Back to Archived Journals » ChronoPhysiology and Therapy » Volume 4
Role of corticosteroids in the antidepressant response
Authors Pierscionek T, Adekunte O, Watson S, Ferrier IN, Alabi A
Received 6 May 2014
Accepted for publication 8 July 2014
Published 4 November 2014 Volume 2014:4 Pages 87—98
DOI https://doi.org/10.2147/CPT.S67272
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Tomasz Pierscionek, Oluyemi Adekunte, Stuart Watson, I Nicol Ferrier, Akintunde Alabi
Wolfson Research Institute, Institute of Neuroscience, Newcastle University, Campus for Ageing and Health, Newcastle upon Tyne, UK
Abstract: Anything that engenders a homeostatic response in the tightly regulated hypothalamic–pituitary–adrenal (HPA) axis may be thought of as a stressor and may exert an allostatic load, engendering a sustained change in the regulation of this system. Genetic, epigenetic, endocrine, post mortem, and animal studies suggest that dysregulation of the HPA axis plays a part in the pathophysiology of mood disorders and negatively impacts the antidepressant response and prognosis. Neuropsychological impairment, which is a common and disabling concomitant of depression, has been linked to disturbance of the HPA axis. A number of HPA axis-mediated treatment strategies have shown benefit in open or small-scale preliminary trials, and there are ongoing studies seeking both to replicate these initial findings and to develop new targets. HPA axis-based treatments are a fertile area of research, and much current thought pertains to the optimum targets, optimum population (including the potential for stratified medicine), and optimum outcome measures. We have, for instance, argued here that neuropsychological performance may be more sensitive and robust than scores on traditional depression rating scales.
Keywords: hypothalamic–pituitary–adrenal axis, cortisol, corticotrophin-releasing hormone, arginine vasopressin, depression, bipolar disorder, antidepressant response
Introduction
Depression is one of the major debilitating illnesses facing the modern world and the leading chronic mental health condition.1 The global disease burden is shifting away from premature deaths and towards chronic conditions, hence depression and depressive conditions cause a progressively greater disability adjusted life-years burden.1 Moreover, societies in the developed and increasingly in the developing world are ageing, so chronic conditions are more prevalent and confer a greater financial burden. In England in 2007, the cost to health services of managing patients with depression was £1.7 billion. Adding the cost of lost employment, the figure becomes £7.5 billion and is projected to rise to £12.2 billion by 2026.2
Current treatments provide full remission after first-line antidepressants for only one third of patients.3 The physiological and biological causes of depression are not fully understood, and this is at least part of the reason why treatment is not successful in a substantial number of cases. In this paper, we review the evidence that the hypothalamic–pituitary–adrenal (HPA) axis is dysregulated in depression and outline the potential impact and its consequences. We also look into the evidence base for drugs that target the HPA axis.
Physiology of the stress response
Depression has been identified as one of the major outcomes of stressful situations, and Hans Selye (1907–1982) defined stress as “the non-specific response of the body to any demand placed upon it”.4 This definition was further expanded by Bruce McEwen, who described stress as “experiences that are challenging emotionally and physiologically”. He also stated that “a hallmark of the stress response is the activation of the autonomic nervous system and hypothalamic–pituitary–adrenal axis”.5 He argued that these biological factors, which mediate the effects of stress on the body, have protective and adaptive effects but can become detrimental in the long run. He thus introduced the concept of allostasis.6
The HPA axis, which comprises the hypothalamus, pituitary gland, adrenal cortex, and associated releasing factors and hormones, has both neuronal and endocrine functions (Figure 1). It is influenced by afferent projections from numerous structures, including the limbic system, and it contributes to regulation of the immune system, energy release and storage, sleep, and sexual function. Corticotrophin-releasing hormone (CRH) and arginine vasopressin (AVP), released from the paraventricular nucleus of the hypothalamus into the hypophyseal portal system, act on membrane type I CRH and on vasopressin receptors in the anterior pituitary lobe to stimulate the release of adrenocorticotropic hormone (ACTH). ACTH in turn stimulates the production of glucocorticoids in the zona fasciculata of the adrenal cortex.7 The rate-limiting step in cortisol synthesis is the transfer of cholesterol into the mitochondria.8 The subsequent metabolism of cholesterol relies on a number of adrenal enzymes. For example, 21-hydroxylase catalyses the conversion of progesterone to 11-deoxycorticosterone and 17 hydroxyprogesterone to 11- deoxycortisol that is further metabolized by 11β-hydroxylase to cortisol (Figure 2).
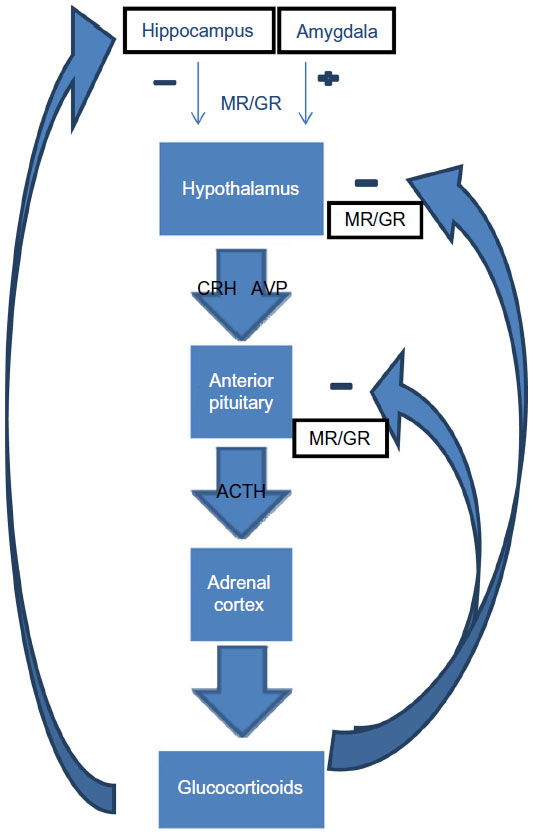 | Figure 1 Hypothalamic–pituitary–adrenal axis. |
 | Figure 2 Glucocorticoid biosynthetic pathway. |
Cortisol is released into the circulatory system in a diurnal fashion. The level peaks early in the morning around 8 am with a concentration of approximately 20 μg/dL. It reaches its trough level of approximately 5 μg/dL around 4 pm.9 A number of factors impact receptor availability of cortisol. The majority of cortisol in the circulation is bound to corticosteroid binding globulin.10 Permeability glycoprotein-1 acts as an efflux pump and actively transports cortisol out of cells.10 It has a relatively high expression in the adrenal gland.11 Two isoenzymes, 11β-hydroxysteroid dehydrogenase type 1 (11β-HSD1) and type 2 (11β-HSD2) regulate glucocorticoids at the tissue level.12 11β-HSD1 converts cortisone to cortisol, whereas 11β-HSD2 converts cortisol to the relatively inactive cortisone.
