Back to Journals » International Journal of Nephrology and Renovascular Disease » Volume 9
Rodent models of diabetic nephropathy: their utility and limitations
Authors Kitada M, Ogura Y, Koya D
Received 13 August 2016
Accepted for publication 4 October 2016
Published 14 November 2016 Volume 2016:9 Pages 279—290
DOI https://doi.org/10.2147/IJNRD.S103784
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Editor who approved publication: Professor Pravin Singhal
Munehiro Kitada,1,2 Yoshio Ogura,2 Daisuke Koya1,2
1Division of Anticipatory Molecular Food Science and Technology, Medical Research Institute, 2Department of Diabetology and Endocrinology, Kanazawa Medical University, Uchinada, Ishikawa, Japan
Abstract: Diabetic nephropathy is the most common cause of end-stage renal disease. Therefore, novel therapies for the suppression of diabetic nephropathy must be developed. Rodent models are useful for elucidating the pathogenesis of diseases and testing novel therapies, and many type 1 and type 2 diabetic rodent models have been established for the study of diabetes and diabetic complications. Streptozotocin (STZ)-induced diabetic animals are widely used as a model of type 1 diabetes. Akita diabetic mice that have an Ins2+/C96Y mutation and OVE26 mice that overexpress calmodulin in pancreatic β-cells serve as a genetic model of type 1 diabetes. In addition, db/db mice, KK-Ay mice, Zucker diabetic fatty rats, Wistar fatty rats, Otsuka Long-Evans Tokushima Fatty rats and Goto-Kakizaki rats serve as rodent models of type 2 diabetes. An animal model of diabetic nephropathy should exhibit progressive albuminuria and a decrease in renal function, as well as the characteristic histological changes in the glomeruli and the tubulointerstitial lesions that are observed in cases of human diabetic nephropathy. A rodent model that strongly exhibits all these features of human diabetic nephropathy has not yet been developed. However, the currently available rodent models of diabetes can be useful in the study of diabetic nephropathy by increasing our understanding of the features of each diabetic rodent model. Furthermore, the genetic background and strain of each mouse model result in differences in susceptibility to diabetic nephropathy with albuminuria and the development of glomerular and tubulointerstitial lesions. Therefore, the validation of an animal model reproducing human diabetic nephropathy will significantly facilitate our understanding of the underlying genetic mechanisms that contribute to the development of diabetic nephropathy. In this review, we focus on rodent models of diabetes and discuss the utility and limitations of these models for the study of diabetic nephropathy.
Keywords: diabetic nephropathy, rodent model, albuminuria, mesangial matrix expansion, tubulointerstitial fibrosis
Introduction
The prevalence of diabetes mellitus has been increasing worldwide in recent years. Long-term diabetes results in vascular changes and dysfunction, and complications of diabetes are the major causes of morbidity and mortality in diabetic patients. Among the vascular complications of diabetes, diabetic nephropathy develops in 40% of patients and remains the leading cause of end-stage renal disease (ESRD) worldwide.1 Multifactorial management, including diet therapy and glycemic, blood pressure (BP), and lipid control, is recommended for diabetic nephropathy.2 Some patients with advanced diabetic nephropathy rapidly progress to ESRD, despite having received adequate multifactorial treatment. Therefore, a novel and more beneficial treatment to suppress diabetic nephropathy has to be developed.
In humans, diabetic nephropathy manifests as a clinical syndrome consisting of albuminuria, a progressive decline in renal function, and an increased risk of developing cardiovascular disease.2 Clinically, albuminuria is the hallmark biomarker for the diagnosis of diabetic nephropathy, and it is also used to stage diabetic nephropathy. The histopathological features of diabetic nephropathy in humans include glomerular hypertrophy, a thickening of the glomerular basement membrane, and mesangial matrix expansion, including the formation of glomerular nodular lesions known as Kimmelstiel-Wilson nodules.3,4 In addition to glomerular lesions, tubulointerstitial lesions contribute significantly to the decline in renal function in human diabetic nephropathy.The decline in renal function is associated with both glomerulosclerosis and tubulointerstitial fibrosis. In particular, the degree of tubulointerstitial fibrosis, rather than the number of glomerular lesions, is recognized as the most important predictive factor for progressive renal decline.5,6 Therefore, an animal model of diabetic nephropathy should exhibit this characteristic of renal histology in the glomeruli and tubulointerstitial area, which is associated with increased albuminuria and a decline in renal function.
The nephropathy subcommittee of the Animal Models of Diabetic Complications Consortium (AMDCC) has published the following validation criteria for rodent models of diabetic nephropathy based on the clinical and pathological features of human diabetic nephropathy: 1) >50% decrease in renal function, 2) >10-fold increase in albuminuria, and 3) pathological features, including advanced mesangial matrix expansion (± nodules), thickening of the glomerular basement membrane, arteriolar hyalinosis, and tubulointerstitial fibrosis.7 A rodent model that completely develops all of these features of human diabetic nephropathy is not currently available. However, the available rodent models of diabetes can be useful in the study of diabetic nephropathy by increasing our understanding of the features of each diabetic rodent model. In this review, we focus on streptozotocin (STZ)-induced type 1 diabetic and genetically modified type 1 or type 2 diabetic rodent models, and we discuss the utility and limitations of these models for the study of diabetic nephropathy.
Rodent models of type 1 diabetes
STZ-induced type 1 diabetes in mice
STZ, which is toxic to pancreatic β-cells, results in absolute insulin deficiency and is widely used for the induction of experimental type 1 diabetes in rodents.8 In addition to pancreatic β-cells, STZ may also have toxic effects on other organs, including the kidney.9 Therefore, the administration of high doses of STZ (150–200 mg/kg) to mice should be avoided. To minimize the nonspecific toxicity of STZ, multiple injections of low doses of STZ have been used to induce diabetes by causing repetitive low-grade cell damage accompanied by the lymphocytic infiltration of the pancreatic islets.10 This method consists of daily intraperitoneal injections of 40–50 mg/kg of STZ for 5 consecutive days and is recommended in the AMDCC protocol. At later stages, STZ-induced diabetic mice may exhibit significant weight loss, possibly as a result of the catabolic effects of insulin deficiency and severe hyperglycemia, as well as the volume depletion associated with osmotic diuresis. Furthermore, STZ-induced diabetic mice do not develop hypertension. For long-term studies, intermittent treatment with small amounts of insulin that are sufficient to reverse weight loss without reversing hyperglycemia may be considered.
The STZ-induced mouse model of diabetes is widely used for numerous studies in the field of diabetes and diabetic complications. However, previous reports have shown that differences in susceptibility to diabetic nephropathy exist in mice based on the genetic background and strain.7 Therefore, when the STZ-induced mice model of diabetes is used to study diabetic nephropathy, we must consider the genetic background and strain of the mice and the features of kidney injury, including albuminuria/proteinuria and histological changes.
C57BL/6
C57BL/6 mice are the most commonly used strain in preclinical research, and many genetic modifications have been performed on this background because these mice breed well, have long life spans, and have a low susceptibility to tumors.7 However, this strain is relatively resistant to the development of renal injury in experimental models of kidney diseases, including diabetic nephropathy.7 C57BL/6 mice with STZ-induced diabetes exhibit mild to moderate albuminuria at 6 months after the induction of diabetes, but the increase is less than 10-fold compared to non-diabetic control mice.11 Qi et al12 have also shown that control C57BL/6J mice exhibit a urinary albumin/creatinine (Cr) ratio of 45.6 ± 12.4 before vehicle injection, 20.1 ± 1.6 at 5 weeks, 48.11 ± 16.5 at 15 weeks, and 53.2 ± 24.3 μg/mg Cr at 25 weeks after vehicle injection; in contrast, STZ-induced diabetic C57BL/6J mice exhibit a higher urinary albumin/Cr ratio of 36.9 ± 7.6 before the induction of diabetes, and the ratios are 35.4 ± 5.7 at 5 weeks, 29.5 ± 12.4 at 15 weeks, and 72.1 ± 20.4 μg/mg Cr at 25 weeks after the induction of diabetes, which were higher than those of the control C57BL/6J mice.12 Renal pathological changes, including glomerular hypertrophy, a thickening of the glomerular basement membrane, and mild to moderate mesangial matrix expansion, were observed in STZ-induced diabetic C57BL/6 mice at more than 6 months after the induction of diabetes.11,12 Oxidative stress and inflammation, which are strongly implicated in the pathogenesis of diabetic nephropathy, have also been observed in the kidneys of STZ-induced diabetic C57BL/6 mice.13,14 However, albuminuria and renal pathological changes are less commonly observed in diabetic C57BL/6 mice than in other strains, such as the DBA/2 and CD1 mice that are described in the later text.7,11,12 In addition, although renal tubular damage, such as enlargement of the tubule and tubular cell atrophy, has been observed in diabetic C57BL/6 mice, tubulointerstitial fibrosis does not occur.11 Thus, STZ-induced diabetic C57BL/6 mice may be useful for elucidating the early pathogenesis of diabetic nephropathy. However, C57BL/6 mice are usually less susceptible to STZ-induced, diabetes-associated renal injury than other strains, such as DBA/2 and CD1 mice.
