, et al. SIRT3-dependent mitochondrial dynamics remodeling contributes to oxidative stress-induced melanocyte degeneration in vitiligo. Theranostics. 2019;9(6):1614–1633. doi:10.7150/thno.30398
59. Wang Q, Li L, Li C, Pei Z, Zhou M, Li N. SIRT3 protects cells from hypoxia via PGC-1α- and MnSOD-dependent pathways. Neuroscience. 2015;286:109–121. doi:10.1016/j.neuroscience.2014.11.045
60. Chen Y, Pitzer A.L, Li X, Li P.L, Wang L, Zhang Y. Instigation of endothelial Nlrp3 inflammasome by adipokine visfatin promotes inter-endothelial junction disruption: role of HMGB1. J Cell Mol Med. 2015;19(12):2715–27.
61. Kurundkar D, Kurundkar AR, Bone NB, et al. SIRT3 diminishes inflammation and mitigates endotoxin-induced acute lung injury. JCI Insight. 2019;4(1). doi:10.1172/jci.insight.120722
62. Fang P, Liang J, Jiang X, et al. Quercetin attenuates d-GaLN-induced L02 cell damage by suppressing oxidative stress and mitochondrial apoptosis via inhibition of HMGB1. Front Pharmacol. 2020;11:608. doi:10.3389/fphar.2020.00608
63. Liu B, Gan X, Zhao Y, Gao J, Yu H. Inhibition of HMGB1 reduced high glucose-induced BMSCs apoptosis via activation of AMPK and regulation of mitochondrial functions. J Physiol Biochem. 2021;77(2):227–235. doi:10.1007/s13105-021-00784-2
64. Dong WW, Liu YJ, Lv Z, et al. Lung endothelial barrier protection by resveratrol involves inhibition of HMGB1 release and HMGB1-induced mitochondrial oxidative damage via an Nrf2-dependent mechanism. Free Radic Biol Med. 2015;88(Pt B):404–416. doi:10.1016/j.freeradbiomed.2015.05.004
65. Huang CY, Chiang SF, Chen WT, et al. HMGB1 promotes ERK-mediated mitochondrial Drp1 phosphorylation for chemoresistance through RAGE in colorectal cancer. Cell Death Dis. 2018;9(10):1004. doi:10.1038/s41419-018-1019-6
66. Park E, Kim Y, Chang K. Hemin reduces HMGB1 release by UVB in an AMPK/HO-1-dependent pathway in human keratinocytes HaCaT cells. Arch Med Res. 2017;48(5):423–431. doi:10.1016/j.arcmed.2017.10.007
67. Nygaard U, van den Bogaard E, Niehues H, et al. The ”Alarmins” HMBG1 and IL-33 downregulate structural skin barrier proteins and impair epidermal growth. Acta Derm Venereol. 2017;97(3):305–312. doi:10.2340/00015555-2552
68. Chen X, Guo W, Chang Y, et al. Oxidative stress-induced IL-15 trans-presentation in keratinocytes contributes to CD8 T cells activation via JAK-STAT pathway in vitiligo. Free Radic Biol Med. 2019;139:80–91. doi:10.1016/j.freeradbiomed.2019.05.011
69. Toki Y, Takenouchi T, Harada H, et al. Extracellular ATP induces P2X7 receptor activation in mouse Kupffer cells, leading to release of IL-1beta, HMGB1, and PGE2, decreased MHC class I expression and necrotic cell death. Biochem Biophys Res Commun. 2015;458(4):771–776. doi:10.1016/j.bbrc.2015.02.011
70. Chan H, Chou H, Duran M, et al. Major role of epidermal growth factor receptor and Src kinases in promoting oxidative stress-dependent loss of adhesion and apoptosis in epithelial cells. J Biol Chem. 2010;285(7):4307–4318. doi:10.1074/jbc.M109.047027
71. Li S, Kang P, Zhang W, et al. Activated NLR family pyrin domain containing 3 (NLRP3) inflammasome in keratinocytes promotes cutaneous T-cell response in patients with vitiligo. J Allergy Clin Immunol. 2020;145(2):632–645. doi:10.1016/j.jaci.2019.10.036
72. Jia C, Zhang J, Chen H, et al. Endothelial cell pyroptosis plays an important role in Kawasaki disease via HMGB1/RAGE/cathepsin B signaling pathway and NLRP3 inflammasome activation. Cell Death Dis. 2019;10(10):778. doi:10.1038/s41419-019-2021-3
73. Xie WH, Ding J, Xie XX, et al. Hepatitis B virus X protein promotes liver cell pyroptosis under oxidative stress through NLRP3 inflammasome activation. Inflamm Res. 2020;69(7):683–696. doi:10.1007/s00011-020-01351-z
74. Hegab DS, Attia MAS. Decreased circulating T regulatory cells in Egyptian patients with nonsegmental vitiligo: correlation with disease activity. Dermatol Res Pract. 2015;2015:1–7.