Cortisol has a wide range of both central and peripheral effects, including effects on energy metabolism and on immune and inflammatory responses.13,14 These effects are mediated by at least two intracellular, specialized steroid receptor subtypes, ie, the high-affinity type I mineralocorticoid receptor (MR) and the low-affinity type II glucocorticoid receptor (GR).15 GR is a member of the superfamily of nuclear hormone receptors and is a ligand-dependent transcription factor, expressed as an intracellular protein.10,16,17 After ligand binding, the GR translocates from the cytosol to the nucleus where it modulates the activity of glucocorticoid response elements and thus regulates gene transcription. The GR binds to and inactivates other transcription factors, such as nuclear factor kappa beta and AP-1, and thus exerts an anti-inflammatory response.18–21 This activation and translocation process and subsequent GR-mediated gene transcription is regulated by heat shock proteins 70 and 90 and their cochaperones, such as FK binding protein 51 (FKBP5) and BCL2 associated athanogene.9 FKBP5 impedes GR activation, and when FKBP5 is bound to the receptor complex, cortisol binds with lower affinity and the nuclear translocation of the receptor is less efficient.22
The HPA axis is under homeostatic control, so activation of MR and GR causes feedback inhibition of the HPA axis at the hippocampus, hypothalamus, and the anterior pituitary gland, and facilitation by the amygdala.23 In addition, GR activation induces FKBP5 mRNA and protein expression and this provides an ultrashort feedback loop to rapidly decrease GR sensitivity. Variants in the FKBP5 gene are associated with differential protein expression. High induction alleles are associated with relative GR resistance.24
There is a reciprocal relationship between the HPA axis and the monoamine systems.25 Serotonin neurons originate in the raphe nuclei, which are located in the midline of the brain stem. Serotonergic neurons ramify widely and terminate throughout the brain and in the spinal cord. The hippocampus expresses corticosteroid and serotonin (5-HT) receptors,26–28 and is considered to be a focal point of the interaction.29 Both 5-HT1A and 5-HT2C are highly expressed in the hippocampus30,31 and are involved in HPA axis regulation.32 Hippocampal expression of 5-HT1A and 5-HT2C receptors have been shown to be altered by glucocorticoids.33–35 Altered 5-HT function in the hippocampus has been suggested as a cause of abnormal changes in the negative feedback mechanism to glucocorticoids in depression.36 Hippocampal cultures from fetal guinea pigs exposed to 5-HT showed a significant increase in GR mRNA levels,37 suggesting that early exposure to 5-HT can affect GR expression within the hippocampus during development and can lead to permanent reprogramming of the HPA axis.37
The HPA axis plays a central role in regulating a range of physiological functions. The system has inbuilt negative feedback loops that prevent excessive release of the primary stress hormone cortisol from the adrenal cortex. Whilst the human body has the capacity to manage and maintain an allostatic response, it is believed that permanent structural changes can occur in the HPA axis, leading to a dysfunction which is manifested in an inability to adapt to stress and dysregulated hormonal levels. HPA dysfunction is in turn believed to be associated with a reduced response to antidepressant medication and a worse prognosis.
HPA axis and monoamine function in patients with mood disorder
The role of the HPA axis in the pathogenesis of depression and mood disorders in general has been extensively investigated. Cross-sectional endocrinology studies suggest that HPA axis dysregulation is a common although not inevitable finding in depressed patients. However, its prevalence is greater amongst in-patients and individuals with severe episodes of melancholia or psychosis.38 Studies have been performed to measure basal cortisol levels in urine, blood, and saliva at single time points, as well as sequentially in order to determine whether diurnal variation or a wakening response exists. Saliva assays of HPA axis hormones have proven reliable and noninvasive, less likely to induce a stress response, and more consistent in response to suppression testing.39 The so-called “activated tests” are more sensitive than basal measures. Examples are measurements of cortisol before and/or after a pharmacological challenge with agonists or antagonists of CRH, AVP, ACTH, MR, or GR. This could be done either individually or in combination (eg, the dexamethasone/CRH test) or after psychological stressors, eg, the Trier Social Stress Test.40 The most reliable finding to emerge from studies of the HPA is the presence of reduced GR functional activity,38 often referred to as glucocorticoid resistance. The interindividual variability in the outcome of HPA axis function tests in patients with psychiatric syndromes has led some to argue for the use of HPA axis-defined subgroups with the prediction that this development may lead to a more rational choice of treatment strategy.41
The relevance of HPA axis dysregulation in mood disorders and especially in depression is exemplified by the results of studies investigating patients with primary endocrine abnormalities.42 Neuropsychiatric symptoms, including depressed mood, are common in both Cushing’s disease43 and Addison’s disease.44 Cushing’s disease is characterized by increased secretion of ACTH from the anterior pituitary. Pituitary adenomas or elevated production of hypothalamic CRH may cause this pathology. Addison’s disease is a disorder of the adrenal cortex resulting in inadequate secretion of glucocorticoids and mineralocorticoids. The mood symptoms associated with both these conditions respond well to normalization of steroid production.
Post mortem studies of individuals with a history of depression45 and those who committed suicide46 have been used to investigate the hypothesis that depression is associated with HPA dysfunction. Studies have shown reduced MR expression in the hippocampus,45,46 increased MR expression in the hypothalamus,47 as well as reduced GR expression in the cingulate gyrus, inferior frontal gyrus,45 prefrontal cortex, and frontal and inferior temporal cortices.48 This concurs with animal studies that showed downregulation of GR mRNA expression in the hippocampus and frontal cortex in rodents with hypercortisolemia.49 Interestingly, one study showed that hippocampal GR mRNA in depressed patients did not differ from matched controls that reported chronic stress.46 Selective abnormalities of GR mRNA expression in the orbitofrontal cortex and alterations in protein isoforms have been noted in a range of psychiatric conditions.50 Post mortem studies of depressed patients who committed suicide have also shown a reduction in CRF receptor sites in the cerebral cortex.51
Whether HPA axis dysregulation is a cause, consequence, or an epiphenomenon of depression can be determined by considering an individual’s genetic profile. The observation that depression tends to cluster in families suggests that genes play an etiological role.52 It is possible that some of this risk is attributable to the genes coding for HPA axis proteins, given a 62% heritability of basal cortisol concentrations.53 This is in addition to the finding that apparently healthy first-degree relatives of probands with major depressive episodes exhibit HPA dysregulation similar to that seen in affected relatives and different to the response seen in healthy controls.54 The failure of genome wide association studies to identify relevant genetic polymorphisms (or for that matter any polymorphisms) associated with the HPA axis55 is likely attributable to the complex relationship between the environmental and genetic factors that predispose to depression. However, candidate gene approaches yield interesting results. For example, polymorphisms of the FKBP5 gene affect GR sensitivity and hence the stress hormone response.22,24 Individuals carrying alleles and haplotypes associated with GR resistance have been shown to have increased vulnerability to depression,56 bipolar disorder,57 and post traumatic stress disorder,24,58 as well being at increased risk of suicide.59 Functional allelic variations in the GR,60,61 CRH receptor 1,57 AVP1b receptor,62 11β-HSD1 receptor,63 amino peptidase N (which controls AVP release),55 and P-glycoprotein genes64 also show an association with depression and response to antidepressant treatment.65
Epigenetic changes that include DNA methylation are responsible for a reversible functional impact66 causing changes in the expression of genes coding for AVP,67 and FKBP5,22 and can arise following trauma or adversity during critical developmental periods. Increased cytosine methylation of an NR3C1 (GR) promoter has been seen in post mortem hippocampi obtained from victims of suicide who had been abused as children (compared with suicide victims with no history of childhood abuse and well as healthy controls).68 Moreover, the interaction between genetic predisposition and childhood trauma has been elegantly demonstrated by Zimmermann et al, who showed that individuals who are homozygous for the high induction alleles of the FKBP5 gene are particularly sensitive to the effects of childhood trauma.56 It is likely, but not established, that such epigenetic changes will impact on the response to antidepressants.