DBA/2
STZ-induced diabetic DBA/2 mice exhibit albuminuria at 5 weeks after the induction of diabetes and develop more marked albuminuria after 25 weeks more compared to C57BL/6 mice.12 In detail, control DBA/2 mice exhibit urinary albumin/Cr ratios of 19.7 ± 5.1 before vehicle injection, 48.7 ± 7.0 at 5 weeks, 71.1 ± 15.2 at 15 weeks, and 65.8 ± 13.2 μg/mg Cr at 25 weeks after vehicle injection. In contrast, STZ-induced diabetic DBA/2 mice exhibit higher urinary albumin/Cr ratios of 26.6 ± 6.6 before the induction of diabetes, 424.4 ± 89.4 at 5 weeks, 608.0 ± 220.8 at 15 weeks, and 421.4 ± 167.3 μg/mg Cr at 25 weeks after induction of diabetes. Additionally, diabetic DBA/2 mice exhibit some of the pathological features of human diabetic nephropathy including nodular glomerulosclerosis, arteriolar hyalinosis, thickening of the glomerular basement membrane, and severe mesangial matrix expansion at 25 weeks after the induction of diabetes. Furthermore, renal tubular damage, including enlargement of the tubule and tubular cell atrophy, is observed in STZ-induced diabetic DBA mice. However, tubulointerstitial fibrosis does not occur.11 Although diabetic C57BL/6 mice can live at more than 45 weeks after the onset of hyperglycemia, the mortality rate of diabetic DBA/2 mice is markedly increased after 25 weeks of hyperglycemia, with 40% of these mice dying by this time.12 Thus, STZ-induced diabetic DBA/2 mice are highly susceptible to renal injury compared with C57BL/6 mice, and they may be useful as a model of diabetic nephropathy.
CD1
Sugimoto et al11,15 have reported that STZ-induced diabetic CD1 mice develop chronic renal injury associated with tubulointerstitial fibrosis and decreased renal function within 6 months after a single intraperitoneal injection of STZ (200 mg/kg). They also compared the development of renal histology and albuminuria in the STZ-induced diabetic CD1 mice to the development of these conditions in the C57BL/6 and 129/Sv strains. All these mice strains treated with STZ exhibited glomerular lesions, including moderate mesangial matrix expansion, increased glomerular surface area, and tubular cell damage. However, nodular lesions in the glomeruli were not observed. The levels of albuminuria, which were estimated using the urinary albumin/Cr ratio, were moderately and more than 10-fold higher in the STZ-induced CD1 mice than in the control CD1 mice. In contrast, STZ-injected C57BL/6J and 129/Sv mice exhibited a less than 10-fold increase in the urinary albumin/Cr ratio compared with the control mice.11 Tubulointerstitial fibrosis developed in the STZ-induced diabetic CD1 mice, but not in the C57BL/6 and 129/Sv mice. Although the reasons why the diabetic CD1 mice exhibited tubulointerstitial fibrosis are not clear, genetic background is an important factor in the utility of an animal model for elucidating the mechanisms of advanced diabetic nephropathy. Therefore, STZ-induced diabetic CD1 mice may be a useful model for investigating the pathologies associated with advanced human diabetic nephropathy.
129/Sv
Together with C57BL/6 mice, 129/Sv mice are one of the most commonly used strains for studying diseases. The 129/Sv strain develops more significant glomerulosclerosis, proteinuria, increases in BP, and apparent renal failure compared with C57BL/6 mice after 5/6 nephrectomy.16 In studies of hypertension induced with deoxycorticosterone acetate (DOCA) salt, 129/Sv mice were more susceptible to elevations in BP, glomerular sclerosis, tubulointerstitial fibrosis, and albuminuria than C57BL/6 mice.17 Susceptibility to the development of hypertension and further glomerulosclerosis in 129/Sv and C57BL/6 strains may be influenced by renin gene polymorphisms because some mouse strains, including C57BL/6, have one renin gene (Ren-1c), whereas other strains, including 129/Sv, have two renin genes (Ren-1d> and Ren-2).18 Therefore, the accelerated renal injuries, such as glomerular sclerosis and tubulointerstitial damage after 5/6 nephrectomy or the induction of DOCA salt hypertension, that are observed in 129/Sv mice may be mediated through both the hemodynamic and nonhemodynamic effects of the increased renin levels and the activation of the renin–angiotensin–aldosterone system. However, no reports have examined whether this susceptibility to renal injury and hypertension in 129/Sv contributes to the advanced progression of diabetic nephropathy compared to C57BL/6 and other strains of mice. Previous reports have shown that 129/Sv mice with STZ-induced diabetes exhibit mild to moderate albuminuria, but there is a less than 10-fold increase in the urinary albumin/Cr ratio compared with control mice.11 Renal histological changes observed in STZ-induced diabetic 129/Sv mice include a mild to moderate increase in mesangial matrix accumulation and tubular damage, which were also observed in STZ-induced diabetic C57BL/6 mice.11 Tubulointerstitial fibrosis was not observed in STZ-induced diabetic 129/Sv mice, which was also similar to the C57BL/6 and DBA/2 mice with diabetes. Thus, 129/Sv mice exhibit a lower susceptibility to STZ-induced diabetic renal injury with histological changes than DBA/2 and CD1 mice.
STZ-induced diabetic rats
STZ-induced diabetic rats have also been used to study diabetic nephropathy. Diabetes is usually induced in male Sprague-Dawley (S-D) or other rats weighing 170–200 g by a single intravenous injection of STZ (50–55 mg/kg body weight).19,20 Mima et al21 demonstrated that urinary albumin excretion in STZ-induced diabetic S-D rats (12 ± 1.3 mg/day) was higher than in control rats (1.0 ± 0.5 mg/day) at 24 weeks after the induction of diabetes.21 Renal function evaluated by creatinine clearance (Ccr) showed an increase in diabetic rats (26 ± 9.0 mL/day/100 g body wt) compared with control rats (8.3 ± 0.2 mL/day/100 g body wt). The BPs were not significantly different between the STZ-induced diabetic and control rats, and the STZ-induced diabetic rats did not develop hypertension. Morphologically, the diabetic rats exhibited an increase in mesangial matrix proteins, mesangial matrix fraction, and accumulation of type 4 collagen compared to the control rats. However, no severe mesangial matrix accumulation, nodular lesions in the glomeruli, severe tubular cell damage, or tubulointerstitial fibrosis was observed. Additionally, oxidative stress and inflammation were observed in the kidneys of diabetic rats.19,20,22 Thus, STZ-induced diabetic rats may be useful as a model of early changes in diabetic nephropathy. However, the hyperglycemia-induced morphological changes in the kidneys of this rat model were less significant than those observed in the STZ-induced diabetic mice.