75. Klarquist J, Denman CJ, Hernandez C, et al. Reduced skin homing by functional Treg in vitiligo. Pigment Cell Melanoma Res. 2010;23(2):276–286. doi:10.1111/j.1755-148X.2010.00688.x
76. Dwivedi M, Laddha N, Arora P, Marfatia Y, Begum R. Decreased regulatory T-cells and CD4 (+) /CD8 (+) ratio correlate with disease onset and progression in patients with generalized vitiligo. Pigment Cell Melanoma Res. 2013;26(4):586–591. doi:10.1111/pcmr.12105
77. Zhang J, Chen L, Wang F, et al. Extracellular HMGB1 exacerbates autoimmune progression and recurrence of type 1 diabetes by impairing regulatory T cell stability. Diabetologia. 2020;63(5):987–1001. doi:10.1007/s00125-020-05105-8
78. Esquivel D, Mishra R, Srivastava A. Stem cell therapy offers a possible safe and promising alternative approach for treating vitiligo: a review. Curr Pharm Des. 2020;26(37):4815–4821. doi:10.2174/1381612826666200730221446
79. Paudel YN, Angelopoulou E, Piperi C, Othman I, Shaikh MF. HMGB1-mediated neuroinflammatory responses in brain injuries: potential mechanisms and therapeutic opportunities. Int J Mol Sci. 2020;21(13):4609. doi:10.3390/ijms21134609
80. Andersson U, Yang H, Harris H. Extracellular HMGB1 as a therapeutic target in inflammatory diseases. Expert Opin Ther Targets. 2018;22(3):263–277. doi:10.1080/14728222.2018.1439924
81. Satoh T. The role of HMGB1 in inflammatory skin diseases. J Dermatol Sci. 2022;107:58–64. doi:10.1016/j.jdermsci.2022.07.005
82. Lamore S, Cabello C, Wondrak G. HMGB1-directed drug discovery targeting cutaneous inflammatory dysregulation. Curr Drug Metab. 2010;11(3):250–265. doi:10.2174/138920010791196337
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Endoplasmic Reticulum Dysfunction: An Emerging Mechanism of Vitiligo Pathogenesis
Xie Y, Wu N, Tang S, Zhou Z, Chen J, Li J, Wu F, Xu M, Xu X, Liu Y, Ma X
Clinical, Cosmetic and Investigational Dermatology 2024, 17:1133-1144
Published Date: 17 May 2024
Does Bilirubin Have a Causal Relationship With Vitiligo? A Mendelian Randomization Study and Bioinformatics Analysis
Xu D, Yin Y, Teng Y, Huang Y, Yu Y, Tao X, Fan Y, Ding X
Clinical, Cosmetic and Investigational Dermatology 2025, 18:1107-1119
Published Date: 6 May 2025
Identification of Oxidative Stress-Related Shared Biomarkers in Vitiligo and Periodontitis: A Bioinformatics and Machine Learning Study
Zeng H, Luo Z, Tian W, Xiang J, Liao W, Cao L, Zhang C, Wang X
Clinical, Cosmetic and Investigational Dermatology 2025, 18:2719-2737
Published Date: 23 October 2025
Autologous Serum and Non-Cultured Epidermal Cell Suspension for Stable Vitiligo: A 30-Patient Case Series
Liang J, Liang J, Jiang Y, Li W, Feng W, Yang Y, Liu B, Chen N, Wang H, Zhang X
Clinical, Cosmetic and Investigational Dermatology 2025, 18:2995-3004
Published Date: 12 November 2025