There has been much debate about the primary site of HPA axis dysregulation, but both CRH69 and AVP overdrive have been suggested.70 The latter is supported by animal studies showing that under conditions of chronic stress, the expression of AVP in the CRH-secreting neurons of the paraventricular nucleus increases and AVP becomes the predominant regulator of ACTH secretion.70 However, others suggest dysregulation of GR38 or the MR/GR balance as being a cause.71 Altered regulation of GR may be tissue-specific (eg, it may differ between the immune system and other tissues) and it may be mediated by altered number or sensitivity of GR.72,73 GR resistance may be secondary to an increase in endogenous (chronic stress)74–76 or exogenous glucocorticoids.10 In depression,77 it may be secondary to ligand-independent processes, such as signal transduction pathways, regulated by nonsteroidal compounds,78 including cytokines such as interleukin-1 and protein kinase A.79–81 The GR theory of depression is supported by studies which suggested that mice with an experimentally induced acquired forebrain-specific disruption of GR showed behavioral features consistent with depression as well as impaired negative feedback regulation of the HPA axis similar to that seen in mood disorder patients.39
Glucocorticoids also have a mediating effect on neurogenesis.82 The impact of neurogenesis, neurodegeneration, and impaired neuronal networks on the precipitation and maintenance of depression has previously been demonstrated83 and are supported by findings that brain-derived neurotrophic factor (BDNF) and signaling of its receptor, TrkB, play a role in recovery from depression.84 The antidepressant response to medication may be mediated by BDNF signaling through TrkB, causing brain reorganization and neurogenesis, a process that takes time and results in a delayed response.85 Serum levels of BDNF are reduced in depressed patients compared with controls,86 and a reduction in the release of BDNF84 and 5-HT uptake in platelets is seen in depression.87 Animal models have shown that the effect of antidepressants can be simulated by injecting BNDF into the hippocampus or midbrain.88,89 However, drug treatment is ineffective when levels of BDNF or TrkB are reduced.90,91
The evidence for monoamines having a role in the pathophysiology of depression comes partly from the demonstration that symptoms of depression may be induced, in vulnerable individuals, by depleting 5-HT92 and by blocking the synthesis of norepinephrine.93 The efficacy of drugs that inhibit serotonin and noradrenaline reuptake further supports this notion. It is argued that the ability of antidepressants to increase the activity of forebrain postsynaptic 5-HT1A receptors mediates the clinical response, especially for serotonin receptor reuptake inhibitors. This is driven by desensitization of somatodendritic 5-HT1A receptors in the dorsal raphe nuclei in combination with attenuated synaptic reuptake in the forebrain. There have been a number of studies demonstrating that HPA axis dysregulation is a poor prognostic factor for depressed patients receiving serotonergic antidepressants.94 The mechanism for this is suggested by rodent studies showing that even subtle alterations in HPA axis rhythm (for instance, a flattened rhythm induced by implantation of a low-dose corticosteroid pellet) can attenuate 5-HT1A autoreceptor function95 and reduce the ability of long-term fluoxetine treatment to elevate forebrain 5-HT.96 This effect is reversed by administration of GR antagonists. Importantly, it has also been shown that GR antagonists enhance the serotonergic response to serotonin receptor reuptake inhibitors97 even in rats with a normal HPA axis rhythm.98 It has been argued that the efficacy of antidepressants may be mediated directly by their effects on the HPA axis. Animal studies showed that the tricyclic imipramine and the monoamine oxidase inhibitor phenelzine have opposing effects on the activity of the HPA axis and on expression of the GR in the hypothalamus and prefrontal cortex forebrain of adrenalectomized mice.99 Similarly, imipramine and phenelzine also have different effects on brainstem GR. Phenelzine decreased GR expression in the locus coeruleus and imipramine decreased GR expression in the dorsal raphe nuclei.100
Additionally, the role of sleep in HPA axis dysfunction and the development of depression have been explored. For example, there is an existing hypothesis that obstructive sleep apnea through repeated arousals and subsequent cortisol release leads to HPA axis activation101 and this has been linked to the development of depression.102 Further, an animal model has been applied and shown the consequences of chronically disrupted and restricted sleep on HPA axis dysfunction.103
Overall, HPA dysfunction is commonly seen in patients with depression, and although not always present in such individuals, it does have links with increased severity. Post mortem studies of patients with a history of depression who later committed suicide reveal an association with both reduced and increased MR expression in different areas of the brain as well as reduced expression in other areas. Such findings may pave the way for future studies in vivo, allowing early detection of individuals with dysregulated HPA. HPA dysfunction does not have a clear genetic basis but is believed to be attributable to complex relationships between the environmental and genetic factors.
Neuropsychological dysfunction in mood disorders and the HPA axis
Neuropsychological impairments are a common concomitant of Cushing’s syndrome104 and resolve with successful endocrinology treatment.105 Such deficits are also seen following acute or chronic exogenous administration of steroids14,106 and have been identified in patients with unipolar107 and bipolar108 mood disorders. Cognitive dysfunction has been linked to the duration of illness, number of previous manic episodes, hospitalizations, and suicide attempts.109 However, cognitive deficits do not appear to be restricted to patients in acute phases of illness, but have been found in euthymia,108 suggesting persistence into recovery and raising the possibility that these deficits may predate the emergence of symptoms.110 The deficits may be attributed to HPA axis dysregulation, which has been shown in some111,112 but not all113 studies, to be associated with neuropsychological impairment. Moreover, reduced cortisol levels and effective treatment of depression have been shown to correlate with improvement in several domains of neuropsychological performance.111 The effect of cortisol on verbal memory is possibly independent of diagnosis in patients.114 Patients who have recovered from a major depressive episode may be especially vulnerable to the detrimental effects of subtle HPA disturbances on cognitive performance.
Cognitive deficits have been identified in patients with mood disorders and appear related to HPA dysfunction and raised cortisol levels. Although treatment tends to ameliorate such neuropsychological impairment, individuals who have recovered from a major depressive episode may be especially vulnerable to cognitive impairment secondary to small disturbances in HPA dysfunction. Such individuals would appear to have a reduced tolerance to future stress.
Treatments that target the HPA axis
The putative role of stress in the etiology of depression, the evidence of HPA axis dysregulation, and the negative effects of this dysregulation on the response to antidepressant therapy supports the use of drugs targeting the HPA axis as novel therapeutic strategies. There are a number of potential sites within the HPA axis that could be targeted as monotherapy approaches or as augmenting agents for traditional antidepressants.
Cortisol synthesis inhibitors
Aminoglutethimide attenuates the conversion of cholesterol to pregnenolone (Figure 2) and hence would be expected to reduce the synthesis of a wide range of adrenal steroids. It has shown promise in open trials of depression in combination with other drugs that affect cortisol biosynthesis.115–118
Ketoconazole is an antifungal agent with antiglucocorticoid effects; specifically, it inhibits the side chain cleavage in the conversion of cholesterol to pregnenolone and inhibits the action of 11β-hydroxylase.119 Efficacy in depression was suggested by case reports120,121 and by small open studies.122,123 Malison et al124 did not demonstrate benefit in a 6-week, double-blind, placebo-controlled trial in 16 adult patients with treatment-refractory major depressive disorder. However, a subsequent study by Wolkowitz et al reported efficacy in a double-blind trial of depressed patients who were hypercortisolemic at baseline.125
Metyrapone inhibits 11β-hydroxylase and 11β-HSD2 (see Figure 2). Antidepressant properties of metyrapone have been demonstrated in small clinical trials in treatment-resistant depression with coadministration of hydrocortisone126 and imipramine.127 Jahn et al128 have demonstrated a beneficial effect on mood symptoms in a double-blind, randomized trial involving 63 in-patients with a DSM-IV (Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition) diagnosis of major depressive disorder. These patients were randomized to receive either metyrapone 1 g/day or placebo augmentation of the serotonergic drugs nefazodone or fluvoxamine for 3 weeks. The ADD (Antiglucocorticoid augmentation of anti-Depressants in Depression) study currently being carried out by our group129 is a randomized, controlled trial of metyrapone augmentation of serotonergic antidepressants in 165 patients with treatment-resistant depression and will evaluate the efficacy of this cortisol synthesis inhibitor. The results of this study are not yet available.
Glucocorticoid receptor antagonists
The abortifacient, mifepristone, is a progesterone receptor antagonist and also a powerful glucocorticoid receptor antagonist. There have been a number of treatment studies involving the use of mifepristone for psychotic major depression, predominantly by the group based at Stanford.130,131 Initial open130 and small, double-blind, placebo-controlled131 trials in patients with psychotic depression suggested efficacy. Subsequent, larger, randomized controlled trials by the same group showed a benefit for psychotic but not mood symptoms in one trial132 and no benefit on primary outcome measures in a second.133 A further trial has shown the importance of plasma levels of mifepristone, with a benefit seen in patients whose levels were above a cut-off point.134 The group is conducting a further clinical trial of mifepristone in psychotic major depression using the higher (1,200 mg) dose.135
Our group136 reported selective improvements in spatial working memory performance, verbal fluency, and spatial recognition memory following treatment with mifepristone 600 mg/day in a 3-week, double-blind, crossover design in bipolar depression. In a parallel-design replication, using spatial working memory as the primary outcome, mifepristone was associated with a sustained improvement, which was evident 7 weeks after cessation of treatment. The improvement in depressed mood seen in the first study136 was not confirmed in the replication study.137
Mifepristone is also being assessed in ongoing trials in a number of other disorders to assess its benefits, ie, in post-traumatic stress disorder,138 alcoholism (for craving),139 cognitive impairment,140 depression in Gulf war veterans,141 and in borderline personality disorder.142
Dehydroepiandrosterone and treatment of affective disorders
Dehydroepiandrosterone (DHEA) is an endogenous steroid produced in the adrenal gland. It is an intermediate in the production of the sex hormones. Open143 and randomized, double-blind144 trials of DHEA in patients with depression have shown benefits in depressive symptoms143,144 and memory performance.143 The efficacy of DHEA in the treatment of dysthymia145 as well as in human immunodeficiency virus-positive cohorts146 has also been demonstrated.
Dexamethasone
Open147,148 and small-scale, randomized, controlled trials149 have demonstrated a benefit of dexamethasone in the treatment of depressive symptoms. Dinan et al150 reported a significant improvement in Hamilton scores in six of ten patients in response to dexamethasone augmentation of previously ineffective standard antidepressant therapy.