Genetic models of type 1 diabetes in mice
Akita Ins2+/C96Y mutant mice
Akita mice have Ins2+/C96Y mutation, which is a single nucleotide substitution in the Ins2 gene.23 The Ins2+/C96Y mutation causes abnormal folding of the insulin protein and toxic injury to pancreatic β-cells, as well as a diminished capacity to secrete insulin, which results in the development of type 1 diabetes. Originally, Akita diabetic mice of the C57BL/6 strain developed modest levels of albuminuria and modest structural changes, including an increase in the mesangial matrix, thickening of glomerular basement membrane, and depletion of podocytes, which were partially due to increased apoptosis.24,25 Additionally, many studies have reported oxidative stress and inflammation in the kidneys of Akita Ins2+/C96Y C57BL/6 mice, which play an important role in the pathogenesis of diabetic nephropathy.26–30 However, the genetic background of Akita mutation mice contributes to the severity of albuminuria and histological changes. Gurley et al31 studied the effect of breeding the Ins2+/C96Y mutation into the DBA/2 and 129/SvEv strains on susceptibility to diabetic nephropathy.31 They found that C57BL/6, DBA/2, and 129/SvEv mice with the Akita mutation developed similar degrees of hyperglycemia, but they differed in their manifestations of diabetes-induced renal injury. The DBA/2-Ins2+/C96Y mice exhibited more albuminuria (345 ± 110 μg/day) than the other strains (129/SvEv-Ins2+/C96Y: 151 ± 77 μg/day; C57BL/6-Ins2+/C96Y: 40 ± 3 μg/day), but only the C57BL/6 and 129/SvEv mice exhibited a moderate increase in mesangial matrix accumulation at 6 months of age.31 Significant strain differences in baseline BPs also existed in the nondiabetic controls. Systolic BPs were higher in both the nondiabetic 129/SvEv-Ins2+/+ (130 ± 4 mmHg) and DBA/2-Ins2+/+ animals (134 ± 2 mmHg) compared with the C57BL/6-Ins2+/+ group (103 ± 2 mmHg). The effects of diabetes on BP also varied among the strains. Systolic BPs were significantly higher in the C57BL/6-Ins2+/C96Y (112 ± 4 vs. 103 ± 2 mmHg) and 129/SvEv-Ins2+/C96Y groups (143 ± 3 vs. 130 ± 4 mmHg) compared to their respective Ins2+/+ wild-type mice. In contrast, there were no differences in BP between the 6-month-old DBA/2-Ins2+/C96Y mice and the DBA/2-Ins2+/+ wild-type controls (134 ± 3 vs. 134 ± 2 mmHg).31 Regardless of the strain, the Akita mice exhibited none of the structural alterations that are observed in advanced human diabetic nephropathy, such as mesangiolysis, widespread marked or nodular mesangial sclerosis, and tubulointerstitial fibrosis. Thus, Akita Ins2+/C96Y mutation mice may be a useful model of early to moderately advanced renal morphological changes in diabetic nephropathy. However, the susceptibility to diabetic nephropathy is dependent on the strain of the mouse.
Furthermore, Haseyama et al32 reported that Akita mice with a C57BL/6 genetic background develop diffuse granular mesangial deposits of immunoglobulin A (IgA) starting at 20 weeks of age.32 Compared to controls, male Akita diabetic mice, but not female mice, develop impaired renal function with an elevation of serum IgA after 30 weeks of age. However, Akita mice with other genetic backgrounds have not been evaluated for mesangial depositions of IgA. The mesangial deposition of IgA is a major cause of mesangial proliferative glomerulonephritis. Therefore, using Akita mice of the C57BL/6 strain may be problematic and may limit the evaluation of diabetic nephropathy-induced alterations in mesangial area and mesangial matrix accumulation. Therefore, Akita mice are a unique mouse model that exhibits diabetes-related glomerular lesions, including mesangial sclerosis, and possibly IgA nephropathy.
OVE26 FVB mice
OVE26 mice are characterized by the transgenic overexpression of calmodulin in pancreatic β-cells, which results in the development of type 1 diabetes due to deficient insulin production within the first week of life.33 Male heterozygous OVE26 diabetic mice are bred with female wild-type FVB mice, and the resulting OVE26 mice exhibit significant albuminuria by 2 months of age. Albuminuria increases progressively with age and is markedly increased at 9 months of age (average urinary albumin excretion exceeding 15 mg/24 hr). The glomerular filtration rate (GFR) of OVE26 mice significantly increases from 2 to 3 months of age and then decreases significantly from 5 to 9 months, and the GFR of 9-month-old diabetic mice is significantly lower than that of 9-month-old control mice. Diastolic BP, as measured with a tail cuff, has been shown to be elevated in conscious OVE26 mice at 3 months of age compared to non-diabetic mice. By 8 months of age, OVE26 mice exhibit significant elevations in both diastolic and systolic BP compared to non-diabetic mice (approximately 130/90 mmHg vs. 120/80 mmHg). The kidney weights of OVE26 mice show an almost twofold increase between 2 and 5 months. Diabetic OVE26 mice also exhibit progressively enlarged glomeruli, a thickening of the glomerular basement membrane, podocyte loss, and a marked increase in mesangial area with diffuse and nodular expansion of the mesangial matrix.34 Additionally, tubulointerstitial fibrosis has been observed in diabetic OVE26 mice. Diabetes-induced oxidative stress and inflammation in the kidneys of OVE26 mice have also been observed and are related to the pathogenesis of diabetic nephropathy.30 OVE26 mice exhibit most of the characteristics of advanced human diabetic nephropathy with chronic hyperglycemia. Therefore, this mouse model may increase our understanding of the pathophysiology of diabetes and lead to treatments for diabetic nephropathy. However, difficulties with the viability of these mice have been reported.35
Rodent models of type 2 diabetes
Mouse model
db/db mice
db/db mice are the most widely used model of type 2 diabetes.36 They have a deletion mutation in the leptin receptor (LepRdb/db) that results in abnormal splicing and a defective receptor for the adipocyte-derived hormone leptin.37 The defect in leptin signaling produced by the LepRdb/db deletion affects hypothalamic responses, leading to the development of hyperphagia, obesity, hyperlipidemia, hyperinsulinemia, insulin resistance, and diabetes. Diabetes is more severe in male mice than in female mice. The LepRdb/db mutation was originally recognized in the C57BLKS/J strain and was modified to contain part of the DBA/2 strain,38 which exhibits several features of human diabetic nephropathy, because the identical mutation in mice of the C57BL/6 strain does not result in significant diabetic nephropathy. Male C57BLKS/J db/db mice rapidly develop hyperglycemia at 6–10 weeks of age and moderate to severe albuminuria at 8–25 weeks of age. Moderate to marked albuminuria at 8–25 weeks ranges from 68 to 600 μg/24 hr in the db/db male mouse model,39–46 whereas it is between 4 and 44 μg/24 hr in age-matched, heterozygous littermates, non -diabetic db/m (m+/+ LepRdb).7 The degree of albuminuria does not consistently increase with the duration of diabetes, as similar levels of albuminuria have been observed between 8 and 25 weeks.38,43 Renal function in these diabetic mice declines at 15–18 weeks, and their islet cells are sensitive to the toxic effects of hyperglycemia. These mice eventually become dependent on insulin administration to control their increasing blood glucose levels and survive. db/db mice at 17 weeks of age show no obvious hypertension, but the mean blood pressures of db/db mice (102.8 ± 6.56 mmHg) are significantly higher than those of db/m mice (94.7 ± 5.3 mmHg).44 db/db mice of the C57BLKS/J strain develop progressive histological changes in their kidneys, including an increase in glomerular basement thickening, podocyte loss,25 and moderate mesangial matrix expansion.43,44 Additionally, numerous studies have reported oxidative stress and inflammation in the kidneys of db/db mice.44,47,48 However, db/db mice do not develop mesangiolysis, nodular mesangial sclerosis, or severe tubulointerstitial fibrosis, which are features of advanced diabetic nephropathy in humans. They also do not develop progressive renal insufficiency. Therefore, db/db mice of the C57BLKS/J strain are a useful model of early to moderately advanced morphological changes in the kidney that are associated with diabetic nephropathy.
KK and KK-Ay mice
The KK strain was originally established from the inbreeding of a Japanese mouse by Kondo et al.49 They exhibit mild insulin resistance and obesity, which is more severe in male mice than in female mice.50,51 KK mice develop albuminuria after 10–15 weeks of age,52 but the factors contributing to albuminuria in KK mice have not yet been fully elucidated. STZ-induced diabetic KK/H1J mice exhibit a further increase in albuminuria, which indicates that this strain is also susceptible to albuminuria in a diabetic state. Control KK/H1J mice exhibit urinary albumin/Cr ratios of 72.8 ± 11.4 before vehicle injection, 177.0 ± 50.7 at 5 weeks, 224.4 ± 80.8 at 15 weeks, and 349.1 ± 39.7 µg/mg Cr at 25 weeks. In contrast, STZ-induced diabetic KK/H1J mice exhibit higher urinary albumin/Cr ratios of 72.9 ± 12.1 before STZ injection, 509.9 ± 87.6 at 5 weeks, 586.9 ± 84.9 at 15 weeks, and 635.8 ± 122.0 μg/mg Cr at 25 weeks after STZ injection.12 KK mice exhibit a mild increase in mesangial matrix expansion and glomerular basement membrane thickening. They also appear to be predisposed to the development of renal lesions of diabetic nephropathy. Regardless, these mice exhibit only mild insulin resistance and a small elevation in blood glucose levels.52 The phenotypic characteristics of KK/H1J mice are not particularly significant, but STZ-induced diabetic KK/H1J mice exhibit severe mesangial matrix expansion, nodular glomerulosclerosis, and arteriolar hyalinosis 35 weeks after the induction of diabetes that is more severe than in the uninjected KK/H1J mice.12 The KK-Ay mouse was developed by Nishimura et al in 1969 by transferring the yellow obese gene (Ay allele) into the KK mouse.53 KK-Ay mice are more severely obese and are more likely to develop hyperglycemia and albuminuria (median urinary albumin/Cr ratio: 1,086 mg/g Cr, 25th percentile: 942, 75th percentile 1,310 mg/g Cr) at 16 weeks of age than KK mice (median urinary albumin/Cr ratio: 23 mg/g Cr, 25th percentile: 18, 75th percentile: 28 mg/g Cr) at 16 weeks of age.54 Therefore, these mice are widely used as an experimental model of type 2 diabetes.54,55 Previous reports have also demonstrated an increase in systolic BP in KK-Ay mice (123 ± 3 mmHg) compared with non-diabetic KK mice (112 ± 4 mmHg) at 16 weeks of age, but no obvious hypertension in KK-Ay mice.54 The histological changes observed in the kidneys of KK-Ay mice include diffuse and moderate to severe mesangial matrix expansion with mesangial cell proliferation, segmental sclerosis, nodular mesangial lesions, and decreased podocyte numbers,54,55 and these changes are more severe than in KK mice. Additionally, oxidative stress and inflammation have been observed in the kidneys of KK-Ay mice,56,54 and this strongly contributes to the pathogenesis of kidney injury. However, STZ-induced diabetic KK mice and KK-Ay mice exhibit no tubulointerstitial fibrosis. Therefore, although albuminuria progressively increases with age in the KK-Ay mouse model, these mice do not develop renal insufficiency or failure. The pathological changes in the glomeruli of KK-Ay mice were consistent with those observed in the early stages of human diabetic nephropathy. It appears that KK-Ay mice are a suitable animal model of the early to moderate stages of type 2 diabetic nephropathy, particularly regarding the histopathological findings in glomeruli.
High-fat diet-induced type 2 diabetic mice
High-fat diet (HFD) feeding is known to induce various systemic metabolic alterations in mice, including obesity, insulin resistance, hyperglycemia, and abnormal lipid profiles. These alterations are similar to those observed in patients with metabolic syndrome, and type 2 diabetes is closely related to metabolic syndrome. One study has shown that there are increases in the body weights and blood glucose levels of C57BL/6 mice on a HFD compared with those of mice on a low-fat diet (LFD) at 4, 8, and 12 weeks after the initiation of the diet intervention.57 The systolic BPs were also significantly higher in the HFD-fed mice than in the LFD-fed mice at 8 and 12 weeks after initiation of the diet intervention, but the HFD-fed mice exhibited no hypertension. After the onset of metabolic syndrome at 12 weeks, C57BL/6 mice on a HFD exhibited a mild increase in urinary albumin excretion and histological changes in their kidneys, such as the accumulation of extracellular matrix proteins, glomerular basement thickening, oxidative stress, and inflammation. However, systemic lipid overload (lipotoxicity), rather than hyperglycemia, may be related to the renal lipid accumulation and renal injuries observed in this model, including mesangial area expansion, interstitial fibrosis, and albuminuria. Therefore, attention should be paid to these factors when using HFD-induced type 2 diabetic mice as a model of diabetic nephropathy.
Rat models
Zucker diabetic fatty rats and Wistar fatty rats
The Zucker fatty (ZF) rat harbors a homozygous missense mutation (fatty, fa) in the leptin receptor gene (Lepr) and develops obesity without diabetes. Zucker diabetic fatty (ZDF) rats, which are derived from the ZF strain, exhibit obesity with diabetes and are widely used in the study of type 2 diabetes.58–60 The Wistar fatty (WF) rat is a congenic strain of the Wistar Kyoto (WKY) rat that also has a fa/fa homozygous missense mutation in the Lepr gene. This strain develops obesity and obesity-related features, such as hyperinsulinemia and hyperlipidemia, in the same manner as ZF rats.61–63 The diabetes-induced changes appear to be caused by the combination of a predisposition for diabetes in the WKY rat and fa-induced obesity. Because fa/fa homozygous animals are infertile, mating between fa/+ heterozygous animals is required to maintain the production of fa/fa homozygous strains.
ZDF rats
ZDF rats develop progressive insulin resistance and glucose intolerance between 3 and 8 weeks of age and become overtly diabetic between 8 and 10 weeks of age. The level of albuminuria in male ZDF rats is slightly higher than in control lean rats at 6 weeks of age (1.4 ± 0.4 [control lean rats] vs. 0.5 ± 0.1 mg/day [ZDF rats]).64,65 After that, albuminuria in the ZDF rats increases progressively with aging (reaching 418 ± 57 [range 107–761] mg/day by 36 weeks of age). Systolic BPs are within the normal range in both control lean and ZDF rats from 6 weeks to 36 weeks of age.65 The GFR increases until 12 weeks of age but then falls to the level of the control lean rats by 28 weeks of age. Slight renal hypertrophy is observed at 12 weeks of age, and renal hypertrophy is more prominent by 16 weeks of age. Pathological changes, such as glomerulosclerosis, tubular cell damage, tubulointerstitial fibrosis, and inflammation, are observed in male ZDF rats as they age. Although ZDF rats do not exhibit glomerulosclerosis, tubular cell damage, or tubulointerstitial fibrosis at 8 weeks of age, the glomeruli of ZDF rats appear slightly hypertrophic at 8 weeks of age compared with the control lean rats. Thereafter, they exhibit a further increase in glomerular lesions that is accompanied by moderate to severe mesangial matrix expansion at 22 weeks of age.66 Furthermore, tubular cell damage, moderate to severe tubulointerstitial fibrosis, inflammation, and hypoxia are observed in the kidneys of ZDF rats from 22 to 39 weeks of age.67 Therefore, the ZDF rat is a useful rodent model of diabetic nephropathy. However, ZDF rats do not exhibit glomerular nodular lesions or mesangiolysis, which are observed in advanced cases of human diabetic nephropathy.
Additionally, ZDF rats and lean control rats exhibit hydronephrosis.68,69 Baynes and Murray68 reported that 14% of ZDF rats show signs of moderate levels of hydronephrosis at 16 weeks of age. At 36 weeks of age, a shift from mostly mild (28%) to moderate (77%) hydronephrosis is also apparent in the lean control group. Furthermore, 36-week-old ZDF rats exhibit predominately moderate (60%) to severe (30%) signs of hydronephrosis.68 However, it is not clear whether hydronephrosis in ZDF rats is associated with diabetes-induced renal injuries, including albuminuria and histological changes.
WF rats
Similar to the ZDF rat, WF rats develop progressive insulin resistance, glucose intolerance, and obesity between 3 and 8 weeks of age and become overtly diabetic between 8 and 10 weeks of age. However, the diabetic state of WF rats is mild compared to ZDF rats. In WF rats, increased albuminuria (a urinary albumin/Cr ratio of 2.3 ± 3.1 [control lean rats] vs. 118.5 ± 161.9 mg/mmol [WF rats]) and urinary liver-type fatty acid-binding protein excretion are observed by 24 weeks, and a markedly progressive increase in these urinary biomarkers develops through 44 weeks of age (a urinary albumin/Cr ratio of 52.9 ± 27.4 [control lean rats] vs. 1,377.0 ± 968.0 mg/mmol [WF rats]),62 which is accompanied by increased plasma cystatin C levels. The mean BPs indicate no obvious hypertension in both control lean and WF rats at 44 weeks of age.62 Kidney and glomerular hypertrophy has been observed at 24 and 44 weeks of age, respectively. Histologically, WF rats exhibit increased glomerular basement thickening, foot process effacement of the podocytes, mesangial area expansion, tubulointerstitial inflammation, tubular cell damage, and tubulointerstitial fibrosis at 24 weeks. These histological changes in the kidneys of WF rats progressively increase through 44 weeks of age.62 Therefore, diabetes-induced renal injuries in WF rats are progressive for moderate to severe glomerular lesions and severe tubulointerstitial fibrosis in advanced stages of diabetic nephropathy. Thus, WF rats may be a useful rodent model for advanced stages of diabetic nephropathy. However, the nodular lesions in the mesangial area and the mesangiolysis that are observed in human diabetic nephropathy cases do not occur in the kidneys of WF rats, which is similar to observations from ZDF rats.