Corticotrophin-releasing hormone antagonists
The selective CRH 1 receptor antagonist, R121919, attenuates the endocrine stress response in rodents151 and was shown to reduce depression scores in a dose-escalation, open-label Phase II study in depressed patients152 without affecting weight or leptin levels.153 A reversible increase in liver enzymes in a separate Phase I study154 led to discontinuation of its clinical development. There has been a series of other compounds that have generated promising animal data, but a number of these have led to abandoned155 or negative Phase II trials,156 eg, CP-316,311 in a recurrent depression trial,157 pexacerfont (BMS-562,086) in a generalized anxiety disorder trial,158 and GSK561679 in a depression study.159 The results of a depression trial160 using SSR125543, the Sanofi-Aventis compound, are not yet available. There are ongoing trials looking at the benefits of pexacerfont in anxiety-related alcohol craving,161 of GSK561679 on the human startle response in normal volunteers,162 and in post traumatic stress disorder,163 and of a CRF1 antagonist in social anxiety.164
The CRF2 receptor has been suggested to mediate an anxiolytic response.165 This, in addition to recent work elucidating the structure of the CRH 1 receptor and the nature of its ligand interactions,166 may support the further development of novel CRH antagonists.
Vasopressin receptor antagonist
Vasopressin serves as a regulator of the HPA axis, potentiating the stimulatory effects of corticotrophin releasing hormone as well as the relative refractoriness to glucocorticoid feedback in chronic stress.167 It exerts its effects on the brain mainly via V1a receptors168 and V1b receptors which are located in the anterior pituitary, through which it mediates corticotrophin secretion.169 V1a receptors are abundant in the cerebral cortex, limbic system, hypothalamus, and brain stem.170 There are a number of antagonists of V1a, V1b, and V2 in development, particularly for the treatment of hyponatremia. The non-peptide V1b receptor antagonist, SSR149415, has demonstrated anxiolytic and antidepressant properties in animal models. However, the lack of efficacy demonstrated in clinical trials in patients with generalized anxiety disorder and depression171 led to discontinuation of development of the compound by Sanofi.172 Abbott Laboratories has made a strategic decision to discontinue a depression trial, although a study using heavy drinking days in patients with alcohol dependence as the outcome measure is still recruiting.173 V1a receptor antagonists being examined in Phase I trials include the Azevan Pharmaceuticals Inc compounds SRX 251 and SRX 246. Roche also has RG7314 in a Phase II trial in autism spectrum disorder.156
A number of novel medications that target the HPA axis have recently been developed. Because HPA axis dysfunction is believed to be associated with a reduced response to antidepressant treatment as well as a poorer prognosis, the HPA itself has been identified as a therapeutic target. Cortisol synthesis inhibitors, glucocorticoid receptor antagonists, DHEA, dexamethasone, CRH receptor antagonists, and vasopressin receptor antagonists are medications that may in time prove beneficial in treating refractory depression. Larger studies with a greater duration of follow-up are required to determine whether these novel therapies are both safe and effective.
Outcomes measures and prediction of response
The hope that antiglucocorticoid strategies would revolutionize psychiatric treatment has not been realized. This does not necessarily mean that this approach should be discarded, rather that thought needs to be given to appropriate outcome measures and populations.
In the studies above, it appears there was some benefit of mifepristone augmentation, particularly where higher concentrations were achieved. In mifepristone studies of psychotic major depression, the psychotic symptoms may respond preferentially. The randomized controlled trial of metyrapone102 is promising, as are the preliminary studies with the CRH receptor antagonists, but all these findings require replication.
It is noteworthy that the bipolar mifepristone studies demonstrated a significant effect on spatial working memory, with no appreciable benefit on depressive symptoms.136,137 It could be argued that future HPA efficacy studies should focus on improving neuropsychological performance.
HPA axis dysregulation is not invariable in mood disorder patients. Studies may benefit from identifying those who are more likely to respond and hence examining efficacy in enriched samples. In the future, endocrinology and genetic measures of the HPA axis may inform individual treatment plans.
Future perspectives
We look forward to results of further trials targeting the HPA axis in the treatment of affective disorders with the hope of developing novel, safe, and effective treatments that overcome the negative effects of dysregulation of the HPA axis on the antidepressant response. Such strategies may deliver a rapid and sustained response, either as monotherapy agents or as augmenters of other antidepressant strategies. The roles of the infralimbic cortex, which has the mesocortical dopamine system as its afferent modulator,174 and iron deficiency anemia in infancy175 in regulating behavioral and physiological responses via the HPA axis need further exploration.
Disclosure
The authors report no conflicts of interest in this work.
References
Murray CJL, Vos T, Lozano R, et al. Disability-adjusted life years (DALYs) for 291 diseases and injuries in 21 regions, 1990–2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet. 2012;380:2197–2223. | |
National Institute for Health and Clinical Excellence. Depression in adults: the treatment and management of depression in adults. National Clinical Practice Guideline 90. National Collaborating Centre for Mental Health; 2010. Available from: https://www.google.co.nz/?gfe_rd=cr&ei=-Y6iU8LFIObC8geQ1oDQAg&gws_rd=ssl. Accessed August 7, 2014. | |
Rush AJ, Trivedi MH, Wisniewski SR, et al. Acute and longer-term outcomes in depressed outpatients requiring one or several treatment steps: a STAR*D report. Am J Psychiatry. 2006;163:1905–1917. | |
Pasqualini CD, Pasqualini RQ. [Hans Selye: the stress of his life]. Medicina (B Aires). 1983;43:109–111. Spanish. | |
McEwen BS. Physiology and neurobiology of stress and adaptation: central role of the brain. Physiol Rev. 2007;87:873–904. | |
McEwen BS. Stress, adaptation, and disease. Allostasis and allostatic load. Ann N Y Acad Sci. 1998;840:33–44. | |
Ben-Zvi A, Vernon SD, Broderick G. Model-based therapeutic correction of hypothalamic-pituitary-adrenal axis dysfunction. PLoS Comput Biol. 2009;5:e1000273. | |
Colasanti A, Owen DR, Grozeva D, et al. Bipolar disorder is associated with the rs6971 polymorphism in the gene encoding 18 kDa translocator protein (TSPO). Psychoneuroendocrinology. 2013;38:2826–2829. | |
Itoi S, Terao M, Murota H, Katayama I. 11beta-Hydroxysteroid dehydrogenase 1 contributes to the pro-inflammatory response of keratinocytes. Biochem Biophys Res Commun. 2013;440:265–270. | |
Silverman MN, Sternberg EM. Glucocorticoid regulation of inflammation and its behavioral and metabolic correlates: from HPA axis to glucocorticoid receptor dysfunction. Ann N Y Acad Sci. 2012;1261:55–63. | |
Thiebaut F, Tsuruo T, Hamada H, Gottesman MM, Pastan I, Willingham MC. Cellular localization of the multidrug-resistance gene product P-glycoprotein In normal human tissues. Proc Natl Acad Sci U S A. 1987;84:7735–7738. | |