Otsuka Long-Evans Tokushima Fatty rats
The Otsuka Long-Evans Tokushima Fatty (OLETF) rat is an established model of type 2 diabetes.70 In male OLETF rats, impaired glucose tolerance is observed from 8 weeks of age, and the plasma glucose levels increase from 18 weeks of age. Hyperglycemia and hyperinsulinemia are exhibited in the early phases of the disease as a result of islet cell hyperplasia and peripheral insulin resistance. Nearly 100% of male OLETF rats develop a diabetic state by 25 weeks of age.71 OLETF rats eventually develop hypoinsulinemia with continued aging (>40 weeks of age) as a result of the deterioration of islet beta cells.72,73 Urinary albumin and protein excretion in male OLETF rats is higher than in lean rats at 10 weeks of age, and there is a progressive two to threefold increase in albuminuria and proteinuria from 20 to 30 weeks of age. At 36 weeks of age, albuminuria in OLETF rats reaches a level of ~200 mg/day.74 Moreover, the GFR is increased compared to that of control lean rats at 30 weeks of age. Male OLETF rats exhibit pathological changes, including diffuse glomerulosclerosis and nodular lesions, which progress with age and the duration of the diabetic state.75 There are no obvious alterations in the glomeruli at 15 weeks of age, and mesangial cell proliferation is observed by 25 weeks of age. After 40 weeks of age, moderate mesangial matrix expansion accompanied by accumulation of the extracellular matrix and thickening of the glomerular capillary walls are observed, which suggests diffuse glomerulosclerosis. Moreover, renal hypertrophy is observed at 55 weeks of age. Thereafter, expansion of the glomerular nodular lesions and a severe increase in mesangial matrix expansion are observed at 65 weeks of age.70 Tubulointerstitial damages, such as dilated and flat tubular cells, mononuclear cell filtration, and fibrosis around atrophic tubules, is also observed at this time. Thus, OLETF rat may be a useful type 2 diabetic model for investigating advanced diabetic nephropathy.
Goto-Kakizaki rats and a genetically modified GK substrain (T2DN/Mcwi) of rats
GK rats
The Goto-Kakizaki (GK) rat is a non-obese model of type 2 diabetes.76 The GK strain was developed from a colony of Wistar rats through the selective breeding over many generations of rats with elevated blood glucose levels during a glucose tolerance test. GK rats exhibit glucose intolerance as early as 2 weeks of age and exhibit elevated plasma glucose levels after the administration of a glucose load by 4 weeks of age. This is due to the impaired development of islet cells that secrete insulin in response to glucose, as well as insulin resistance.77 By 12 weeks of age, GK rats develop type 2 diabetes characterized by elevated fasting glucose and insulin levels and a prolonged elevation of plasma glucose levels.
Previous studies have demonstrated that GK rats are relatively resistant to the development of diabetic nephropathy.78 Several studies have reported that GK rats exhibit the common histological changes in the kidneys that are observed in other animal models of diabetes, including thickening of glomerular basement membranes, mild to moderate mesangial matrix expansion, glomerular hypertrophy, oxidative stress, and inflammation.79–81 However, even older GK rats do not exhibit progressive renal damage, which is characterized by the development of global glomerulosclerosis, marked mesangial matrix expansion, nodular formation, progressive proteinuria, tubulointerstitial fibrosis, and renal dysfunction.82
T2DN/Mewi rats
Nobrega et al83 reported on a genetically modified substrain of GK (T2DN/Mcwi) that was developed from a cross between GK and Fawn-Hooded Hypertensive (FHH) rats.83 The T2DN/Mcwi rats develop diabetes and progressive proteinuria by 6 months of age, which is accompanied by histological abnormalities in the kidneys, such as focal glomerulosclerosis, severe mesangial matrix expansion, and thickening of the glomerular basement membranes. These changes progress over time, and nearly all type 2 diabetic rats exhibit diffuse global glomerulosclerosis with nodule formation and arteriolar hyalinosis by 18 months of age.82 Protein excretion also increases from 41 ± 2 to 524 ± 50 mg/day in T2DN/Mcwi rats from 3 to 18 months of age. The mean arterial pressure (MAP) of T2DN/Mcwi rats is ~150 mmHg at both 6 and 18 months of age. In contrast, the MAP of GK rats is ~140–145 mmHg at the same age, but there is no significant difference in MAP between the T2DN/Mcwi and GK rats.82 The histological changes in the kidneys of T2DN/Mcwi rats closely mimic the changes in the kidneys of patients with diabetes. These results indicate that the T2DN/Mcwi rat may be a suitable model for investigating advanced diabetic nephropathy.
Conclusion
Many animal models of diabetes are currently available for elucidating the pathophysiology of diabetes and testing novel therapies for complications, including diabetic nephropathy. The described rodent models of diabetes exhibit hyperglycemia-induced early glomerular lesions, such as mesangial matrix expansion and glomerular basement membrane thickening, as well as albuminuria, oxidative stress, and inflammation. Therefore, these rodent models may be useful for the study of early stages of diabetic nephropathy. However, animal models have limited utility for understanding the pathogenesis of advanced diabetic nephropathy, in part because there is no ideal model that exhibits all of the key histological features of human diabetic nephropathy, such as glomerular nodular lesions and tubulointerstitial fibrosis with progressive renal insufficiency. Features such as albuminuria and histological changes, including mesangial expansion, tubulointerstitial fibrosis, and nodular lesions in type 1 and 2 diabetic rodent models, are summarized in Figures 1 and 2. Among the described animal models of diabetes, STZ-induced diabetic CD1 mice, OVE26 type 1 diabetic mice, ZDF rats, WF rats, OLETF rats, and T2DN/Mcwi rats exhibit tubulointerstitial fibrosis accompanied by glomerular lesions. A recent clinical trial of empagliflozin, a sodium–glucose cotransporter (SGLT) 2 inhibitor, in type 2 diabetic patients clearly demonstrated that the SGLT2 inhibitor slowed a decline in renal function compared with the placebo group.84 Although the renoprotective effects of SGLT2 inhibition may be due to an improvement in diabetes-induced hyperfiltration85 or a reduction in the overloading of renal tubular cells through an increase in ketone body production,86 the detailed mechanisms are still unclear. Therefore, rodent models of diabetes that exhibit both glomerular lesions and tubulointerstitial damage/fibrosis should be useful for elucidating the mechanisms of the renal protective effects of SGLT2 inhibitors. Furthermore, the genetic background and strain of the mice result in differences in their susceptibility to diabetic nephropathy, as described earlier. Therefore, studying the differences in susceptibility to diabetic nephropathy and the pathophysiology in various animal models may lead to a better understanding of the genetic mechanisms that contribute to the development of diabetic nephropathy. Additionally, several specific genetically modified animals that progress to diabetic nephropathy, such as the eNOS knockout in STZ-induced diabetic C57BL/687 or db/db mice88 and the ApoE knockout in STZ-induced C57BL/6 mice,89 have been established. The following models may also be useful for research of advanced diabetic nephropathy: TTRhRen mice with STZ-induced diabetes or FVB mice with the OVE26 mutation, which exhibit type 1 diabetes and hypertension induced by human renin overproduction;90 Cyp1a1mRen2 rats of the Fisher strain in which STZ-induced diabetes is combined with severe hypertension induced by renin overproduction;91 Dahl salt-sensitive hypertensive rats with STZ-induced diabetes;92 and the T2DN/Mcwi rats described earlier. Therefore, in addition to the classic rodent models of diabetes, more refined rodent models that target additional genes for knockout, either in isolation or in combination, may be useful for the research of diabetic nephropathy.
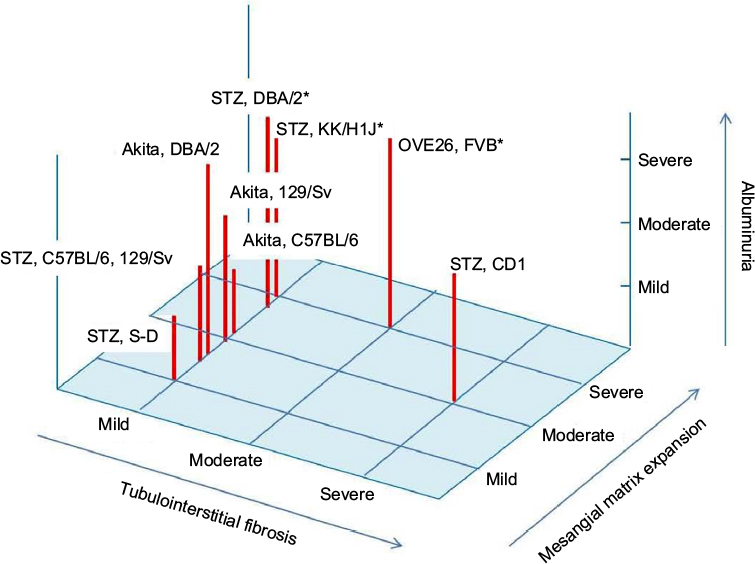 | Figure 1 Albuminuria and renal histology in type 1 diabetic mice model. Notes: The level of albuminuria and mesangial matrix expansion is exhibited as mild, moderate, and severe in each type 1 diabetic animal model. The level of tubulointerstitial fibrosis is also shown as mild, moderate, and severe in each type 1 diabetic animal model. *Animals with nodular glomerular lesion. Abbreviations: STZ, streptozotocin; S-D, Sprague Dawley. |
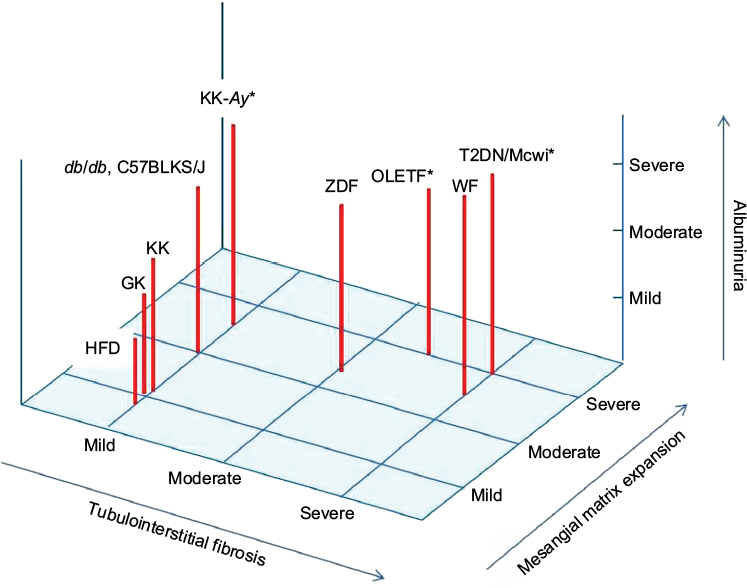 | Figure 2 Albuminuria and renal histology in type 2 diabetic mice model. Notes: The level of albuminuria and mesangial matrix expansion is exhibited as mild, moderate, and severe in each type 2 diabetic animal model. The level of tubulointerstitial fibrosis is also shown as mild, moderate, and severe in each type 2 diabetic animal model. *Animals with nodular glomerular lesion. Abbreviations: ZDF, Zucker diabetic fatty rats; WF, Wistar fatty rats; OLTF, Otsuka Long-Evans Tokushima Fatty rats; GK, Goto-Kakizaki rats; T2DN/Mcwi, a genetically modified substrain of GK rats; HFD, high fat diet. |
Acknowledgments
This work was financially supported by the Grant for Promoted Research from Kanazawa Medical University (S2015-10) to MK and by the Grant-in-Aid for Challenging Exploratory Research (16K15472) and the Grant for Collaborative Research from Kanazawa Medical University (C2015-2) to DK. Boehringer Ingelheim, Mitsubishi Tanabe Pharma, Kyowa Hakko Kirin, Taisho Toyama Pharmaceutical Co., and Ono Pharmaceutical Co. contributed to establishing the Division of Anticipatory Molecular Food Science and Technology.
Disclosure
The authors report no conflicts of interest in this work.
References
Packham DK, Alves TP, Dwyer JP, et al. Relative incidence of ESRD versus cardiovascular mortality in proteinuric type 2 diabetes and nephropathy: results from the DIAMETRIC (Diabetes Mellitus Treatment for Renal Insufficiency Consortium) database. Am J Kidney Dis. 2012;59(1):75–83. | ||
Kitada M, Kanasaki K, Koya D. Clinical therapeutic strategies for early stage of diabetic kidney disease. World J Diabetes. 2014;5(3):342–356. | ||
Mauer SM, Lane P, Zhu D, Fioretto P, Steffes MW. Renal structure and function in insulin-dependent diabetes mellitus in man. J Hypertens Suppl. 1992;10(1):S17–S20. | ||
Mauer SM, Steffes MW, Ellis EN, Sutherland DE, Brown DM, Goetz FC. Structural-functional relationships in diabetic nephropathy. J Clin Invest. 1984;74(4):1143–1155. | ||
Risdon RA, Sloper JC, De Wardener HE. Relationship between renal function and histological changes found in renal-biopsy specimens from patients with persistent glomerular nephritis. Lancet. 1968;2(7564):363–366. | ||
Gilbert RE, Cooper ME. The tubulointerstitium in progressive diabetic kidney disease: more than an aftermath of glomerular injury? Kidney Int. 1999;56(5):1627–1637. | ||
Breyer MD, Bottinger E, Brosius FC 3rd, et al. Mouse models of diabetic nephropathy. J Am Soc Nephrol. 2005;16(1):27–45. | ||
Wilson GL, Leiter EH. Streptozotocin interactions with pancreatic beta cells and the induction of insulin-dependent diabetes. Curr Top Microbiol Immunol. 1990;156:27–54. | ||
Bolzan AD, Bianchi MS. Genotoxicity of streptozotocin. Mutat Res. 2002;512(2–3):121–134. | ||
Like AA, Appel MC, Williams RM, Rossini AA. Streptozotocin-induced pancreatic insulitis in mice. Morphologic and physiologic studies. Lab Invest. 1978;38(4):470–486. | ||
Sugimoto H, Grahovac G, Zeisberg M, Kalluri R. Renal fibrosis and glomerulosclerosis in a new mouse model of diabetic nephropathy and its regression by bone morphogenic protein-7 and advanced glycation end product inhibitors. Diabetes. 2007;56(7):1825–1833. | ||
Qi Z, Fujita H, Jin J, et al. Characterization of susceptibility of inbred mouse strains to diabetic nephropathy. Diabetes. 2005;54(9):2628–2637. | ||
Zheng H, Whitman SA, Wu W, et al. Therapeutic potential of Nrf2 activators in streptozotocin-induced diabetic nephropathy. Diabetes. 2011;60(11):3055–3066. | ||
Okada S, Shikata K, Matsuda M, et al. Intercellular adhesion molecule-1-deficient mice are resistant against renal injury after induction of diabetes. Diabetes. 2003;52(10):2586–2593. | ||
Sugimoto H, LeBleu VS, Bosukonda D, et al. Activin-like kinase 3 is important for kidney regeneration and reversal of fibrosis. Nat Med. 2012;18(3):396–404. | ||
Ma LJ, Fogo AB. Model of robust induction of glomerulosclerosis in mice: importance of genetic background. Kidney Int. 2003;64(1):350–355. | ||
Hartner A, Cordasic N, Klanke B, Veelken R, Hilgers KF. Strain differences in the development of hypertension and glomerular lesions induced by deoxycorticosterone acetate salt in mice. Nephrol Dial Transplant. 2003;18(10):1999–2004. | ||
Wang Q, Hummler E, Nussberger J, et al. Blood pressure, cardiac, and renal responses to salt and deoxycorticosterone acetate in mice: role of renin genes. J Am Soc Nephrol. 2002;13(6):1509–1516. | ||
Kitada M, Koya D, Sugimoto T, et al. Translocation of glomerular p47phox and p67phox by protein kinase C-beta activation is required for oxidative stress in diabetic nephropathy. Diabetes. 2003;52(10):2603–2614. | ||
Hayashi K, Haneda M, Koya D, Maeda S, Isshiki K, Kikkawa R. Enhancement of glomerular heme oxygenase-1 expression in diabetic rats. Diabetes Res Clin Pract. 2001;52(2):85–96. | ||
Mima A, Arai H, Matsubara T, et al. Urinary Smad1 is a novel marker to predict later onset of mesangial matrix expansion in diabetic nephropathy. Diabetes. 2008;57(6):1712–1722. | ||
Kodera R, Shikata K, Takatsuka T, et al. Dipeptidyl peptidase-4 inhibitor ameliorates early renal injury through its anti-inflammatory action in a rat model of type 1 diabetes. Biochem Biophys Res Commun. 2014;443(3):828–833. | ||
Wang J, Takeuchi T, Tanaka S, et al. A mutation in the insulin 2 gene induces diabetes with severe pancreatic beta-cell dysfunction in the Mody mouse. J Clin Invest. 1999;103(1):27–37. | ||
Alpers CE, Hudkins KL. Mouse models of diabetic nephropathy. Curr Opin Nephrol Hypertens. 2011;20(3):278–284. | ||
Susztak K, Raff AC, Schiffer M, Bottinger EP. Glucose-induced reactive oxygen species cause apoptosis of podocytes and podocyte depletion at the onset of diabetic nephropathy. Diabetes. 2006;55(1):225–233. | ||