Tomlinson JW, Walker EA, Bujalska IJ, et al. 11beta-hydroxysteroid dehydrogenase type 1: a tissue-specific regulator of glucocorticoid response. Endocr Rev. 2004;25:831–866. | |
Pariante CM, Miller AH. Glucocorticoid receptors in major depression: relevance to pathophysiology and treatment. Biol Psychiatry. 2001;49:391–404. | |
MacLachlan L, Watson S. Evidence for the role of hypothalamic-pituitary-adrenal axis dysfunction in the pathogenesis of mood disorders. European Psychiatric Review. 2010;3(1):54–57. | |
Herman JP, Spencer R. Regulation of hippocampal glucocorticoid receptor gene transcription and protein expression in vivo. J Neurosci. 1998;18:7462–7473. | |
Silverman MN, Pearce BD, Miller AH. Cytokines and HPA axis regulation. In: Kronfol Z, editor. Cytokines and Mental Health. Norwell, MA, USA: Kluwer Academic Publishers; 2003. | |
Nicolaides NC, Galata Z, Kino T, Chrousos GP, Charmandari E. The human glucocorticoid receptor: molecular basis of biologic function. Steroids. 2010;75:1–12. | |
Brasier AR. The NF-κB regulatory network. Cardiovasc Toxicol. 2006;6:111–130. | |
O’Riordan KJ, Huang I-C, Pizzi M, et al. Regulation of nuclear factor kB in the hippocampus by group I metabotropic glutamate receptors. J Neurosci. 2006;26:4870–4887. | |
Albensi BC, Mattson MP. Evidence for the involvement of TNF and NF-κB in hippocampal synaptic plasticity. Synapse. 2000;35:151–159. | |
Ameyar M, Wisniewska M, Weitzman JB. A role for AP-1 in apoptosis: the case for and against. Biochimie. 2003;85:747–752. | |
Binder EB. The role of FKBP5, a co-chaperone of the glucocorticoid receptor in the pathogenesis and therapy of affective and anxiety disorders. Psychoneuroendocrinology. 2009;34 Suppl 1:S186–S195. | |
Pariante CM. The glucocorticoid receptor: part of the solution or part of the problem? J Psychopharmacol. 2006;20(Suppl 4):79–84. | |
Binder EB, Bradley RG, Liu W, et al. Association of FKBP5 polymorphisms and childhood abuse with risk of posttraumatic stress disorder symptoms in adults. JAMA. 2008;299:1291–1305. | |
Porter RJ, Gallagher P, Watson S, Young AH. Corticosteroid-serotonin interactions in depression: a review of the human evidence. Psychopharmacology (Berl). 2004;173:1–17. | |
Palacios J, Waeber C, Hoyer D, Mengod G. Distribution of serotonin receptors. Ann N Y Acad Sci. 1990;600:36–52. | |
de Kloet ER. Brain corticosteroid receptor balance and homeostatic control. Front Neuroendocrinol. 1991;12:95–164. | |
Wright D, Seroogy K, Lundgren K, Davis B, Jennes L. Comparative localization of serotonin1A,1C and 2 receptor subtype mRNAs in rat brain. J Comp Neurol. 1995;351:357–373. | |
Holmes MC, French KL, Sekl JR. Dysregulation of diurnal rhythms of serotonin 5-HT2C and corticosteroid receptor gene expression in the hippocampus with food restriction and glucocorticoids. J Neurosci. 1997;17:4056–4065. | |
Mengod G, Pompeiano M, Martinez-Mir M, Palacios J. Localization of the mRNA for the 5-HT2 receptor by in situ hybridization histochemistry: correlation with the distribution of receptor sites. Brain Res. 1990;524:139–143. | |
Chalmers D, Watson S. Comparative anatomical distribution of 5-HT1A receptor mRNA and 5-HT1A binding in rat brain: a combined in situ hybridization/in vitro receptor autoradiographic study. Brain Res. 1991;561:51–60. | |
Fuller R. The involvement of serotonin in regulation of pituitary adrenocortical function. Front Neuroendocrinol. 1992;13:250–270. | |
Chalmers D, Kwak S, Mansour A, Akil H, Watson S. Corticosteroids regulate brain hippocampal 5-HT1A receptor mRNA expression. J Neurosci. 1993;13:914–923. | |
Zhong P, Ciaranello RD. Transcriptional regulation of hippocampal 5-HT1A receptors by corticosteroid hormones. Mol Brain Res. 1995;29:23–34. | |
Holmes MC, Yau JLW, French KL, Seckl JR. The effect of adrenalectomy on 5HT and corticosteroid receptor subtype mRNA expression in rat hippocampus. Neuroscience. 1995;64:327–337. | |
Neumaier JF, Sexton TJ, Hamblin MW, Beck SG. Corticosteroids regulate 5-HT1A but not 5-HT1B receptor mRNA in rat hippocampus. Brain Res Mol Brain Res. 2000;20:65–73. | |
Erdeljan P, Andrews MH, MacDonald JF, Matthews SG. Glucocorticoids and serotonin alter glucocorticoid receptor mRNA levels in fetal guinea-pig hippocampal neurons, in vitro. Reprod Fertil Dev. 2005;17:743–749. | |
McQuade R, Young AH. Future therapeutic targets in mood disorders: the glucocorticoid receptor. Br J Psychiatry. 2000;177:390–395. | |
Guilliams T, Edwards L. Chronic stress and the HPA axis: clinical assessment and therapeutic considerations. Standard. 2010;9:1–12. | |
Lauren BO, Charles BN. HPA axis modulation in the treatment of mood disorders. In: Schoepf D, editor. Psychiatric Disorders – New Frontiers in Affective Disorders. 2013. Available from: http://www.intechopen.com/books/psychiatric-disorders-new-frontiers-in-affective-disorders. Accessed August 7, 2014. | |
Gold PW, Gabry KE, Yasuda M, Chrousos GP. Divergent endocrine abnormalities in melancholic and atypical depression: clinical and pathophysiological implications. Endocrinol Metab Clin North Am. 2002;31:37–62. | |
Starkman MN, Giordani B, Berent S, Schork MA, Schteingart DE. Elevated cortisol levels in Cushing’s disease are associated with cognitive decrements. Psychosom Med. 2001;63:985–993. | |
O’Keane V. Evolving model of depression as an expression of multiple interacting risk factors. Br J Psychiatry. 2000;177:482–483. | |
Anglin RE, Rosebush PI, Mazurek MF. The neuropsychiatric profile of Addison’s disease: revisiting a forgotten phenomenon. J Neuropsychiatry Clin Neurosci. 2006;18:450–459. | |
Klok MD, Alt SR, Irurzun Lafitte AJ, et al. Decreased expression of mineralocorticoid receptor mRNA and its splice variants in postmortem brain regions of patients with major depressive disorder. J Psychiatr Res. 2011;45:871–878. | |
Lopez JF, Chalmers DT, Little KY, Watson SJ. AE Bennett Research Award. Regulation of serotonin1A, glucocorticoid, and mineralocorticoid receptor in rat and human hippocampus: implications for the neurobiology of depression. Biol Psychiatry. 1998;43:547–573. | |
Wang SS, Kamphuis W, Huttinga I, Zhou JN, Swaab DF. Gene expression analysis in the human hypothalamus in depression by laser microdissection and real-time PCR: the presence of multiple receptor imbalances. Mol Psychiatry. 2008;13:786–799. | |
Webster MJ, Knable MB, O’Grady J, Orthmann J, Weickert CS. Regional specificity of brain glucocorticoid receptor mRNA alterations in subjects with schizophrenia and mood disorders. Mol Psychiatry. 2002;7:985–994. | |
Sapolsky RM, McEwen BS. Down-regulation of neural corticosterone receptors by corticosterone and dexamethasone. Brain Res. 1985;339:161–165. | |
Sinclair D, Webster MJ, Fullerton JM, Weickert CS. Glucocorticoid receptor mRNA and protein isoform alterations in the orbitofrontal cortex in schizophrenia and bipolar disorder. BMC Psychiatry. 2012;12:84. | |
Watson S, Gallagher P, Ritchie JC, Ferrier IN, Young AH. Hypothalamic-pituitary-adrenal axis function in patients with bipolar disorder. Br J Psychiatry. 2004;184:496–502. | |
McGuffin P, Katz R. The genetics of depression and manic-depressive disorder. Br J Psychiatry. 1989;155:294–304. | |
Bartels M, Van den Berg M, Sluyter F, Boomsma DI, de Geus EJ. Heritability of cortisol levels: review and simultaneous analysis of twin studies. Psychoneuroendocrinology. 2003;28:121–137. | |
Holsboer F, Lauer CJ, Schreiber W, Krieg JC. Altered hypothalamic-pituitary-adrenocortical regulation in healthy subjects at high familial risk for affective disorders. Neuroendocrinology. 1995;62:340–347. | |