Shi Y, Lo CS, Padda R, et al. Angiotensin-(1-7) prevents systemic hypertension, attenuates oxidative stress and tubulointerstitial fibrosis, and normalizes renal angiotensin-converting enzyme 2 and Mas receptor expression in diabetic mice. Clin Sci (Lond). 2015;128(10):649–663. | ||
Fang F, Bae EH, Hu A, et al. Deletion of the gene for adiponectin accelerates diabetic nephropathy in the Ins2 (+/C96Y) mouse. Diabetologia. 2015;58(7):1668–1678. | ||
Liu GC, Fang F, Zhou J, et al. Deletion of p47phox attenuates the progression of diabetic nephropathy and reduces the severity of diabetes in the Akita mouse. Diabetologia. 2012;55(9):2522–2532. | ||
Fujita H, Fujishima H, Morii T, et al. Modulation of renal superoxide dismutase by telmisartan therapy in C57BL/6-Ins2(Akita) diabetic mice. Hypertens Res. 2012;35(2):213–220. | ||
Proctor G, Jiang T, Iwahashi M, Wang Z, Li J, Levi M. Regulation of renal fatty acid and cholesterol metabolism, inflammation, and fibrosis in Akita and OVE26 mice with type 1 diabetes. Diabetes. 2006;55(9):2502–2509. | ||
Gurley SB, Mach CL, Stegbauer J, et al. Influence of genetic background on albuminuria and kidney injury in Ins2 (+/C96Y) (Akita) mice. Am J Physiol Renal Physiol. 2010;298(3):F788–F795. | ||
Haseyama T, Fujita T, Hirasawa F, et al. Complications of IgA nephropathy in a non-insulin-dependent diabetes model, the Akita mouse. Tohoku J Exp Med. 2002;198(4):233–244. | ||
Xu J, Huang Y, Li F, Zheng S, Epstein PN. FVB mouse genotype confers susceptibility to OVE26 diabetic albuminuria. Am J Physiol Renal Physiol. 2010;299(3):F487–F494. | ||
Teiken JM, Audettey JL, Laturnus DI, Zheng S, Epstein PN, Carlson EC. Podocyte loss in aging OVE26 diabetic mice. Anat Rec (Hoboken). 2008;291(1):114–121. | ||
Brosius FC 3rd, Alpers CE, Bottinger EP, et al. Mouse models of diabetic nephropathy. J Am Soc Nephrol. 2009;20(12):2503–2512. | ||
Tesch GH, Lim AK. Recent insights into diabetic renal injury from the db/db mouse model of type 2 diabetic nephropathy. Am J Physiol Renal Physiol. 2011;300(2):F301–F310. | ||
Hummel KP, Dickie MM, Coleman DL. Diabetes, a new mutation in the mouse. Science. 1966;153(3740):1127–1128. | ||
Hummel KP, Coleman DL, Lane PW. The influence of genetic background on expression of mutations at the diabetes locus in the mouse. I. C57BL-KsJ and C57BL-6J strains. Biochem Genet. 1972;7(1):1–13. | ||
Sharma K, McCue P, Dunn SR. Diabetic kidney disease in the db/db mouse. Am J Physiol Renal Physiol. 2003;284(6):F1138–F1144. | ||
Cohen MP, Lautenslager GT, Shearman CW. Increased urinary type IV collagen marks the development of glomerular pathology in diabetic d/db mice. Metabolism. 2001;50(12):1435–1440. | ||
Arakawa K, Ishihara T, Oku A, et al. Improved diabetic syndrome in C57BL/KsJ-db/db mice by oral administration of the Na(+)-glucose cotransporter inhibitor T-1095. Br J Pharmacol. 2001;132(2):578–586. | ||
Mishra R, Emancipator SN, Miller C, Kern T, Simonson MS. Adipose differentiation-related protein and regulators of lipid homeostasis identified by gene expression profiling in the murine db/db diabetic kidney. Am J Physiol Renal Physiol. 2004;286(5):F913–F921. | ||
Koya D, Haneda M, Nakagawa H, et al. Amelioration of accelerated diabetic mesangial expansion by treatment with a PKC beta inhibitor in diabetic db/db mice, a rodent model for type 2 diabetes. FASEB J. 2000;14(3):439–447. | ||
Kitada M, Kume S, Imaizumi N, Koya D. Resveratrol improves oxidative stress and protects against diabetic nephropathy through normalization of Mn-SOD dysfunction in AMPK/SIRT1-independent pathway. Diabetes. 2011;60(2):634–643. | ||
Ha TS, Barnes JL, Stewart JL, et al. Regulation of renal laminin in mice with type II diabetes. J Am Soc Nephrol. 1999;10(9):1931–1939. | ||
Ziyadeh FN, Hoffman BB, Han DC, et al. Long-term prevention of renal insufficiency, excess matrix gene expression, and glomerular mesangial matrix expansion by treatment with monoclonal antitransforming growth factor-beta antibody in db/db diabetic mice. Proc Natl Acad Sci U S A. 2000;97(14):8015–8020. | ||
Liu W, Yang Y, Liu Y, et al. Exogenous kallikrein protects against diabetic nephropathy. Kidney Int. Epub 2016 Aug 18. | ||
Yiu WH, Wong DW, Wu HJ, et al. Kallistatin protects against diabetic nephropathy in db/db mice by suppressing AGE-RAGE-induced oxidative stress. Kidney Int. 2016;89(2):386–398. | ||
Kondo K, Nozawa K, Tomita T, et al. Inbred strains resulting from Japanese mice. Bull Exp Anim. 1957;6:107–112. | ||
Taketomi S, Ikeda H, Ishikawa E, Iwatsuka H. Determination of overall insulin sensitivity in diabetic mice, KK. Horm Metab Res. 1982;14(1):14–18. | ||
Matsuo T, Shino A. Induction of diabetic alterations by goldthioglucose-obesity in KK, ICR and C57BL mice. Diabetologia. 1972;8(6):391–397. | ||
Tomino Y, Tanimoto M, Shike T, et al. Pathogenesis and treatment of type 2 diabetic nephropathy: lessons from the spontaneous KK/Ta mouse model. Curr Diabetes Rev. 2005;1(3):281–286. | ||
Nishimura M. Breeding of mouse strains for diabetes mellitus. Exp Anim. 1969;18:147–157. | ||
Omote K, Gohda T, Murakoshi M, et al. Role of the TNF pathway in the progression of diabetic nephropathy in KK-A(y) mice. Am J Physiol Renal Physiol. 2014;306(11):F1335–F1347. | ||
Ito T, Tanimoto M, Yamada K, et al. Glomerular changes in the KK-Ay/Ta mouse: a possible model for human type 2 diabetic nephropathy. Nephrology (Carlton). 2006;11(1):29–35. | ||
Matsumoto M, Tanimoto M, Gohda T, et al. Effect of pitavastatin on type 2 diabetes mellitus nephropathy in KK-Ay/Ta mice. Metabolism. 2008;57(5):691–697. | ||
Deji N, Kume S, Araki S, et al. Structural and functional changes in the kidneys of high-fat diet-induced obese mice. Am J Physiol Renal Physiol. 2009;296:F118–F126. | ||
Shiota M, Printz RL. Diabetes in Zucker diabetic fatty rat. Methods Mol Biol. 2012;933:103–123. | ||
Clark JB, Palmer CJ, Shaw WN. The diabetic Zucker fatty rat. Proc Soc Exp Biol Med. 1983;173(1):68–75. | ||
Phillips MS, Liu Q, Hammond HA, et al. Leptin receptor missense mutation in the fatty Zucker rat. Nat Genet. 1996;13(1):18–19. | ||