Hek K, Demirkan A, Lahti J, et al. A genome-wide association study of depressive symptoms. Biol Psychiatry. 2013;73:667–678. | |
Zimmermann P, Bruckl T, Nocon A, et al. Interaction of FKBP5 gene variants and adverse life events in predicting depression onset: results from a 10-year prospective community study. Am J Psychiatry. 2011;168:1107–1116. | |
Willour VL, Chen H, Toolan J, et al. Family-based association of FKBP5 in bipolar disorder. Mol Psychiatry. 2009;14:261–268. | |
Mehta D, Binder EB. Gene x environment vulnerability factors for PTSD: the HPA-axis. Neuropharmacology. 2012;62:654–662. | |
Roy A, Hodgkinson CA, Deluca V, Goldman D, Enoch MA. Two HPA axis genes, CRHBP and FKBP5, interact with childhood trauma to increase the risk for suicidal behavior. J Psychiatr Res. 2012;46:72–79. | |
Manenschijn L, van den Akker EL, Lamberts SW, van Rossum EF. Clinical features associated with glucocorticoid receptor polymorphisms. An overview. Ann N Y Acad Sci. 2009;1179:179–198. | |
van Rossum EF, Binder EB, Majer M, et al. Polymorphisms of the glucocorticoid receptor gene and major depression. Biol Psychiatry. 2006;59:681–688. | |
Leszczynska-Rodziewicz A, Szczepankiewicz A, Dmitrzak-Weglarz M, Skibinska M, Hauser J. Association between functional polymorphism of the AVPR1b gene and polymorphism rs1293651 of the CRHR1 gene and bipolar disorder with psychotic features. J Affect Disord. 2012;138:490–493. | |
Dekker MJ, Tiemeier H, Luijendijk HJ, et al. The effect of common genetic variation in 11beta-hydroxysteroid dehydrogenase type 1 on hypothalamic-pituitary-adrenal axis activity and incident depression. J Clin Endocrinol Metab. 2012;97:E233–E237. | |
Santos M, Carvalho S, Lima L, et al. Common genetic polymorphisms in the ABCB1 gene are associated with risk of major depressive disorder in male Portuguese individuals. Genet Test Mol Biomarkers. 2014;18:12–19. | |
Velders FP, Kuningas M, Kumari M, et al. Genetics of cortisol secretion and depressive symptoms: a candidate gene and genome wide association approach. Psychoneuroendocrinology. 2011;36:1053–1061. | |
Weaver IC, Cervoni N, Champagne FA, et al. Epigenetic programming by maternal behavior. Nat Neurosci. 2004;7:847–885. | |
Murgatroyd C, Patchev AV, Wu Y, et al. Dynamic DNA methylation programs persistent adverse effects of early-life stress. Nat Neurosci. 2009;12:1559–1566. | |
McGowan PO, Sasaki A, D’Alessio AC, et al. Epigenetic regulation of the glucocorticoid receptor in human brain associates with childhood abuse. Nat Neurosci. 2009;12:342–338. | |
Keller PA, McCluskey A, Morgan J, O’Connor SM. The role of the HPA axis in psychiatric disorders and CRF antagonists as potential treatments. Arch Pharm (Weinheim). 2006;339:346–355. | |
Scott LV, Dinan TG. Vasopressin and the regulation of hypothalamic-pituitary-adrenal axis function: implications for the pathophysiology of depression. Life Sci. 1998;62:1985–1998. | |
De Kloet ER, Vreugdenhil E, Oitzl MS, Joels M. Brain corticosteroid receptor balance in health and disease. Endocr Rev. 1998;19:269–301. | |
Cotter P, Mulligan O, Landau S, Papadopoulos A, Lightman S, Checkley S. Vasoconstrictor response to topical beclomethasone in major depression. Psychoneuroendocrinology. 2002;27:475–487. | |
Maguire TM, Thakore J, Dinan TG, Hopwood S, Breen KC. Plasma sialyltransferase levels in psychiatric disorders as a possible indicator of HPA axis function. Biol Psychiatry. 1997;41:1131–1136. | |
Bauer M, Vedhara K, Perks P, Wilcock G, Lightman S, Shanks N. Chronic stress in caregivers of dementia patients is associated with reduced lymphocyte sensitivity to glucocorticoids. J Neuroimmunol. 2000;103:84–92. | |
Miller GE, Cohen S, Ritchey AK. Chronic psychological stress and the regulation of proinflammatory cytokines: a glucocorticoid-resistance model. Health Psychol. 2002;21:531–541. | |
Miller GE, Chen E, Sze J, et al. A functional genomic fingerprint of chronic stress in humans: blunted glucocorticoid and increased NF-kappa B signaling. Biol Psychiatry. 2008;64:266–272. | |
Avissar S, Nechamkin Y, Roitman G, Schreiber G. Reduced G protein functions and immunoreactive levels in mononuclear leukocytes of patients with depression. Am J Psychiatry. 1997;154:211–217. | |
O’Malley BW, Schrader WT, Mani S, et al. An alternative ligand independent pathway for activation of steroid receptors. Recent Prog Horm Res. 1995;50:333–347. | |
Rangarajan PN, Umesono K, Evans RM. Modulation of glucocorticoid receptor function by protein kinase A. Mol Endocrinol. 1992;6:1451–1457. | |
Miller AH, Pariente CM, Pearce BD. Effects of cytokines on glucocorticoid receptor expression and function: glucocorticoid resistance and relevance to depression. Adv Exp Med Biol. 1999;461:107–116. | |
Pariante CM, Pearce BD, Pisell TL, et al. The proinflammatory cytokine, interleukin-1 alpha, reduces glucocorticoid receptor translocation and function. Endocrinology. 1999;140:4359–4366. | |
Lehmann ML, Brachman RA, Martinowich K, Schloesse RJ, Herkenham M. Glucocorticoids orchestrate divergent effects on mood through adult neurogenesis. J Neurosci. 2013;33:2961–2972. | |
Feder A, Nestler EJ, Charney DS. Psychobiology and molecular genetics of resilience. Nat Rev Neurosci. 2009;10:446–457. | |
Castren E, Rantamaki T. The role of BDNF and its receptors in depression and antidepressant drug action: reactivation of developmental plasticity. Dev Neurobiol. 2010;70:289–297. | |
Castren E. Is mood chemistry? Nat Rev Neurosci. 2005;6:241–246. | |
Karege F, Bondolfi G, Gervasoni N, Schwald M, Aubry JM, Bertschy G. Low brain-derived neurotrophic factor (BDNF) levels in serum of depressed patients probably results from lowered platelet BDNF release unrelated to platelet reactivity. Biol Psychiatry. 2005;57:1068–1072. | |
Tuomisto J, Tukiainen E. Decreased uptake of 5-hydroxytryptamine in blood platelets from depressed patients. Nature. 1976;262:596–598. | |
Siuciak JA, Boylan C, Fritsche M, Altar CA, Lindsay RM. BDNF increases monoaminergic activity in rat brain following intracerebroventricular or intraparenchymal administration. Brain Res. 1996;710:11–20. | |
Koponen E, Rantamaki T, Voikar V, Saarelainen T, MacDonald E, Castren E. Enhanced BDNF signaling is associated with an antidepressant-like behavioral response and changes in brain monoamines. Cell Mol Neurobiol. 2005;25:973–980. | |
Saarelainen T, Hendolin P, Lucas G, et al. Activation of the TrkB neurotrophin receptor is induced by antidepressant drugs and is required for antidepressant-induced behavioral effects. J Neurosci. 2003;23:349–357. | |
Monteggia LM, Barrot M, Powell CM, et al. Essential role of brain-derived neurotrophic factor in adult hippocampal function. Proc Natl Acad Sci U S A. 2004;101:10827–10832. | |
Schatzberg AF. Pharmacological principles of antidepressant efficacy. Hum Psychopharmacol. 2002;17:S17–S22. | |
Delgado PL, Moreno FA. Role of norepinephrine in depression. J Clin Psychiatry. 2000;61 Suppl 1:5–12. | |
Appelhof BC, Huyser J, Verweii M, et al. Glucocorticoids and relapse of major depression (dexamethasone/corticotropin-releasing hormone test in relation to relapse of major depression). Biol Psychiatry. 2006;59:696–701. | |
Leitch MM, Ingram CD, Young AH, McQuade R, Gartside SE. Flattening the corticosterone rhythm attenuates 5HT1A autoreceptor function in the rat: relevance for depression. Neuropsychopharmacology. 2003;28:119–125. | |
Gartside SE, Leitch MM, Young AH. Altered glucocorticoid rhythm attenuates the ability of a chronic SSRI to elevate forebrain 5-HT: implications for the treatment of depression. Neuropsychopharmacology. 2003;28:1572–1578. | |
Johnson DA, Grant EJ, Ingram CD, Gartside SE. Glucocorticoid receptor antagonists hasten and augment neurochemical responses to a selective serotonin reuptake inhibitor antidepressant. Biol Psychiatry. 2007;62:1228–1235. | |
Johnson DA, Ingram CD, Grant EJ, Craighead M, Gartside SE. Glucocorticoid receptor antagonism augments fluoxetine-induced downregulation of the 5-HT transporter. Neuropsychopharmacology. 2009;34:399–409. | |
Heydendael W, Jacobson L. Differential effects of imipramine and phenelzine on corticosteroid receptor gene expression in mouse brain: potential relevance to antidepressant response. Brain Res. 2008;1238:93–107. | |
Heydendael W, Jacobson L. Glucocorticoid status affects antidepressant regulation of locus coeruleus tyrosine hydroxylase and dorsal raphe tryptophan hydroxylase gene expression. Brain Res. 2009;1288:69–78. | |
Buckley TM, Schatzberg AF. On the interactions of the hypothalamic-pituitary-adrenal (HPA) axis and sleep: normal HPA axis activity and circadian rhythm, exemplary sleep disorders. J Clin Endocrinol Metab. 2005;90:3106–3114. | |
Carotenuto M, Esposito M, Parisi L, et al. Depressive symptoms and childhood sleep apnea syndrome. Neuropsychiatr Dis Treat. 2012;8:369–373. | |
Novati A, Roman V, Cetin T, et al. Chronically restricted sleep leads to depression-like changes in neurotransmitter receptor sensitivity and neuroendocrine stress reactivity in rats. Sleep. 2008;31:1579–1585. | |
Starkman MN, Schteingart DE, Schork MA. Depressed mood and other psychiatric manifestations of Cushing’s syndrome: relationship to hormone levels. Psychosom Med. 1981;43:3–18. | |
Nehra R, Grover S, Bhansali ADM, Khehra N. Effect of corrective surgery on neurocognitive functions in endogenous cushing syndrome. Endocrinologist. 2009;19:205–207. | |
Lupien SJ, Maheu F, Tu M, Fiocco A, Schramek TE. The effects of stress and stress hormones on human cognition: implications for the field of brain and cognition. Brain and Cogn. 2007;65:209–237. | |
Austin MP, Ross M, Murray C, O’Carroll RE, Ebneier KP, Goodwin GM. Cognitive function in major depression. J Affect Disord. 1992;25:21–29. | |
Robinson LJ, Thompson JM, Gallagher P, et al. A meta-analysis of cognitive deficits in euthymic patients with bipolar disorder. J Affect Disord. 2006;93:105–115. | |
Martinez-Aran A, Vieta E, Reinares M, et al. Cognitive function across manic or hypomanic, depressed, and euthymic states in bipolar disorder. Am J Psychiatry. 2004;16:262–270. | |
Ferrier IN, Chowdhury R, Thompson JM, Watson S, Young AH. Neurocognitive function in unaffected first-degree relatives of patients with bipolar disorder: a preliminary report. Bipolar Disord. 2004;6:319–322. | |
Hinkelmann K, Moritz S, Muhtz C, et al. Cognitive deficits and salivary cortisol in major depression: effects of treatment. Eur Neuropsychopharmacol. 2010;20:S356–S357. | |
Watson S, Gallagher P, Ferrier IN, Young AH. Post-dexamethasone arginine vasopressin levels in patients with severe mood disorders. J Psychiatr Res. 2006;40:353–359. | |
van der Werf-Eldering MJ, Riemersma-van der Lek RF, Burger H, Holfhausen EA, Aleman A, Nolen WA. Can variation in hypothalamic-pituitary-adrenal (HPA)-axis activity explain the relationship between depression and cognition in bipolar patients? PLoS One. 2012;7:e37119. | |
Gomez RG, Posener JA, Keller J, DeBattista C, Solvason B, Schatzberg AF. Effects of major depression diagnosis and cortisol levels on indices of neurocognitive function. Psychoneuroendocrinology. 2009;34:1012–1018. | |
Murphy BE. Antiglucocorticoid therapies in major depression: a review. Psychoneuroendocrinology. 1997;22 Suppl 1:S125–S132. | |
Murphy BE, Ghadirian AM, Dhar V. Neuroendocrine responses to inhibitors of steroid biosynthesis in patients with major depression resistant to antidepressant therapy. Can J Psychiatry. 1998;43:279–286. | |
Ghadirian AM, Engelsmann F, Dhar V, et al. The psychotropic effects of inhibitors of steroid biosynthesis in depressed patients refractory to treatment. Biol Psychiatry. 1995;37:369–375. | |
Murphy BE, Dhar V, Ghadirian AM, Chaouinard G, Keller R. Response to steroid suppression in major depression resistant to antidepressant therapy. J Clin Psychopharmacol. 1991;11:121–126. | |
Loose DS, Kan PB, Hirst MA, Marcus RA, Feldman D. Ketoconazole blocks adrenal steroidogenesis by inhibiting cytochrome P450-dependent enzymes. J Clin Invest. 1983;71:1495–1499. | |
Anand A, Malison R, McDougle CJ, Price LH. Antiglucocorticoid treatment of refractory depression with ketoconazole: a case report. Biol Psychiatry. 1995;37:338–340. | |
Sonino N, Fava GA. Tolerance to antidepressant treatment may be overcome by ketoconazole. Report of two cases. J Psychiatr Res. 2003;37:171–173. | |
Thakore JH, Dinan TG. Cortisol synthesis inhibition – a new treatment strategy for the clinical and endocrine manifestations of depression. Biol Psychiatry. 1995;37:364–368. | |
Wolkowitz OM, Reus VI, Manfredi F, Ingbar J, Brizendine L, Weingartner H. Ketoconazole administration in hypercortisolemic depression. Am J Psychiatry. 1993;150:810–812. | |
Malison RT, Anand A, Pelton GH, et al. Limited efficacy of ketoconazole in treatment-refractory major depression. J Clin Psychopharmacol. 1999;19:466–470. | |
Wolkowitz OM, Reus VI, Chan T, et al. Antiglucocorticoid treatment of depression: double-blind ketoconazole. Biol Psychiatry. 1999;45:1070–1074. | |
Odwyer AM, Lightman SL, Marks MN, Checkley SA. Treatment of major-depression with metyrapone and hydrocortisone. J Affect Disord. 1995;33:123–128. | |
Rogoz Z, Skuza G, Wójcikowski J, et al. Effect of metyrapone supplementation on imipramine therapy in patients with treatment-resistant unipolar depression. Pol J Pharmacol. 2004;56:849–855. | |
Jahn H, Schick M, Kiefer F, Kellner M, Yassouridis A, Wiedemann K. Metyrapone as additive treatment in major depression – a double-blind and placebo-controlled trial. Arch Gen Psychiatry. 2004;61:1235–1244. | |
McAllister-Williams RH, Smith E, Anderson I, et al. Study protocol for the randomised controlled trial: Antiglucocorticoid augmentation of anti-Depressants in Depression (The ADD Study). BMC Psychiatry. 2013;13:205. | |
Belanoff JK, Rothschild AJ, Cassidy F, et al. An open label trial of C-1073 (mifepristone) for psychotic major depression. Biol Psychiatry. 2002;52:386–392. | |
Belanoff JK, Flores BH, Kalezhan M, et al. Rapid reversal of psychotic depression using mifepristone. J Clin Psychopharmacol. 2001;21:516–521. | |
DeBattista C, Belanoff J, Glass S, et al. Mifepristone versus placebo in the treatment of psychosis in patients with psychotic major depression. Biol Psychiatry. 2006;60:1343–1349. | |
Blasey CM, Debattista C, Roe R, Block T, Belanoff JK. A multisite trial of mifepristone for the treatment of psychotic depression: a site-by-treatment interaction. Contemp Clin Trials. 2009;30:284–288. | |
Blasey CM, Block TS, Belanoff JK, Roe RL. Efficacy and safety of mifepristone for the treatment of psychotic depression. J Clin Psychopharmacol. 2011;31:436–440. | |
Clinical Trials.gov. A study of mifepristone vs placebo in the treatment of patients with major depression with psychotic features. Trial identifier NCT00637494. Available from: http://clinicaltrials.gov/ct2/show/NCT00637494. Accessed August 7, 2014. | |
Young AH, Gallagher P, Watson S, Del-Estal D, Owen BM, Ferrier IN. Improvements in neurocognitive function and mood following adjunctive treatment with mifepristone (RU-486) in bipolar disorder. Neuropsychopharmacology. 2004;29:1538–1545. | |
Watson S, Gallagher P, Porter RJ, et al. A randomized trial to examine the effect of mifepristone on neuropsychological performance and mood in patients with bipolar depression. Biol Psychiatry. 2012;72:943–949. | |