Ikeda H, Shino A, Matsuo T, Iwatsuka H, Suzuoki Z. A new genetically obese-hyperglycemic rat (Wistar fatty). Diabetes. 1981;30(12):1045–1050. | ||
Kitada M, Ogura Y, Suzuki T, et al. A very-low-protein diet ameliorates advanced diabetic nephropathy through autophagy induction by suppression of the mTORC1 pathway in Wistar fatty rats, an animal model of type 2 diabetes and obesity. Diabetologia. 2016;59(6):1307–1317. | ||
Kitada M, Takeda A, Nagai T, Ito H, Kanasaki K, Koya D. Dietary restriction ameliorates diabetic nephropathy through anti-inflammatory effects and regulation of the autophagy via restoration of Sirt1 in diabetic Wistar fatty (fa/fa) rats: a model of type 2 diabetes. Exp Diabetes Res. 2011;2011:908185. | ||
Katsuda Y, Ohta T, Miyajima K, et al. Diabetic complications in obese type 2 diabetic rat models. Exp Anim. 2014;63(2):121–132. | ||
Vora JP, Zimsen SM, Houghton DC, Anderson S. Evolution of metabolic and renal changes in the ZDF/Drt-fa rat model of type II diabetes. J Am Soc Nephrol. 1996;7(1):113–117. | ||
Chander PN, Gealekman O, Brodsky SV, et al. Nephropathy in Zucker diabetic fat rat is associated with oxidative and nitrosative stress: prevention by chronic therapy with a peroxynitrite scavenger ebselen. J Am Soc Nephrol. 2004;15(9):2391–2403. | ||
Takiyama Y, Harumi T, Watanabe J, et al. Tubular injury in a rat model of type 2 diabetes is prevented by metformin: a possible role of HIF-1alpha expression and oxygen metabolism. Diabetes. 2011;60(3):981–992. | ||
Baynes J, Murray DB. Cardiac and renal function are progressively impaired with aging in Zucker diabetic fatty type II diabetic rats. Oxid Med Cell Longev. 2009;2(5):328–334. | ||
Marsh SA, Powell PC, Agarwal A, Dell’Italia LJ, Chatham JC. Cardiovascular dysfunction in Zucker obese and Zucker diabetic fatty rats: role of hydronephrosis. Am J Physiol Heart Circ Physiol. 2007;293(1):H292–H298. | ||
Kawano K, Hirashima T, Mori S, Saitoh Y, Kurosumi M, Natori T. Spontaneous long-term hyperglycemic rat with diabetic complications. Otsuka Long-Evans Tokushima Fatty (OLETF) strain. Diabetes. 1992;41(11):1422–1428. | ||
Nagai N, Ito Y. Therapeutic effects of sericin on diabetic keratopathy in Otsuka Long-Evans Tokushima Fatty rats. World J Diabetes. 2013;4(6):282–289. | ||
Nagai N, Murao T, Ito Y, Okamoto N, Sasaki M. Enhancing effects of sericin on corneal wound healing in Otsuka Long-Evans Tokushima fatty rats as a model of human type 2 diabetes. Biol Pharm Bull. 2009;32(9):1594–1599. | ||
Yabuki A, Tahara T, Taniguchi K, Matsumoto M, Suzuki S. Neuronal nitric oxide synthase and cyclooxygenase-2 in diabetic nephropathy of type 2 diabetic OLETF rats. Exp Anim. 2006;55(1):17–25. | ||
Lee EY, Kim GT, Hyun M, et al. Peroxisome proliferator-activated receptor- δ activation ameliorates albuminuria by preventing nephrin loss and restoring podocyte integrity in type 2 diabetes. Nephrol Dial Transplant. 2012;27(11):4069–4079. | ||
Kawano K, Mori S, Hirashima T, Man ZW, Natori T. Examination of the pathogenesis of diabetic nephropathy in OLETF rats. J Vet Med Sci. 1999;61(11):1219–1228. | ||
Portha B, Serradas P, Bailbe D, Suzuki K, Goto Y, Giroix MH. Beta-cell insensitivity to glucose in the GK rat, a spontaneous nonobese model for type II diabetes. Diabetes. 1991;40(4):486–491. | ||
Ostenson CG, Khan A, Abdel-Halim SM, et al. Abnormal insulin secretion and glucose metabolism in pancreatic islets from the spontaneously diabetic GK rat. Diabetologia. 1993;36(1):3–8. | ||
Yagihashi S, Goto Y, Kakizaki M, Kaseda N. Thickening of glomerular basement membrane in spontaneously diabetic rats. Diabetologia. 1978;15(4):309–312. | ||
Feng B, Yan XF, Xue JL, Xu L, Wang H. The protective effects of alpha-lipoic acid on kidneys in type 2 diabetic Goto-Kakisaki rats via reducing oxidative stress. Int J Mol Sci. 2013;14:6746–6756. | ||
Chen S, Yang T, Liu F, et al. Inflammatory factor-specific sumoylation regulates NF-kappaB signalling in glomerular cells from diabetic rats. Inflamm Res. 2014;63:23–31. | ||
Wang Y, Nie M, Lu Y, et al. Fucoidan exerts protective effects against diabetic nephropathy related to spontaneous diabetes through the NF-kappaB signaling pathway in vivo and in vitro. Int J Mol Med. 2015;35(4):1067–1073. | ||
Kojima N, Slaughter TN, Paige A, Kato S, Roman RJ, Williams JM. Comparison of the development diabetic induced renal disease in strains of Goto-Kakizaki rats. J Diabetes Metab. 2013;(Suppl 9) (5):S9–005. | ||
Nobrega MA, Fleming S, Roman RJ, et al. Initial characterization of a rat model of diabetic nephropathy. Diabetes. 2004; 53(3):735–742. | ||
Wanner C, Inzucchi SE, Lachin JM, et al. Empagliflozin and progression of kidney disease in type 2 diabetes. N Engl J Med. 2016;375:323–334. | ||
Cherney DZ, Perkins BA, Soleymanlou N, et al. Renal hemodynamic effect of sodium-glucose cotransporter 2 inhibition in patients with type 1 diabetes mellitus. Circulation. 2014;129(5):587–597. | ||
Fioretto P, Zambon A, Rossato M, Busetto L, Vettor R. SGLT2 inhibitors and the diabetic kidney. Diabetes Care. 2016;39(Suppl 2):S165–S171. | ||
Nakagawa T, Sato W, Glushakova O, et al. Diabetic endothelial nitric oxide synthase knockout mice develop advanced diabetic nephropathy. J Am Soc Nephrol. 2007;18(2):539–550. | ||
Zhao HJ, Wang S, Cheng H, et al. Endothelial nitric oxide synthase deficiency produces accelerated nephropathy in diabetic mice. J Am Soc Nephrol. 2006;17:2664–2669. | ||
Lassila M, Seah KK, Allen TJ, et al. Accelerated nephropathy in diabetic apolipoprotein e-knockout mouse: role of advanced glycation end products. J Am Soc Nephrol. 2004;15(8):2125–2138. | ||
Thibodeau JF, Holterman CE, Burger D, Read NC, Reudelhuber TL, Kennedy CR. A novel mouse model of advanced diabetic kidney disease. PLoS One. 2014;9(12):e113459. | ||
Hartner A, Cordasic N, Klanke B, Wittmann M, Veelken R, Hilgers KF. Renal injury in streptozotocin-diabetic Ren2-transgenic rats is mainly dependent on hypertension, not on diabetes. Am J Physiol Renal Physiol. 2007;292(2):F820–F827. | ||
Korner A, Jaremko G, Eklof AC, Aperia A. Rapid development of glomerulosclerosis in diabetic Dahl salt-sensitive rats. Diabetologia. 1997;40(4):367–373. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.