Clinical Trials.gov. A randomized clinical trial of mifepristone in PTSD. Trial identifier NCT01946685. Available from: http://clinicaltrials.gov/ct2/show/NCT01946685. Accessed August 7, 2014. | |
Clinical Trials.gov. Medication development in alcoholism: investigating glucocorticoid antagonists. Trial identifier NCT01548417. Available from: http://clinicaltrials.gov/show/NCT01946685. Accessed August 7, 2014. | |
King’s College London. Double-blind, 36 month, placebo-controlled trial of mifepristone on cognition in alcoholics. London: King’s College London; 2014. Available from: http://www.kcl.ac.uk/ioppn/depts/addictions/research/alcohol/MIFCOG.aspx. Accessed October 15, 2014. | |
Clinical Trials.gov. A controlled trial of mifepristone in Gulf war veterans with chronic multisymptom illness. Trial identifier NCT00691067. Available from: http://clinicaltrials.gov/ct2/show/NCT00691067. Accessed August 7, 2014. | |
Clinical Trials.gov. Preliminary trial of the effect of glucocorticoid receptor antagonist on borderline personality disorder (BPD). Trial identifier NCT01212588. Available from: http://clinicaltrials.gov/ct2/show/NCT01212588. Accessed August 7, 2014. | |
Wolkowitz OM, Reus VI, Roberts E, et al. Dehydroepiandrosterone (DHEA) treatment of depression. Biol Psychiatry. 1997;41:311–318. | |
Wolkowitz OM, Reus VI, Keebler A, et al. Double-blind treatment of major depression with dehydroepiandrosterone. Am J Psychiatry. 1999;156:646–649. | |
Bloch M, Schmidt PJ, Danaceau MA, Adams LF, Rubinow DR. Dehydroepiandrosterone treatment of midlife dysthymia. Biol Psychiatry. 1999;45:1533–1541. | |
Rabkin JG, McElhiney MC, Rabkin R, McGrath PJ, Ferrando SJ. Placebo-controlled trial of dehydroepiandrosterone (DHEA) for treatment of nonmajor depression in patients with HIV/AIDS. Am J Psychiatry. 2006;163:59–66. | |
Arana GW, Forbes RA. Dexamethasone for the treatment of depression: a preliminary report. J Clin Psychiatry. 1991;52:304–306. | |
Bodani M, Sheehan B, Philpot M. The use of dexamethasone in elderly patients with antidepressant-resistant depressive illness. J Psychopharmacol. 1999;13:196–197. | |
Arana GW, Santos AB, Laraia MT, et al. Dexamethasone for the treatment of depression: a randomized, placebo-controlled, double-blind trial. Am J Psychiatry. 1995;152:265–267. | |
Dinan TG, Lavelle E, Cooney J, et al. Dexamethasone augmentation in treatment-resistant depression. Acta Psychiatr Scand. 1997;95:58–61. | |
Gutman DA, Owens MJ, Skelton KH, Thrivikraman KV, Nemeroff CB. The corticotropin-releasing factor 1 receptor antagonist R121919 attenuates the behavioral and endocrine responses to stress. J Pharmacol Exp Ther. 2003;304:874–880. | |
Zobel AW, Nickel T, Kunzel HE, et al. Effects of the high-affinity corticotropin-releasing hormone receptor 1 antagonist R121919 in major depression: the first 20 patients treated. J Psychiatr Res. 2000;34:171–181. | |
Kunzel HE, Ising M, Zobel AW, et al. Treatment with a CRH-1-receptor antagonist (R121919) does not affect weight or plasma leptin concentration in patients with major depression. J Psychiatr Res. 2005;39:173–177. | |
Kunzel HE, Zobel AW, Nickel T, et al. Treatment of depression with the CRH-1-receptor antagonist R121919: endocrine changes and side effects. J Psychiatr Res. 2003;37:525–533. | |
EvaluateTM [homepage on the Internet]. Available from: http://www.evaluategroup.com/default.aspx. Accessed September 10, 2014. | |
[No authors listed]. Should Neurocrine get stressed over CRF1 failure? Available from: http://www.epvantage.com/Universal/ View.aspx?type=Story&id=224081&isEPVantage=yes. Accessed August 7, 2014. | |
Binneman B, Feltner D, Kolluri S, Shi Y, Qiu R, Stiger T. A 6-week randomized, placebo-controlled trial of CP-316,311 (a selective CRH1 antagonist) in the treatment of major depression. Am J Psychiatry. 2008;165:617–620. | |
Coric V, Feldman HH, Oren DA, et al. Multicenter, randomized, double-blind, active comparator and placebo-controlled trial of a corticotropin-releasing factor receptor-1 antagonist in generalized anxiety disorder. Depress Anxiety. 2010;27:417–425. | |
[No authors listed]. NBI-77860. Neurocrine Biosciences. Available from: http://www.neurocrine.com/index.cfm?navId=24. Accessed August 7, 2014. | |
Clinical Trials.gov. A trial evaluating the efficacy and tolerability of SSR125543 in outpatients with major depressive disorder (AGATE). Trial identifier NCT01034995. Available from: http://clinicaltrials.gov/ct2/show/NCT01034995. Accessed August 7, 2014. | |
Clinical Trials.gov. Corticotropin-releasing hormone receptor 1 (CRH1) antagonism in anxious alcoholics. Trial identifier NCT01227980. Available from: http://clinicaltrials.gov/ct2/show/NCT01227980. Accessed August 7, 2014. | |
Clinical Trials.gov. Effects of a CRF1 receptor antagonist on human startle in normal female volunteers. Trial identifier NCT01059227. Available from: http://clinicaltrials.gov/ct2/show/NCT01059227. Accessed August 7, 2014. | |
Clinical Trials.gov. Evaluation of GSK561679 in women with post-traumatic stress disorder. Trial identifier NCT01018992. Available from: http://clinicaltrials.gov/ct2/show/NCT01018992. Accessed August 7, 2014. | |
Clinical Trials.gov. A study to compare the putative anxiolytic effect of 2 new drugs in subjects with social anxiety disorder. Trial identifier NCT00555139. Available from: http://clinicaltrials.gov/show/NCT00555139. Accessed August 7, 2014. | |
Kishimoto T, Radulovic J, Radulovic M, et al. Deletion of CRHR2 reveals an anxiolytic role for corticotropin-releasing hormone receptor-2. Nat Genet. 2000;24:415–419. | |
Hollenstein K, Kean J, Bortolato A, et al. Structure of class B GPCR corticotropin-releasing factor receptor 1. Nature. 2013;499:438–443. | |
Aguilera G, Rabadan-Diehl C. Vasopressinergic regulation of the hypothalamic-pituitary-adrenal axis: implications for stress adaptation. Regul Pept. 2000;96:23–29. | |
Raggenbass M. Vasopressin- and oxytocin-induced activity in the central nervous system: electrophysiological studies using in-vitro systems. Prog Neurobiol. 2001;64:307–326. | |
Hernando F, Schoots O, Lolait SJ, Burbach JP. Immunohistochemical localization of the vasopressin V1b receptor in the rat brain and pituitary gland: anatomical support for its involvement in the central effects of vasopressin. Endocrinology. 2001;142:1659–1668. | |
Simon NG, Guillon C, Fabio K, et al. Vasopressin antagonists as anxiolytics and antidepressants: recent developments. Recent Pat CNS Drug Discov. 2008;3:77–93. | |
Griebel G, Beeske S, Stahl SM. The vasopressin V(1b) receptor antagonist SSR149415 in the treatment of major depressive and generalized anxiety disorders: results from 4 randomized, double-blind, placebo-controlled studies. J Clin Psychiatry. 2012;73:1403–1411. | |
Sanofi Aventis. Press release. Available from: http://en.sanofi.com/Images/14212_08-07-31_T2_Results_EN.pdf. Accessed August 7, 2014. | |
ClinicalTrials.gov. Study of EVP-6124 (alpha-7 nAChR) as an adjunctive pro-cognitive treatment in schizophrenia subjects on chronic stable atypical antipsychotic therapy. Trial identifier NCT01716975. Available from: http://clinicaltrials.gov/ct2/show/NCT01716975. Accessed August 7, 2014. | |
Sullivan RM, Dufresne MM. Mesocortical dopamine and HPA axis regulation: role of laterality and early environment. Brain Res. 2006;1076:49–59. | |
Felt BT, Peirano P, Algarín C, et al. Long-term neuroendocrine effects of iron-deficiency anemia in infancy. Pediatr Res. 2012;71:707–712. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.